









-
Mesembryanthemum crystallinum (ice plant, 2n = 2x = 18) is a member of the Aizoaceae family, characterized by its robust adaptability to drought and high salinity conditions. M. crystallinum has a widespread cultivation I[1]. M. crystallinum is a kind of green vegetable renowned for its nutritional richness, containing a variety of amino acids, flavonoids, calcium, and other related elements. In addition, it boasts a noteworthy content of pinoresinol and inositol, constituents with documented potential for exerting anti-diabetic effects and conferring beneficial health properties upon consumers[2]. This species can transfer photosynthesis from C3 to CAM under drought or salt stress[3]. It is an excellent model plant for studying the transition from the C3 pathway to the CAM pathway.
Photosynthesis stands as the fundamental process driving plant growth and sustenance[4,5]. Through this intricate metabolic pathway, green plants harness solar energy, converting it into bioenergy that serves as the lifeblood for nearly all terrestrial organisms[3]. According to the differences in the pathway of carbon assimilation in photosynthesis, there are three pathways (C3, C4, and CAM). C3 photosynthesis serves as a predominant pathway wherein plants uptake and fix CO2 to synthesize carbohydrates. Remarkably, C3 photosynthesis typifies the metabolic strategy of approximately 95% of plant species on Earth[6]. Most crops and vegetables belong to C3 plants, such as wheat, rice, soybeans, spinach, and tomatoes. C4 and CAM species only account for about 3% and 6% of flowering plants, respectively[7,8]. Maize, sugarcane, sorghum, and other C4 plants exhibit a remarkable adaptation to arid environments characterized by high light intensity. CAM plants, prevalent in arid ecosystems, include succulent families such as Crassulaceae, Cactaceae, and Bromeliaceae. These plants demonstrate remarkable adaptability to exceedingly arid environments and exhibit significantly enhanced water use efficiency (WUE) in CO2 assimilation, compared to C3 and C4 plants[9]. CAM plants are mainly divided into two types. One is the plants that only use the CAM pathway to fix CO2, which are termed constitutive CAM plants, such as Opuntia dilleniid and Ananas comosus. Another type encompasses facultative CAM plants, also known as 'C3/CAM plants', which mainly carry out C3 photosynthesis under favorable environmental conditions. However, CO2 is mainly fixed through the CAM pathway when encountering abiotic stressors, and is reverted to C3 photosynthesis when the external stress disappears[10]. The representative plants are M. crystallinum and Talinum triangulare[10,11].
The CAM pathway exhibits an obvious circadian rhythm, which can be divided into four distinct phases according to the physiological and biochemical differences in the process of carbon assimilation[12,13]. The first phase occurs at night. The typical feature is that the stomata are open to uptake CO2, then converted into bicarbonate (HCO3−). HCO3− combines with phosphoenolpyruvate (PEP) to produce oxaloacetic acid (OAA), which is then reduced to malic acid and stored in the vacuole. The second phase occurs at dawn, when CAM plants undergo a shift from phosphoenolpyruvate carboxykinase (PEPC) carboxylation to Ribulose Bisphosphate Carboxylase Oxygenase (Rubisco) carboxylation. In this stage, CO2 is fixed by both Rubisco and PEPC enzymes. The fixed CO2 comes from two parts, which are the decarboxylation release of malic acid and absorption in the atmosphere. The C4 pathway and C3 pathway operate simultaneously, resulting in a peak of CO2 absorption and the cessation of malic acid accumulation. Fundamental attributes of this stage encompass a reduction in PEPC activity and the activation of Rubisco. The third phase occurs on the subsequent day. In this phase, the stomata are closed, the absorption of CO2 from the atmosphere is stopped, and a large amount of malic acid in the vacuole is continuously transferred to the cytoplasmic matrix for decarboxylation. The decarboxylation reaction catalyzes the conversion of malic acid into CO2 and pyruvate (PYR). The released CO2 enters the chloroplast for assimilation via the C3 pathway, causing an intercellular CO2 concentration surrounding the Rubisco enzyme approximately 60-fold higher than atmospheric levels. Elevated CO2 concentration effectively inhibits the interaction between the Rubisco enzyme and O2, thereby augmenting the carboxylation activity of Rubisco. Consequently, photorespiration is inhibited, and the efficiency of photosynthesis is heightened. The fourth phase occurs in the evening, characterized by a transition from Rubisco carboxylation to PEPC carboxylation. This phase is distinguished by a decline in malic acid concentration to the lowest point. Additionally, the rate of decarboxylation reaction diminishes, intercellular CO2 concentration decreases, stomatal aperture initiates, CO2 absorption increases, and the cycle from the C3 pathway to the C4 pathway recommences in the subsequent diurnal cycle[8,14], as depicted in Fig. 1.
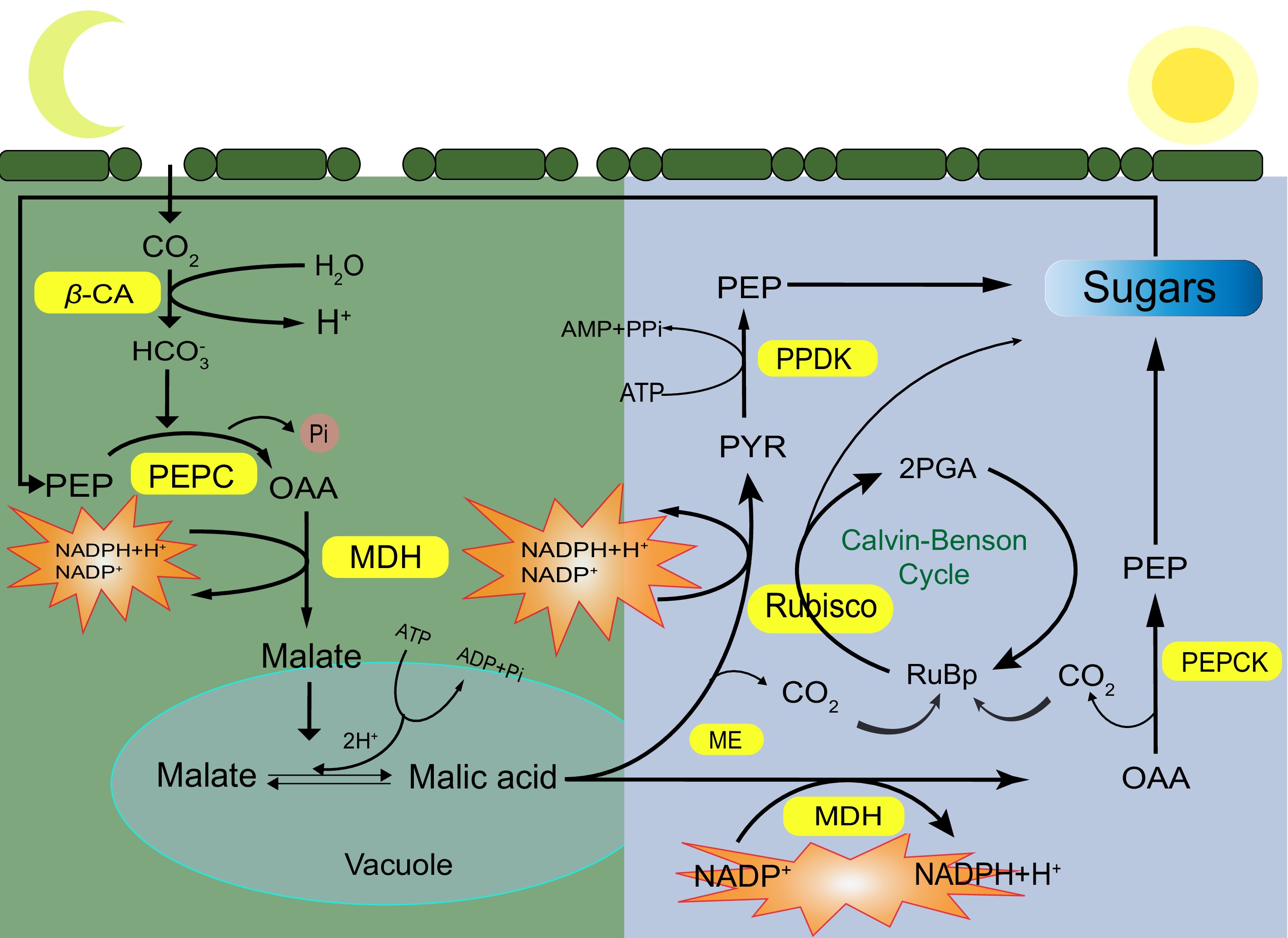
Figure 1.
The specific process of crassulacean acid metabolism in plants. Yellow is the key enzyme in the CAM pathway. CA: carbonic anhydrase; PEPC: phosphoenolpyruvate carboxylase; PEPCK: phosphoenolpyruvate carboxylase kinase; MDH: malic dehydrogenase; ME: malic acid enzyme; PPDK: pyruvate phosphokinase; Rubisco: ribulose 1,5-diphosphate carboxylase/oxygenase.
Here, based on the 'telomere-centered genome recombination model'[15], we curated a comprehensive dataset comprising eight well-assembled chromosome-level genomes of Caryophyllales plants. Leveraging homologous fragments derived from both polyploidization events and species divergence, we systematically reconstructed the ancestral karyotype of Caryophyllales. Furthermore, CAM genes in ice plants were identified by bioinformatics methods, and phylogenetic tree analysis, gene domain analysis, and chromosome localization analysis were performed on these genes.
-
The establishment of homologous relationships is crucial for the inference of ancestral chromosomes based on the framework of the 'Telomere-centric genome recombination model'[15,16]. Fusion emerges as the primary mechanism driving chromosome rearrangements, encompassing various types such as reciprocally translocated chromosome arms (RTA), end-to-end joining (EEJ) of two different chromosomes, and nested chromosome fusion (NCF)[17]. Notably, the latter two forms of fusion, EEJ and NCF, precipitate a reduction in the number of chromosomes and a concomitant gene loss[18,19]. Within the fusion state, ancestral chromosomes may either persist as independent chromosomes or become entirely nested within the fused chromosomes within the extant genome.
