










-
Tea polyphenols (TPs), mainly including catechins, anthocyanins, flavones, phenolic acids and their derivatives, are important components for quality and function of tea. Galloylglucoses (GGs), belonging to phenolic acid derivatives and the subclass of gallotannins, are composed of glucose and gallic acid moieties through so-called esterification or O-acylation, and can further be divided into mono-, di-, tri-, tetra-, penta-galloylglucose and so on, according to the number of gallic acid groups. It is well known that GGs, available in the brew, can extensively and intensively interact with proteins, thus, they will greatly impact on the mouthfeel[1], and have been considered as a natural defense against herbivores[2]. In recent decades, studies revealed that GGs have many biological activities in vitro and in vivo, including excellent anti-oxidation[3,4], anti-tumor[5], cardiovascular protective effect[6], human erythrocytes protective effect[7], radioprotection activity[8], anti-aggregation effects on Alzheimer's amyloid beta proteins[9], myocardium protective effect[10], anti-inflammatory activities[11], antibacterial properties[12], antidiabetic activity[13], alpha-amylase and alpha-glucosidase inhibitory activity[14]. Therefore, GGs can be potentially applied in the development of functional foods and pharmaceuticals in the future because of their proven health beneficial effects.
GGs, a kind of widely distributed secondary metabolites, had been identified qualitatively and quantitatively from many plant materials by occupying modern analysis technologies. Mämmelä et al.[15] analyzed the extractable tannins of North American white oak (Quercus alba) and European red oak (Quercus robur) by using high performance liquid chromatography-electrospray ionization mass spectrometry (HPLC-ESI/MS) technology, and partially determined the structures of monogalloyl glucose, digalloyl glucose and trigalloyl glucose of the two oaks. Vu et al.[16] employed HPLC-MS/MS technology and identified penta-O-galloyl-β-D-glucose and 1,3,6-trigalloylglucose from 11 different black walnut cultivars according to the ESI− fragments. Meyers et al.[17] employed HPLC-DAD-ESI/MS technology and identified 14 GGs (including mono-, di-, tri-, tetra- and penta- type) from acorns of the tanoak (Lithocarpus densiflorus) according to the ESI− fragments and UV absorbance of the target components. Wang et al.[18] purified 12 phenolic compounds from leaf extract of the Toona sinensis through column chromatography on Sephadex LH-20, MCI-gel CHP20P and Chromatorex ODS, and characterized 4 GGs, i.e., 6-O-galloyl-D-glucose, 1,2,3-tri-O-galloyl-β-D-glucopyranose, 1,2,3,6-tetra-O-galloyl-β-D-glucopyranose, 1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose, after comprehensive examination with UV, IR, MS and NMR spectroscopies. Later, characterization of 1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose separated from Elaeocarpus sylvestris[19] and T. sinensis[20] had been confirmed through similar study strategies. After preparation of gallotannins from Galla chinensis through sephadex LH20 chromatography and semi-preparative HPLC, Tian et al.[21] determined more than 30 GGs (3 mono-, 5 di-, 8 tri-, 7 tetra-, 5 penta-, 5 hexa-, 3 hepta-galloylglucose isomers) according to the LC/MS data. Nearly 20 GGs could be found in seeds of the Vitis rotundifolia (muscadine grapes), but only 2−3 were detected in the skin and pulp[22]. Fourteen GGs (1 di-, 5 tri-, 7 tetra-, and 1 penta-galloyl glucopyranose isomers) were identified from heartwood extracts of the Castanea sativa Mill, and most of them significantly decreased after toasting of the wood[23]. Engels et al.[24] applied the high-speed counter-current chromatography with hexane/ethyl acetate/methanol/water solvent system (0.5/5/1/5, v/v/v/v) to separate the gallotannins from mango (Mangifera indica L.) materials, characterized the compounds using HPLC-ESI-QTOF-MS in the negative ionization mode, and obtained tri-, tetra-, penta-, hexa-, hepta-, octa-, nona- and deca-O-galloylglucose and found the content of the GGs in peels was higher than that in seeds. Sixty eight phenolic compounds were identified from the leaf of the Phyllagathis rotundifolia by HPLC separation and MS/MS detection, of which seven were GGs, including 6-O-galloyl-D-glucose, 3,6-di-O-galloyl-D-glucose, 1,2,3-tri-O-galloyl-β-D-glucose, 1,4,6-tri-O-galloyl-β-D-glucose, 3,4,6-tri-O-galloyl-D-glucose, 1,2,3,6-tetra-O-galloyl-β-D-glucose, and 1,2,3,4,6-penta-O-galloyl-β-D-glucose[25]. GGs could also be tracked from seeds of the Oenothera paradoxa[26], peels of the jocote fruits[27], wood extracts of the Eucalyptus[28], fruits of the Phyllanthus emblica[29], exocarps of the Schinus terebinthifolius Raddi[30], seed kernels of the Juglans regia L.[31], fruits of the Euterpe edulis Mart., Psidium cattleianum Sabine, Eugenia pyriformis Cambess[32], bark of the Sclerocarya birrea[33], the extract of Trapa natans L. pericarps[34], the small leaves of Geranium sylvaticum[35], jaboticaba fruit peel[36], parasitic Sapria himalayana f. albovinosa and S. myanmarensis (Rafflesiaceae) in Myanmar[37], baru nuts[38].
Three GGs (mono-, 1,6-di-, and 1,2,6-tri-) were identified from pu-erh, green, and white teas by UPLC-DAD/MS tech, but they were not the major components of tea polyphenols because of their low concentration[39]. In the past several years, we found four new clones with a high concentration of galloylglucose from more than 430 tea plant germplasms during a large-scale chemical screening. In order to deeply understand the mechanism of the distinctive polyphenols accumulation pattern of these clones and comprehensively utilize them, it is necessary to figure out the physic-chemical characteristics of the galloylglucose. In this study, the abundant galloylglucose was purified and identified as 1,4,6-tri-O-galloyl-β-D-glucopyranose (1,4,6-TGG), and its antioxidant capacity was also investigated.
