








-
Arachidonic acid (AA), an ω-6 polyunsaturated fatty acid (PUFA), is a ubiquitous endogenous active substance found especially in brain and nervous tissue[1]. AA and its metabolites have complex and diverse biological roles and are involved in regulating numerous physiological processes in the body, as well as the development of major diseases[2]. Abnormal AA metabolism is implicated in many metabolic diseases, including fatty liver, insulin resistance, and hyperlipidemia, that disrupt hepatic glucose and lipid metabolism homeostasis[3,4].
Ferroptosis is a newly recognized mode of cell death driven by iron-dependent accumulation of lethal lipid peroxides[5−7]. Ferroptosis is characterized by peroxidation of phospholipids, impaired glutathione peroxidase 4 (GPX4) activity, and accumulation of redox-active iron, which are closely linked to the development of various diseases[6,8]. Fe2+ plays a significant role in regulating oxidative stress and metabolic processes and can bind to ferritin to form a complex for storage[9]. Increased intracellular Fe2+ levels promote ferroptosis by promoting iron uptake or impairing iron storage[6,10]. Lipid metabolomics indicate that AA is particularly susceptible to lipid peroxidation and induces ferroptosis, but the underlying mechanism of iron metabolism dysfunction remains unclear[11]. Several types of autophagy have been reported to influence ferroptosis by modulating iron accumulation, lipid peroxidation, and antioxidant protein degradation[12−14].
Ferritinophagy, in particular, mediated by nuclear receptor coactivator 4 (NCOA4), degrades ferritin and releases Fe2+, thereby increasing intracellular iron levels and promoting ferroptosis[9]. Moreover, endoplasmic reticulum stress (ER stress) plays a crucial role in ferroptosis regulation, but its function in ferroptosis seems to be context-dependent[15−17]. There are reports showing that in some disease conditions, the ER stress promotes the occurence of ferroptosis. In addition, autophagy activation is also closely related to ER stress through the ATF4-CHOP signaling pathway[18].
In this study, we explored the role and association of ER stress, ferritinophagy, and ferroptosis in AA-induced hepatocyte death. Our study reveals that AA-induced ferroptosis in hepatocytes is linked to iron overload due to impaired iron metabolism. AA upregulates the expression of transferrin receptor 1 (TfR1) on the cell membrane, leading to greater cellular iron uptake. Additionally, AA induces ER stress and activates ferritinophagy, causing the degradation of ferritin and releasing Fe2+. Our study provides new insights into the mechanism by which ER stress-ferrintinophagy regulates ferroptosis and participates in AA-induced liver injury, and serves as the basis for searching for potential strategies for lipid metabolism disorder-related diseases based on ferroptosis process intervention.
-
Arachidonic acid (AA) (purity ≥ 99%), chloroquine (CQ), IRE1α inhibitor 4µ8C, PERK inhibitor GSK2606414 and ferroptosis inhibitor liproxstatin-1 (Lip-1) were purchased from MedChemExpress (Monmouth Junction, NJ, USA). Cycloheximide (CHX) was obtained from Sigma-Aldrich (St Louis, MO, USA). Primary antibodies specific for phospho-PERK (#3192), phospho-eIF2α (#3398), Bip (#3183), phospho-JNK (#4668), FTH (#4393) and β-actin (#4970) were purchased from Cell Signaling Technology (Beverly, MA, USA). Phospho-IRE1α (#ab48187) was purchased from Abcam (Cambridge, MA, USA). NOCA4 (#abs134557) was purchased from Absin (Shanghai, China). Transferrin receptor (#ab84036) was purchased from Proteintech (Rosemont, IL, USA). ATF4 (#60035-1-Ig) was purchased from Proteintech (Rosemont, IL, USA). And antibodies for p62 (#PM045), LC3 (#PM036), ATG5 (#M153-3) and the second-antibody specific for Rabbit and Mouse were purchased from MBL International Corporation (Woburn, MA, USA).
Cell culture and treatments
-
AML-12 cells were grown in DMEM/F12 medium (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum and 1% ITS without antibiotics. L02 cells were grown in RPMI 1640 medium (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum without antibiotics. All cells were cultured in a humidified incubator with 5% CO2 at 37 °C. Cells were grown in 6-well plates for 24 h to 40%−50% confluence. The medium was then changed and cells were incubated in fresh medium with different agents.