We reconstructed the ancestral karyotype of Caryophyllales species through comparative genomic analysis using WGDI software (v0.6.5). First, the whole genome alignment was performed using the 'blast' module (E < 1e-5), and conserved syntenic blocks were then identified using the 'collinearity' module (parameters: maximum gap value mg = 40). The synonymous substitution rates (Ks) of homologous gene pairs were calculated based on the 'ks' module, and the block information statistics were performed using the 'blockinfo' module. Genome duplication events were detected using the 'polyploidy classification' module, enabling final ancestral chromosome reconstruction through the 'ancestral karyotype' module. The identification of orthologous fragments was guided by the following criteria: (1) they must be derived from a common ancestor; (2) they must exhibit conserved gene blocks and arrangement order. Through dotplot analysis, we were able to identify highly conserved collinearity blocks between species. These blocks represent genomic regions that have remained stable throughout the evolutionary process. The gene arrangement patterns within these conserved regions were then systematically analyzed using comparative genomics methods. This analysis allowed us to infer the possible structure of the ancestral genome. It is notable that C. pallidicaule did not experience additional doubling events subsequent to the core eudicot common hexaploidization (ECH) event. This underscores the evolutionary stability of its karyotypic architecture following the pivotal genome duplication event, highlighting the distinctive genomic dynamics within Caryophyllales plants (Supplementary Figs S1−S4). Therefore, nine chromosomes inherent to Chenopodium album have been identified as putative ancestral chromosomes of C. pallidicaule, while the genomes of other Caryophyllales plants may be obtained by recombination. Given the relatively conservative nature of grape evolution, the inference of ancestral chromosome evolutionary trajectories within Caryophyllales plants is facilitated through the utilization of homologous gene dotplots comparing grapes with C. pallidicaule. In parallel, the evolutionary trajectory of the ice plant genome can be discerned by employing homologous dotplot juxtaposing C. pallidicaule with the ice plant. The specific process is shown in Supplementary Fig. S5.
Identification of gene family members
-
CAM-related genes were identified based on Pfam annotation (version 37.3) using accession numbers with E-value < 1e-5, including PEPC (PF003119.11), CA (α-type: PF00484, β-type: PF00194), PEPCK (PF01293), and NADP-ME (PF00390) according to previous reports[1,20,21]. Candidate genes from M. crystallinum were further validated through HMMER 3.0 and BLASTP analyses (E-value ≤ 1e-5), retaining only those with complete functional domains confirmed by InterProScan 5.52-86.0[22]. Subsequently, the protein sequences corresponding to CAM-related genes were aligned using the Mafft program[23]. Following alignment, a maximum likelihood tree was constructed using FastTree, with bootstrap values at 1,000 iterations[24,25].
Conserved amino acid sequence analysis of gene family
-
Protein-conserved motifs were discerned utilizing the MEME suite (
https://meme-suite.org/meme/ ) and subsequently visualized through iTOL (https://itol.embl.de/ ), offering insights into the structural conservation of gene families. Additionally, TBtools[26], facilitated the analysis of gene family structures, including the determination of gene positions on chromosomes and their subsequent visualization. -
The reconstruction of ancestral chromosomes serves as a valuable tool for elucidating the evolutionary process of polyploidization and the potential effects of chromosome rearrangements[27]. After the polyploidization event, the species will undergo interchromosomal fusion and translocation, leading to significant changes in chromosomal architecture. It provides important reference resources for speculating on the evolution of modern genomes. We selected eight Caryophyllales plants to infer ancestral chromosomes. The species selected are M. crystallinum, Amaranthus cruentus, Atriplex hortensis, Chenopodium pallidicaule, Spinacia oleracea, Beta vulgaris, Hylocereus undatus, and Simmondsia chinensis. They belong to the Aizoaceae, Amaranthaceae, Cactaceae, and Simmondsiaceae families. In these eight Caryophyllales plants, all species have experienced the core ECH event. Additionally, H. undatus and A. cruentus have experienced an additional independent whole genome duplication (WGD) event, and S. chinensis has experienced an independent whole genome triplication (WGT) event[1]. After clarifying the polyploidization events of the species, the orthologous fragments among these species were used to infer the ancestral karyotype of Caryophyllales plants. The homologous gene dotplot between eight Caryophyllales plant species was constructed (Fig. 2a−c, Supplementary Figs S1−S4). Analysis of the dotplot comparison between C. pallidicaule and other closely related species revealed that its genome was relatively intact. The evolutionary structure exhibited a remarkable degree of conservation, and no complex chromosome rearrangement or gene loss occurred. For instance, analysis of the dotplot comparing C. pallidicaule and B. vulgaris revealed that the nine chromosomes of B. vulgaris were mapped to the nine chromosomes of C. pallidicaule, displaying complete correspondence. This observation suggests a potential common ancestral origin for these chromosomes. (Supplementary Fig. S1). A. cruentus has experienced an independent WGD event, establishing a homologous relationship between A. cruentus and C. pallidicaule at a ratio of 2:1. Examination of the dotplot revealed that each chromosome of C. pallidicaule can be matched with two chromosomes of A. cruentus, with the fragments displaying relatively complete alignment. This observation suggests a potential shared ancestral origin between these species (Fig. 2a). S. chinensis also experienced a WGT event. As evident from the dot plot analysis, one chromosome of C. pallidicaule corresponds to the three chromosomes of S. chinensis, aligning with the 3:1 homologous relationship between the two species (Fig. 2b). Furthermore, dotplots depicting A. hortensis, M. crystallinum, S. oleracea, and C. pallidicaule exhibited a consistent 1:1 homologous relationship (Supplementary Figs S2−4). Consequently, nine chromosomes of C. pallidicaule were identified as the ancestral chromosomes of Caryophyllales.
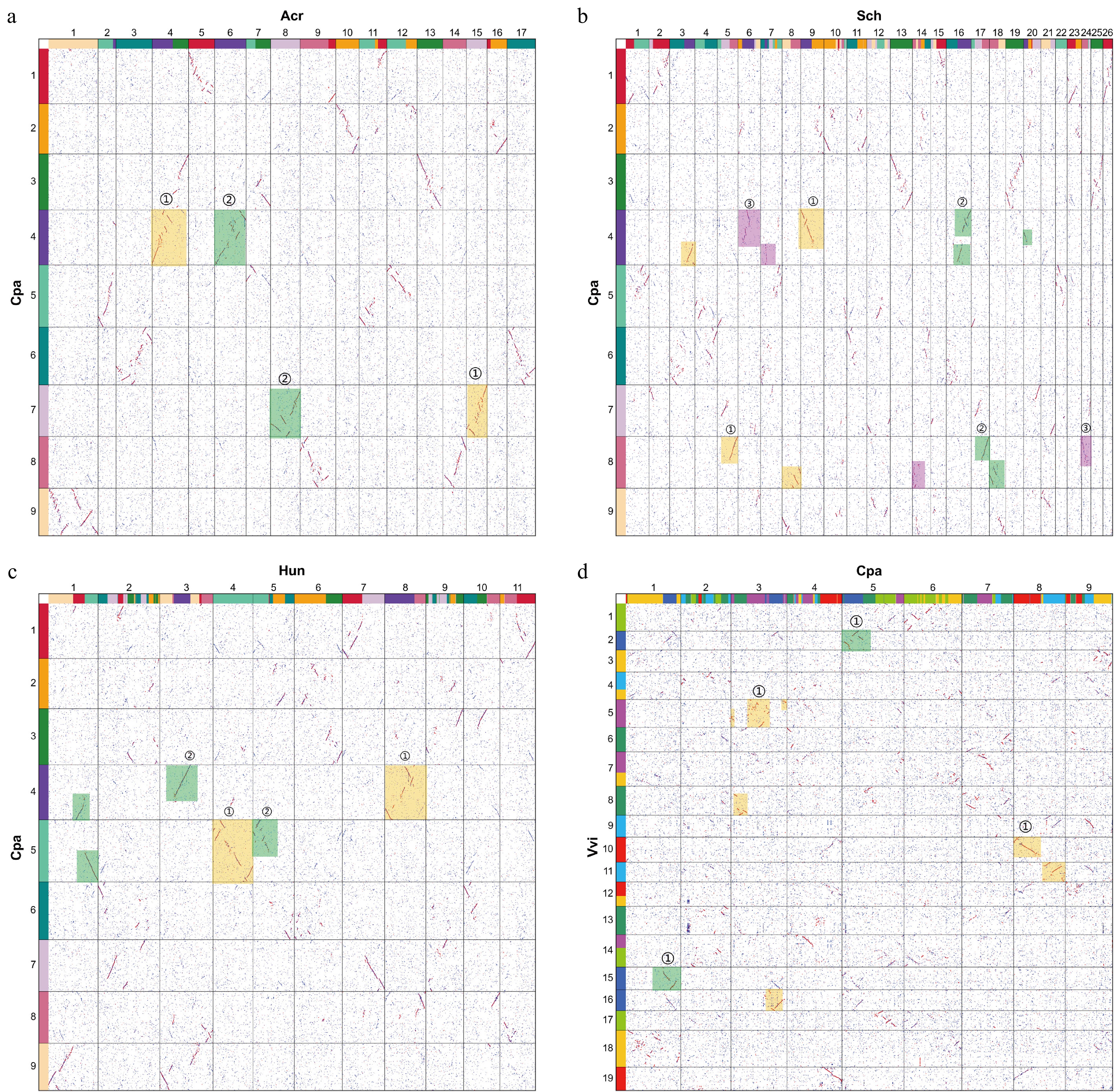
Figure 2.
