










-
Endophytic bacteria are defined as a group of bacteria that are present in plants without causing any detrimental impact to the plants[1]. Since the discovery of endophytic bacteria in 1926, there are many studies focussing on its potential as plant growth-promoting bacteria, biocontrol bacteria, microbial pesticides and so on[2]. The peanut endophytic bacterium LDO2 exhibited growth-promoting ability and promote peanut root growth[3]. Five endophytic bacteria showed antagonism against Pseudomonas syringae pv. actinidiae (Psa), a bacterial canker in kiwifruit in vitro[4]. Bacteria with plant growth-promoting ability are important for green development of crops. Recently, the growth-promoting mechanisms of plant growth-promoting endophytic bacteria has gained considerable attention. There two direct and indirect main growth-promoting mechanisms, endophytic bacteria themselves provide soluble phosphorus, fix nitrogen, produce phytohormones IAA, and ACC deaminase for promoting plant growth directly and endophytic bacteria promote plant growth indirectly through inducing systemic resistance (ISR) and producing siderophore and antibiotics[5]. Based on the functional properties of endophytic bacteria for crop growth, it is important to apply them in sustainable agricultural development.
There are many studies on the genome analysis of endophytic bacteria for understanding the mechanisms of interaction between endophytic bacteria and crops. Enterobacter sp. 638 genome sequence showed the mechanism of plant growth promotion, interaction with host plant and endophytic colonization[6]. Genomic analysis of Gluconacetobacter diazotrophicus Pal5 revealed genes related with plant growth promotion, transport systems and so on[7]. The plant growth–promoting bacterium Bacillus amyloliquefaciens FZB42 genome revealed potential secondary metabolites production capacity and more than 8.5% of the genome was related to plant growth promotion[8]. Therefore, analysis of the endophytic bacterial genome will contribute to a comprehensive characterization of endophytic bacteria and explore interactions with their hosts; it will also help to screen endophytic bacteria suitable for different crops and growing environments.
Tea is one of the most popular non-alcoholic beverages in the world. China is the world's largest tea producer. The health and quality of tea has greatly challenged tea export in China. The traditional use of chemical fertilizers and pesticides causes gradual acidification of the soil, pesticide residues in tea and inevitably other quality problems. Therefore, biological pesticides and fertilizers instead of chemicals will help to improve the quality of tea and reduce pesticide residues and heavy metal pollution in tea cultivation.
We isolated one endophytic bacterium from albino tea leaf, which was named as V4. In a previous study, the V4 strain has been shown to have plant growth promoting ability through plate identification and pot inoculation experiments[9]. Inoculation of tea seedlings with V4 endophytic bacteria helps new shoot germination and growth under greenhouse conditions. However, the mechanisms of plant growth promotion and the interaction with the host plant have not been clarified. In the present study, the physiological and biochemical evaluation, as well as the appearance of V4 were analyzed. The aims of V4 endophytic bacterium genome analysis were to deepen our knowledge of its phylogenetic classification, the plant growth promotion ability, colonization capacity, and potential ability on interaction with host plants. Also, the genome of V4 bacterium is a valuable source to study the value of agricultural applications as a biofertilizer of tea plants and other economically important crops.
-
The endophytic bacterium V4 was isolated from albino tea plant (Camellia sinensis) leaf. Standard gram-staining was performed to evaluate physical properties of bacterial cell wall of V4. Biochemical characteristics were tested using HBI Rapid ID Panel for Enterobacteriaceae Bacteria according to the manufacturer's instructions, also, 100 μg/mL IAA standard were used as positive control for indole tests using this biochemical assay. Morphological observations were examined using scanning electron microscopy (Hitachi S-4800, Tokyo, Japan) and transmission electron microscope (Hitachi HT-7700, Tokyo, Japan). For SEM (scanning electron microscopy) observation, 10 h of V4 bacterium grown in LB solid medium were harvested as a suspension by using sterile water. After standing for 30 min, the 2 μL suspension were added in silicon wafer and stood for 1 h. Then, the silicon wafer was fixed with 5% glutaraldehyde for 8 h, dehydrated by using an ethanol series (10%, 30%, 50%, 70%, 90%, 100%) for 10 min per gradient step. Finally, the silicon wafer was soaked in acetone for 10 min and dried in the natural environment basing on the methods available, with some modifications[10]. The prepared V4 bacterium was observed by scanning electron microscopy operated at an accelerating voltage of 3.0 KV for SEM (scanning electron microscopy) observation, 10 h of V4 bacterium grown in LB solid medium were harvested as a suspension by using sterile water. After standing for 5 min, a part of the suspension was transferred to a new centrifuge tube for morphological observation. Then, 20 μL suspension was adsorbed by copper screening for 5 min, followed by dyeing though 20 μL, 10 g/L Salkowski's solution for 50 s, finally dried in the natural environment. The prepared V4 bacterium was observed at 80.0 KV with a transmission electron microscope[11].
Genomic DNA extraction, sequencing, assembly, and annotation
-
The V4 bacterium was grown in 50 mL LB liquid medium at 28 °C and 200 rpm for 7.5 h. Bacterial cells were harvested by centrifugation at 2,627× g and 4 °C for 2 min and the genomic DNA was performed using HiPure Bacterial DNA Kits (Magen, Guangzhou, China) according to the manufacturer's instructions. The DNA libraries were sequenced on the PacBio sequencing platform by Genedenovo Biotechnology Co., Ltd (Guangzhou, China). Specifically, qualified genomic DNA was fragmented with G-tubes (Covaris, Woburn, MA, USA) and end-repaired to prepare SMRTbell DNA template libraries (with fragment size of > 10 Kb) according to the manufacturer's specification (PacBio, Menlo Park, CA, USA).
Also, the qualified genomic DNA was sonicated randomly and then end-repaired, a-tailed, and adaptor ligated using NEBNext® ΜLtra™ DNA Library Prep Kit for Illumina (NEB, USA). Genome sequencing was performed on the Illumina Novaseq 6000 sequencer using the pair-end technology (PE 150). Continuous long reads were attained from SMRT sequencing runs and were used for de novo assembly using Falcon (version 0.3.0). The specific parameters were as follows: length_cutoff = 10000, length_cutoff_pr = 4000, pa_HPCdaligner_option = -v -B4 -t16 -e.70 -l1000 -s1000, ovlp_HPCdaligner_option = -v -B4 -t32 -h60 -e.96 -l500 -s1000, falcon_sense_option = --output_multi --min_idt 0.75 --min_cov 6 --max_n_read 200 --n_core 6[12].
Raw data from Illumina platform were acquired and filtered to obtain clean reads. Then clean reads were used to correct the genome sequences using Pilon (version 1.23). Finally, the genome sequences were annotated by aligning with the deposited ones in diverse protein databases including National Center for Biotechnology Information (NCBI) non-redundant protein sequence database using diamond V2.0.9 (diamond blastp -k20 -f 6), UniProt/Swiss-Prot using diamond V2.0.9 (diamond blastp -k20 -f 6), Kyoto Encyclopedia of Genes and Genomes (KEGG) using diamond V2.0.9 (diamond blastp -k20 -f 6 -e 1e-5 --salltitles), Gene Ontology using diamond V2.0.9 (diamond blastp –sensitive -e 1e-5 --max-hsps 1 --min-score 50 --query-cover 20 --max-target-seqs 1 --outfmt 6), hmmsearch v3.3.2 (hmmsearch -E 0.1 --domE 0.01 --incE 0.01 --incdomE 0.01), eggNOG-mapper v2.1.4 (emapper.py --no_file_comments --go_evidence all --tax_scope Bacteria --tax_scope_mode broadest --target_orthologs one2one), Cluster of Orthologous Groups of proteins using diamond V2.0.9 (diamond blastp -k20 -f 6 -e 1e-5 --salltitles)[13]. Protein families annotation was applied with Pfam_Scan (version 1.6) basing on Pfam database (version 32.0)[14]. Gene clusters of secondary metabolites and Two-component systems (TCSs) were predicted using antiSMASH (version 4.1.0) based on their structural characteristics respectively[15,16].