Western blotting
-
Cell lysates were prepared in ice-cold RIPA lysis buffer containing protease inhibitors and phosphatase inhibitors (Sigma-Aldrich). After the samples were centrifuged, their protein concentrations were determined using a BCA protein quantitative assay kit (Solarbio Life Science, Beijing, China). Equal amounts of protein samples were separated by SDS-PAGE and transferred to nitrocellulose membranes (0.2 μm, Merck Millipore, USA). Membranes were blocked with 5% skim milk PBS for 1 h at room temperature, and then performed with the primary antibody at 4 °C overnight with gentle shaking. Signals were visualized by enhanced chemiluminescence (Fisher/Pierce, Rockford, IL, USA; 32106) and recorded on X-ray film (Eastman Kodak Company, Rochester, NY, USA; XBT-1). Bands were quantified with Image J software, normalized to β-actin.
Crystal violet staining
-
To assess cell viability, after drug treatment, the medium was removed and cells were fixed in 1% glutaraldehyde solution in PBS for 15 min, and then stained with 0.02% aqueous crystal violet solution for 30 min. After washing with PBS, stained cells were solubilized with 75% ethanol. The absorbance at 570 nm with the reference filter 405 nm was evaluated using a microplate reader (Thermo).
Measurement of intracellular Fe2+ and lipid peroxides
-
The intracellular Fe2+ and lipid peroxides were assessed using FerroOrange (#F374, DojinDo, Japan) and Liperfluo (#L248, DojinDo, Japan), respectively. In brief, the cells were treated with drugs for a certain period of time and washed twice with PBS to remove residues. Then cells were treated with 1 μM FerroOrange or 1 μM Liperfluo with PBS for 30 min at 37 °C. After incubation, FerroOrange or Liperfluo was removed by washing three times with PBS, followed by measurement with a fluorescence microplate reader, respectively.
RNA interference
-
siRNAs targeting PERK (sc-36213) and IRE1α (sc-40705) were purchased from Santa Cruz Biotechnologies. The cells were transfected with specific or non-targeting siRNA for 24 h using INTERFER in siRNA transfection reagent (polyplus-transfection, Inc. 409-10) according to the manufacturer's instructions, and then were used for subsequent experiments.
Statistical analysis
-
Statistical analysis was performed using GraphPad Prism. Data are presented as mean ± standard deviation. Statistical analysis was assessed by means of one-way analysis of variance (ANOVA) with appropriate post hoc comparisons. p < 0.05 (*) was considered statistically significant.
-
In this study, we evaluated the effect of exogenous AA on cell viability in L02 and AML12 cell lines. A dose-dependent reduction was observed in cell viability upon treatment with increasing concentrations of AA (Fig. 1a & b). To further investigate the role of ferroptosis in AA-induced cell death, we used the ferroptosis inhibitor liproxstatin-1 (lip-1) and detected that pretreatment with lip-1 significantly alleviated AA-induced cytotoxicity and increased cell viability (Fig. 1c & d, ** p < 0.01). Additionally, lip-1 pretreatment was found to reduce the elevation of iron content and lipid peroxides caused by AA (Fig. 1e−h). Taken together, these findings support the involvement of ferroptosis in AA-induced hepatocyte death.

Figure 1.
AA induces ferroptosis in hepatocytes at different levels. (a) L02 and (b) AML12 were treated with indicated AA for 24 h. The relative viability of cells was determined by crystal violet staining. Cell viability obtained after (c) L02 and (d) AML12 cells were pretreated with 1 μM Lip-1 for 6 h and then treated with the indicated AA for 24 h. The data are presented as mean ± SD. ** p < 0.01 versus the control group. # p < 0.05 versus the AA treatment group. Lip-1 blocked AA-induced upregulation of Fe2+, quantified in (e) L02 and (f) AML12 cells using the FerroOrange kit. Lip-1 blocked AA-induced upregulation of lipid peroxide levels, quantified in (g) L02 and (h) AML12 cells using the Liperfluo kit. Values are presented as mean ± SD. * p < 0.05, ** p < 0.01 and *** p < 0.001 versus the control group. # p < 0.05, ## p < 0.01 and ### p < 0.01 versus the AA treatment group.