-
Among the four different developing solvents, the solvent system of acetonitrile/acetic acid/water/n-butanol (69.5/20/10/0.5, v/v/v/v) and n-butanol/water (3/1, v/v) could successfully separate the target compound at RF 0.55−0.70, as shown in Fig. 1. The n-butanol/water solvent was chosen as the developing solvent for preparing the target compound because of its relative simplicity and high separation efficiency. When the developed plate was treated with vanillin, only the part at RF > 0.70 could be stained, indicating the target compound (RF 0.55−0.70) does not belong to catechins and possesses relatively higher hydrophilicity than that of catechins. Results also showed that the target compound could be sufficiently recovered by 50% acetonitrile from gels instead of 50% methanol and 50% ethanol (Supplemental Fig. S1).
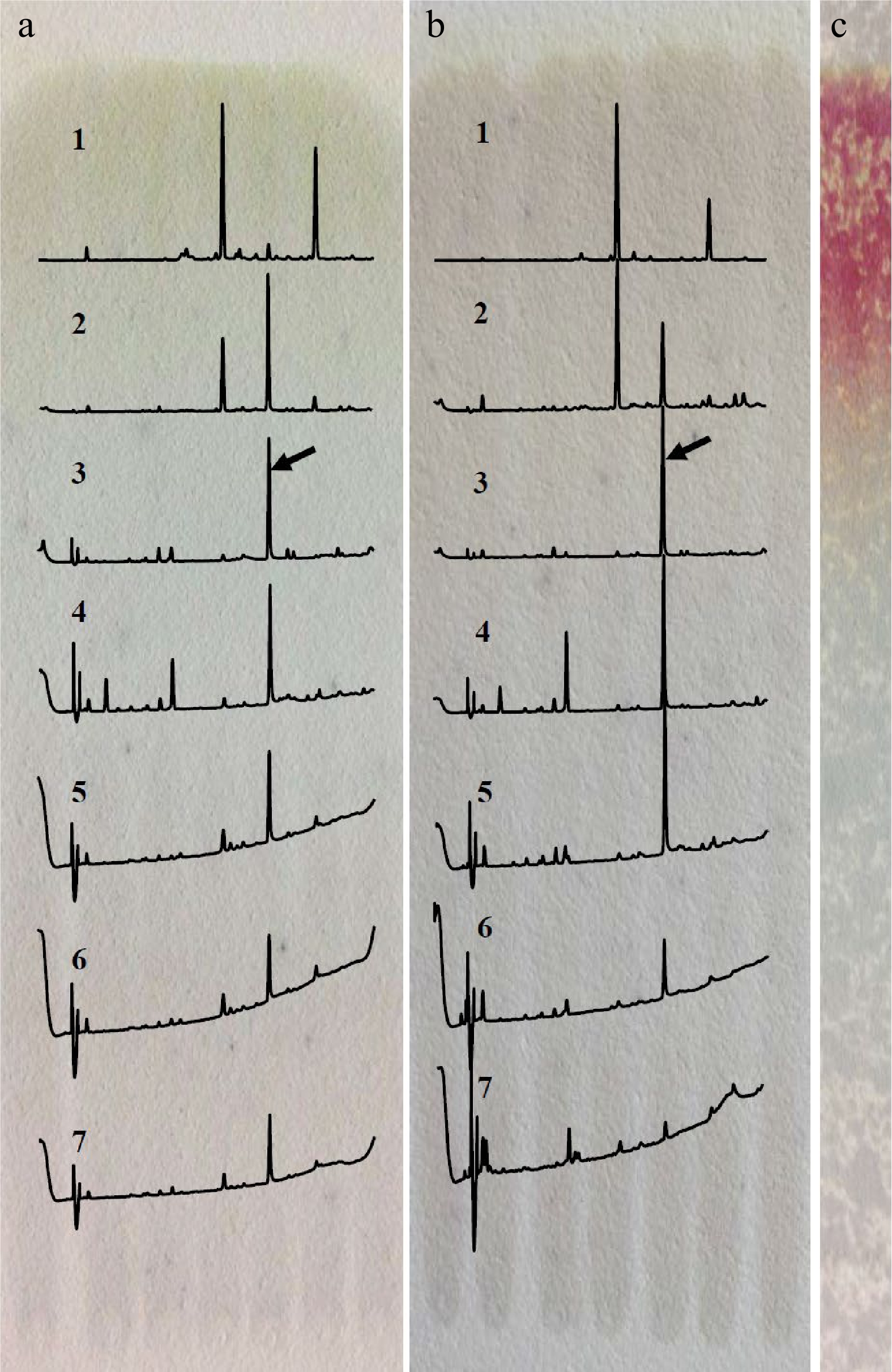
Figure 1.
Separation of the target compound by TLC developed with different solvents. (a) Acetonitrile/acetic acid/water/n-butanol (69.5/20/10/0.5, v/v/v/v). (b) n-butanol/water (3/1, v/v). (c) Staining with vanillin after development with n-butanol/water. 1. RF > 0.85, 2. RF 0.70−0.85, 3. RF 0.55−0.70, 4. RF 0.40−0.55, 5. RF 0.25−0.40, 6. RF 0.10−0.25, 7. RF < 0.10; the array showed the peak of target compound.
According to the analysis result of UPLC-DAD-MS/MS (Fig. 2), the separated target compound possessed a negative parent ion at m/z 635.10 ([M-H]−) with daughter ions at m/z 483.08 ([M-galloy-H]−), 465.08 ([M-GA-H]−), 313.02 ([M-GA-galloyl-H]−) and 168.98 ([GA-H]−) under ES− mode, indicating the molecular weight of the target compound was 636Da with more than one galloyl group; the result of UV diode array showed that the target compound had two absorbance peaks at 221 nm and 278 nm. After a comparison of MS[15,21,23,25,28,31,32,40−42] and UV data[23] from published papers, the target compound was considered as trigalloyglucose. A reference compound of 1,3,6-tri-O-galloyl-β-D-glucose (1,3,6-TGG) was purchased for comparative study. Confirmation tests showed that the target might be an isomer of 1,3,6-TGG because their MS and UV data were very similar (Fig. 2a & b), but could not be the 1,3,6-TGG since their retention times were quite distinctive in HPLC spectrum (Fig. 2c). Fresh shoot with two leaves and a bud plucked from the four special tea clones ('FZ1', 'FZ2', '1-17' and '1-18', all of them belong to Camellia sinensis var. sinensis) was ground and extracted with 75% ethanol (material/liquor = 1/20, w/v). The extract was monitored using ions pair m/z at 635/483, five peaks were detected and abundance of the target compound was the highest among them (Fig. 3), indicating at least four isomers of trigalloyglucose were synthesized besides the target compound in these tea clones. In order to characterize the molecular structure clearly, the target compound purified through TLC was subjected to NMR analysis, but an unsatisfactory result was obtained because the separated target was not pure enough.