Taxonomic characterization and phylogenetic analyses
-
To investigate the taxonomical classification of V4, DNA was extracted by using bacterial gDNA kit (Biomiga), and was used as the template for amplifying 16S rRNA gene following the method of Jia et al.[9]. The 16S rRNA gene sequence was first identified though BLAST analysis in the National Center for Biotechnology Information (NCBI) database.
For the phylogenetic analysis, two bioinformatic approaches were performed: the sequences of 16S rRNA gene of V4 were compared with the corresponding sequences of 34 Erwinia genus strains and one Herbaspirillum seropedicae Z67T strain 16S rRNA genes with over 1200 bp sequence length acquired from the NCBI database. The nucleotide sequences were aligned using Mafft, and the maximum likelihood phylogenetic tree were constructed using FastTree Version 2.1.10 double precision (No SSE3) tool, with Jukes-Cantor model[17,18]. The V4 whole-genome phylogenetic analysis was performed using protein sequences. It was conducted with 28 genomic protein sequences of Erwinia genus representative strains and an outgroup H. seropedicae Z67T acquired from the NCBI database. The phylogenetic tree was constructed though OrthoFinder in the default mode[19].
Comparative genomic analysis
-
Five complete bacteria genomes were used to compare with the V4 genome according to the results of V4 taxonomical classification. E. aphidicola 18B1 was the closest to the V4 phylogenetic and had not been shown to have the ability to promote plant growth or plant pathogenicity[20]. E. rhapontici BY21311 and E. persicina B64 were phytopathogenic bacteria showing celery stem rot and onion rot diseases[21,22]. E. tasmaniensis Et1/99 and H. seropedicae Z67 were plant growth promoting bacteria[23,24]. The five complete genomes were retrieved from NCBI and analyzed together with the V4 genome for gene family analysis by using the BBH (bidirectional best-hit) criterion (80% of the length of the shortest protein sequence has 40% amino acid similarity). Specifically, the amino acid sequences of all bacteria involved in the analysis were compared using diamond[25], similarity clustering was performed using OrthoMCL[26], a list of homologous genes clustered as clusters was obtained, and the species distribution of each protein cluster was counted.
Genbank accession number
-
The V4 genomic sequence reported in this article has been deposited in the NCBI database, the accession number is PRJNA855316. Furthermore, the V4 bacterium culture was preserved in China center for type culture collection (CCTCC), with the accession number M 2021027.
-
The V4 bacterial cells were Gram-negative, with the red phenotype under standard gram-staining method (Supplemental Fig. S1a). The results of biochemical characteristics showed that the following substrates were utilized: mannitol, inositol, melibiose and raffinose. Other substrates unavailable: sorbitol, ribol, phenylalanine, ornithine and lysine. Tests were negative for methyl red, indole, urease, H2S and citrate utilization, except for V-P test. Also, the test was positive for motility test medium (semisolid agar) (Supplemental Fig. S1b). The 100 μg/mL IAA standard showed negative for indole tests using this biochemical assay (Supplemental Fig. S1c). The SEM image showed that the appearance of V4 was rod shaped, with average dimensions of 1.34−1.5 μm long and 0.32−0.39 μm wide. The TEM observation showed pure cultures of strain V4 in LB medium revealed flagellum at both ends, showing an average up to 5 μm long (Fig. 1).
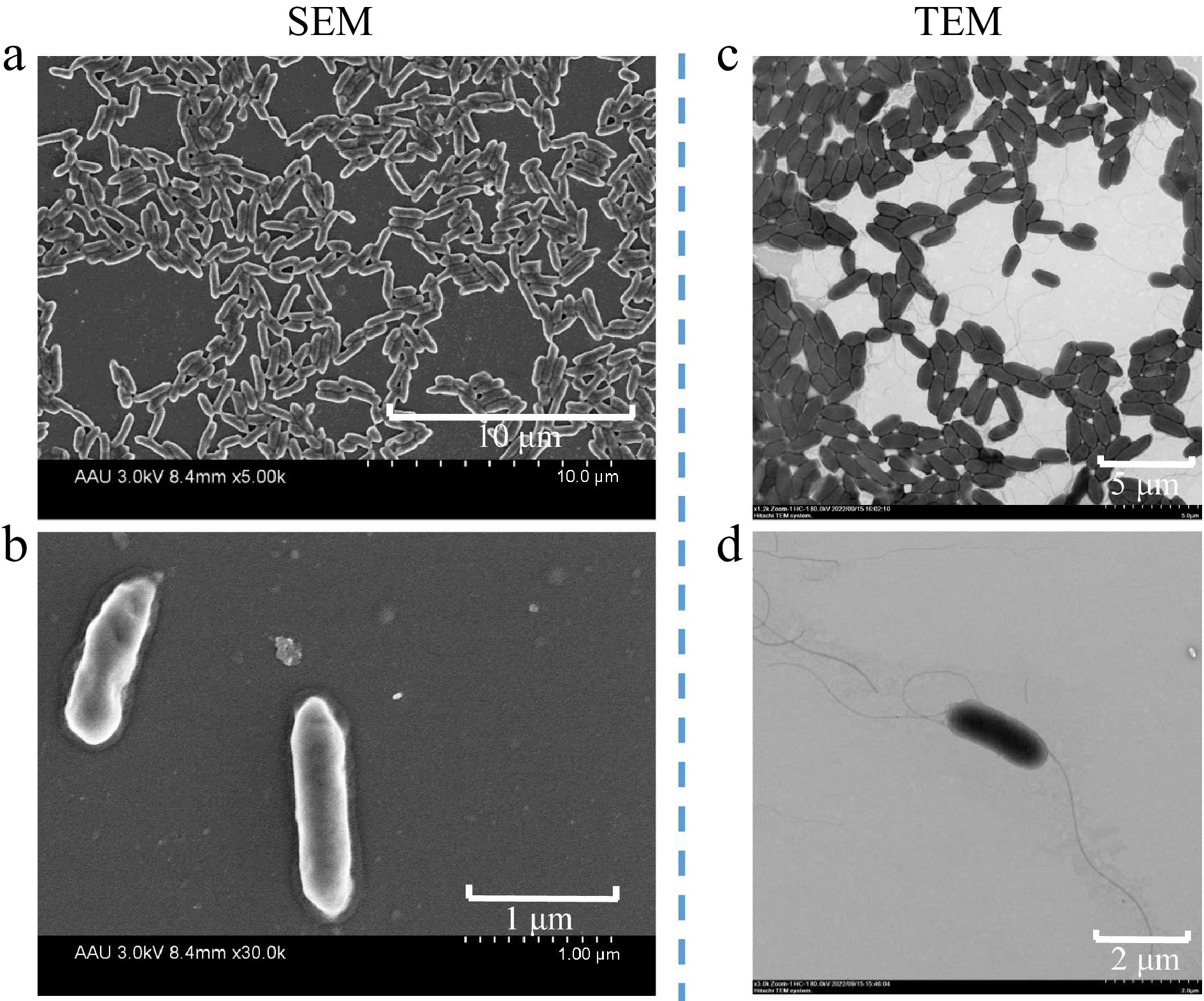
Figure 1.
Scanning electron micrograph (SEM) of strain V4 grown for 10 h in LB solid medium, acceleration voltage of 3.0 kv, (a) 8.4 mm × 5.00 k, scale bar 10 μm, (b) 8.4 mm × 30.0 k, scale bar 1 μm. Transmission electron micrograph (TEM) showing cells of strain V4 grown for 10 h in LB solid medium, with flagella at both ends, acceleration voltage of (c) 80 kv, × 1.2 k, scale bar 5 μm, (d) × 3.0 k, scale bar 2 μm.