AA upregulates TfR1, thereby transporting more iron into cells
-
Recent research suggests that TfR1 could be a specific marker for ferroptosis[19]. To determine the source of iron during AA-induced ferroptosis, we first examined TfR1 expression in cells. As shown in Fig. 2, compared to untreated cells, AA treatment significantly increased TfR1 expression in L02 and AML12 cells, indicating that AA upregulates TfR1 to facilitate iron uptake. GPX4 is the primary enzyme that protects cell membranes from peroxidative damage, and its inactivation can induce ferroptosis[7,20,21]. Therefore, GPX4 expression changes in cells was also examined. Consistently, AA treatment caused a significant decrease in GPX4 expression (Fig. 2, * p < 0.05, ** p < 0.01). These results suggest that AA increases intracellular iron levels by upregulating TfR1 expression and alters cellular redox homeostasis, leading to decreased GPX4 expression.

Figure 2.
AA up-regulates TfR1 expression to transport more iron into cells, while also down-regulating GPX4. Western blotting analysis of TfR1 and GPX4 were performed 24 h after AA treatment in (a) L02 and (b) AML12 cells. The relative intensity of TfR1 was analyzed after AA treatment. Values are presented as means ± SD. * p < 0.05, ** p < 0.01, *** p < 0.001 versus the control group.
NCOA4-mediated ferrintinophagy exacerbates AA-induced ferroptosis by degrading ferritin
-
To investigate the mechanism of Fe2+ release, we next detected the expression of FTH. It was significantly increased in L02 and AML12 cells treated with AA, along with NCOA4, potentially due to the upregulation of TfR1 and the regulation of NCOA4 transcriptional level by AA (Fig. 3a & b). To further explain whether the degradation of ferritin lead to the increase of Fe2+ in the labile iron pool of cells, we examined the changes in autophagy-related markers and found that AA treatment upregulated the expression of ATG5 and increased the conversion of LC3 I to II (Fig. 3a & b). Furthermore, the autophagy flux inhibitor CQ was observed to reverse the decrease in cell viability caused by AA (Fig. 3c & d), indicating that AA activated autophagy. In addition, the levels of p62, LC3 II, NCOA4, and FTH were increased to a greater extent in AA-treated cells in the presence of CQ (Fig. 3e & f). Although CQ has been reported to disrupt transferrin flux[22], it caused a similar, or even greater degree of FTH accumulation in AA-treated L02 and AML12 cells, suggesting AA activates ferrintinophagy, leading to the degradation of FTH in both cell lines. To further demonstrate our findings, we examined the effect of AA on FTH expression in the presence of the protein synthesis inhibitor cycloheximide (CHX). AA treatment reduced FTH in the presence of CHX, supporting its role in activating ferritinophagy. These results suggest that AA-induced ferroptosis involves NCOA4-mediated ferritinophagy by accelerating FTH degradation.