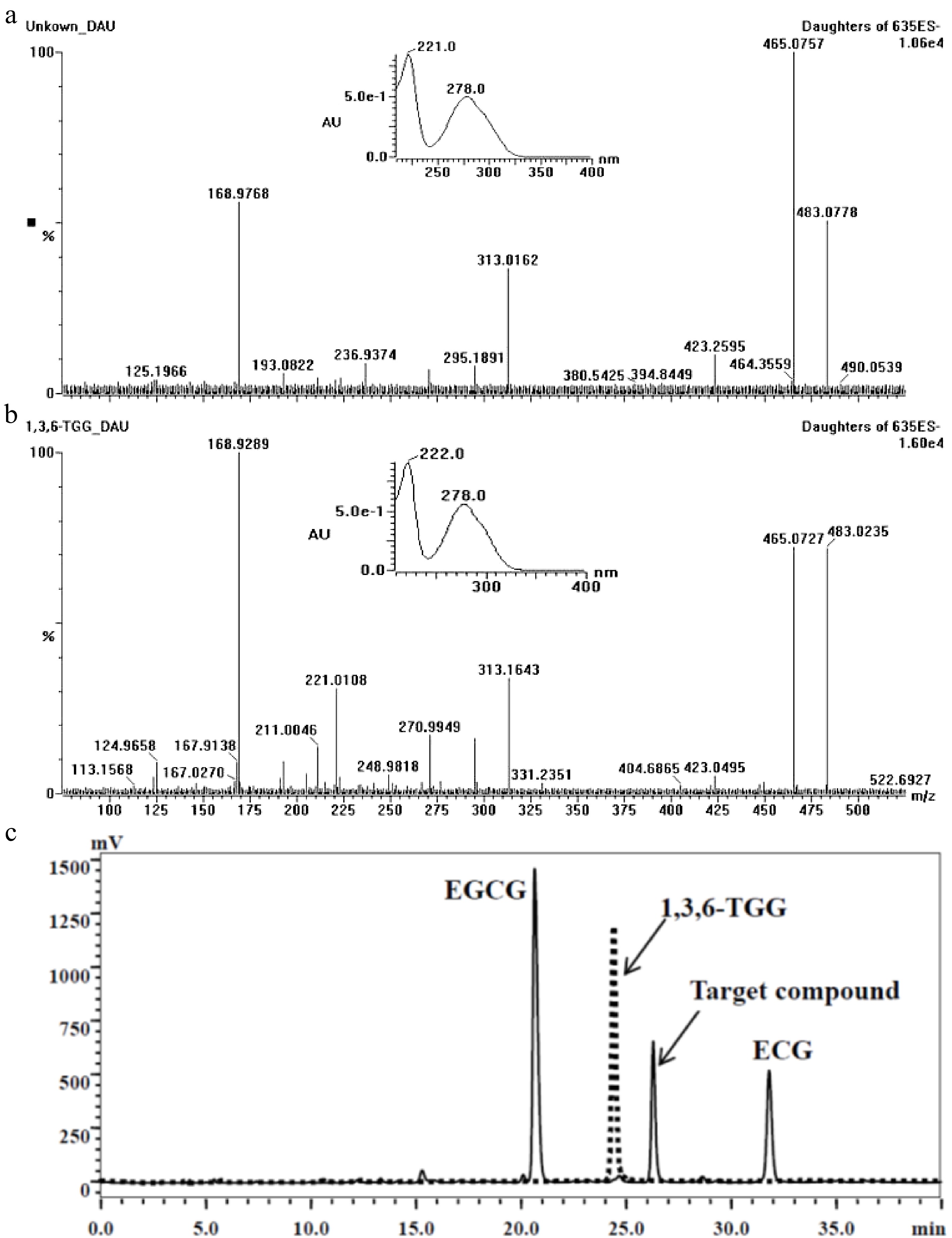
Figure 2.
Comparison of the target compound with 1,3,6-TGG. (a) Daughter ions and UV absorbance of the target compound (m/z 635). (b) Daughter ions and UV absorbance of the 1,3,6-TGG. (c) Different retention time between target compound (solid line) and 1,3,6-TGG (dash line) in HPLC spectrum.
