









-
Grapevines are among the most widely grown and economically important fruit crops globally. Grapes are used not only for wine making and juice, but also are consumed fresh and as dried fruit[1]. Additionally, grapes have been increasingly recognized as an important source of resveratrol (trans-3, 5,4'-trihydroxystilbene), a non-flavonoid stilbenoid polyphenol that in grapevine may act as a phytoalexin. In humans, it has been widely reported that dietary resveratrol has beneficial impacts on various aspects of health[2, 3]. Because of the potential value of resveratrol both to grapevine physiology and human medicine, resveratrol biosynthesis and its regulation has become an important avenue of research.
Similar to other stilbenoids, resveratrol synthesis utilizes key enzymes of the phenylpropanoid pathway including phenylalanine ammonia lyase (PAL), cinnamate 4-hydroxylase (C4H), and 4-coumarate-CoA ligase (4CL). In the final steps, stilbene synthase (STS), a type II polyketide synthase, produces trans-resveratrol from p-coumaroyl-CoA and malonyl-CoA, while chalcone synthase (CHS) synthesizes flavonoids from the same substrates[4, 5]. Moreover, trans-resveratrol is a precursor for other stilbenoids such as cis-resveratrol, trans-piceid, cis-piceid, ε-viniferin and δ-viniferin[6]. It has been reported that stilbenoid biosynthesis pathways are targets of artificial selection during grapevine domestication[7] and resveratrol accumulates in various structures in response to both biotic and abiotic stresses[8−12]. This stress-related resveratrol synthesis is mediated, at least partialy, through the regulation of members of the STS gene family. Various transcription factors (TFs) participating in regulating STS genes in grapevine have been reported. For instance, MYB14 and MYB15[13, 14] and WRKY24[15] directly bind to the promoters of specific STS genes to activate transcription. VvWRKY8 physically interacts with VvMYB14 to repress VvSTS15/21 expression[16], whereas VqERF114 from Vitis quinquangularis accession 'Danfeng-2' promotes expression of four STS genes by interacting with VqMYB35 and binding directly to cis-elements in their promoters[17].
Aided by the release of the first V. vinifera reference genome assembly[18], genomic and transcriptional studies have revealed some of the main molecular mechanisms involved in fruit ripening[19−24] and stilbenoid accumulation[8, 25] in various grapevine cultivars. Recently, it has been reported that a root restriction treatment greatly promoted the accumulation of trans-resveratrol, phenolic acid, flavonol and anthocyanin in 'Summer Black' (Vitis vinifera × Vitis labrusca) berry development during ripening[12]. However, most of studies mainly focus on a certain grape variety, not to investigate potential distinctions in resveratrol biosynthesis among different Vitis genotypes.
In this study, we analyzed the resveratrol content in seven grapevine accessions and three berry structures, at three stages of fruit development. We found that the fruits of two wild, Chinese grapevines, Vitis amurensis 'Tonghua-3' and Vitis davidii 'Tangwei' showed significant difference in resveratrol content during development. These were targeted for transcriptional profiling to gain insight into the molecular aspects underlying this difference. This work provides a theoretical basis for subsequent systematic studies of genes participating in resveratrol biosynthesis and their regulation. Further, the results should be useful in the development of grapevine cultivars exploiting the genetic resources of wild grapevines.
-
For each of the seven cultivars, we analyzed resveratrol content in the skin, pulp, and seed at three stages of development: Green hard (G), véraison (V), and ripe (R) (Table 1). In general, we observed the highest accumulation in skins at the R stage (0.43−2.99 µg g−1 FW). Lesser amounts were found in the pulp (0.03−0.36 µg g−1 FW) and seed (0.05−0.40 µg g−1 FW) at R, and in the skin at the G (0.12−0.34 µg g−1 FW) or V stages (0.17−1.49 µg g−1 FW). In all three fruit structures, trans-resveratrol showed an increasing trend with development, and this was most obvious in the skin. It is worth noting that trans-resveratrol was not detectable in the skin of 'Tangwei' at the G or V stage, but had accumulated to 2.42 µg g−1 FW by the R stage. The highest amount of extractable trans-resveratrol (2.99 µg g−1 FW) was found in 'Tonghua-3' skin at the R stage.