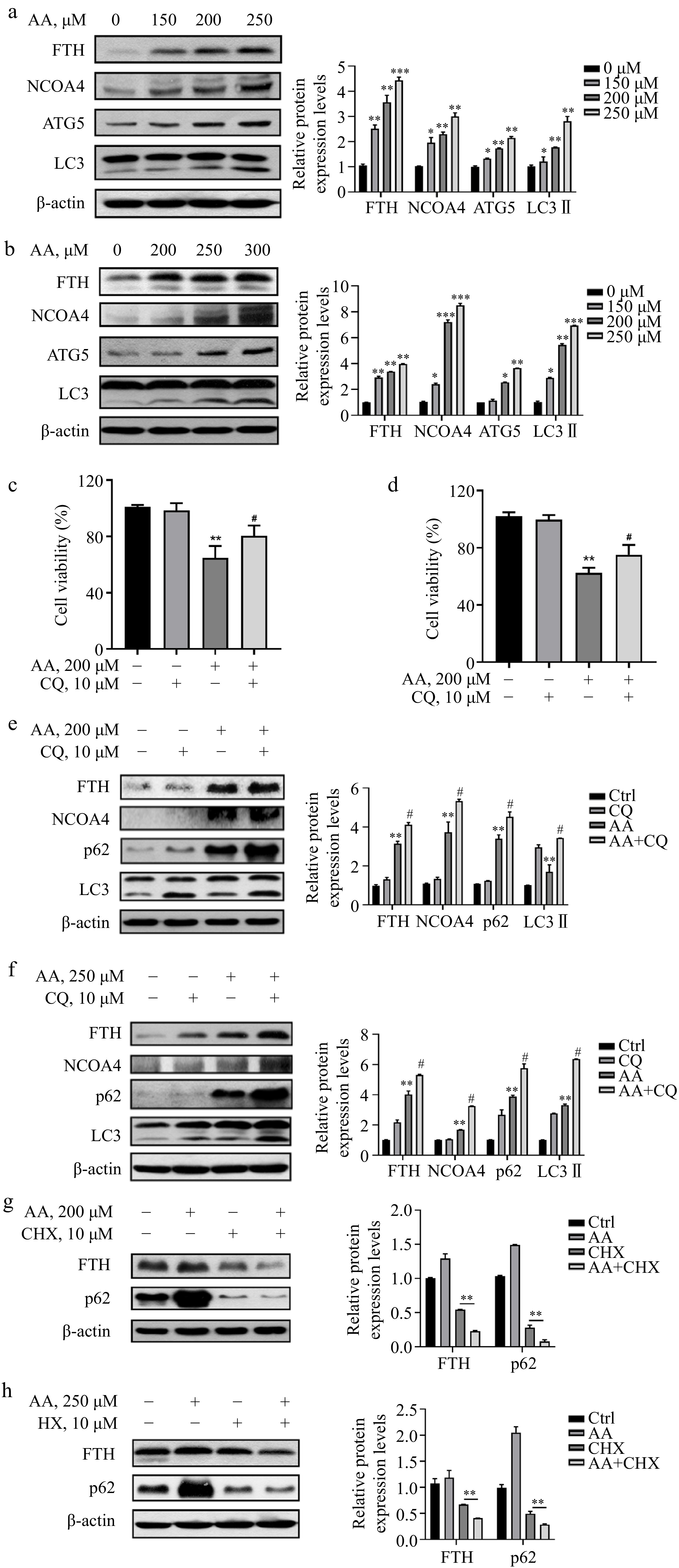
Figure 3.
AA increases free iron levels in labile iron pools by activating ferritinophagy. AA activated the expression of ferritinophagy-related proteins. The cells were treated with AA for 24 h, and the expression of FTH, NCOA4, ATG5 and LC3 in (a) L02 and (b) AML12 cells were detected by Western blotting. Inhibition of autophagy alleviated AA-induced ferroptosis. Cells were treated with AA and/or the autophagy flux inhibitor CQ, and the cell viability of (c) L02 and (d) AML12 was measured. Expression of ferritinophagy-related proteins. The cells were treated with AA and/or CQ for 24 h, and the lysates of (e) L02 and (f) AML12 cells were subjected to Western blot analysis to analyze the changes of FTH, NCOA4, p62 and LC3 protein expression. Expression of FTH and p62 after inhibition of protein synthesis. After the cells were treated with CHX, they were treated with AA for a certain period of time. Changes in FTH and p62 expression in (g) L02 and (h) AML12 cells were detected by Western blot to further demonstrate the activation of ferritinophagy. β-actin served as a loading control. The intensity of each protein expression band was quantified by densitometry normalized to β-actin. * p < 0.05, ** p < 0.01 and *** p < 0.001 versus the control group. # p < 0.05 and ## p < 0.01 versus the AA treatment group.