Assembly and annotation of the V4 genome sequence
-
The assembled genome sequence of V4 was composed of one circular chromosome of 4,697,109 base pairs (bp) and two plasmids of 160,141 and 71,044 bp, respectively. The GC content of the complete genome was 56.88%. The chromosome genome sequence was predicted to contain 4189 protein-coding genes, 22 tRNA, 84 rRNA and 31 sRNA. It also harbored four gene islands and two prophages. The larger plasmid 1 had 131 protein-coding genes without noncoding RNA genes. Also, one putative coding sequence encoded putative components of the Type IV secretion system (T4SS). The small plasmid 2 had only 64 protein-coding genes (Table 1). The GC depth map and reads comparison map showed the quality of the assembly. The reads obtained from Illumina among the original sequencing were used to compare to our assembly results to obtain GC depth map. The GC content showed a concentrated distribution indicating the absence of species contamination (Supplemental Fig. S2a). Then, the first and last 800 bp of the assembly results were joined together, and then the reads obtained from Illumina sequencing were compared to the joined sequences to assess whether they were looped or not. The sign of loop formation was that complete reads could cross the joining point, which meant that assembly results could form a loop with the first and the last reads connected. Most of the reads in the reads comparison map were well connected to the end and the first end, indicating that the assembly result was not missing at the end and had formed a loop (Supplemental Fig. S2b). In addition, the number of CDS, GC content, and total length of our assembly were comparable to that of the E. rhapontici BY21311 complete genome (PRJNA773578) published in 2022[21]. In the V4 and E. rhapontici BY21311 genomes, the number of CDS were 4,189 and 4,612, GC content were 56.68% and 54.12%, and total length were 4,697,109 bp and 5.16 Mb respectively, which demonstrated that the level of completeness of V4 genome was similar to that of same genus Erwinia.
Table 1. General characteristics of the strain V4 genome.
Chromosome Plasmid 1 Plasmid 2 Size (bp) 4,697,109 160,141 71,044 G+C content (%) 56.68% 53.46% 54.25% Total protein-coding genes 4189 131 64 Total length (bp) 4,080,951 119,871 55,104 G + C content (%) 57.94% 54.65% 56.67% No. of rRNA operons 22 0 0 No. of tRNAs 84 0 0 No. of sRNAs 31 0 0 Gene islands 4 0 0 Prophage 2 0 0 TNSS II, III, IV IV 0 A total of 4,189 putative coding genome sequences were annotated though diverse protein databases. The 4,162, 3,372 and 2,865 coding genome sequences were annotated though the NR, Swissport and KEGG databases, respectively. Therefore, three type secretion systems II, III, IV were acquired based on the annotated result of KEGG and NR databases (Fig. 2a, b). Go enrichment analysis showed characteristics of gene function distribution in molecular function, biological process, cellular component (Fig. 2c). In addition, the function of 3,357 coding genome sequences representing 80.14% of all the sequences were categorized by comparison with the COGs. These functional sequences located in 21 functional categories as showing in Fig. 2d. The function category R (representing general functions) was the largest category, followed by E (amino acid transport and metabolism), G (carbohydrate transport and metabolism), S (function unknown), K (transcription), and P (inorganic ion transport and metabolism). AntiSMASH predicted that strain V4 contained six secondary metabolic biosynthetic gene clusters, including a cluster of siderophore, hserlactone, thiopeptide and three clusters of non-ribosomal peptide synthetases (Supplemental Fig. S3). The circular chromosome and plasmids of the V4 are shown in (Fig. 3). Also, GC skew was used to measure the relative amounts of G and C and mark start and end points in ring chromosomes, GC skew = (G − C)/(G + C), the window size was 10 kb.
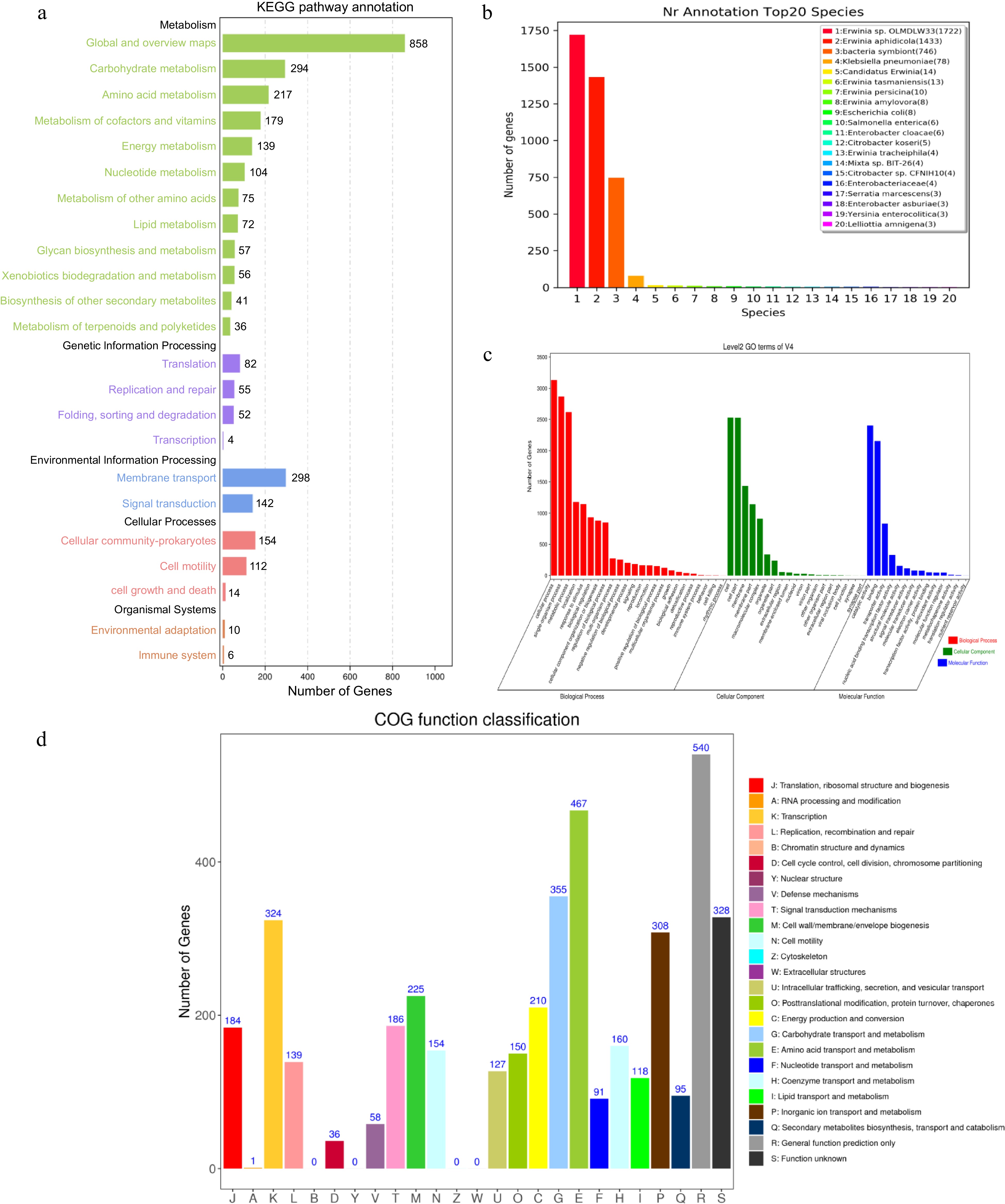
Figure 2.
The V4 chromosome genomic annotation information of gene function based on (a) KEGG, (b) NR, (c) GO and (d) COG databases.