Homologous dotplots between Chenopodium pallidicaule and other related species. The numbers in the dotplot denote the chromosome numbers of each species. Red fragments indicate the primary species-to-species alignment, while blue segments denote the secondary alignments. (a) C. pallidicaule (Cpa) vs A. cruentus (Acr). The orange and green rectangular boxes represent the two distinct sets of subgenomes identified. (b) C. pallidicaule (Cpa) vs S. chinensis (Sch). The orange, purple, and green rectangular boxes represent the three distinct sets of subgenomes identified. (c) C. pallidicaule (Cpa) vs H. undatus (Hun). The orange and green rectangular boxes represent the two distinct sets of subgenomes identified. (d) C. pallidicaule (Cpa) vs V. vinifera (Vvi). The contiguous line segments comprising red dots within the dotplot exhibit a distinct absence of overlap along both the horizontal and vertical axes. This characteristic pattern reflects a one-to-one correspondence in the ratio of best-matched homologous regions between the compared genomes.
Through scrutiny of the homologous structure among Caryophyllales plants, it was discerned that the nine chromosomes of C. pallidicaule were closest to the ancestral chromosomes of Caryophyllales plants. Subsequent genomes of other Caryophyllales plants are presumed to have arisen through recombination events. Leveraging the homologous gene dotplot of C. pallidicaule alongside other Caryophyllales plants as a reference, the karyotype evolution pathway from Caryophyllales ancestors to extant species was reconstructed and depicted in the form of the phylogenetic tree (Fig. 3a). The genomes of B. vulgaris and A. hortensis are relatively conservative. For example, compared with the ancestral chromosomes, only three of the nine chromosomes of B. vulgaris had reciprocally translocated chromosome arms, and the remaining six were entirely preserved. The nine chromosomes of A. hortensis were formed by two chromosome inversions and two chromosome crossovers of the ancestral chromosomes. The karyotype alterations observed in S. oleracea exhibited a higher level of complexity. Following multiple chromosome translocations, the current configuration of six chromosomes emerged through two instances of EEJ and one NCF. Conversely, H. undatus has recently experienced a WGD event. Its 11 chromosomes evolved from nine ancestral chromosomes of the Caryophyllales order via a sequence of multiple chromosome translocations resulting in interchromosomal rearrangements, and then through five EEJs and two NCFs. The 26 chromosomes observed in S. chinensis emerged from the ancestral chromosomes through a series of multiple chromosome translocations along with one instance of EEJ. Similarly, A. cruentus underwent a WGD event, resulting in its 17 chromosomes arising from a combination of alterations in ancestral chromosomes, alongside one occurrence of EEJ.
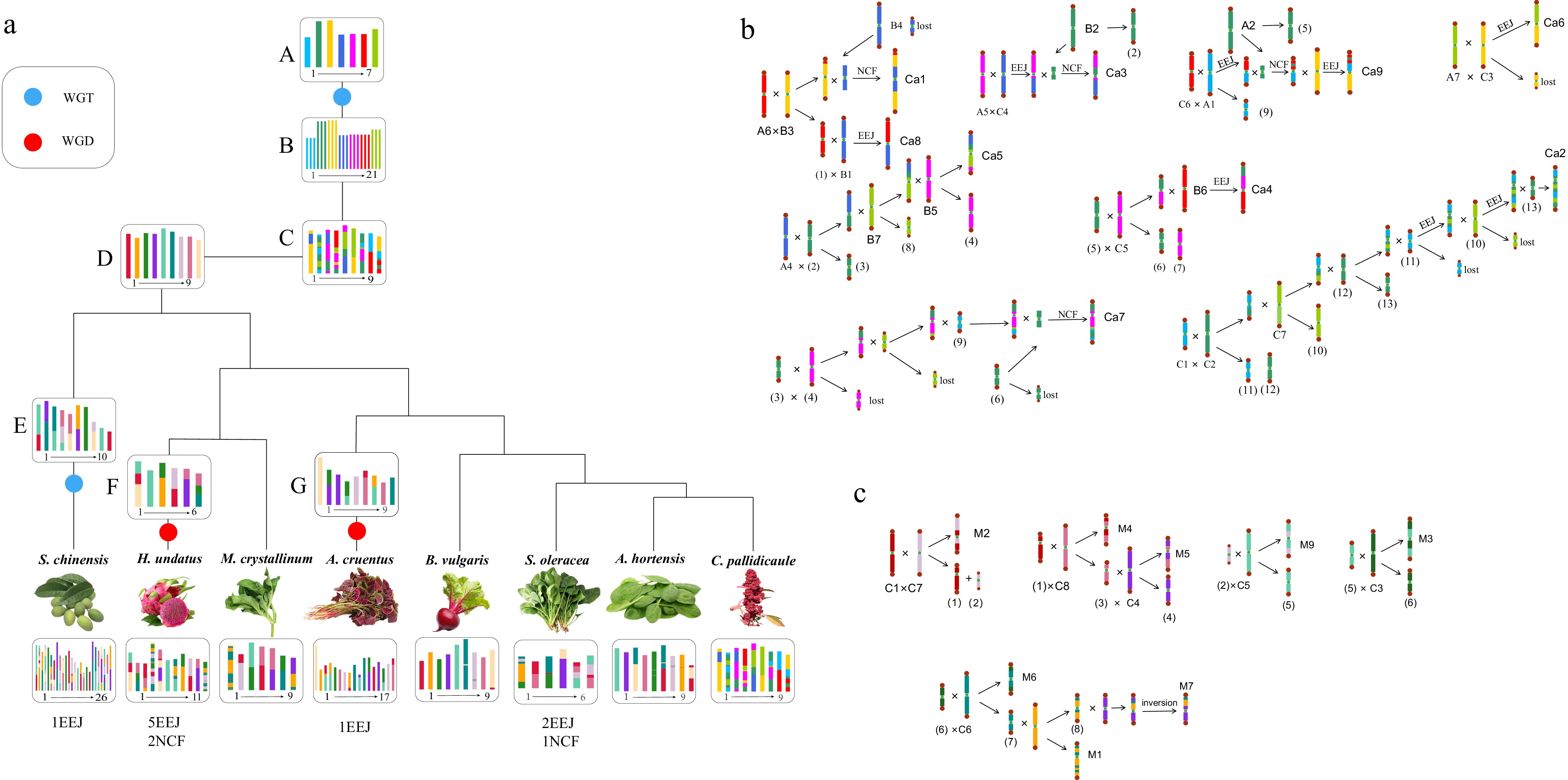
Figure 3.
Chromosome karyotype reconstruction and evolution in Caryophyllales species. (a) The blue circle denotes the WGT event, while the red circle represents the WGD event. The chromosome karyotype of the key nodes and the chromosome numbers of each species are depicted in the diagram, with species denoted by their Latin names. (b) The schematic illustrates the formation process of the ancestral chromosome of Caryophyllales. A1–A7, B1–B7, and C1–C7 represent the ancestral chromosomes of the core eudicot species post-WGT event. Ca1–Ca9 represent the ancestral chromosome of Caryophyllales, with 'lost' indicating the loss of the resulting B chromosome. (c) The diagram portrays the formation process of M. crystallinum chromosome. C1–C9 represent the ancestral chromosome of Caryophyllales, while M1–M9 denote the M. crystallinum chromosome.
Grapes, characterized by their evolutionary conservatism, serve as a pivotal reference point in elucidating the evolutionary trajectory of ancestral chromosomes within Caryophyllales plants. Leveraging the homologous gene dotplot between grapes and C. pallidicaule, it can be discerned that the evolutionary trajectory of the ancestral chromosomes of Caryophyllales plants. The ancestral eudicot chromosomes, designated as G1–G7, underwent a series of ECH events, resulting in 21 chromosomes denoted as A1–A7, B1–B7, and C1–C7. Meanwhile, the ancestral chromosomes of the ancestors of Caryophyllales are labeled as CA1–CA7. Analysis of the dot plot comparing C. pallidicaule and grape (Fig. 2d) revealed that grape chromosome 10 (A6) and chromosome 18 (B3) produced an intermediate chromosome through chromosome translocation. Subsequently, grape chromosome 15 (B4) was inserted into the intermediate chromosome, resulting in the production of the chromosome CA1, with the loss of a B chromosome. Chromosome 5 (A5) and 16 (C4) of grape form an intermediate chromosome through end-to-end joining. Subsequently, a segment of grape chromosome 8 is inserted into this intermediate chromosome, giving rise to CA3 and forming another intermediate chromosome (2). During the process leading to the formation of the nine chromosomes of Dianthus, a total of eight end-to-end joinings and four nested chromosome fusions occur. Additionally, seven B chromosomes are produced and subsequently lost. Ultimately, nine chromosomes representing the ancestors of Caryophyllales are formed. The formation pathways of the remaining chromosomes are depicted in Fig. 3b.
M. crystallinum has no additional recent duplication event, so it has a one-to-one homologous relationship with C. pallidicaule. Consequently, the homologous dotplot of C. pallidicaule and M. crystallinum is used to infer the evolutionary trajectory of M. crystallinum (Fig. 3c). For clarity, the color scheme of the nine chromosomes of C. pallidicaule is redefined, with C1–C9 representing the nine chromosomes of C. pallidicaule, and M1–M9 designating the nine chromosomes of M. crystallinum. During the transition from the nine ancestral chromosomes of Caryophyllales to the nine chromosomes of M. crystallinum, neither EEJ nor NCF events occurred. Instead, multiple chromosome translocations and inversions were experienced. For instance, chromosome 2 of M. crystallinum is derived from chromosome 4 of grapes (C1) and chromosome 14 of grapes (C7) through chromosome translocations, resulting in the formation of two intermediate chromosomes (1) and (2). These two intermediate chromosomes subsequently contributed to the formation of chromosomes 4 and 9 of M. crystallinum. Detailed accounts of the formation processes for other chromosomes are delineated in the trajectory Fig. 3c. Notably, examination of the dot plot of M. crystallinum and C. pallidicaule indicates relatively intact chromosomes in M. crystallinum, with minimal gene loss attributed to the conservative evolution of grapes.