Table 1. Resveratrol concentrations in the skin, pulp and seed of berries from different grapevine genotypes at green hard, véraison and ripe stages.
Structures Species Accessions or cultivars Content of trans-resveratrol (μg g−1 FW) Green hard Véraison Ripe Skin V. davidii Tangwei nd nd 2.415 ± 0.220 V. amurensis Tonghua-3 0.216 ± 0.041 0.656 ± 0.043 2.988 ± 0.221 Shuangyou 0.233 ± 0.062 0.313 ± 0.017 2.882 ± 0.052 V. amurensis × V. Vinifera Beibinghong 0.336 ± 0.076 1.486 ± 0.177 1.665 ± 0.100 V. vinifera Red Global 0.252 ± 0.051 0.458 ± 0.057 1.050 ± 0.129 Thompson seedless 0.120 ± 0.025 1.770 ± 0.032 0.431 ± 0.006 V. vinifera × V. labrusca Jumeigui 0.122 ± 0.016 0.170 ± 0.021 0.708 ± 0.135 Pulp V. davidii Tangwei 0.062 ± 0.006 0.088 ± 0.009 nd V. amurensis Tonghua-3 0.151 ± 0.066 0.324 ±0.104 0.032 ± 0.004 Shuangyou 0.053 ± 0.008 0.126 ± 0.044 0.041 ± 0.017 V. amurensis × V. Vinifera Beibinghong 0.057 ± 0.014 0.495 ± 0.068 0.087 ± 0.021 V. vinifera Red Global 0.059 ± 0.018 0.159 ± 0.013 0.027 ± 0.004 Thompson seedless 0.112 ± 0.016 0.059 ± 0.020 nd V. vinifera × V. labrusca Jumeigui 0.072 ± 0.010 0.063 ± 0.017 0.359 ± 0.023 Seed V. davidii Tangwei 0.096 ± 0.014 0.169 ± 0.028 0.049 ± 0.006 V. amurensis Tonghua-3 0.044 ± 0.004 0.221 ± 0.024 0.113 ± 0.027 Shuangyou nd 0.063 ± 0.021 0.116 ± 0.017 V. amurensis × V. Vinifera Beibinghong nd 0.077 ± 0.003 0.400 ± 0.098 V. vinifera Red Global 0.035 ± 0.023 0.142 ± 0.036 0.199 ± 0.009 Thompson seedless − − − V. vinifera × V. labrusca Jumeigui 0.077 ± 0.025 0.017 ± 0.004 0.284 ± 0.021 'nd' indicates not detected in samples, and '−' shows no samples are collected due to abortion. Analysis of RNA-Seq data
-
To gain insight into gene expression patterns influencing resveratrol biosynthesis in 'Tangwei' and 'Tonghua-3', we profiled the transcriptomes of developing berries at G, V, and R stages, using sequencing libraries representing three biological replicates from each cultivar and stage. A total of 142.49 Gb clean data were obtained with an average of 7.92 Gb per replicate, with average base Q30 > 92.5%. Depending on the sample, between 80.47%−88.86% of reads aligned to the V. vinifera reference genome (Supplemental Table S1), and of these, 78.18%−86.66% mapped to unique positions. After transcript assembly, a total of 23,649 and 23,557 unigenes were identified as expressed in 'Tangwei' and 'Tonghua-3', respectively. Additionally, 1,751 novel transcripts were identified (Supplemental Table S2), and among these, 1,443 could be assigned a potential function by homology. Interestingly, the total number of expressed genes gradually decreased from the G to R stage in 'Tangwei', but increased in 'Tonghua-3'. About 80% of the annotated genes showed fragments per kilobase of transcript per million fragments mapped (FPKM) values > 0.5 in all samples, and of these genes, about 40% showed FPKM values between 10 and 100 (Fig. 1a). Correlation coefficients and principal component analysis of the samples based on FPKM indicated that the biological replicates for each cultivar and stage showed similar properties, indicating that the transcriptome data was reliable for further analyses (Fig. 1b & c).