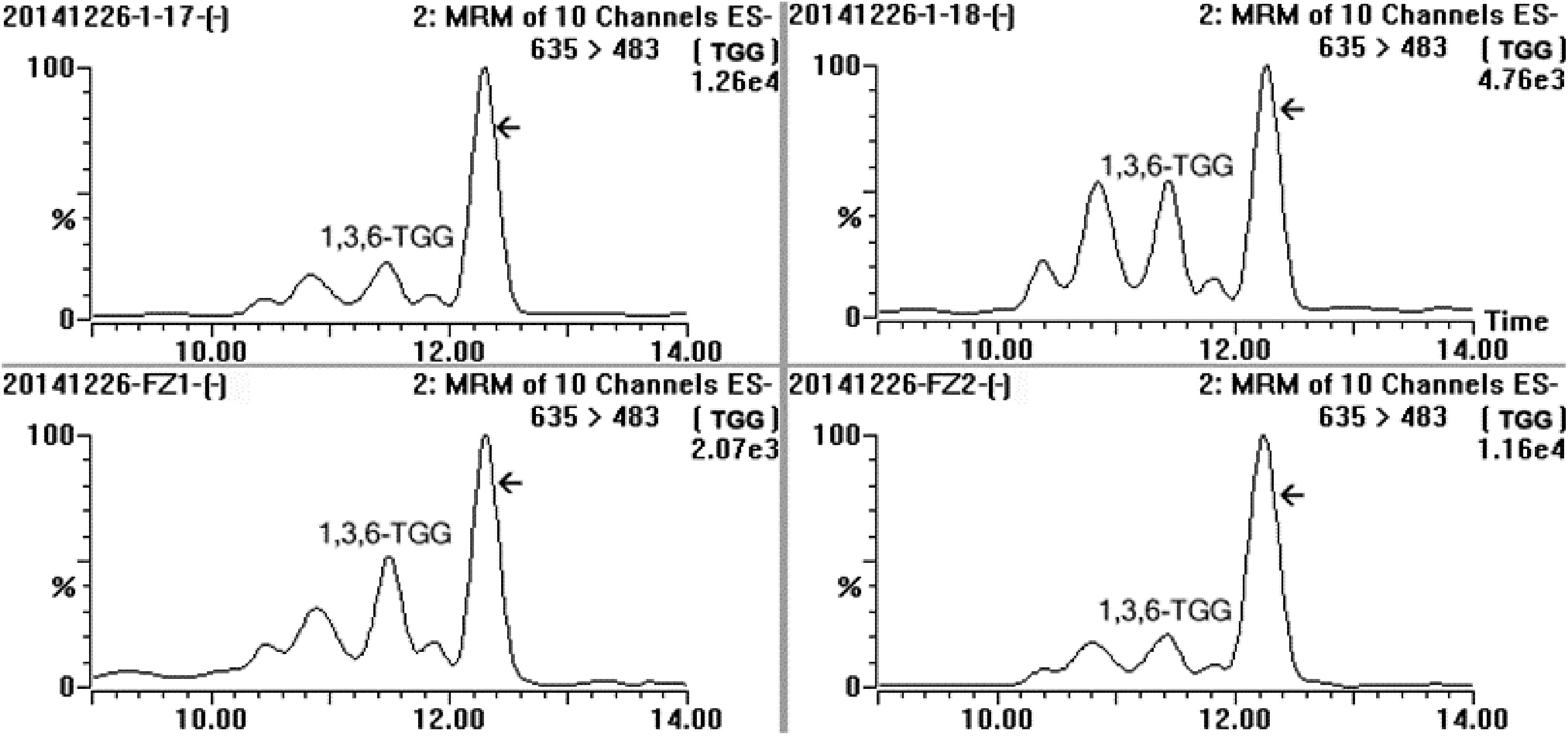
Figure 3.
MRM spectrum of the sample extracted from fresh leaves of four special tea plant clones (‘1-17’, ‘1-18’, ‘FZ1’ and ‘FZ2’) in channel 635/483, and MRM was performed at ES− mode, five distinct compounds were detected and the array indicated the target compound.
Purification and structure identification of the trigalloylglucose
-
Semi-preparative chromatography was applied for obtaining highly purified target compound to meet the requirement of NMR analysis. Many elution time programs were attempted to separate the target trigalloylglucose, the best program was carried out at a total flow of 2.75 ml, and could be described as the following: linearly increasing mobile phase B from 30% to 44% in the early 51 min and then to 100% in the next 1 min, and holding 100% B for another 7 min, finally in the next 3 min linearly decreasing B to 30%. Under this condition the target could successfully achieve baseline separation (Supplemental Fig. S2). UPLC-DAD-MS/MS analysis showed that only one peak in DAD with an ion at m/z 635([M-H]−) was obtained, confirming that the target trigalloyglucose was highly purified in the pooled eluates (Fig. 4).
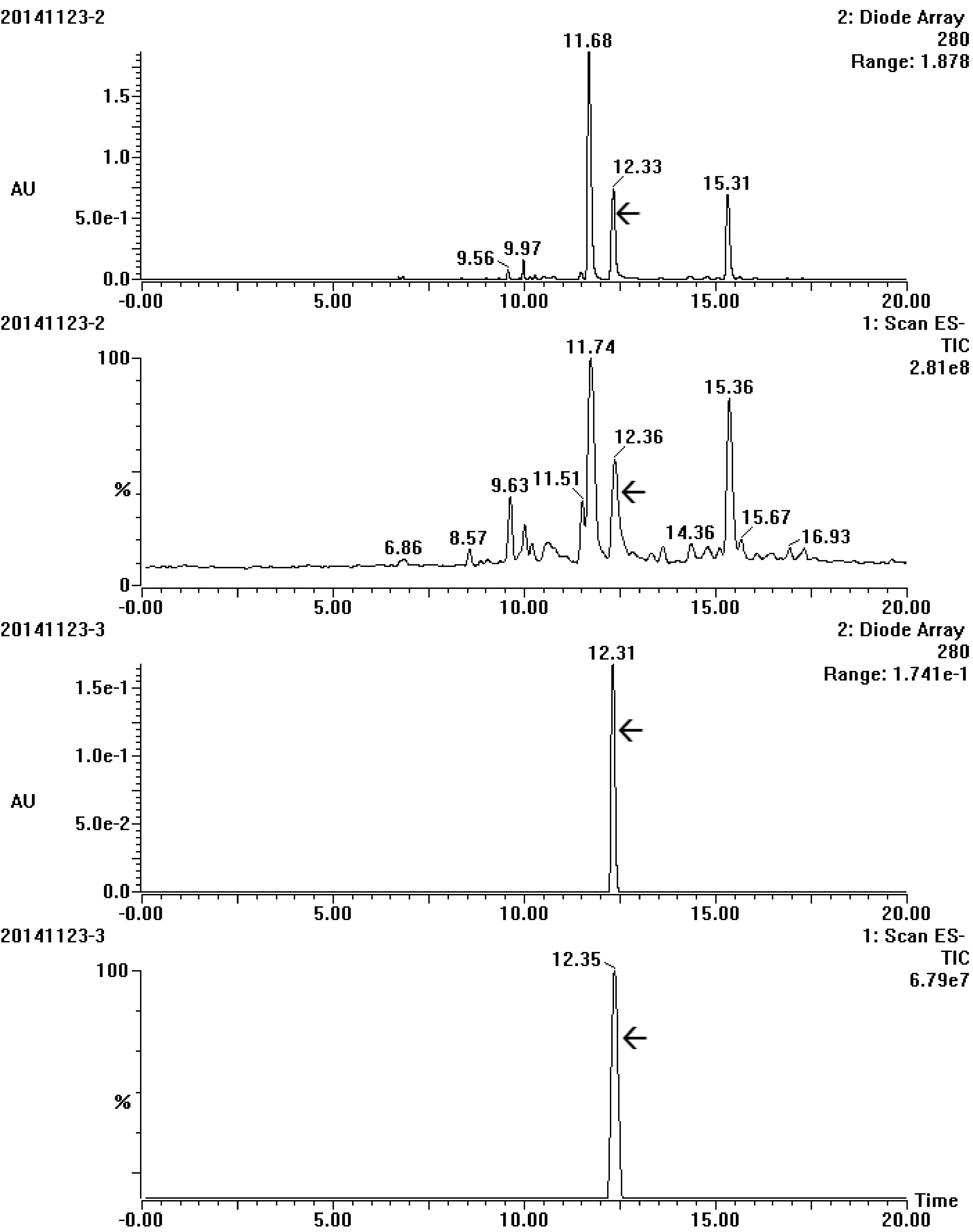
Figure 4.