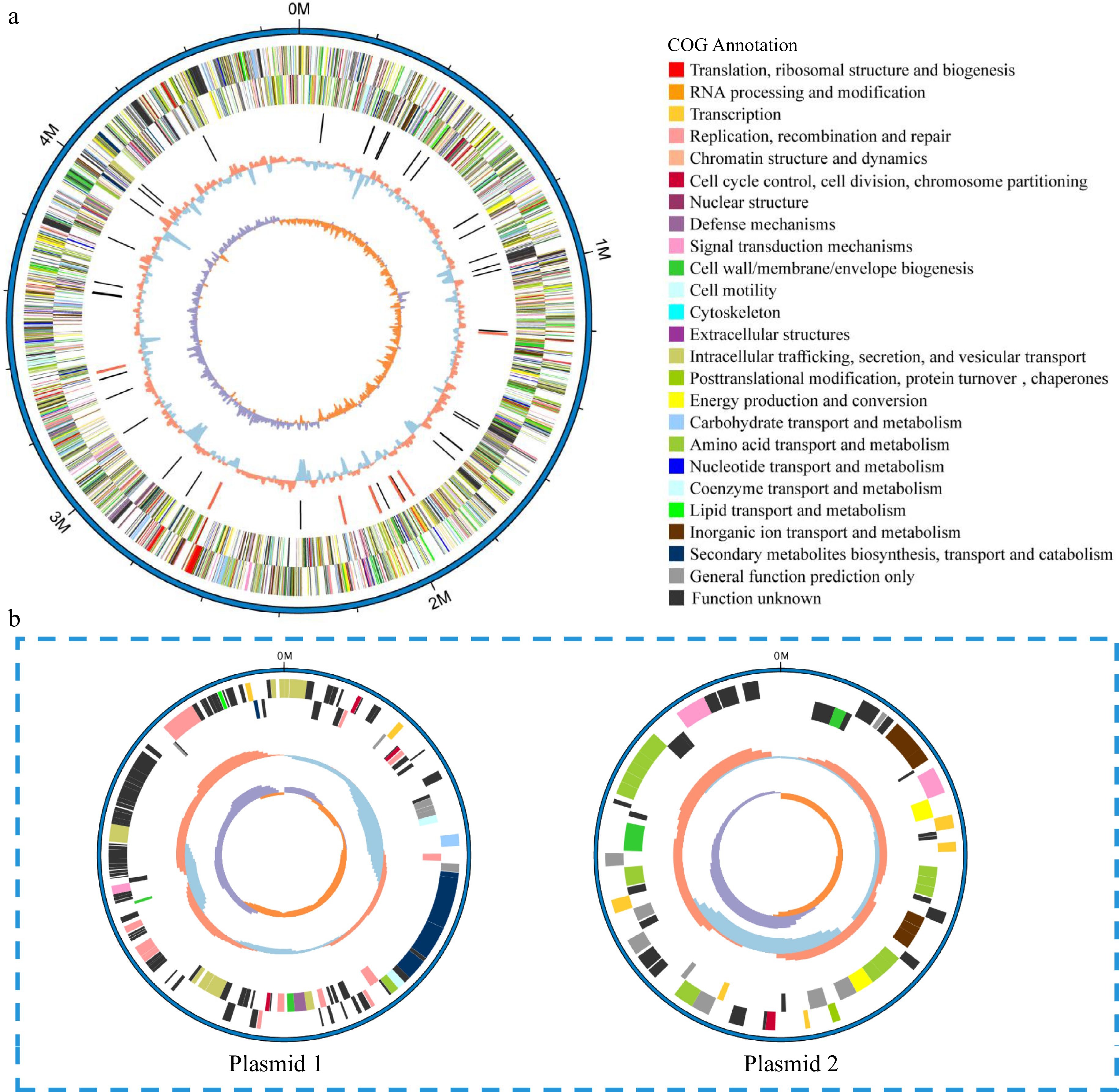
Figure 3.
Circular representation of the (a) chromosome and (b) plasmids of V4 strain. Circles (from outside to inside): forward strand genes and reverse strand genes (annotation information of gene function based on COG databases), ncRNA (black, tRNA, red, rRNA), GC content (red, > mean value of GC content, blue, < mean value of GC content), GC skew (positive and negative values being indicated with purple and orange colors respectively) (purple, > 0, orange, < 0).
Taxonomic classification of V4
-
The Blast result of 16S rRNA gene revealed that V4 strain had high sequence identity with the E. rhapontici strain DQ-03 (99.58%), Erwinia sp. Strain 20TX0058 (99.58%), Erwinia sp. CSQXZR5.2.3. (99.51%), Erwinia sp. IMCC25602 (99.58%), and E. aphidicola strain X 001 (99.58%). Next, a preliminary phylogenetic study was undertaken to clarify the taxonomic classification of V4 using cloned 16S rRNA gene sequence. The 34 genus Erwinia and one Herbaspirillum strains 16S rRNA gene sequences with length > 1,200 bp were obtained from NCBI. In total, there were 34 strains of the genus Erwinia belonging to 19 species, including psidii, papaya, mallotivora, gerundensis, oleae, tracheiphila, typograhi, toletana, iniecta, carotovora, billingiae, rhapontici, aphidicola, persicina, tasmaniensis, piriflorinigrans, amylovora, uzenensis, pyrifoliae. The maximum likelihood phylogenetic tree showed that the V4 strain formed a monophyletic clade with members of the species E. aphidicola with 78.8% 16S rRNA gene sequence similarity (Fig. 4a). This clade was clustered with members of the species E. persicina with 23.4% 16S rRNA gene sequence similarity becoming a branch, and this branch was clustered with members of the species E. rhapontici with 98.9% 16S rRNA gene sequence similarity.
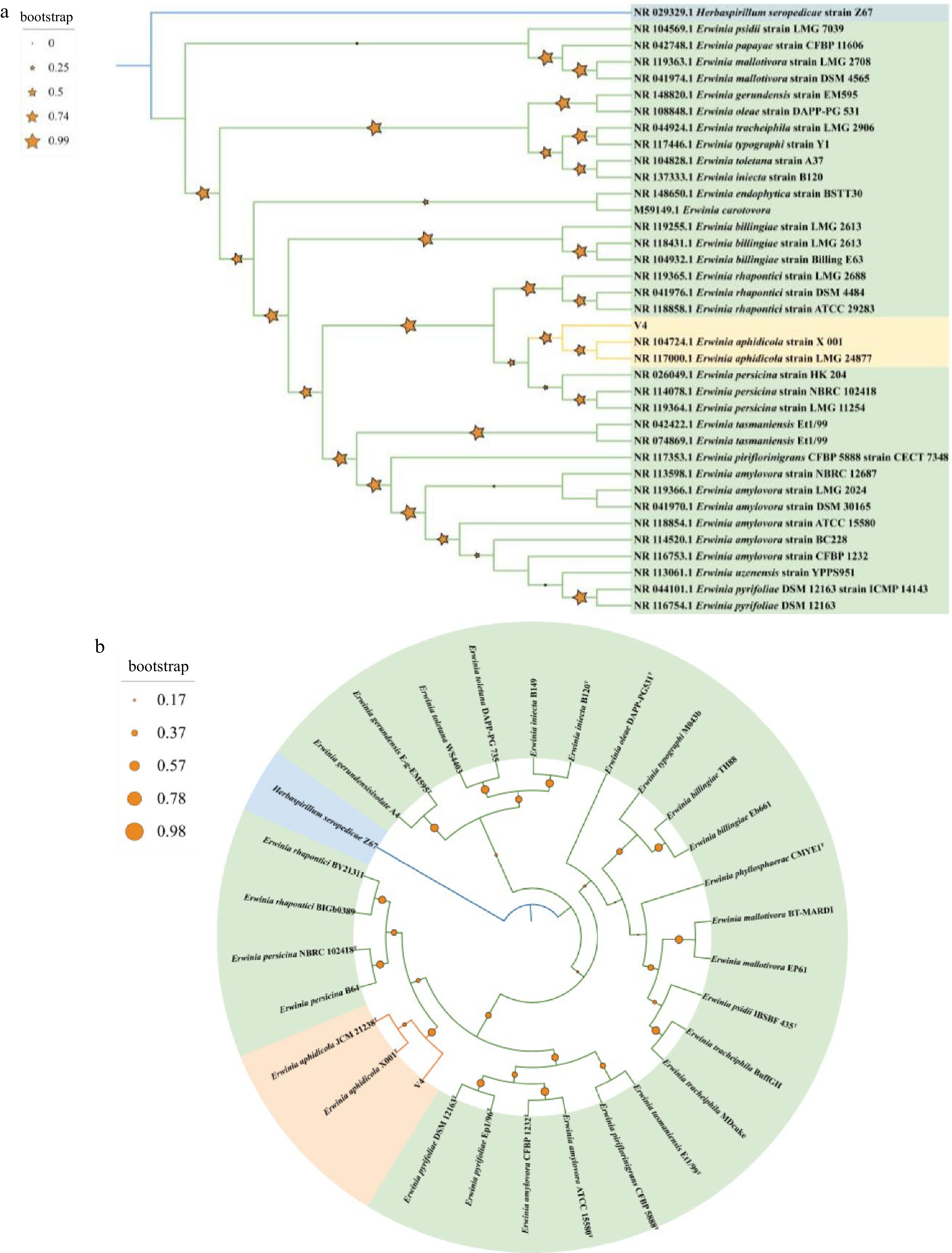
Figure 4.