Exploring the evolutionary imprint of CAM-related genes at a large scale
-
We identified 11,148 CAM-related genes in ice plants and another 287 species with completed whole-genome sequences, representing a diverse array of plant taxa (Figs 4−6, Supplementary Table S1). These species encompassed 174 eudicots, 59 monocots, two magnolias, three basal angiosperms, five gymnosperms, three ferns, six mosses, and 36 algae. Among the identified genes, the highest numbers were MDH genes (3,662), followed by ME genes (2,334), PEPC (1,840), CA (1,780), PEPCK (1,038), and PPDK (494) (Fig. 4, Supplementary Table S1).
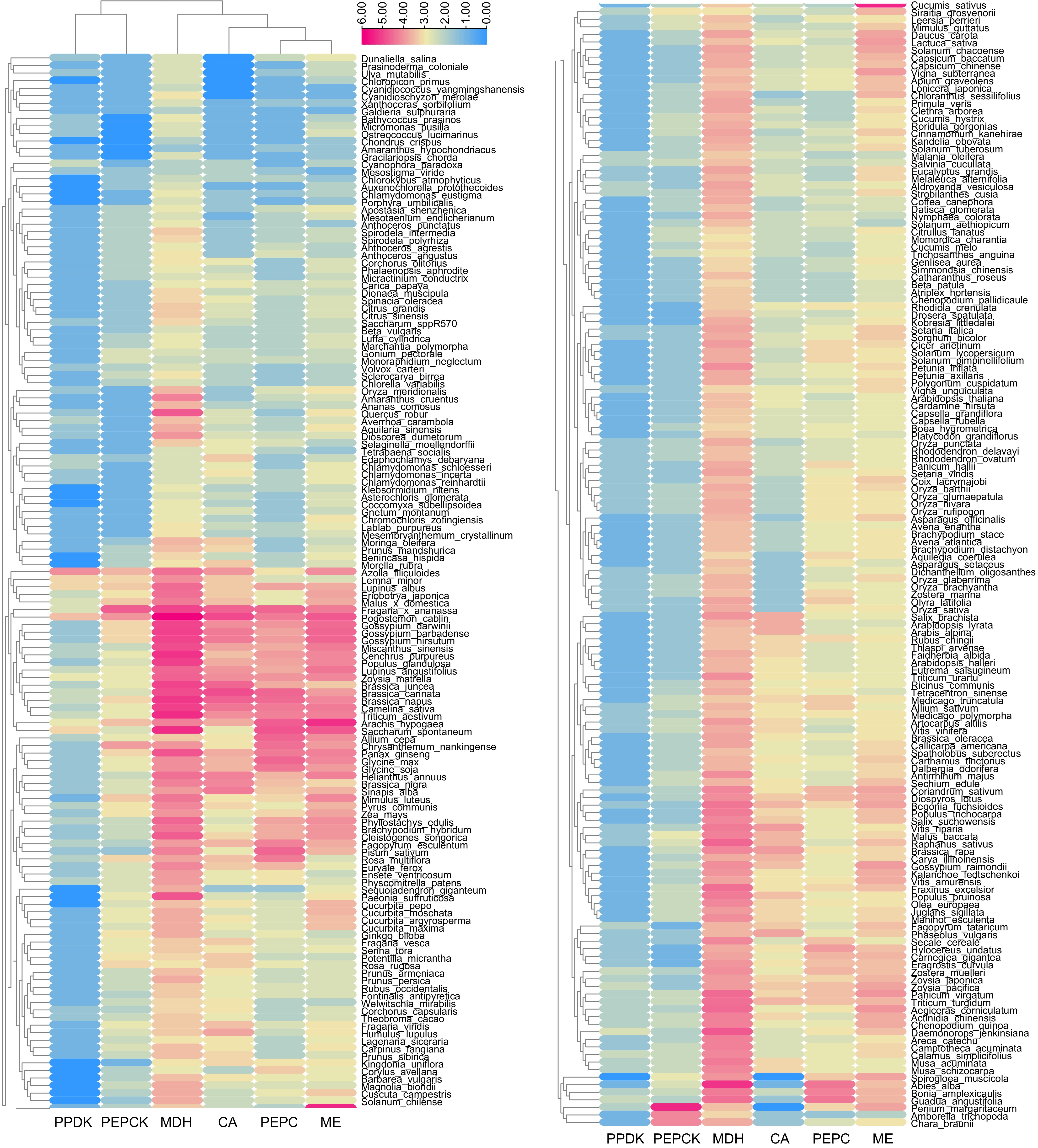
Figure 4.
Heatmap of CAM-related gene number in 288 plants. The numbers were transformed by log2.
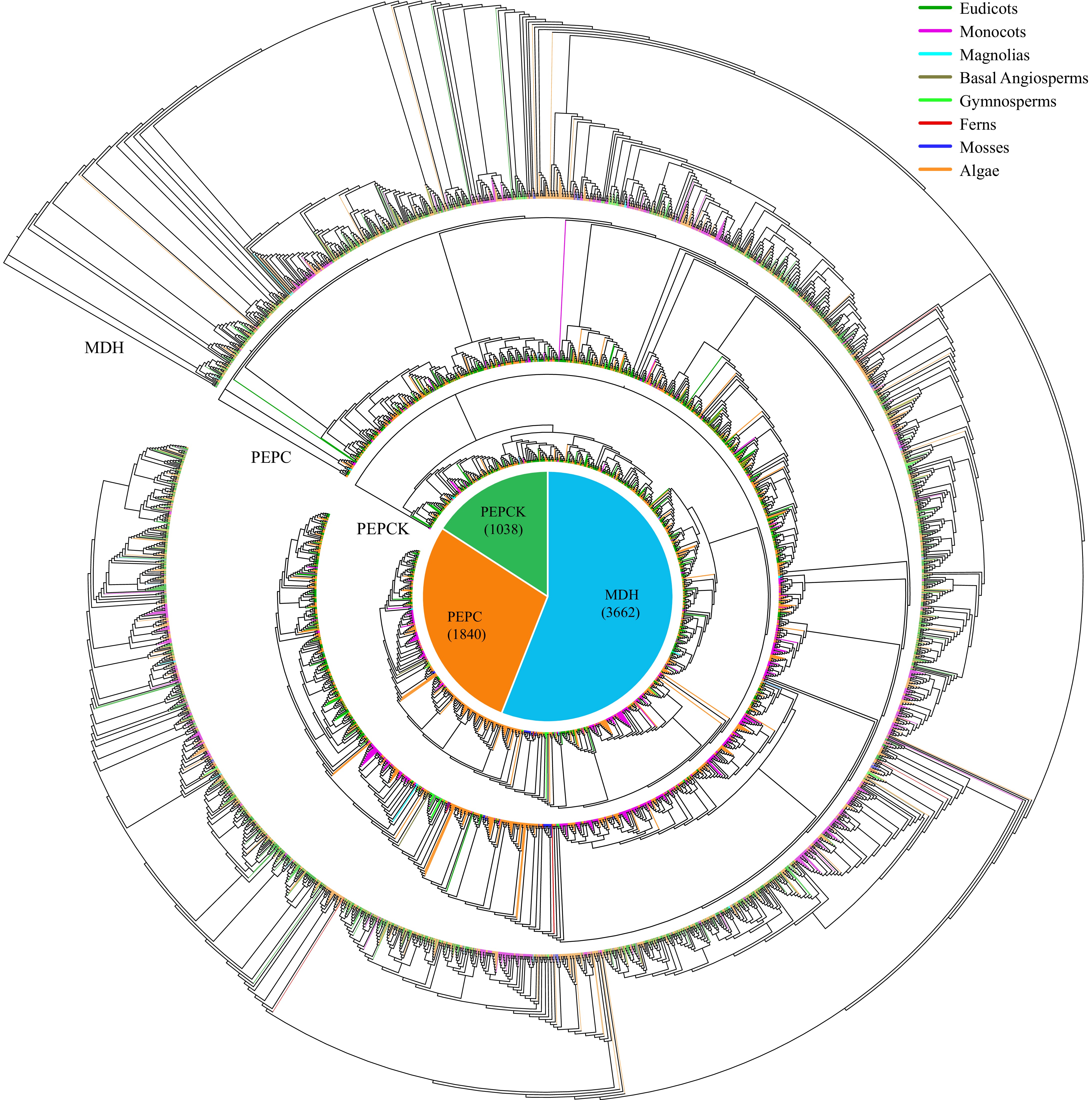
Figure 5.
Maximum-likelihood trees were constructed for CAM-related genes, encompassing families such as MDH, PEPC, and PEPCK, utilizing amino acid sequences from 288 species with 1,000 bootstraps. The pie chart illustrates the overall distribution of each CAM-related gene across the 288 species. Branches within the evolutionary tree are color-coded to denote the taxonomic classification of the respective species.