UV and total ion chromatogram spectrum of the target compound before (20141123-2) and after (20141123-3) purification by semi-preparative chromatography. The array showed the target compound.
The target grayish-white powder was obtained after freeze-drying and dissolved in DMSO-D6. 1H-NMR analysis showed that typical and low-field shifted glucose proton signals appeared in the range δ3.44−5.73 and three strong characteristic absorption peaks of gallic acyl protons were highly visible in the low-field range δ7.04−6.94 (Fig. 5a), indicating that three protons on glucose of the target compound were replaced by gallic acyl[43]. Each signal peak specifically belonged to: 1H-NMR (500 MHz, DMSO) δ: 7.04 (s, 2H,C-3’’H, C-7’’H), 6.97 (s, 2H, C-3’H, C-7’H), 6.94 (s, 2H C-3’’’H, C-7’’’H), 5.73 (d, J = 8.2 Hz, 1H, C-1H), 5.03 (t, J = 9.7 Hz, 1H, C-4H), 4.22 (m, 1H, C-6H-A), 4.10 (dd, J = 12.4, 4.7 Hz, 1H, C-6H-B), 4.03 (ddd, J = 10.0, 4.6, 2.1 Hz, 1H, C-5H), 3.69 (t, J = 9.2 Hz, 1H, C-3H), 3.44 (t, J = 8.6 Hz, 1H, C-2H). In 13C-NMR spectrum, a set of glucose carbon signals (δ: 94.68, 74.25, 73.28, 72.82, 70.56 and 62.59), a group of replaced benzene carbon signals (δ: 146.14, 146.02, 145.99, 139.86, 139.27, 119.44, 118.64, 109.53, 109.37 and 109.21) and 3 carbonyl carbon signals (166.05, 165.42 and 165.00) were clearly present (Fig. 5b). Each signal peak specifically belonged to: 13C-NMR (126 MHz, DMSO) δ: 166.05 (C-1’’’), 165.42 (C-1’’), 165.00 (C-1’), 146.14 (C-4’, C-6’), 146.02 (C-4’’, C-6’’), 145.99 (C-4’’’, C-6’’’), 139.86 (C-5’, C-5’’), 139.27 (C-5’’), 119.44 (C-2’’), 118.64 (C-2’), 109.53 (C-3’’, C-7’’), 109.37 (C-3’’’,C-7’’’), 109.21 (C-3’,C-7’), 94.68 (C-1), 74.25 (C-3), 73.28 (C-2), 72.82 (C-5), 70.56 (C-4), 62.59 (C-6). 2D NMR spectra confirmed that the proton signal δ94.68 (C-1) was linked to the signal δ5.73 (C-1H) in HMQC spectrums (Fig. 5c). According to H1-H1 COSY, the rest of the H protons i.e., C-1H, C-4H, C-6H-A and C-6H-B, shifted to the high field (Fig. 5d), indicating that the H proton of the hydroxyl on C-1, C-4 and C-6 was replaced by the gallic acyl, respectively. After comparing the obtained 1H-NMR, 13C-NMR, H1-H1 COSY and HMQC spectrums with the published papers [5,31,43,44] as well as the predicted data through MestReNova (Mestrelab Research, S.L., Santiago de Compostela, Spain), the target compound was identified as 1,4,6-tri-O-galloyl-β-D-glucopyranose (1,4,6-TGG) with the chemical formula of C27H24O18, as shown in Fig. 5e, and its exact molar mass was 636.0966 g/mol according to the high precision MS detection (Fig. 5f). After quantification by using the purified 1,4,6-TGG as the reference, the concentration of this compound in the shoots of ‘FZ1’, ‘FZ2’, ‘1-17’ and ‘1-18’ ranged from 43.62−56.60 mg/g.