(a) Maximum-likelihood phylogenomic tree based on 16S rRNA gene sequences among V4 strain and Erwinia genus strains. Herbaspirillum seropedicae Z67T was used as outgroup. Bootstrap values are shown in tree branches calculating for 1000 subsets. (b) Whole-genome-based phylogenomic tree of V4 strain, available Erwinia genus strains and Herbaspirillum seropedicae Z67T. Support values are shown in tree branches.
Based on the completion of the V4 strain genome, phylogenetic analysis of the genome was carried out. The peptide sequences of 28 representative strains belonging to 14 Erwinia species including type strains and one H. seropedicae Z67T as outgroup were obtained from NCBI (Supplemental Table S1). The phylogenetic analysis was carried out using homologous gene sequences though OrthoFinder. Phylogenetic analysis of V4 strain and members of the species E. aphidicola (E. aphidicola JCM 21238T and E. aphidicola X001T) also corroborated a close relationship within a single clade with 91.3% similarity (Fig. 4b). This clade was clustered with members of the species E. rhapontici and E. persicina with 70.2% similarity. These analyses allowed us to conclude that V4 was a strain of E. aphidicola.
Genes involved in plant growth promotion traits present in the genome of V4
-
The V4 strain had shown plant growth-promoting ability through plate identification and pot inoculation experiments in a previous study[9]. Plate identification experiments demonstrated that the V4 strain has the ability to produce IAA, ACC deaminase, nitrogen fixation, phosphorus solubilization and siderophores production. Assembly of the genome sequence of V4 therefore provide us with the opportunity to identify key genes and compare their copy number variations associated with plant growth promotion traits (Supplemental Tables S2 & S3).
IAA biosynthesis and ACC deaminase
-
The indole-3-acetic acid (IAA) as a plant hormone was involved in the regulation of plant growth and development. The indole pyruvate decarboxylase, a key rate-limiting enzyme encoded by one copy of ipdC gene and aldehyde dehydrogenase encoded by two copies of dhaS gene, which catalyzed the conversion of indole-3-pyruvic acid to indole-3-acetaldehyde and the dehydrogenation of indole-3-acetaldehyde to indole-3-acetic acid in indole-3-pyruvic acid (IPA) pathway were found in V4 genome. ACC deaminase regulated ethylene production by utilizing the exuded ACC, the immediate precursor of ethylene in higher plants. ACC deaminase was a member of the tryptophan synthase β subunit family of PLP-dependent enzymes, the 1-aminocyclopropane-1-carboxylate (ACC) deaminase and cysteine desulfhydrase were all belonging to PLP-dependent enzymes family with high degree of homology. The ACC deaminase structural gene (acdS) was not found in V4 genome, however, one copy of cysteine desulfhydrase gene (dcyD) which was annotated as 1-aminocyclopropane-1-carboxylate deaminase in COG database present in the genome.
Nitrogen and phosphorus acquisition
-
Nitrogenase was a complex metalloenzyme with conserved structure and biological characteristics, which had the ability to convert nitrogen from air into nitrogenous compounds. It was found that two copies of nitrogen regulation system related genes ntrB (nitrogen regulation protein NR(II)) and ntrC (nitrogen regulation protein NR(I)) were present in the V4 genome. However, the nif family nitrogen fixing genes encoding nitrogenase were not found. The main mechanism of solubilization of insoluble mineral phosphate complexes by gram-negative bacteria was that the direct oxidation of glucose to produce gluconic acid, which was synthesized by glucose dehydrogenase (GDH) and the co-factor pyrroloquinoline quinine (PQQ). The results also showed that one copy of gcd and pqqE gene encoding GDH and PQQ respectively were present in V4 genome. Also, one copy of pstA and two copies of pitA genes related to the high-affinity phosphate transport (Pst) system and low-affinity phosphate transport (Pit) system to obtain effective phosphorus were also present. Phosphorus transport system-related binding protein genes were also identified, including one copy of phnD2, phnC, phnL and phnK genes, respectively. In addition, the one copy of appA and agp genes associated with phytases synthesis, and a phoR-phoP phosphate regulation system regulated phytases to initiate the release of phosphate from phytate were found in V4 genome.
Siderophores production
-
Siderophore biosynthesis occured via two pathways: the non-ribosomal peptide synthetase (NRPS) pathway and the NRPS-independent siderophore synthetase (NIS) pathway[27]. In V4 genome, the siderophore biosynthesis gene cluster belonging to NIS synthetase pathway was found through antiMAST analysis. Core biosynthesis gene iucC and additional biosynthesis genes ddc and alcA were responsible for the production of siderophore, and three transport-related genes mdfA, zunA, zunC were involved in the transport of siderophore, others genes were hexR, pykA, lpxM, mepM in this siderophore biosynthesis gene cluster. The siderophore outer membrane receptor proteins (fhuA, fhuE, fepA and tonB) and ABC-type Fe2+/Fe3+-hydroxamate transport protein (fepB, fhuB, fhuC, fhuD, fepC and fepD) were found in NRPS pathway.
Others
-
Annotation of the V4 genome also identified candidate genes related to plant growth regulator, plant resistance, extracellular polysaccharide production and heavy metal resistance. One copy of hemA gene encoding glutamyl-tRNA reductase involved in 5-Aminolevulinic acid (5-ALA) biosynthesis were found in the V4 genome. One copy of SpeE gene encoded spermidine synthase that associated with plant resistance. Two copies of GalE genes were related with extracellular polysaccharide biosynthesis. The V4 genome carried one copy of genes including copper-transporting ATPase copA, zinc/cadmium/mercury/lead-transporting ATPase zntA, mtnABCDKN encoding metallothionein, which was able to bind metals. One copy of gstB and gst3 genes, two copies of gstA genes encoded glutathione S-transferase that catalyzed the binding of the sulfur group of glutathione. CysC, cysD, cysH, cysK genes possessing one copy gene number showed ability in sulfate assimilation pathway.
In total, the schematic overview of main plant growth-promoting traits in V4 is shown in Fig. 5. These included IAA biosynthesis, phosphorus acquisition, siderophores production and others plant growth-promoting traits, which indicated V4 could promote plant growth though producing soluble phosphate, plant growth regulator, promoting the uptake of iron ions, improving plant resistance and heavy metal resistance.
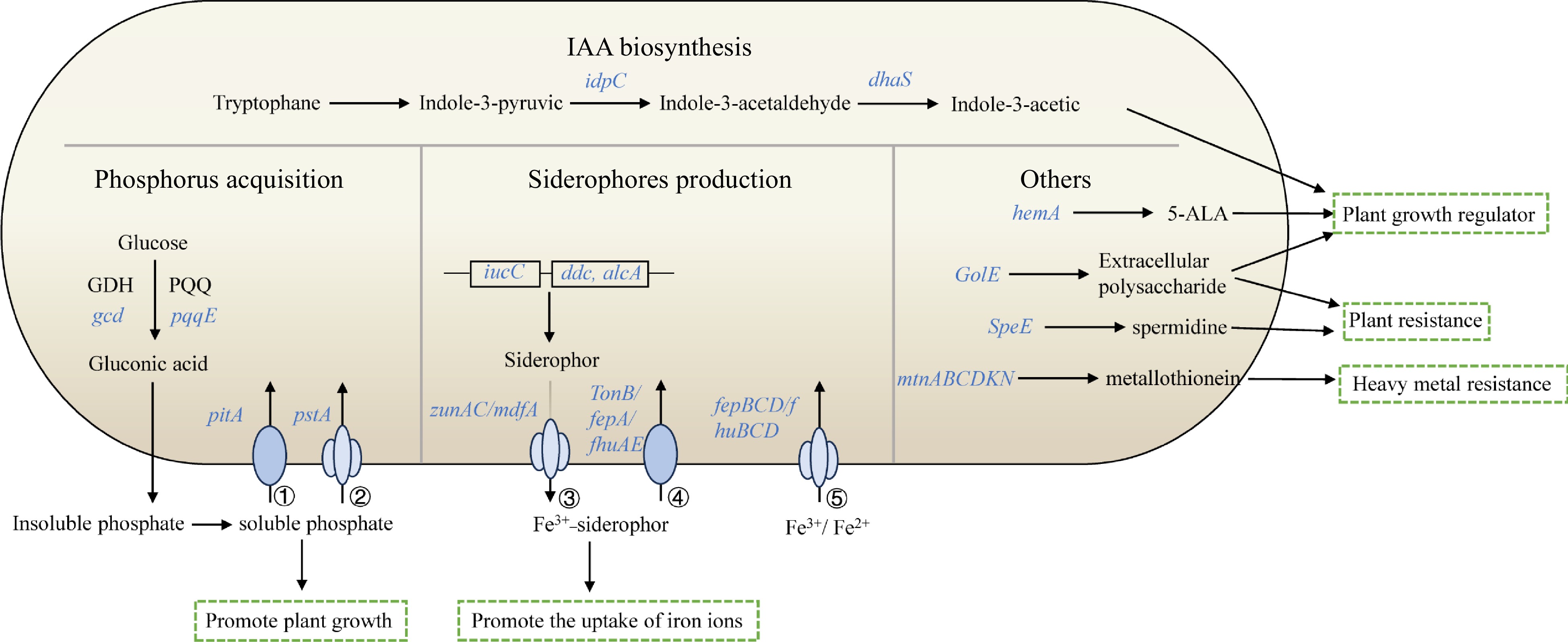
Figure 5.