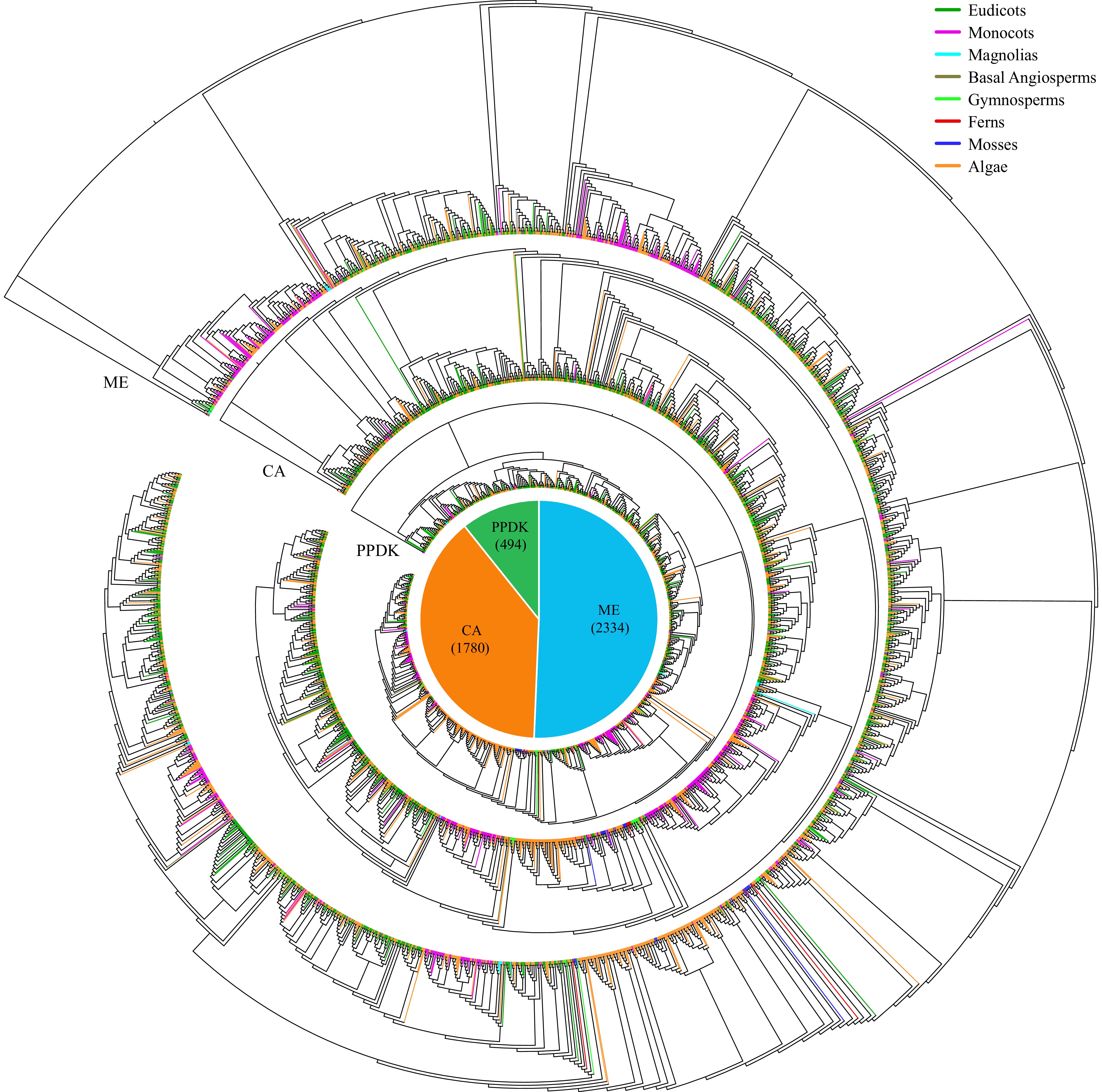
Figure 6.
Maximum-likelihood trees were generated for CAM-related genes, encompassing families such as ME, CA, and PPDK, utilizing amino acid sequences from 288 species with 1,000 bootstraps. The pie chart depicts the total number of each CAM-related gene across the 288 species. Different colored branches within the evolutionary tree denote the taxonomic affiliations of the respective species.
To explore the evolution trajectory of CAM-related genes in plants, we constructed six phylogenetic trees using the protein sequences of CAM-related genes across 288 plant species (Figs 5 and 6). Across all six evolutionary trees, a consistent pattern emerged: CAM-related genes from algae were clustered alongside those from monocots and eudicots, respectively. This intriguing observation suggests an independent evolutionary path for CAM-related genes in monocots and dicots.
Gene family analysis
-
CAM photosynthesis represent a distinctive photosynthetic pathway characterized by two primary phases: carboxylation and decarboxylation. In the carboxylation phase, stomata aperture facilitates the absorption of CO2, which is subsequently converted into HCO3− under the catalytic influence of CAs. HCO3− is then enzymatically converted by PEPC into OAA[28]. Then, MDH reduces OAA to malic acid and it is stored in vacuoles. During the decarboxylation phase, malic acid is transported to the cytoplasmic matrix, and two different reactions occur according to the different enzymes in the catalytic reaction process. The first step involves the decarboxylation of malic acid, facilitated by ME, resulting in the production of pyruvic acid and the release of CO2. Subsequently, pyruvic acid is enzymatically converted to PEP under the action of PPDK[29]. The second step is a PEPCK-mediated decarboxylation process. In this pathway, malate is first converted to OAA by ME. Subsequently, OAA undergoes decarboxylation to yield PEP and CO2 under the catalytic action of PEPCK. PEP is further metabolized into glycogen via gluconeogenesis and stored within plants. These stored sugars can be converted back into PEP through glycolysis, thereby facilitating continued carboxylation during nocturnal periods[6]. The ice plant exhibits facultative capabilities in both C3 and CAM photosynthesis. To elucidate the CAM-related genes in ice plants, a comprehensive analysis was conducted using representatives from various photosynthetic pathways. This included three C3 plants (Oryza sativa, V. vinifera, and A. thaliana), three C4 plants (Z. mays, S. bicolor, and A. cruentus), two CAM plants (H. undatus and A. comosus), and one C3-CAM species (ice plant). Through this systematic approach, the gene families encoding six enzymes crucial to the CAM pathway were thoroughly investigated (Fig. 7a, b).
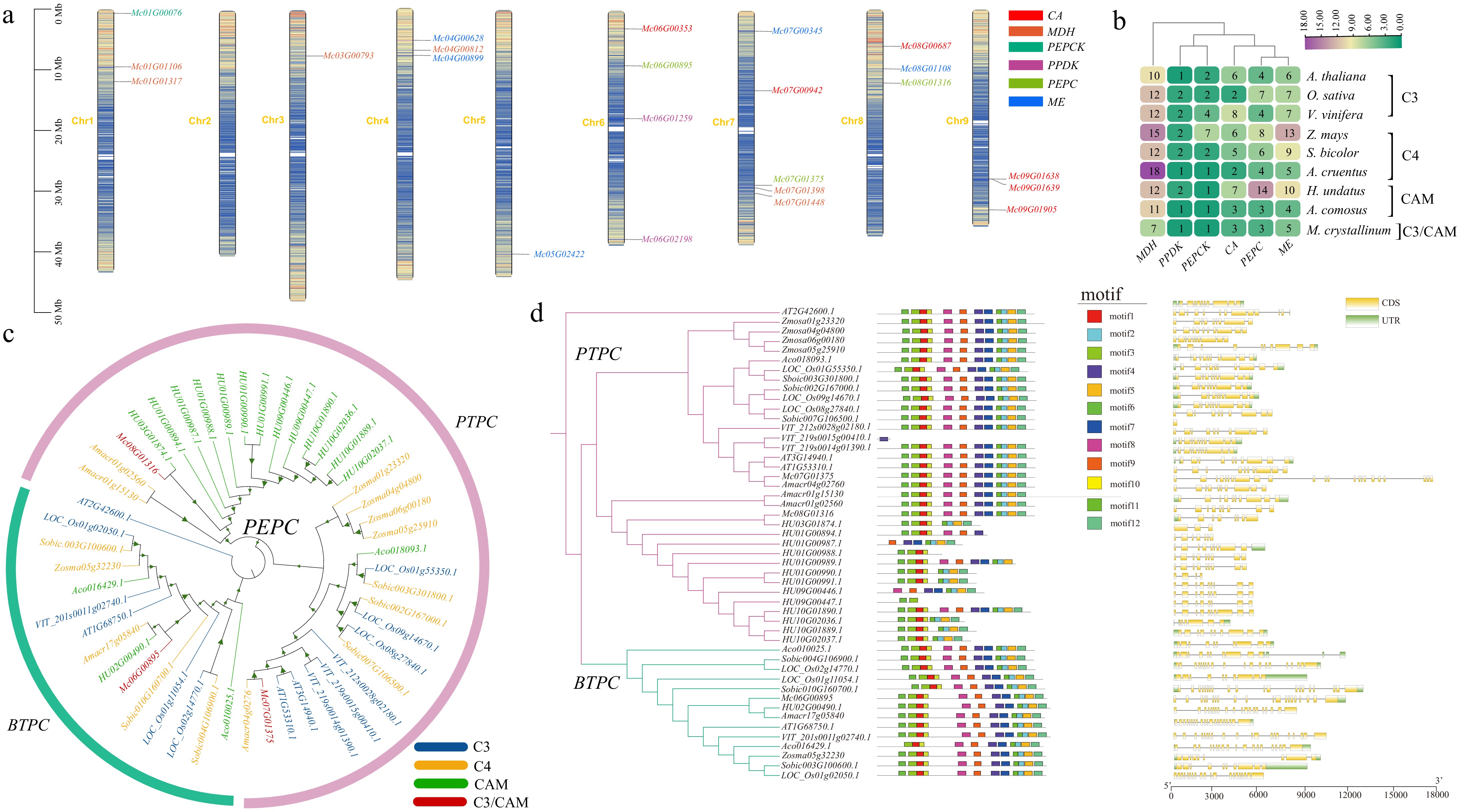
Figure 7.
CAM gene analysis across the ice plant and other eight representative species. (a) Map illustrating the distribution pattern of CAM genes within the genome of the ice plant. (b) Heatmap of CAM gene numbers across ice plants and other eight representative species. (c) Phylogenetic tree of PEPC proteins in ice plants and other species. The color of genes represents C3, C4, CAM, and C3/CAM types. The color of the outer circle denotes the three subfamilies of the PEPC gene family. (d) Phylogenetic tree, conserved motifs, and gene structure of PEPC across the ice plant and other species. To the left of the phylogenetic tree, schematic representations depict conserved motifs within PEPC proteins, as identified through MEME analysis. Different colors are utilized to signify different motifs. On the right side of the tree, a schematic diagram illustrates the gene structure of PEPC proteins using TBtools. Here, the coding sequences (CDS) are visually distinguished in yellow, while the untranslated regions (UTR) are depicted in green, facilitating a comprehensive understanding of the structural organization of PEPC genes across taxa.