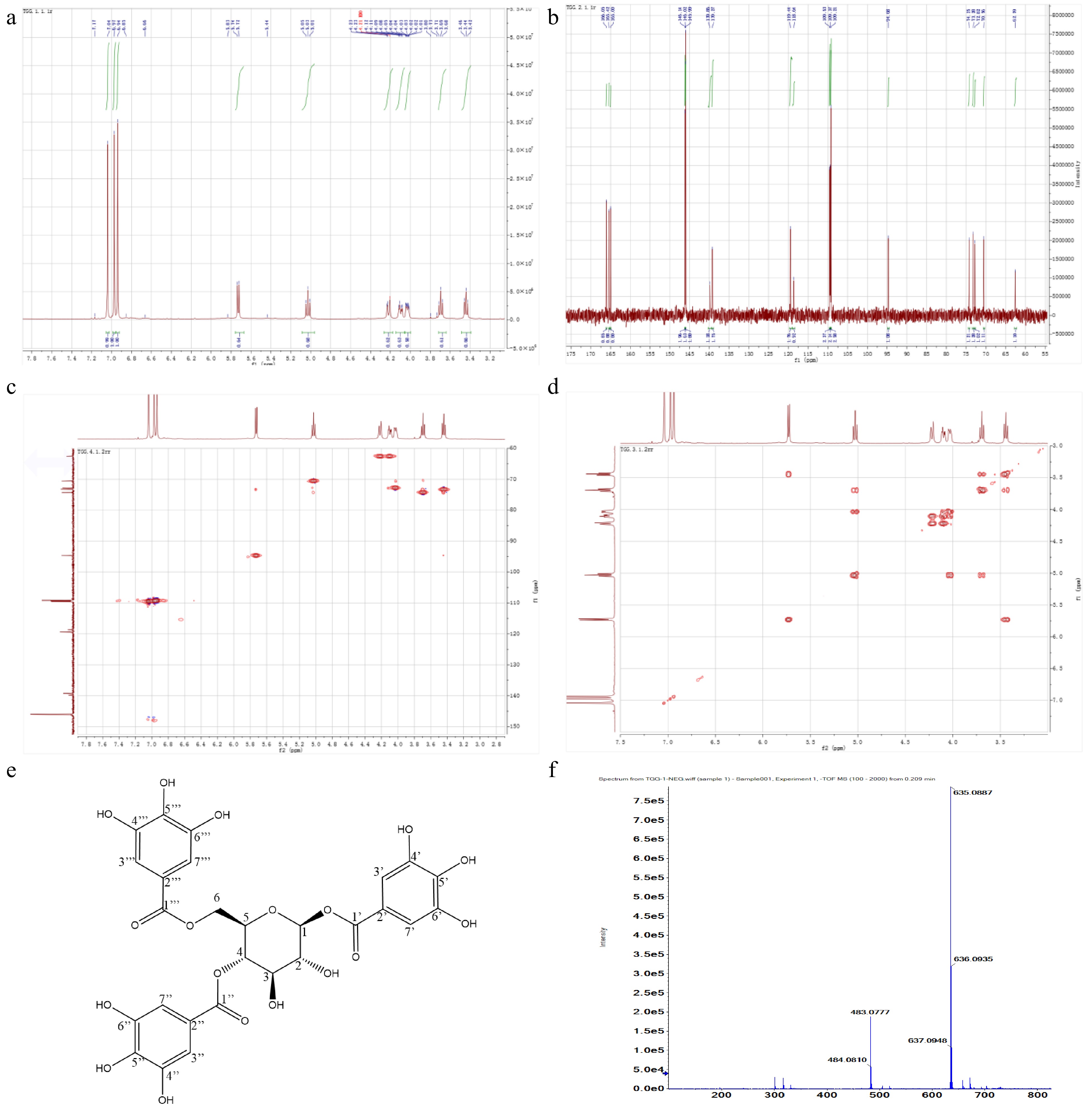
Figure 5.
NMR spectrums and characteristics of target compound. (a) 1H-NMR. (b) 13C-NMR. (c) HMQC. (d) H1-H1 COSY. (e) Chemical structure. (f) Exact molecular weight.
Antioxidant capacity of the 1,4,6-TGG
-
The ORAC, DPPH and FRAP were applied for evaluating the antioxidant capacity of the purified 1,4,6-TGG, the result showed that the capacity of 1,4,6-TGG was around 2.68−5.77 μmol TEAC/μmol (4.21−9.07 μmol TEAC/mg), and significantly higher (42.7%−419.4%) than that of vitamin C but slightly lower (5.5%−11.6%) than that of EGCG (Table 1). EGCG had been considered as one of the most efficient antioxidants in tea[45]. These data showed that 1,4,6-TGG also possessed very strong antioxidant potential close to EGCG. This finding was also consistent with many published reports in which some other TGG isomers were also proved to possess high antioxidant capacity[18]. Similar to other phenolic compounds, effectively quenching free radicals and sufficiently providing reducing power by the 1,4,6-TGG might also originate from its multiple free aromatic hydroxyls on the three galloyl groups.
Table 1. Antioxidant capacity of the 1,4,6-TGG.
Compound ORAC DPPH FRAP 1,4,6-TGG 2.68 ± 0.03b 5.67 ± 0.09b 5.77 ± 0.08b Vitamin C 1.23 ± 0.06c 1.20 ± 0.03c 4.04 ± 0.09c EGCG 2.83 ± 0.02a 6.36 ± 0.12a 6.40 ± 0.13a Antioxidant capacity was expressed as μmol trolox equivalent antioxidant capacity (TEAC) per μmol compound. Different letters in same column showed significant difference (p < 0.05). -
An extraordinary phenolic compound was found during chemical assessment of new tea plant clones. This compound was purified through TLC and semi-preparative chromatography, and identified as a novel trigalloylglucose 1,4,6-TGG according to the analysis of UPLC-DAD-MS/MS and NMR. The level of the 1,4,6-TGG in the shoots of some high abundance tea clones exceeded 40 mg/g. It could be dissolved in the infusion during brewing, and its antioxidant capacity was slightly lower than EGCG, but significantly higher than VC, indicating the 1,4,6-TGG could be applied in the development of functional foods and pharmaceuticals in the future.
-
Tender shoots with two leaves and a bud were plucked from the special clones, i.e., 'FZ1', 'FZ2', '1-17' and '1-18', with high concentration of galloyglucose on the 2nd April. These four clones belong to C. sinensis var. sinensis and were cultivated in experimental tea garden of Zhejiang University (Hanghou, PR China). The collected shoots were divided into two parts, and one was weighted and packed with aluminum foil and stored at −80 °C; another was steamed at 100 °C for 60 s to denature the enzymes, then completely dried at 80 °C in a dryer (Shanghai Fuma Laboratory Instrument Co., Ltd., Shanghai, PR China). The dried shoots were ground in a grinder (Wuyi Yili Tools Co., Ltd., Zhejiang, PR China) and passed through 30 mesh sieve, and stored in a fridge at 4 °C. Dimethyl sulfoxide-D6 was obtained from Sigma products (Sigma-Aldrich, St. Louis, MO, USA). 1,3,6-trigalloy-β-D-glucose (purity > 90%) was ordered from Shanghai Tauto Biotech Co., Ltd (Shanghai, PR China). HPLC grade acetonitrile and methanol were supplied by Merck (Darmstadt, Germany). Ethanol, chloroform and ethyl acetate were of analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd. (Beijing, PR China). Chromatography silica gel (200−300 mesh) was purchased from Anhui Liangchen Silicon Source Co., Ltd. (Anhui, PR China). Ultrapure water used was prepared by EASY Pure II Water System (Barnstead International, Dubuque, IA, USA). Other reagents used were of analytical grade except where stated otherwise.