ER stress participates involved in AA-induced hepatocyte death
-
Studies have demonstrated that excessive consumption of PUFAs in the diet can cause endoplasmic reticulum (ER) stress. In liver diseases such as non-alcoholic steatohepatitis (NASH), the proportion of ω-6 PUFAs is notably increased. Therefore, the regulation of ER stress is deemed an important target for treating NASH[23,24]. Based on this, we postulated that ER stress might be implicated in AA-induced ferroptosis. Consistent with our hypothesis, the result showed that AA prompted ER stress in a dose-dependent manner in hepatocytes L02 and AML12, involving two branches of PERK and IRE1. It was evidenced by the elevated protein expression levels of Bip, p-PERK, p-eIF2α, ATF4, p-IRE1 and p-JNK (Fig. 4a & b). In conclusion, these findings indicate that PERK- and IRE1-mediated ER stress plays a role in AA-induced hepatocyte ferroptosis.
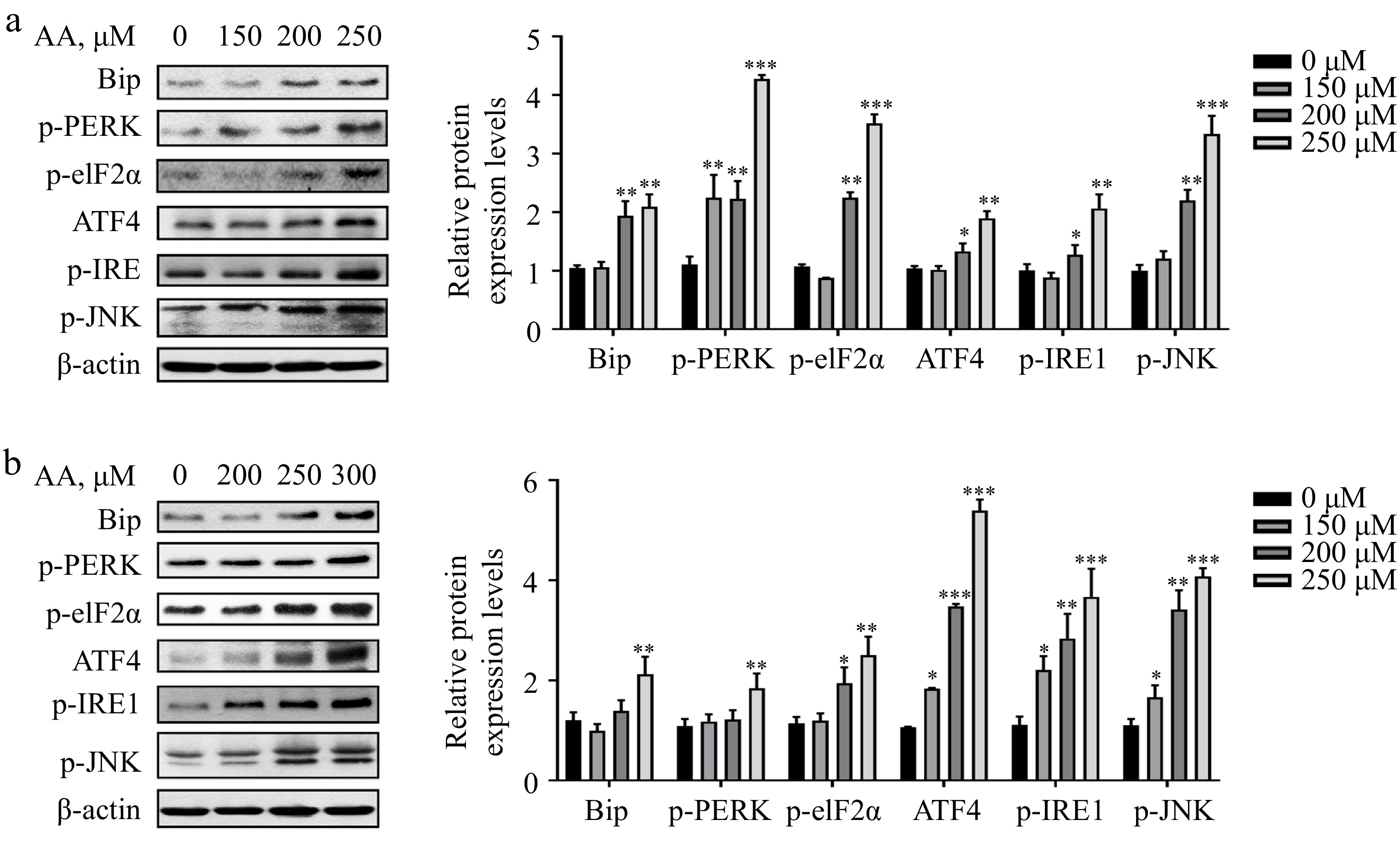
Figure 4.