PEPC enzyme gene family
-
PEPC is an enzyme that is widely present in various plants and plays a crucial role in the process of photosynthesis. It is capable of directly carboxylating atmospheric carbon dioxide to form OAA in C4 photosynthesis and CAM pathways, and OAA is of great significance in the accumulation of substances[30,31]. In the photosynthetic tissues of C3 plants and some non-photosynthetic tissues, PEPC mainly participates in the synthesis of intermediate metabolites in the tricarboxylic acid cycle (TCA), which are the main metabolites for the synthesis of other important components such as carbohydrates, lipids, and proteins[32]. In addition, PEPC is extensively involved in plant development and physiological processes, such as the formation of malate in guard cells during stomatal opening and its unique role in the root nodules of nitrogen-fixing leguminous plants[33]. For instance, four PEPC family members were characterized in A. thaliana, while six PEPC family members were identified in O. sativa[34]. According to the difference in its phosphorylation structure, the PEPC gene family is divided into plant-type PEPC (PTPC) and bacterial-type PEPC (BTPC). The N-terminal region of the BTPC sequence does not contain a phosphorylated structure. A total of 49 members of the PEPC gene family were identified across nine species. In the ice plant, the PEPC gene family includes Mco6G00895, Mc07G01375, and Mc08G01316, which are located on chromosomes 6, 7, and 8, respectively (Fig. 7a). To elucidate the phylogenetic relationships within the PEPC family, protein sequences from the 49 PEPC were analyzed, resulting in the construction of a phylogenetic tree (Fig. 7c). The analysis revealed the division of PEPC family members into two distinct subgroups. Specifically, Mc07G01375 and Mc08G01316 clustered with AT1G53310.1 belonging to the PTPC subgroup, whereas Mc06G00895 and AT1G68750.1 formed a separate group within the BTPC subgroup. Subsequently, MEME was used to analyze the conserved domain of PEPC proteins. The analysis revealed the presence of 12 conserved motifs within the ice plant's protein. Notably, the order of these conserved motifs remained consistent within the two subgroups, with members of the same subgroup exhibiting similar gene structures and functional domains (Fig. 7d).
CA enzyme gene family
-
Carbonic anhydrase (CA) is a zinc-containing metalloenzyme that catalyzes the reversible hydration of CO2 to HCO3−, which is found across eukaryotes and prokaryotes. Based on structural homology, CA is classified into six gene families (α, β, γ, δ, ε, and ζ), though only α, β, and γ types exist in higher plants[35]. Despite their structural divergence, all plant isoforms employ the same catalytic mechanism[36]. Functionally, CA overexpression in A. thaliana enhances cytoplasmic substrate supply for PEPC, promoting malate synthesis to fuel the TCA cycle[37]. Intriguingly, chloroplast-localized CA does not directly provide CO2 for Rubisco but instead supplies HCO3- for biosynthesis, akin to its role in microorganisms. Consequently, while CA deficiency impairs C3 plant development, it does not affect photosynthetic carbon fixation[38,39]. A total of 84 CA gene family members were obtained from nine species, and six CA gene family members, Notably, within the genomic inventory of ice plants, six CA gene family members, namely Mc06G00353, Mc08G00687, Mc07G00942, Mc09G01639, Mc09G01638, and Mc09G01905, have been elucidated. It is noteworthy that these gene family members exhibit an uneven chromosomal distribution, being dispersed across four distinct chromosomes within the genome of the ice plant (Fig. 7a). Multiple sequence alignment of CA protein sequences from the ice plant and eight other species was conducted using MAFFT algorithm. Subsequently, a phylogenetic tree was constructed employing FastTree elucidating the evolutionary relationships among the CA proteins across these diverse taxa (Supplementary Fig. S6). According to the classification of the CA gene family in A. thaliana[40], the 84 identified family members were divided into two distinct categories. Notably, Mc09G01638 and Mc09G01905 belong to α-CA, while the remaining four belong to β-CA. Subsequently, MEME analysis was employed to predict conserved motifs within the protein sequences of the CA gene family. The investigation revealed the presence of 10 conserved motifs, designated as motifs 1 ~ 10 (Supplementary Fig. S7). Remarkably, Mc09G01638 and Mc09G01905 encompassed five conserved motifs, denoted as motif1-motif5, while Mc06G00353, Mc08G00687, Mc07G00942, and Mc09G01639 harbored five additional conserved motifs, designated as motif6-motif10. Subsequently, TBtools was utilized to visualize the gene structure of all CA gene family protein sequences (Supplementary Fig. S7).
MDH enzyme gene family
-
MDH is ubiquitously present in nature and exerts influence over various facets of plant growth and development. Extensive research endeavors have consistently underscored the pivotal involvement of MDH in essential physiological processes, including energy metabolism, respiration, and active oxygen metabolism. Furthermore, MDH assumes a critical role in conferring stress resistance mechanisms within plant systems[41]. At present, the MDH gene family has been identified in a variety of plants based on whole genome data, such as A. thaliana, Populus trichocarpa, and O. sativa[42−44]. In our study, a total of 92 MDH genes were successfully identified across nine species. Among these, six MDH genes were specifically pinpointed within the ice plant genome, namely Mc01G01106, Mc01G01317, Mc03G00793, Mc04G00812, Mc07G01398, and Mc07G01448. Notably, these genes exhibit an uneven distribution pattern across the chromosomes of the ice plant (Fig. 7a). There are two genes located on chromosomes 1 and 7, respectively, while the remaining two are distributed across chromosomes 3 and 4. To explore the diversity and phylogenetic relationship among MDH proteins, a series of analytical procedures were conducted. Firstly, MEME analysis was employed to identify conserved motifs within the MDH amino acid sequences derived from nine distinct plant species. Subsequently, multiple sequence alignment was performed to elucidate the sequence similarities and variations across these MDH proteins. Finally, a phylogenetic tree was constructed based on the aligned sequences, facilitating the delineation of evolutionary relationships among MDH proteins across the diverse taxa under consideration (Supplementary Fig. S8). According to the classification of MDH proteins in rice, as elucidated by Zhang et al.[44], an analysis of motif types and abundance facilitates the subdivision of the 92 identified MDHs into three distinct subgroups: I, II, and III (Supplementary Fig. S9). Group I comprises MDHs characterized by four conserved motifs, while Group II and Group III exhibit six and eight motifs, respectively. Notably, among the six MDH genes identified in the ice plant, there are three assigned to Group I, while the remaining three belong to Group II. Additionally, upon scrutinizing the gene structure, it becomes evident that MDH genes within a given subfamily or chromosome do not exhibit uniformity in terms of their gene structure, particularly concerning the number or length of introns andexons (Supplementary Fig. S9).
PPDK enzyme gene family
-
PPDK is a key enzyme in C4 and CAM photosynthesis, catalyzing the formation of PEP required for CO2 fixation. In the obligate CAM plant Kalanchoë fedtschenkoi, PPDK exhibits light-responsive gene expression and circadian-regulated protein abundance, suggesting its crucial role in modulating daytime decarboxylation under varying light conditions[45]. Subcellular localization classifies PPDK into two isoforms: chloroplast-targeted C4PPDK and cytoplasmic cyPPDK[46]. This compartmentalization is functionally significant in facultative CAM species like Kalanchoë blossfeldiana, where both isoforms show coordinated accumulation (maintaining a stable 2.4-3.0 cytoplasm:chloroplast ratio) upon CAM induction by drought or short-day conditions[47]. Crucially, cyPPDK synergizes with PEP carboxylase in nocturnal CO2 fixation, while chloroplast PPDK facilitates daytime carbon metabolism. These findings establish that PPDK optimizes CAM efficiency through spatiotemporal regulation of isoform partitioning. A comprehensive examination revealed the presence of 20 PPDK genes across nine distinct species. Notably, within the ice plant, two specific PPDK genes, denoted as Mc06G01259 and Mc06G02198, were discerned, both of which were localized on chromosome 6 (Fig. 6a). To elucidate the extent of diversity and ascertain the interrelations among PPDK proteins, an analytical framework was employed. This encompassed the utilization of MEME analysis alongside multiple sequence alignment of the amino acid sequences inherent to PPDK across the aforementioned nine species. Subsequently, a phylogenetic tree was constructed to delineate the evolutionary trajectories and relationships amongst these proteins (Supplementary Fig. S10a). Analysis of the outcomes revealed a discernible categorization of the 20 PPDKs into two distinct subgroups, designated as Groups I and II, predicated upon the nature and abundance of motifs identified. Group I exhibited the presence of nine distinctive motifs, whereas Group II displayed a more limited repertoire with only two motifs identified. There were nine motifs in Group I, and two motifs in Group II. Notably, the two PPDK genes identified within the ice plant exhibited membership in both Group I and Group II. Subsequent investigation into the gene architecture unveiled notable distinctions in the arrangement, quantity, and length of introns/exons between the two PPDK genes inherent to the ice plant (Supplementary Fig. S11a).