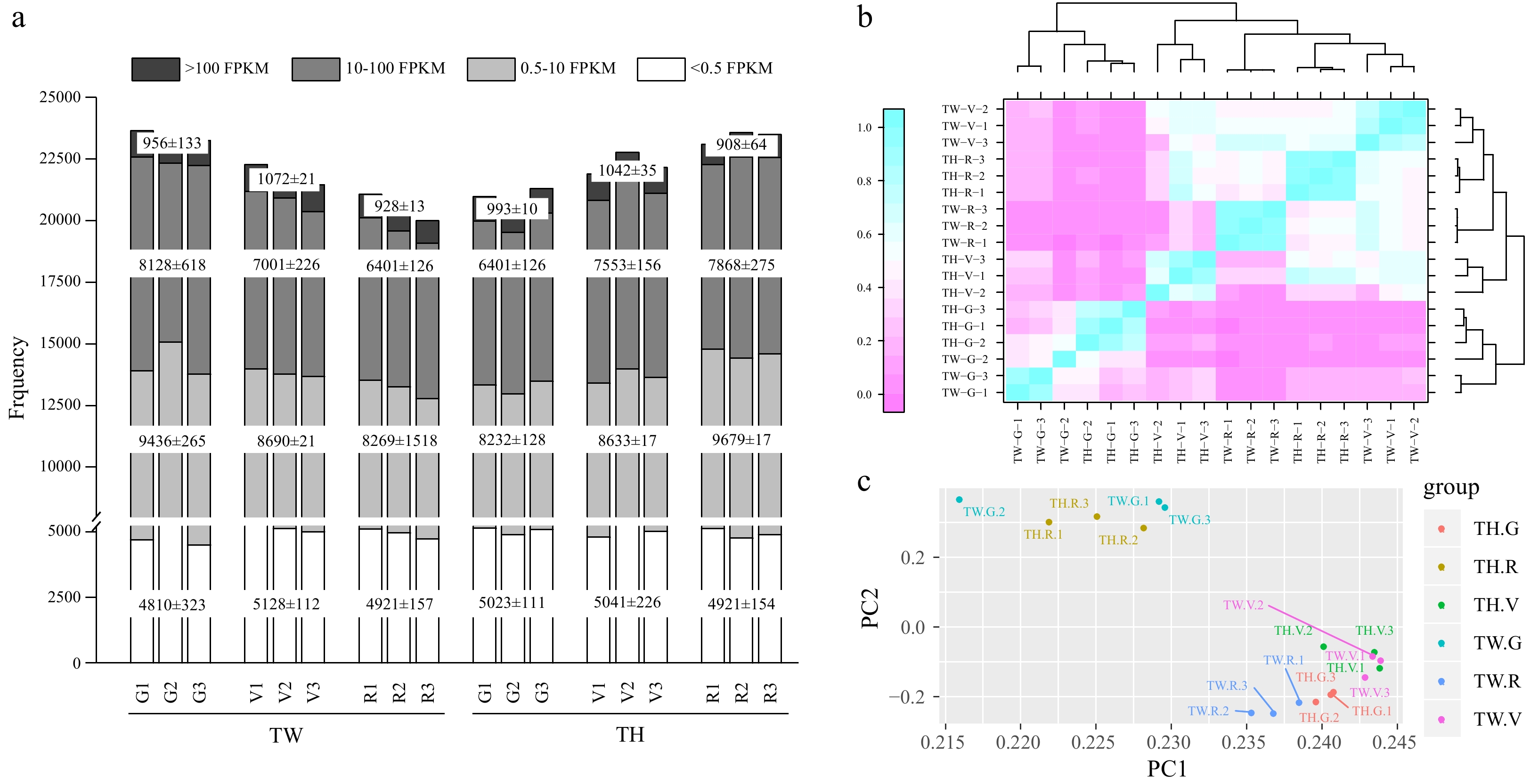
Figure 1. Properties of transcriptome data of 'Tangwei' (TW) and 'Tonghua-3' (TH) berry at green hard (G), véraison (V), and ripe (R) stages. (a) Total numbers of expressed genes with fragments per kilobase of transcript per million fragments mapped (FPKM) values; (b) Heatmap of the sample correlation analysis; (c) Principal component analysis (PCA) showing clustering pattern among TW and TH at G, V and R samples based on global gene expression profiles.