Activated ER stress is involved in AA-induced ferroptosis. Western blotting analysis of Bip, p-PERK, p-eIF2α, ATF4, p-IRE1α and p-JNK were performed in (a) L02 and (b) AML12 cells. Bar chart shows relative quantitative levels of each protein. β-actin was used as a loading control. Data are presented as mean ± SD. * p < 0.05, ** p < 0.01 and *** p < 0.001 versus the control group.
Blockade of PERK-mediated ER stress alleviates AA-induced ferroptosis.
-
We proceeded to investigate the role of ER stress in AA-induced ferroptosis. Firstly, to assess the contribution of PERK signaling in AA-induced ferroptosis, we pre-treated cells with GSK2606414 (an inhibitor of PERK) prior to AA exposure. Figure 5a & b demonstrate that 1 μM GSK pre-treatment reversed AA-induced reduction in cell viability in L02 and AML12 cells. In addition, RNA interference is utilized to evaluate cell viability changes after PERK knockdown in L02 cells, revealing that PERK knockdown similarly reversed AA-induced reduction of cell viability (Fig. 5c). Next, we assessed changes in ferritinophagy markers in cells after inhibiting the PERK signaling pathway. Both GSK pre-treatment or PERK-inhibited led to increased accumulation of FTH and NCOA4, and decreased LC3I to LC3II conversion in AA-treated cells, indicating weakened ferritinophagy (Fig. 5d & e). In conclusion, our findings suggest that ER stress is upstream of ferritinophagy, and inhibiting PERK signaling can alleviate ferritinophagy by reducing ER stress, ultimately alleviating AA-induced cellular ferroptosis.

Figure 5.
AA triggers lethal ferritinophagy by activating the PERK signaling pathway. Cell viability of (a) AML12 and (b) L02 cells pretreated with 1 μM GSK for 2 h and then stimulated with AA for 24 h. (c) Effect of knockdown of PERK on AA-induced ferroptosis in L02 cells. Western blotting analysis of p-eIF2α, ATF4, FTH, NCOA4, p62 and LC3 were performed in (d) AML12 and (e) L02 cells treated with GSK or PERK-si and AA. The data are presented as mean ± SD. ** p < 0.01 and *** p < 0.001 versus the control group. # p < 0.05 and ## p < 0.01 versus the AA treatment group.
Inhibition of IRE1 exacerbates AA-induced ferrintinophagy by further activating the PERK signaling pathway
-
To investigate the specific role of IRE1 in AA-induced ferroptosis, we utilized the IRE1 inhibitor 4μ8C to observe changes in cell viability after IRE1 inhibition. Surprisingly, inhibiting IRE1 not only failed to alleviate cellular ferroptosis, but it further decreased cell viability (Fig. 6a−c). Previous studies have demonstrated that PERK and IRE1 can alleviate ER stress by coordinating the unfolded protein response (UPR)[25]. However, in unresolved ER stress, the PERK signaling pathway is continuously activated while the IRE1 signaling pathway is paradoxically attenuated, ultimately promoting cell death[25]. Therefore, the two branches of ER stress appear to play distinct roles in AA-induced ferroptosis. Specifically, IRE1 is activated as a protective signal of cells, and its inhibition exacerbates ferroptosis. Building on previous findings, we wondered whether IRE1 inhibition could exacerbate ferritinophagy by further activating the PERK signaling pathway. As such, the changes in the expression of related proteins were examined. As depicted in Fig. 6d & e, inhibition of IRE1 did activate the PERK signaling pathway, as evidenced by the down-regulation of p-IRE1 and up-regulation of p-eIF2α. Furthermore, the decreased accumulation of FTH, NCOA4, and p62, as well as the increased conversion of LC3I to LC3II, indicated further activation of ferritinophagy. These findings support the critical role played by IRE1 in ER stress-induced ferroptosis, where it functions as a protective signal in cells. Additionally, inhibiting IRE1 could exacerbate AA-induced ferritinophagy by further activating the PERK signaling pathway.