Schematic overview of main plant growth-promoting traits in V4. The depicted pathways were predicted based on the genomic data of V4. Details are available in Supplemental Table S2. These include IAA biosynthesis, phosphorus acquisition, siderophores production and others. Individual pathways were denoted by single-headed arrows, genes were shown in blue italics. Abbreviations: GDH (glucose dehydrogenase); PQQ (pyrroloquinoline quinine); 5-ALA (5-Aminolevulinic acid); ① low-affinity phosphate transport system, Pit; ② high-affinity phosphate transport system, Pst; ③ siderophore transporter; ④ siderophore outer membrane receptor proteins; ⑤ ABC-type Fe2+/Fe3+-hydroxamate transport protein.
Genome mining for V4 endophytic colonization
-
V4 whole genome sequence analysis revealed functional genes and their copy numbers variations potentially associated with colonization according to KEGG database and COG function analysis (Supplemental Tables S4 & S5). In detail, genes for flagellar assembly, chemotaxis were connected with motility, and pilus assembly were important for attachment to plant surfaces for host plant colonization.
Motility was an important characteristic for bacteria. V4 was well equipped with flagellar to move towards plants actively. Its genome contained three region flagellar biosynthesis genes. Flagellar assembly genes contained flgABCDEFGH1IKLMN, flhABCD, motAB and fliACDEFGHIJKMNOPQRST in region-I, II, and III respectively. All of the genes possessed two copies, except for one copy of flgM, fliK, fliT genes and three copies of fliC genes. The flgABCDEFGH1IKLMN genes were involved in complex basal body component. The elongation of hook was controlled by flgE and fliK genes. The fliC gene involved in the assembly of filament as the last step. The products of motAB and fliGMN genes were responsible for energizing the flagellar motors. Each flagellum was driven by a flagellar motor located at the base, which rotates and drives the cell movement.
Chemotaxis enabled microorganisms to move towards beneficial or away from harmful substances in their environments through flagellar motility. V4 had multiple clusters of chemotaxis genes including cheA, cheB, cheY, cheW, cheV2, cheR, cheZ, and mcpA. The copy numbers of cheABYWRZ and mcpA genes had two copies, in addition to cheV2 gene had one copy. The methyl-accepting chemotaxis protein encoding by mcpA coupled the sensor histidine kinase cheA via cheW protein were conserved as chemotaxis signal transduction system. Then cheA phosphorylated the response regulators cheB and cheY. CheB balanced the activity of the methltransferase, and cheY controled the flagellar motor, cheZ promoted cheY-P dephosphorylation and recovered the bacteria ability to respond to external signals. CheR protein added methyl groups to methyl-accepting chemotaxis protein. Additional chemotaxis genes tar, tsr, tas, tap, tcp, trg, ctpL, dppA, mglB, mocB, and rbsB encoding methyl-accepting chemotaxis proteins were involved in chemotaxis signal transduction system to control flagellar motility directly through the motAB and fliGMN genes controlling flagellar motors. There was one copy of tap, tcp, trg, aer, ctpL, mglB, mocB, rbsB genes, two copies of tsr, dppA genes, four copies of tar genes and six copies of tas genes.
Pilus were involved in adhesion to plant surfaces. V4 also had a large number of pilus biosynthesis genes and possessed multiple copies of genes, including one copy of afaC, cpxP, lpfB, pmfD, pmfC, spy, ppdD, hofB, yggR genes, two copies of yhcA, smf-1 genes, three copies of yadV, yhcD, yfcS, vfcU, smfA genes, and four copies of htrE. MrkC, fimD, and htrE were homologous genes. All of them were located in the genomic chromosome. The plasmid 1 was known as a F plasmid which contained tra gene to produce sex pilus. Therefore, V4 could realize conjugative DNA transfer and plant-bacteria interactions by the F-plasmid though a type IV secretion system (T4SS).
Genome mining for signal transduction mechanisms
-
In general, the intracellular signal transduction mechanisms are used for regulating biological process in microorganism including two-component regulatory systems, quorum sensing and so on. Bacterial signal transduction mechanisms are mainly referred to as 'two-component regulatory systems'. There were many two-component signal-transduction systems (TCSs) in bacteria and the structures contain a histidine protein kinase (HK) as a sensor receptor, a reaction regulatory protein as a response regulator, which contains one or more DNA-binding effector domains that participate in transcriptional regulation to generate various responses to environmental alteration. In the V4 genome, 142 and 74 genes were annotated as functional genes involved in two-component systems and quorum sensing respectively basing on the KEGG database. Also, COG database annotation showed 186 genes belonging to T (signal transduction mechanisms) function classification. Combining the annotation results of the above two databases, the numbers of two-component system and quorum sensing genes belonging to T classification were 76 and 8 respectively (Supplemental Table S6). The copy numbers of these genes are shown in Supplemental Table S7. There were 15 TCSs in V4 genomic sequences, including cheA-cheB/cheY phosphotransfer signaling for flagellar chemotaxis, phoR-phoB, pmrB-pmrA, phoQ-phoP and kdpD-kdpE phosphotransfer signaling for phosphate regulation, iron (Fe3+) regulation, magnesium (Mg2+) regulation and potassium (K+) transport individually, arcB-arcA phosphotransfer signaling for anaerobic metabolism and biofilm formation regulation, envZ-ompR and cpxA-cpxR phosphotransfer signaling for osmotic regulation, rcsC-rcsB and ntrB-ntrC phosphotransfer signaling for capsular polysaccharide synthesis and nitrogen regulation, bygS-bvgA phosphotransfer signaling for the production of virulence factors, qseC-qseB phosphotransfer signaling for flagellar and virulence factors genes expressions, rstB-rstA phosphotransfer signaling for multi-drug resistance, dcuS-dcuR phosphotransfer signaling for controlling genes expression in response to C4-dicarboxylates, baeS-baeR phosphotransfer signaling for regulating genes expression. These two-component systems played a major role in regulating cell activities in V4.
Quorum sensing had been shown to be important in traits such as virulence, biofilm formation and swarming motility in bacteria and involved in communication with host plants. In the V4 genome, the eight quorum sensing genes were one copy of qseC, qseE, qseB, luxS, crp, glrR, kdpE genes and three copies of pdeR genes. The qseC-qseB was a two-component regulatory system involved in the regulation of flagella and motility. LuxS was the gene for synthesizing autoinducer 2 (AI-2), which could mediate expression of virulence genes in response to the bacterial cell density as bioactive small diffusible molecules. The cyclic adenosine monophosphate receptor protein was encoded by crp gene, proquorum sensing CRP agonists could inhibit bacteria virulence. KdpE, a KDP operon transcriptional regulatory protein, it regulated potassium (K+) transport in the stressful conditions and contributed to bacterial survival in the host.