GO and KEGG pathway enrichment analysis of DEGs
-
By comparing the transcriptomes of 'Tangwei' and 'Tonghua-3' at the G, V and R stages, we identified 6,770, 3,353 and 6,699 differentially expressed genes (DEGs), respectively (Fig. 2a). Of these genes, 1,134 were differentially expressed between the two cultivars at all three stages (Fig. 2b). We also compared transcriptional profiles between two adjacent developmental stages (G vs V; V vs R) for each cultivar. Between G and V, we identified 1,761 DEGs that were up-regulated and 2,691 DEGs that were down-regulated in 'Tangwei', and 1,836 and 1,154 DEGs that were up-regulated or down-regulated, respectively, in 'Tonghua-3'. Between V and R, a total of 1,761 DEGs were up-regulated and 1,122 DEGs were down-regulated in 'Tangwei', whereas 2,774 DEGs and 1,287 were up-regulated or down-regulated, respectively, in 'Tonghua-3' (Fig. 2c). Among the 16,822 DEGs between the two cultivars at G, V, and R (Fig. 2a), a total of 4,570, 2,284 and 4,597 had gene ontology (GO) annotations and could be further classified to over 60 functional subcategories. The most significantly represented GO terms between the two cultivars at all three stages were response to metabolic process, catalytic activity, binding, cellular process, single-organism process, cell, cell part and biological regulation (Fig. 2d).
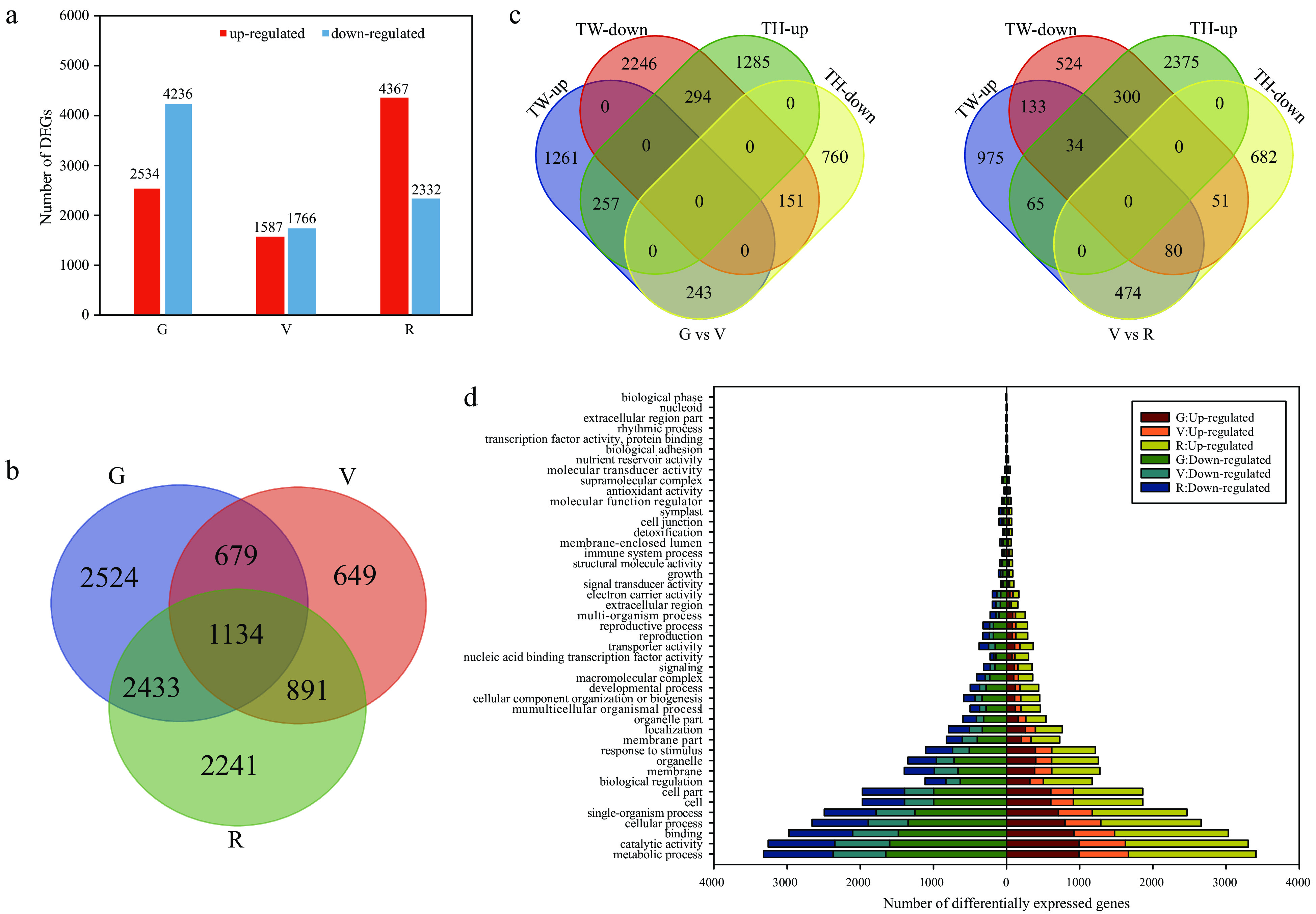
Figure 2. Analysis of differentially expressed genes (DEGs) at the green hard (G), véraison (V), and ripe (R) stages in 'Tangwei' (TW) and 'Tonghua-3' (TH). (a) Number of DEGs and (b) numbers of overlapping DEGs between 'Tangwei' and 'Tonghua-3' at G, V and R; (c) Overlap among DEGs between G and V, and V and R, for 'Tangwei' and 'Tonghua-3'; (d) Gene ontology (GO) functional categorization of DEGs.