Figure 6.
Inhibition of IRE1 instead activates the PERK signaling pathway, which in turn exacerbates AA-induced ferritinophagy. Cell viability of (a) AML12 and (b) L02 cells pretreated with 25 μM 4μ8C for 2 h and then stimulated with AA for 24 h. (c) Effect of knockdown of IRE1 on AA-induced ferroptosis in L02 cells. Western blotting analysis of p-eIF2α, ATF4, FTH, NCOA4, p62 and LC3 were performed in (d) AML12 and (e) L02 cells treated with GSK or PERK-si and AA. The data are presented as mean ± SD. ** p < 0.01 and *** p < 0.001 versus the control group. # p < 0.05 and ## p < 0.01 versus the GSK group or PERK-si group.
-
ω-6 and ω-3 PUFAs are essential fatty acids that play important roles in energy storage and production, as well as being crucial components of biological membranes and signaling molecules[26,27]. Many lipids derived from ω-6 PUFAs have pro-inflammatory functions, while those derived from ω-3 PUFAs possess anti-inflammatory properties[28,29]. AA, as a typical ω-6 PUFA, has a wide range of biological roles but its metabolic abnormalities are implicated in various liver diseases[3,4]. In this study, we conducted an in-depth investigation into the mechanism of iron metabolism disturbance in AA-induced ferroptosis.
Recent studies suggest that ferroptosis may be an autophagy-dependent cell death. Ferroptosis inducers such as erastin and RSL3 lead to the accumulation of autophagosomes, while autophagy-deficient cells exhibit impaired ferroptosis under various stimulations[9]. Multiple forms of selective autophagy, including NCOA4-mediated ferritinophagy, RAB7A-dependent lipophagy, and heat shock protein 90-dependent chaperone autophagy, have been shown to promote ferroptosis by increasing iron load and impairing antioxidant systems[12−14]. Especially for ferrintinophagy, high levels of labile iron ensure rapid accumulation of intracellular ROS, which is critical for ferroptosis[30]. Our investigation into the role of autophagy in AA-induced ferroptosis revealed that inhibition of autophagy by CQ could limit AA-induced ferroptosis in hepatocytes, indicating that AA activates autophagy in hepatocytes. Furthermore, AA increases cellular iron uptake by upregulating TfR1 expression, which results in an increase in intracellular FTH expression. At the same time, AA also accelerates the degradation of ferritin, leading to an increase in Fe2+ levels in cells. These results explain the hyperactivation of autophagy during AA-induced toxicity. However, further investigation is needed to determine if other forms of autophagy are involved in AA-induced ferroptosis.
In recent years, researchers have found that ER stress may play an important role in ferroptosis[15−17], but the relationship between ER stress and ferroptosis and its specific mechanism remain unclear. ER stress can affect different processes in various cells. Pathological conditions such as energy or nutrient deprivation and altered redox state can disrupt the homeostasis of the ER, leading to protein misfolding and activation of a series of signaling pathways, which is called the unfolded protein response (UPR)[31]. The UPR plays a key role in the maintenance of cellular homeostasis and is the key to the normal physiological functions of cells[31]. It has been reported that the expression level of ATF4 is increased in erastin-induced ferroptosis, and ATF4 can enhance the expression of SLC7A11 through a feedback loop, thereby limiting ferroptosis[32]. On the other hand, the production of ROS during ferroptosis is related to the PERK/eIF2α signaling pathway[15]. Additionally, ER stress promotes ferroptosis in ulcerative colitis and exposure to cigarette smoke, a mechanism involving heme oxygenase-1[33−35]. It has also been reported that the activation of ER stress participates in the synergistic effect of ferroptosis and apoptosis through the CHOP-PUMA pathway[36,37]. In addition, under pathological conditions, activated ER stress not only does not inhibit ferroptosis, but may aggravate the occurrence of ferroptosis, indicating that under persistent or severe disease conditions, ER stress can not only initiate apoptosis and autophagy signaling pathways but also cause ferroptosis. Our study shows that AA activates ER stress, including both PERK and IRE1 signaling pathways. Moreover, AA-activated ER stress can affect cellular ferroptosis by regulating ferritinophagy. Inhibition of PERK alleviated AA-induced ferroptosis in hepatocytes by attenuating ferritinophagy, whereas inhibition of IRE1 aggravated AA-induced ferritinophagy by further activating PERK signaling.