Results of comparative genomic analysis
-
Based on the results of gene family analysis, we further compared the copy number variations of genes associated with plant growth promoting traits, colonization, and signal transduction mechanisms. It was shown that V4 and E. tasmaniensis ET1/99 endophytic bacteria both had genes associated with IAA synthesis, and P-solubilization, also had dcyD, ntrB, and ntrC genes associated with ACC deaminase and Nitrogen-fixation abilities. The V4, E. tasmaniensis ET1/99, H. seropedicae Z67 endophytic bacteria and E. aphidicola 18B1 all had genes for production of siderophores. Moreover, V4 has two copies of dhaS, pitA, fhuA and fhuD genes. In others promotion mechanism, V4 had more genes copies than E. tasmaniensis ET1/99 endophytic bacteria, such as, one copy of gstB, gst3 genes and two copies of gstA genes associated with heavy metal resistance, two copies of gale genes associated with extracellular polysaccharide. V4 had core siderophores biosynthesis gene iucC, additional biosynthesis genes ddc, alcA and outer membrane receptor proteins fepA comparing with E. rhapontici BY21311 and E. persicina B64 plant pathogenic bacteria. V4 possessed higher copy numbers of genes associated with flagellar assembly, bacterial chemotaxis, P pilus assembly and two-component system comparing with E. tasmaniensis ET1/99, H. seropedicae Z67, E. rhapontici BY21311 and E. persicina B64 bacteria. Also, both V4 and E. aphidicola 18B1 all had higher gene copy numbers in flagellar assembly.
-
In order to examine the plant growth promoting ability of V4 endophytic bacteria, their basal characteristics were first identified in the study. The result of physiological and biochemical characterization showed that V4 bacterium is Gram-negative bacteria and has the ability to metabolize sugar alcohol compounds. We found that V4 bacterium was rod-shaped with flagella at both ends and the size of the bacterium was 1.34−1.5 μm long and 0.32−0.39 μm wide. The above information showed that the V4 bacterium belonged to the basic category of rod-negative bacteria and it had the capability to move. Since the result of IAA standard in indole test was negative, it could not be considered that the V4 bacterium showed negative results in this indole test.
The V4 bacterium was initially identified as Erwinia genus strain according to the sequence alignment of 16S rRNA[28]. To further determine the phylogenetic classification of V4 bacterium, the 16S rRNA gene maximum likelihood phylogenetic tree and whole-genome phylogenetic analysis two bioinformatic approaches were performed. The 16S rRNA gene sequence was extensively used as a criterion for classifying bacterial systems for it is highly conserved and present in all bacteria[29]. However, it had been shown that genome phylogenetic analysis was more reliable than 16S rRNA alignment[30]. Therefore, both bioinformatic approaches were widely used for bacterial identification to obtain higher accuracy. For example, the phylogenetic tree was constructed by genome sequences and 16S rRNA gene for establishing a stable taxonomy of Pseudomonas syringae strain and Rhodococcus strain IGTS8[13,31]. In the present study, V4 bacterium was identified as Erwinia genus strain according to 16S rRNA gene BLAST analysis in NCBI database. Next, we obtained 16S rRNA gene sequences of all Erwinia species larger than 1,200 bp from NCBI and constructed 16S rRNA gene maximum likelihood phylogenetic tree to make sure the credibility of V4 bacterium classification result. The tree showed that V4 bacterium formed a monophyletic clade with members of the species E. aphidicola with 78.8% 16S rRNA gene sequence similarity. To further clarify the systematic classification of V4 bacterium, phylogenetic analysis of the genome was carried out. There were many bacteria genomes belonging to Erwinia genus, therefore, we chose 28 Erwinia species type strains as representative strains. The result showed that V4 strain and members of the species E. aphidicola (E. aphidicola JCM 21238T and E. aphidicola X001T) corroborated a close relationship within a single clade with 91.3% similarity. In summary, we considered that V4 was a strain of E. aphidicola.
V4 bacterium had shown plant growth promoting ability through plate identification and pot inoculation experiments in a previous study[9]. In this study, it was possible to find genes and examine their copy number variations involved in plant growth promotion traits present in the genome of V4. Many studies had shown that a higher gene copy number would help enhance its gene function. Rhodococcus sp. JG3 survived at subzero growth down to −5°C benefiting from a higher copy number of genes associated with protection from cold shock and stress response[32]. K. radicincitans endophytic bacteria showed more powerful competitiveness owing to the multiple copies of complex genes clusters associated with metabolism[33].
In trp-dependent IAA biosynthesis, there were four synthetic pathways, which were indole-3-acetamide pathway, indole-3-pyruvic acid (IPA) pathway, tryptamine pathway, and indole-3-acetaldxime (IAOX) pathway[34]. Indole-3-pyruvic acid (IPA) pathway to produce indole-3-acetic acid (IAA) as a plant hormone were found in V4 genome. Also, some studies have shown that IAA was a signaling molecule in microorganisms[35]. V4 had two copies of dhaS genes which might enhance IAA biosynthesis capability.
The gcd and pqqE gene encoding GDH and PQQ respectively for producing gluconic acid (GA) to solvent insoluble mineral phosphate complexes and phosphate transport system were necessary for phosphorus acquisition[36]. H. seropedicae Z67 has been shown to have no phosphate solubilization ability as well as to exhibit deletion of gcd and pqqE genes[37]. The gcd and pqqE genes presented in V4 genome showed V4 bacterium could contributes to phosphorus content in soil and plant tissues to promote plant growth. A number of studies had shown that siderophores could promote the uptake of iron ions and induce resistance responses in plants[38]. There were: non-ribosomal peptide synthetase (NRPS) and NRPS-independent siderophore synthetase (NIS) two pathways to synthesize siderophores[27,38]. NRPS-independent siderophore synthetase (NIS) pathways existed in the V4 genome. Research showed that the ability to produce siderophores was critical for bacteria growth and to compete with pathogenic bacteria[39]. Interestingly, V4, E. tasmaniensis ET1/99 plant growth promoting endophytic bacteria and E. aphidicola 18B1 nonpathogenic bacteria had siderophore biosynthesis genes in NIS pathway, H. seropedicae Z67, a plant growth promoting endophytic bacteria had NRPS siderophore pathway, however, E. persicina B64 and E. rhapontici BY21311, plant pathogenic bacteria did not have these genes. The siderophore production of plant growth promoting endophytic bacteria might help their colonization on host plants. Also, V4 had higher copies of fhuA and fhuD genes, it might indicate that siderophores were more helpful for the growth of V4 endophytic bacteria and interaction with host plants.
Otherwise, the nitrogen regulation system related genes ntrB and ntrC were present in the V4 genome without nif family nitrogen fixing genes encoding nitrogenase, showing that it was mainly involved in the transport of nitrogen in itself or in host plants. The ntrB and ntrC proteins were regulators involved in the nitrogen transport mechanism of photosynthetic bacteria[40]. V4 strain had the ability to produce ACC deaminase based on plate identification, however the ACC deaminase structural gene (acdS) was not found in the V4 genome. Analysis of the key sites for enzymatic activity of the acdS and dcyD genes amino acid sequences, it revealed that the amino acid at positions 295 and 322 of the acdS gene were Glu and Leu[41], while those of dcyD were Leu and Phe. Therefore, the dcyD gene had no ACC deaminase function for absence of Glu and Leu in these two positions. On the one hand, due to the limitations of the plate assay, it might be that the bacteria had other abilities to lead to the identification of ACC deaminase ability a false positive. On the other hand, studies had shown that the ability of bacteria was not entirely explained by the presence or absence of genes themselves[31], perhaps other genes played an important role in these two abilities.
The hemA, GalE, copA, zntA, mtnABCDKN, GstA, gstB, gst3, CysC, cysD, cysH, cysK genes were found in V4 genome for contributing to host plants growth, resistance and heavy metal resistance. Previous studies showed that low concentration 5-aminolevulinic acid(5-ALA)as plant growth regulator could promote plant growth[42], extracellular polysaccharide could promote plant growth and improve the ability of resisting extreme environment[43], copper-transporting ATPase, zinc/cadmium/mercury/lead-transporting ATPase, metallothionein were related with heavy metal resistance[44−46], the binding of the sulfur group of glutathione to hydrophobic heterologous substances played a role in excretion and detoxification function[47]. V4 endophytic bacteria had more copies of genes related to heavy metal resistance and extracellular polysaccharide than E. tasmaniensis Et1/99 endophytic bacteria, it might indicate that V4 had much stronger plant growth-promoting ability. To summarize, the V4 bacterium had many genes related to plant growth promotion traits in the genome of V4 and had the potential to promote the host plants growth mainly through the production of IAA, siderophores, and soluble phosphorus.