PEPCK enzyme gene family
-
PEPCK serves as the catalytic linchpin governing the inaugural and rate-limiting step in the gluconeogenic pathway. Specifically, it orchestrates the reversible decarboxylation of OAA into PEP and CO2, leveraging either ATP or GTP as phosphoric donors. It is noteworthy that the enzymes utilizing ATP and those employing GTP delineate into two discrete subfamilies, each characterized by a conspicuous divergence in sequence composition. Despite this dissimilarity, these subfamilies manifest a retention of conserved active site residues, underscoring their functional homology amidst their sequence disparities[48]. ATP-utilizing PEPCKs exhibit a structural configuration comprising monomeric or oligomeric assemblies consisting of identical subunits. This structural archetype is prevalent among certain bacterial strains, yeast species, trypanosomatids, and various plant taxa. Conversely, GTP-utilizing PEPCKs predominantly present as monomeric entities, prevalent among animals and select bacterial species, as elucidated by Villarreal et al.[49]. The identification of a total of 10 PEPCKs across nine distinct species underscores the evolutionary conservation of this crucial enzyme. Notably, within the ice plant, a solitary gene, denoted as Mc01G00076, was mapped to chromosome 1 (Fig. 7a). To delineate the evolutionary relationships and phylogenetic clustering of PEPCKs across diverse species, a comprehensive analysis was conducted. Multiple sequence alignments were conducted on the amino acid sequences intrinsic to PEPCK across the aforementioned nine species, thereby facilitating the construction of a phylogenetic tree (Supplementary Fig. S10b). Conserved domain analysis of PEPCK proteins conducted through the MEME tool revealed that these 10 PEPCKs belonged to ATP-PEPCKs. Notably, with the exception of VIT_200s1995g00010.1, which exhibited a distinct motif composition, the remaining nine genes demonstrated conservation across 10 discernible motifs. Further examination on the gene architecture utilizing Tbtools revealed intriguing differentiations within the structure of these 10 PEPCKs genes. Despite sharing a common motif structure, notable disparities emerged in terms of the number or length of introns/exons. For instance, the gene HU10G00784.1 displayed the lengthiest gene sequence, with pronounced variations observed among the remaining gene lengths (Supplementary Fig. S11b).
NADP-ME enzyme gene family
-
ME plays a pivotal role in metabolic processes, facilitating the oxidative decarboxylation of malate to yield pyruvate. Notably, carbon dioxide released from the reaction may be used in sugar production during the Calvin cycle of photosynthesis[50]. ME manifests in three distinct forms, each characterized by its specific cofactor dependency and substrate specificity. These include three forms: a NAD-dependent form that can decarboxylate oxaloacetate, another NAD-dependent form that cannot decarboxylate oxaloacetate, and a NADPH-dependent form[51]. NADP-ME catalyzes the oxidative decarboxylation of malic acid to pyruvate, CO2, and NADPH. This enzymatic process holds considerable significance within the framework of malic acid metabolism. Notably, the classification of NADP-MEs into distinct photosynthetic and non-photosynthetic categories delineates their diverse functional role[52]. The genomic landscape harboring NADP-ME genes extends across various plant species, including A. thaliana, Z. mays, and P. trichocarpa, among others[53]. We identified the presence of 57 NADP-ME genes across nine species. Within the ice plant species, specifically, five NADP-ME genes were identified, designated as Mc04G00628, Mc04G00899, Mc05G02422, Mc07G00345, and Mc08G01108, respectively. Notably, two of these genes were localized to chromosome 4 of the ice plant genome (Fig. 7a), and the remaining were located to chromosomes 5, 7, and 8 of the ice plant genome. To elucidate the diversity and phylogenetic relationships inherent within the NADP-ME protein family, a comprehensive analysis was conducted. Multiple sequence alignments were conducted on the amino acid sequences of NADP-ME proteins across the nine sampled species, facilitating the construction of a phylogenetic tree (Supplementary Fig. S12). Utilizing the MEME tool, an in-depth analysis was conducted to decipher the conserved domains inherent within the NADP-ME protein family. The findings revealed a discernible division among the members of this protein family within ice plant, segregating them into two distinct subgroups. Notably, Mc07G00345 and AT1G79750.1 congregated within one cluster, indicative of their membership in the photosynthetic type. Conversely, Mc04G00628, Mc04G00899, Mc05G02422, and Mc08G01108 formed a cohesive cluster alongside AT2G19900.1, AT5G11670.1, and AT5G25880.1, characteristic of the non-photosynthetic type. Further analysis through MEME elucidated the conserved domain architecture of NADP-ME protein in the ice plant, revealing the presence of 14 conserved motifs. Intriguingly, the order of these conserved motifs within the two subgroups remained consistent, indicative of structural homogeneity within each subgroup (Supplementary Fig. S13).
Future research directions
-
Based on the in-depth analysis of gene families encoding key enzymes in the CAM pathway, including PEPC, CA, PEPCK, NADP-ME, PPDK, and MDH in M. crystallinum, future research can be carried out from multiple dimensions.
At the level of gene expression regulation, leveraging the characteristics of the identified key enzyme gene families, further investigation is required to elucidate the specific mechanisms by which individual members of the PEPC gene family are linked to circadian rhythms. This involves precisely controlling PEPC activity through nocturnal regulatory elements. Additionally, for other key enzymes such as CA and PEPCK, efforts should focus on exploring the construction of synthetic plant promoters with predictable expression patterns and utilizing ABA-responsive elements to control their expression within the inducible CAM system, thereby enabling systematic regulation of the CAM pathway.
Regarding the development of CAM engineering strategies, considering the functional characteristics of the key enzyme gene families in M. crystallinum, emphasis should be placed on constructing modular CAM engineering systems in C3 crops. Introducing a core CAM module comprising PEPC and its kinase, plastidic NADP-ME, and PPDK, and connecting it to evening elements and ABA-responsive promoters can achieve strict circadian control, preventing futile cycles and initiating nocturnal carbon capture. Meanwhile, in-depth research is needed to understand the synergistic mechanisms of CA, PEPCK, and MDH within this engineering system, optimizing their expression and functions to enhance overall carbon metabolic efficiency.
Furthermore, several critical challenges remain to be addressed. First, based on the analysis of gene retention patterns in M. crystallinum, optimizing the mechanisms of malate storage and release requires in-depth exploration of the collaborative roles of the Aluminum-activated malate transporter 9 (ALMT9) transporter and key enzymes such as MDH in malate metabolism. Second, establishing a stable coupling between starch and malate metabolism necessitates further clarification of the synergistic mechanisms involving key enzymes like PEPC, NADP-ME, and PPDK, given the co-evolutionary patterns indicated by gene retention. Third, developing synthetic promoter systems capable of replicating the phased expression elements observed in CAM-adapted species is crucial for precisely regulating the expression of genes encoding key enzymes. Notably, the redox sensitivity of NADP-ME and the substrate-driven reversibility of enolase/PGlyM may simplify temporal regulation, and their application potential should be further explored in conjunction with the regulatory patterns of key enzyme gene families in M. crystallinum.
-
Polyploidization, ubiquitous in plant taxa, stands as a pivotal mechanism driving swift genomic evolution and bolstering adaptive capabilities, thereby fostering heightened biodiversity[54,55]. However, after polyploidization events occur, it is often accompanied by large-scale chromosome rearrangements and gene loss, thereby intricately complicating the genomic landscape of the plant species[56]. The evolutionary trajectories of ancestral karyotypes in plants reveal both shared and lineage-specific mechanisms of post-polyploid genome reorganization. In grasses, chromosome number reduction from a paleo-ancestor (n = 12) was primarily driven by centric nested chromosome fusions (NCFs) through nonrandom double-strand break repair, with synteny break points (SBPs) acting as evolutionary hotspots[57]. Similarly, Asteraceae ancestors underwent significant karyotype simplification from 21 to nine proto-chromosomes via 11 EEJs and one NCF. Further reductions occurred in descendant species, such as from 27 to 15 in the most recent common ancestor of the genus (MRCAK) via 10 EEJs, and from 15 to nine in Lactuca sativa via five EEJs[58]. These comparative analyses provide an important context for understanding chromosome evolution in Caryophyllales. In this study, we selected eight species that experienced different doubling events in Caryophyllales to infer their ancestral chromosomes. Based on the telomere-center chromosome reconstruction model[15], combined with polyploidization events, the ancestral chromosome karyotypes of important evolutionary nodes of Caryophyllales plants and the evolutionary path from ancestral nodes to modern species chromosomes were reconstructed. The inference of the number of ancestral chromosomes at key evolutionary nodes in each branch of the Caryophyllales order suggests a pattern wherein ancestral chromosomes undergo heterogeneous fusion phases from one doubling event to the subsequent, followed by a period of stability, prior to undergoing iterative fusion events during the divergence into distinct species Through exploring the process of chromosome formation in the ancestors of Caryophyllales, it was found that the number of chromosomes changed from 21 to nine. This process involved a total of eight EEJ and four NCF events, resulting in the production and loss of seven B chromosomes. Furthermore, chromosome translocations frequently accompany this process of transformation. Despite the absence of alterations in chromosome count within the ice plant lineage, a substantial incidence of chromosomal translocation exchanges is evident during the evolutionary transition from Caryophyllales ancestors to the ice plant progenitors. The reconstruction of the ancestral chromosomes of Caryophyllales provides a reference for the study of the origin and evolution of Caryophyllales plants, enabling us to better understand the evolutionary history of Caryophyllales species at the chromosome level. Moreover, based on the inference of the ancestral chromosomes of Caryophyllales species, the karyotype path from pre doubling to post doubling of the ancestral chromosomes of Caryophyllales species was reconstructed, thereby addressing a critical lacuna in the understanding of karyotype evolution within Caryophyllales species.