Extraction of tea samples
-
Ground dried tea samples (50 g) were extracted with 2.5 L 50% ethanol (v/v) at room temperature for 45 min. After filtration, the filtrate was partitioned twice with chloroform (1.0 L for each) for removal of caffeine and the up layer was collected. Ethanol in the collected solution was removed at 50 °C under vacuum conditions in an R-501 Rotary Evaporator (Shanghai Shenshun Biotech Co. Ltd., Shanghai, PR China), the residuum was partitioned twice with ethyl acetate (0.5 L for each), the collected ethyl acetate phases were pooled and subjected to rotary evaporation at 40−45 °C for recovery of the ethyl acetate, viscous residuum was dissolved into a small amount of water and dried in Christ Alpha 1-4 LD plus Freeze Dryer (Marin Christ Co., Osterode, German), and 13.2 g crude extract was obtained.
In order to determine whether high-temperature treatment such as fixation would impact on the target compound, the stored fresh shoots were ground with liquid nitrogen, then extracted with 75% aqueous ethanol at the ratio of 1/20 (the material/the liquid, w/v) and 4 °C for 30 min. The supernatant was obtained through centrifugation at 12,000 r/min and 4 °C for 10 min.
Separation of the target compound by TLC
-
Five hundred milligrams of crude extract was dissolved in 100 mL 50% ethanol, supernatant was obtained after centrifugation at 8,000 r/min and 4 °C for 15 min. The sample was loaded on the silica gel plate which was prepared through mixing gel with water at ratio of 1/2 (w/v), drying at room temperature after extension on the clean glass plate (10 × 20 cm) and activating at 100 °C for 1 h. Development was performed at room temperature in a parafilm sealed beaker in which different developing agents including n-propanol/ water/ acetic acid (20/80/1, v/v/v), n-butanol/ acetic acid/ water agent (3/2/1, v/v/v), acetonitrile/acetic acid/water/n-butanol (69.5/20/10/0.5, v/v/v/v) and n-butanol/water (3/1, v/v) were contained respectively. After development, the plate was dried at room temperature, some of them were used for staining with vanillin, and the others were used for recovery of the compounds. Silica gel was scraped down according to the different migration rates and transferred into different eluent (50% ethanol, 50% methanol and 50% acetonitrile) for elution (at room temperature for 1 h). The supernatant was collected after centrifugation of the eluate at 8,000 r/min and 4 °C for 10 min, and used for HPLC examination. The eluate in which the peak area of the target compound was higher than 95%, was pooled together for freeze-drying.
Purification of the target compound by semi-preparative chromatography
-
Purification of the target compound was also performed on an ÄKTA purifier UPC100 semi-preparative chromatography (GE Healthcare Bio-Sciences, Marlborough, MA, USA) coupled with a YMC-Pack ODS-A C18 column (250 × 10 mm i.d., 5 μm, YMC Co., Ltd., Japan). Gradient elution was carried out at room temperature in which mobile phase A was acetonitrile/acetic acid/water = 3/0.5/96.5 (v/v/v) and mobile phase B was acetonitrile/acetic acid/water = 30/0.5/69.5, (v/v/v). Elution time program was optimized to separate the target compound from the crude extract. Sample (5 mg/mL) was loaded onto the column through a 500-μL loop valve. Flow rate was 2.5−3.0 mL/min, the eluate containing the highly purified target compound was collected according to the UV detection at 280 nm. After evaporating the organic solvent from the eluate through flow of N2, the remaining solution was freeze-dried for obtaining the target compound. After dehydration in a desiccator, the dried powders were sealed and stored at 4 °C.
HPLC analysis
-
A model LC 20AT HPLC (Shimadzu Co., Kyoto, Japan) coupled with Zorbax 5 μm TC-C18(2) column (250 × 4.6 mm, Agilent Technologies Inc., CA, USA) was used to estimate components in the tested solutions. The HPLC analysis conditions were as follows: injection volume, 10 µL; oven temperature, 32 °C; mobile phase A, acetonitrile/acetic acid/water (3/0.5/96.5, v/v/v); mobile phase B, acetonitrile/acetic acid/water (30/0.5/69.5, v/v/v); gradient elution, 0.01min-8% mobile phase B → 18min-28% B → 18.01min-43% B → 35min-65% B → 40 min-65% B; flow rate, 1 ml/min; detecting wavelength, 280 nm. Detailed operation was carried out as described in a previous study[46].
Analysis of UPLC-DAD-MS/MS and high precision MS
-
A Model Quattro Premier XE UPLC-DAD-MS/MS (Waters Corporation, Milford, MA, USA) coupled with HSS T3 1.8 μm (150 mm × 2.1 mm) column (Waters Corporation, Milford, MA, USA) was used for analysis of the target compound in the collected solution during TLC and semi-preparative chromatography according to the modified method reported by Sun et al.[47]. Gradient elution was carried out in which the mobile phase A was a solution of 0.1% formic acid in water, and B was a solution of 0.1% formic acid in acetonitrile; the time program of elution was carried out as described in Supplemental Table S1. The column temperature was set at 35 °C and the flow rate was 0.3 ml/min. UV detection was performed at 200−400 nm by a diode array detector (DAD). A mass range of 60−1,000 m/z was scanned after electrospray ionization (ESI) in both negative and positive (ES−, ES+) mode. Parameters of mass spectrometry were set as follows: capillary voltage, 3.0 kV; extractor voltage, 3.0 V; RF lens, 0.1 V; ion source temperature, 150 °C for ES− and 120 °C for ES+; desolvation temperature, 300 °C; desolvation gas flow, 600 L/h; cone voltage, 25 V; cone gas flow, 50 L/h; dwell time, 0.05 s; collision energy, 20 V. The high precision mass spectrometer, Ultrafletreme (Bruker Co., Wissembourg, France), was used to detect the exact molecular weight of the target compound. The detection conditions were set as follows: capillary voltage, 3.0 kV; ion source temperature, 150 °C; desolvation temperature, 300 °C; desolvation gas flow, 600 L/h; cone voltage, 25 V; collision energy, 20 V.