The ability of V4 bacterium to efficiently colonize surfaces of host plants was a prerequisite for phytostimulation. The colonization process of endophytic bacteria have been widely studied: moving toward the plants tissue, adhesion the plants tissue surface, invasion and colonization of host plants[48,49]. Motility was an important characteristic for endophytic bacteria since they need to move to the selected root area and reach the inside the host plant and chemotaxis determined the movement direction[50,51]. There were many genes involved in flagellar assembly and chemotaxis in the V4 genome. Bacteria pilus contributed to adherence to host plant surfaces[52]. The genes related to pilus assembly were also found in the V4 genome. Then, V4 bacterium could enter the plant through cellulase lysis of the cell wall or natural gaps with the plant-bacteria interactions. Also, cell-surface components capsular polysaccharides (CPS) were commonly involved in plant-bacteria interactions[27]. RcsC-rcsB two-component system for capsular polysaccharide synthesis was found in the V4 genome. Otherwise, V4 endophytic bacteria possessed a higher copy number of genes for flagellar assembly, bacterial chemotaxis and P-pilus assembly. Therefore, V4 endophytic bacteria had the stronger ability to colonize the host plant and could communicate with the host plant during colonization. For example, Shewanella putrefaciens CN-32 possessed a complete secondary flagellar system showing stronger motility in different environments[53]. Also, it might be useful as a vector for exogenous genes and other biological functions.
Signal transduction mechanisms were crucial for survival of bacteria. It provided an adaptive advantage for bacteria to adapt to changing environments. Two component systems were the most sensitive regulatory system in bacteria, for example, Pho regulon was controlled by a two component system involved in bacterial Pi regulation for coping with the scarcity of Pi nutrients[54]. There were many two component systems in the V4 genome including phosphate regulation, iron (Fe3+) regulation, magnesium (Mg2+) regulation, potassium (K+) transport, biofilm formation regulation, nitrogen regulation and so on for helping V4 survival in a variety of environments. Also, V4 endophytic bacteria possessed a higher copy number of cheA-cheB/cheY genes for flagellar chemotaxis enhancing chemotaxis in vitro and host colonization ability. Campylobacter jejuni regulated bacterial chemotaxis in vitro and colonization in vivo through the CheA/CheY signaling system[55].
Otherwise, quorum sensing were likely involved in the colonization process and communication with plants, it was supported by a strain Bukholderia phytofirmans PsJN with quorum sensing mutant which could not colonize plants efficiently and influence the ability to promote plant growth[56]. The eight quorum sensing genes were found in the V4 genome, for example, qseC-qseB was involved in the regulation of flagella and motility. Therefore, V4 had the ability to rely on signal transduction systems to sense and respond to environmental changes in order to survive, promote host plant growth, and interact with host plants.
-
In this study, basic physiological, biochemical and morphological characteristics of V4 endophytic bacterium are shown. It was clear that V4 bacteria belonged to the E. aphidicola species according to 16S rRNA gene and whole-genome phylogenetic analysis. There were many genes related to plant growth promoting properties and plant colonization in the V4 genome. V4 had key synthetic genes associated with IAA synthesis, and P-solubilization, siderophores consistent with E. tasmaniensis ET1/99 and H. seropedicae Z67 plant growth-promoting endophytic bacteria. V4 had siderophore biosynthesis genes compared with E. persicina B64 and E. rhapontici BY21311, plant pathogenic bacteria. V4 possessed a higher copy number for genes in flagellar assembly, bacterial chemotaxis, P pilus assembly and two-component system compared with E. tasmaniensis ET1/99, H. seropedicae Z67, E. rhapontici BY21311 and E. persicina B64 bacteria. Also, both V4 and E. aphidicola 18B1 all had higher gene copy numbers in flagellar assembly. Analysis of the V4 endophytic bacterium complete genome sequences provided important insights into the endophytic bacteria-host plant relationship, and suggested many interesting candidate genes for post-genomic experiments.
-
The authors confirm contribution to the paper as follows: Experiment preparation: Jia H; data analysis: Jia H, Yan Y, Ma J, Xia E, Ma R, Li Y, Wang M; draft manuscript preparation: Jia H; Figure and table modification: Yan Y, Ma J, Xia E; manuscript revise: Deng WW, Xia E; Research Guidance: Deng WW. All authors have read and approved the final manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. The V4 genomic sequence reported has been deposited in NCBI database (accession number, PRJNA855316).
This study was supported by the Natural Science Foundation of Anhui Province, Outstanding Youth Project (2008085J18), National Natural Science Foundation of China (NSFC, Grant No. 31870679).
-
The authors declare that they have no conflict of interest.
- Supplemental Fig. S1 The standard gram-staining showed strain V4 cells are Gram-stain negative, with the red phenotype (a). The results of strain V4 biochemical characteristics were shown in after reaction picture. From left to right, they are semisolid agar, ornithine, lysine, amino acid control, citrate utilization, H2S, urease, indole, methyl red, V-P test, phenylalanie, mannitol, inositol, sorbitol, melibiose, ribol and raffinose in HBI Rapid ID Panel For Enterobacteriaceae Bacteria of before reaction and after reaction picture(b). The result of 100 μg/mL IAA standard in indole tests were shown (c).
- Supplemental Fig. S2 The GC depth map and reads comparison map showed the quality of the assembly. The horizontal axis showed the GC content and the vertical axis showed the sequencing depth, with the sequencing depth distribution on the right and the GC content distribution on the top in GC depth map (a), Most of the reads in the reads comparison map were well connected to the end and the first end (b).
- Supplemental Fig. S3 Six predicted secondary metabolite biosynthetic gene clusters in strainV4, including a cluster of siderophore, hserlactone, thiopeptide and three clusters of NRPS.
- Supplemental Table S1 Genomic information of strains in the phylogenetic tree.
- Supplemental Table S2 Plant growth promoting traits associated genes in the genome of V4 strain.
- Supplemental Table S3 Copy numbers of plant growth promoting traits associated genes in the genome of V4 strain and other strains.
- Supplemental Table S4 Bacterial colonization associated genes in the genome of V4 strain.
- Supplemental Table S5 Copy numbers of bacterial colonization associated genes in the genome of V4 strain and other strains.
- Supplemental Table S6 Signal transduction mechanisms associated genes in the genome of V4 strain.
- Supplemental Table S7 Copy numbers of signal transduction mechanisms associated genes in the genome of V4 strain and other strains.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Jia H, Yan Y, Ma J, Xia E, Ma R, et al. 2023. Complete genome sequence of a plant growth-promoting endophytic bacterium V4 isolated from tea (Camellia sinensis) leaf. Beverage Plant Research 3:24 doi: 10.48130/BPR-2023-0024 

Complete genome sequence of a plant growth-promoting endophytic bacterium V4 isolated from tea (Camellia sinensis) leaf
- Received: 13 July 2023
- Revised: 06 September 2023
- Accepted: 07 September 2023
- Published online: 08 October 2023
Abstract: V4 is a Gram-negative, plant growth promoting endophytic bacterium that promotes the growth of tea plants. The appearance of V4 is rod shaped, with average dimensions of 1.34−1.5 × 0.32−0.39 μm and flagellum at both ends. The complete genome contains one circular chromosome and two plasmids. It is 4,697,109 bp in size, and contains 4,189 protein-coding genes, four gene islands and two prophages. Taxonomic classification suggested that V4 was a strain of Erwinia aphidicola. It was possible to find genes involved in plant growth promotion traits present in the genome of V4. Meanwhile, V4 was consistent with plant growth-promoting endophytic bacteria containing key synthetic genes associated with IAA synthesis, and P-solubilization, siderophores. V4 has siderophore biosynthesis genes compared with plant pathogenic bacteria showing stronger survival ability and the ability to interaction with the host plant. In addition, V4 endophytic bacteria possess a higher copy number of genes for flagellar assembly, bacterial chemotaxis and P-pilus assembly indicating stronger colonization and communication ability with host plants compared with five other bacteria in comparative genomic analysis. Analysis of the V4 endophytic bacterium complete genome sequence provides novel insights into the endophytic bacteria-host plant relationship, and suggests many candidate genes for post-genomic experiments.
-
Key words:
- Endophytic bacteria /
- Genome /
- Plant growth-promoting /
- Colonization