The abnormal metabolism of AA is related to the occurrence and development of many metabolic diseases in the human body, which will destroy the homeostasis of lipid metabolism and further lead to inflammation or cancer. It remains to be determined to what extent other cell types produce the AA-induced ferroptosis we observed in hepatocytes by activating ER stress and ferrinrinophagy. Therefore, future studies should focus on evaluating the efficacy of inhibiting ER stress alone, or in combination with ferroptosis inhibitors, and even related validation in animal models to induce the best and beneficial results.
-
In conclusion, our study confirmed that AA induces hepatocyte ferroptosis in a dose-dependent manner, which is closely related to the activation of ER stress and ferritinophagy. Specifically, exogenous AA induces ferroptosis by upregulating the expression of TfR1 on the cell membrane and accelerating the degradation of ferritin, resulting in iron overload. The investigation into the effect of different branches of ER stress on ferritinophagy provides new insights into AA-induced ferroptosis (Fig. 7).

Figure 7.
Potential signaling pathways for AA-induced ferroptosis in hepatocytes. The polyunsaturated fatty acid AA upregulates the expression of TfR1 on the cell membrane surface and activates ferritinophagy, thereby increasing the level of Fe2+ in the cellular labile iron pool. In addition, AA-induced ER stress is located upstream of ferritinophagy, and its two branches, PERK and IRE1 signaling pathways, play different roles in regulating ferritinophagy.
This study was supported by a grant from the National Key Research and Development Program of China (2022YFF1100205).
-
The authors declare that they have no conflict of interest.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press on behalf of China Agricultural University, Zhejiang University and Shenyang Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang H, Han K, Yin S, Fan L, Hu H, et al. 2023. Distinct roles of the IRE1α arm and PERK arm of unfolded protein response in arachidonic acid-induced ferroptosis in hepatocytes. Food Innovation and Advances 2(3):184−192 doi: 10.48130/FIA-2023-0020 

Distinct roles of the IRE1α arm and PERK arm of unfolded protein response in arachidonic acid-induced ferroptosis in hepatocytes
- Received: 31 March 2023
- Accepted: 13 June 2023
- Published online: 05 July 2023
Abstract: Ferroptosis is a distinct form of cell death that is driven by iron-dependent phospholipid peroxidation. Polyunsaturated fatty acids (PUFAs), particularly arachidonic acid (AA) and adrenal acid (AdA), are most prone to lipid peroxidation, which induces ferroptosis and affects the function of cell membranes. In this study, we discovered that AA induces ferritinophagy in hepatocytes, a selective form of autophagy that degrades ferritin, triggering unstable iron overload. Mechanistically, AA enhances cellular uptake of bound iron by up-regulating transferrin receptor 1 (TfR1). Additionally, AA induces endoplasmic reticulum stress (ER stress) and simultaneously activates two of its branches, pancreatic ER kinase (PERK) and inositol-requiring enzyme 1 (IRE1). Notably, PERK and IRE1 appear to play distinct roles in inducing ferritinophagy. Inhibition of PERK reduced the AA-induced increase of Fe2+ by alleviating ferritinophagy, while inhibition of IRE1 further exacerbated ferroptosis by activating ferritinophagy. Furthermore, there seems to be an interaction between the signaling pathways of ER stress, and inhibition of IRE1 exacerbates AA-induced ferritinophagy by further activating the PERK signaling pathway, thereby exacerbating the extent of cell death. Collectively, our findings suggest that iron overload is involved in AA-induced hepatocyte ferroptosis and that this process is regulated by ER stress-mediated ferritinophagy. This study suggests potential therapeutic strategies for treating liver diseases related to lipid metabolism disorders by intervening in the ferroptosis process.
-
Key words:
- Hepatocyte /
- ER stress /
- Ferroptosis /
- Ferritinophagy /
- Polyunsaturated fatty