Based on the whole genome data of the ice plant, this study identified members of six gene families in the ice plant, totaling 23 genes, with the highest number of members in the CA and MDH gene families. CA is necessary for the CO2 concentration mechanism[59], divided into α, β, and γ types. Among the six genes identified in the ice plant, two pertain to α-CAs, while the remaining four are attributed to β-CAs. Notably, β-CAs demonstrate a predilection for heightened expression levels within leaf tissues. Investigations have revealed the localization of β-CAs across diverse cellular compartments, including chloroplasts, mitochondria, cytoplasm, and plasma membrane, within A. thaliana[40]. The CA gene assumes a crucial role in various carboxylation and decarboxylation biological processes such as photosynthesis and respiration, plant growth, and response to environmental stressors[40]. MDH is a highly active enzyme within the plant kingdom, exerting a significant influence in the cellular response to various abiotic stressors[60]. As delineated by the phylogenetic analysis, MDH genes can be categorized into distinct clades denoted as I, II, and III across both monocotyledonous and dicotyledonous plant lineages. This observed phylogenetic clustering underscores the enduring conservation of the MDH gene family throughout extensive periods of evolutionary selection[44]. We identified six MDH genes in the ice plant. These six genes belong to clades I and II. These two clades contain different types and numbers of motifs, but the genes in each subfamily exhibit an identical type and number of motifs. This observation prompts speculation regarding potential functional similarities among MDH members within individual subfamilies.
The phylogenetic segregation of PEPC into PTPC and BTPC subfamilies in M. crystallinum (Fig. 7c) reflects evolutionary specialization analogous to obligate CAM species. Recent functional characterization in K. fedtschenkoi demonstrates this divergence unequivocally: CRISPR-mediated knockout of nocturnal-specific PEPC2 (orthologous to PTPC) completely abolished CAM functionality, whereas constitutive PEPC1 (BTPC-type) knockdown primarily affected stomatal regulation without disrupting the CAM cycle[32]. Strikingly, our motif analysis reveals that the ice plant PTPC-clustered Mc07G01375 preserves critical phosphorylation sites identical to those governing diel regulation of KfPEPC2, strongly implicating its specialized role in nocturnal carbon fixation.
The conserved role of β-CAs in stomatal CO2 sensing, as demonstrated in A. thaliana[61], may have been evolutionarily co-opted to facilitate CAM-specific gas exchange rhythms. Our phylogenetic analysis reveals that ice plant β-CAs (e.g., Mc07G00942) cluster with stomatal CO2 regulators AtβCA1 (AT3G01500.2) (Supplementary Fig. S6), while maintaining complete conservation of catalytically essential zinc-binding residues. This suggests functional convergence in CO2/HCO3- conversion capacity, albeit with potential CAM-specific modifications. During nocturnal phase I, elevated β-CA activity in guard cells could enhance CO2-triggered stomatal opening, optimizing dark-period CO2 uptake; while daytime stomatal closure under elevated internal CO2 may involve complementary α-CA activity (Mc09G01638, Mc09G01639), analogous to AtαCA1-mediated closure in A. thaliana[61].
The facultative CAM species M. crystallinum, whose water deficit-induced CAM activation is distinct from that of CAM species with weaker developmental control, serves as a unique model for identifying the molecular switch in the C3 to CAM transition. Our systematic characterization of CAM-associated enzymes in iceweed revealed a key evolutionary adaptation: due to a WGT event in iceweed, a gene family expansion occurred, allowing for functional diversification of paralogs, as evidenced by PTPC/BTPC-type PEPC isozymes with unique regulatory features, similar to the pattern in obligate CAM plants such as Kalanchoe[14,62]. These findings support an evolution-guided engineering strategy: transferring drought-responsive regulatory modules from ice plant's CAM genes (PEPC promoters with phased expression elements) to crops while retaining native C3 enzyme copies to maintain baseline metabolism, as demonstrated by gene dosage balance in Agave[63]. This approach has significant potential for crop improvement and plant stress resistance breeding. For example, introducing CAM-related genes into major crops such as wheat, corn, and rice could enhance their drought tolerance by enabling more efficient water use and reducing water loss through stomatal closure. The CAM pathway allows plants to perform photosynthesis more efficiently under water-limited conditions, thereby maintaining productivity even in arid environments. This not only increases crop yields but also reduces the need for irrigation, leading to significant economic and environmental benefits.
-
In this study, we reconstructed the ancestral karyotype of Caryophyllales by comparing the genomes of the ice plant and seven additional Caryophyllales plants. Our inference suggests that the most recent common ancestor of Caryophyllales bore nine chromosomes. Furthermore, we elucidated the evolutionary trajectory spanning from the ancestral chromosomes of core eudicots to those of Caryophyllales, and subsequently delineated the evolutionary path from the ancestral chromosomes of Caryophyllales to those observed in ice plants. Subsequently, we aim to investigate the CAM genes within the ice plant using a selection of three C3, three C4, two CAM plants, and one C3-CAM species. Our analysis identified a total of 23 CAM genes in the ice plant, distributed across six gene families and localized on eight chromosomes. These genes encode pivotal enzymes integral to the CAM pathway. Employing phylogenetic tree analysis, chromosome localization, gene domain identification, and gene structure analysis, we further categorized these genes into distinct subfamilies. Notably, upon delineating gene structure and conserved domains, certain gene families demonstrated consistent ordering of conserved motifs among ice plant proteins within respective subgroups, with members within the same subgroup exhibiting analogous gene structures and functional domains, exemplified by the NADP-ME and PEPC gene families. Conversely, in other gene families, subdivision into subfamilies did not yield uniformity in gene structures regarding the number or length of introns/exons among genes within the subfamily.
-
Not applicable.
The authors thank all colleagues and collaborators for their contribution to the work described here. This work was supported by the Tangshan Science and Technology Plan Project (24130219C), the Basic research expenses for provincial universities (JJC2024001), Basic Research Funds for Provincial Universities Basic Research Projects of North China University of Science and Technology (JQN2023036), the Youth Scholars Promotion Plan of North China University of Science and Technology (QNTJ202308), Central Guidance and Support for Local Science and Technology Development Fund (246Z2509G), the S&T Program of Hebei (23372505D), and the Hebei Natural Science Foundation (H2023209084).
-
The authors confirm their contributions to the paper as follows: conception and initiation of the project: Song X; supervision and management of project and research: Song X, Zhang Y, Guo D; data generation and bioinformatic analysis: Song X, Shen S, Meng Z, Zhang X, Zhang C; manuscript organization, writing, and revision: Song X, Shen S, Zhang Y, Guo D, Gaicia P, Zhou R. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Shaoqin Shen, Zipeng Meng
- Supplementary Table S1 The identified genes involved in crassulacean acid metabolism (CAM) pathway in the 288 species.
- Supplementary Figs S1 The Homologous dotplot between Chenopodium pallidicaule and Beta vulgaris.
- Supplementary Fig. S2 The Homologous dotplot between Chenopodium pallidicaule and Atriplex hortensis.
- Supplementary Fig. S3 The Homologous dotplots between Chenopodium pallidicaule and Mesembryanthemum crystallinum.
- Supplementary Figs S4 The Homologous dotplot between Chenopodium pallidicaule and Spinacia oleracea.
- Supplementary Fig. S5 A phylogenetic tree of carbonic anhydrase proteins in ice plant and other species.
- Supplementary Fig. S6 Phylogenetic tree, conserved motifs and gene structure of ice plant and other species carbonic anhydrases.
- Supplementary Fig. S7 A phylogenetic tree of MDH proteins in ice plant and other species.
- Supplementary Fig. S8 The phylogenetic tree, conserved motifs, and gene structure of MDH in ice plant and other species.
- Supplementary Fig. S9 A phylogenetic tree of PPDK and PEPCK proteins in ice plant and other species.
- Supplementary Fig. S10 The phylogenetic tree, conserved motifs, and gene structure of PPDK and PEPCK in ice plant and other species.
- Supplementary Fig. S11 A phylogenetic tree of NADP-ME proteins in ice plant and other species.
- Supplementary Fig. S12 The phylogenetic tree, conserved motifs, and gene structure of NADP-ME in ice plant and other species.
- Supplementary Fig. S13 Flowchart of the ancestral karyotype construction of the Caryophyllales.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Shen S, Meng Z, Garcia P, Zhang X, Zhang C, et al. 2025. Ancestral chromosome karyotype construction and large-scale analysis of CAM biosynthesis genes in ice plant and representative plants. Vegetable Research 5: e040 doi: 10.48130/vegres-0025-0033 

Ancestral chromosome karyotype construction and large-scale analysis of CAM biosynthesis genes in ice plant and representative plants
- Received: 04 May 2025
- Revised: 30 June 2025
- Accepted: 24 July 2025
- Published online: 03 November 2025
Abstract: Caryophyllales is one of the evolutionary branches of core eudicots, comprising numerous economically important species. However, comprehensive investigations into the genomic evolution of Caryophyllales remain scarce. Recently, we conducted de novo genome sequencing, yielding a high-quality genome assembly of ice plant, a typical representative of Caryophyllales. Here, we reconstructed the ancestral karyotype of Caryophyllales plants through a comparison of genomic data. Our analysis unveiled that the most recent common ancestor of Caryophyllales harbored a karyotype comprised of nine chromosomes. Subsequent analysis elucidated the evolutionary trajectory of ancestral chromosomes of Caryophyllales. Additionally, the ice plant characterized by its facultative adoption of Crassulacean acid metabolism (CAM) and C3 photosynthesis, epitomizes a halophytic species renowned for its salt tolerance resilience. In pursuit of understanding CAM biosynthesis mechanisms in ice plant, this investigation employed bioinformatic methodologies to identify 23 candidate genes. A comprehensive analysis was then performed on these genes. The findings showed that 23 identified genes within ice plant are categorized into six distinct gene families, namely CA, PEPC, MDH, PPDK, PEPCK, and ME. Based on the evolutionary tree and gene domain analysis, the identified genes were further subdivided into several distinct subfamilies. Furthermore, an extensive exploration ensued into the phylogenetic relationships among 11,148 CAM-related genes from an array of 287 species possessing well-annotated plant genomes, thereby encompassing major taxonomic groups within the plant kingdom. Consequently, this study furnishes a valuable reference for understanding both the evolutionary dynamics of chromosome karyotypes within Caryophyllales and the intricate network of CAM biosynthesis genes across diverse plant species.