NMR identification
-
The purified target compound (18 mg) was dissolved in 0.6 mL DMSO-D6. 1H-NMR, 13C-NMR, 1H-1H COSY and HMQC spectrums were recorded on an Avance III HD NMR Spectrometer (Bruker Co., Wissembourg, France) at 500 MHz with a 5 mm DCH cryoprobe. Integration of the spectra was performed with software of MestReNova v14.0.0-23239 (Mestrelab Research, S.L., Santiago de Compostela, Spain). Identification was carried out according to the references and relevant literature as well as the predicted spectrum from MestReNova software.
Antioxidation capacity of the purified target compound
-
ORAC assay of the target compound was carried out according to Huang et al.[48] and OxiSelect™ kit protocol (Cell Biolabs, Inc., San Diego, CA, USA). Twenty microlitres series of Trolox standard working solution (0−100 μM) and target compound solution (39.7 mg/L) as well as control compound (vitamin C, VC; epigallocatechin gallate, EGCG) solution (21.2 mg/L VC and 27.5 mg/L EGCG) were transferred into 96-well plate respectively, then 20 μL phosphate buffer (pH 7.4), 20 μL fluorescein probe (200 nM) and 140 μL 2,2'-Azobis(2-amidinopropane) dihydrochloride (633.57 nM) was added in the correct order. After mixing completely, the fluorescence value was recorded in a Synergy H1 multi-mode reader ( BioTek Instruments, Inc., Winooski, VT, USA ) at 37 ºC with an excitation wavelength of 485 nm and an emission wavelength of 527 nm every 2 min for 120 min. The collected data were transferred into the sheet of software Origin Pro v8.5.1 (OriginLab Corporation, Northampton, MA, USA). According to kinetic curve area of the Trolox standard, antioxidant capability of the target compound and control (VC and EGCG) was calculated and expressed as μmol Trolox equivalent antioxidant capacity (TEAC)/μmol. The experiment was repeated three times.
DPPH radical scavenging assay was conducted according to the method of Brand-Williams et al.[49] and kit manual (Shanghai Beyotime Biotechnology Co. Ltd., China). Twenty five microlitres series of Trolox (0−480 μM), target compound (39.7 mg/L), VC (21.2 mg/L) and EGCG (27.5 mg/L) were transferred into 5-mL glass tubes respectively, and then 2 mL DPPH (25 mg/L) methanol solution was added into each tube. Reaction was carried out at 25 °C for 20 min after thoroughly mixing. The absorbance was recorded at 517 nm by a UV-2892S spectrophotometer (Unico (Shanghai) Instrument Co., Ltd., Shanghai, PR China). Antioxidant capability of the target compound, VC and EGCG was calculated according to the standard curve and expressed as μmol Trolox equivalents antioxidant capacity (TEAC)/μmol.
FRAP assay was carried out according to the total antioxidant capacity assay kit manual (Shanghai Beyotime Biotechnology Co. Ltd., PR China). FRAP working solution (180 μL) were transferred into different holes of 96-well microplate respectively, 5 μL distilled water (for blank), FeSO4 (0.15−1.50 mM, for standard curve), Trolox (0.15−1.50 mM, for positive control) and sample solution (target compound, VC and EGCG) were appended, respectively. After mixing gently and incubation at 37 °C for 5 min, the absorbance was recorded at 595 nm in an iMark Microplate Reader (Bio-Rad Co., USA). According to the standard curve, the antioxidant capability was calculated and expressed as μmol Trolox equivalent antioxidant capacity (TEAC)/μmol.
This work was financially supported by the Zhejiang Science and Technology Major Program on Agricultural New Variety Breeding-Tea Plant (No. 2021C02067-6), the National Natural Science Foundation of China (No. 31770728) and China Agriculture Research System of MOF and MARA.
-
The authors declare that they have no conflict of interest.
- Supplemental Fig. S1 Recovery of the target compound from silica gel by 50% ethanol (E1), 50% methanol (E2) and 50% acetonitrile (E3).
- Supplemental Fig. S2 Separation of the target compound through semi-preparative chromatography with the optimized elution time program and the arrow showed the peak of the target compound.
- Supplemental Table S1 Elution time program for UPLC/MS/MS.
- Copyright: © 2022 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yang Y, Zhao M, Liu Y, Fang Z, Li Q, et al. 2022. Separation and identification of an abundant trigalloylglucose from special tea genetic resources. Beverage Plant Research 2:11 doi: 10.48130/BPR-2022-0011 

Separation and identification of an abundant trigalloylglucose from special tea genetic resources
- Received: 13 June 2022
- Accepted: 01 July 2022
- Published online: 15 July 2022
Abstract: Galloylglucoses can extensively and intensively interact with proteins and possess many biological activities and exhibit optimal health benefits. A novel galloylglucose had been observed and separated from fresh shoots of some special tea genetic resources, and was characterized as a trigalloylglucose isomer with mass parent ion [M-H]− at m/z 635.10 and daughter ions at m/z 483.08, 465.08, 313.02 and 168.98 and with UV absorbance peaks at 221 nm and 278 nm after being measured with UPLC-DAD-MS/MS. The trigalloylglucose was highly purified by semi-preparative chromatography and identified as 1,4,6-tri-O-galloyl-β-glucopyranose (1,4,6-TGG) by 1H-NMR, 13C-NMR, H1-H1 COSY and HMQC. Antioxidant capacity of the 1,4,6-TGG was 2.68−5.77 μmol TEAC/μmol, and significantly higher (42.7%−419.4%) than that of ascorbic acid, but similar to another well-known powerful antioxidant epigallocatechin gallate in tea, indicating that 1,4,6-TGG could, in the future, be applied in the development of functional foods and pharmaceuticals.