We also identified 57 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways that were enriched, of which 32, 28, and 31 were enriched at the G, V, and R stages, respectively. Seven of the KEGG pathways were enriched at all three developmental stages: photosynthesis-antenna proteins (ko00196); glycine, serine and threonine metabolism (ko00260); glycolysis/gluconeogenesis (ko00010); carbon metabolism (ko01200); fatty acid degradation (ko00071); cysteine and methionine metabolism (ko00270); and valine, leucine and isoleucine degradation (ko00280) (Supplemental Tables S3−S5). Furthermore, we found that the predominant KEGG pathways were distinct for each developmental stage. For example, phenylpropanoid biosynthesis (ko00940) was enriched only at the R stage. Overall, the GO and KEGG pathway enrichment analysis showed that the DEGs in 'Tangwei' and 'Tonghua-3' were enriched for multiple biological processes during the three stages of fruit development.
DEGs related to phenylalanine metabolism
-
We then analyzed the expression of genes with potential functions in resveratrol and flavonoid biosynthesis between the two cultivars and three developmental stages (Fig. 3 and Supplemental Table S6). We identified 30 STSs, 13 PALs, two C4Hs and nine 4CLs that were differentially expressed during at least one of the stages of fruit development between 'Tangwei' and 'Tonghua-3'. Interestingly, all of the STS genes showed increasing expression with development in both 'Tangwei' and 'Tonghua-3'. In addition, the expression levels of STS, C4H and 4CL genes at V and R were significantly higher in 'Tonghua-3' than in 'Tangwei'. Moreover, 25 RESVERATROL GLUCOSYLTRANSFERASE (RSGT), 27 LACCASE (LAC) and 21 O-METHYLTRANSFERASE (OMT) DEGs were identified, and most of these showed relatively high expression at the G and V stages in 'Tangwei' or R in 'Tonghua-3'. It is worth noting that the expression of the DEGs related to flavonoid biosynthesis, including CHS, FLAVONOL SYNTHASE (FLS), FLAVONOID 3′-HYDROXYLASE (F3'H), DIHYDROFLAVONOL 4-REDUCTASE (DFR), ANTHOCYANIDIN REDUCTASE (ANR) and LEUCOANTHOCYANIDIN REDUCTASE (LAR) were generally higher in 'Tangwei' than in 'Tonghua-3'at G stage.
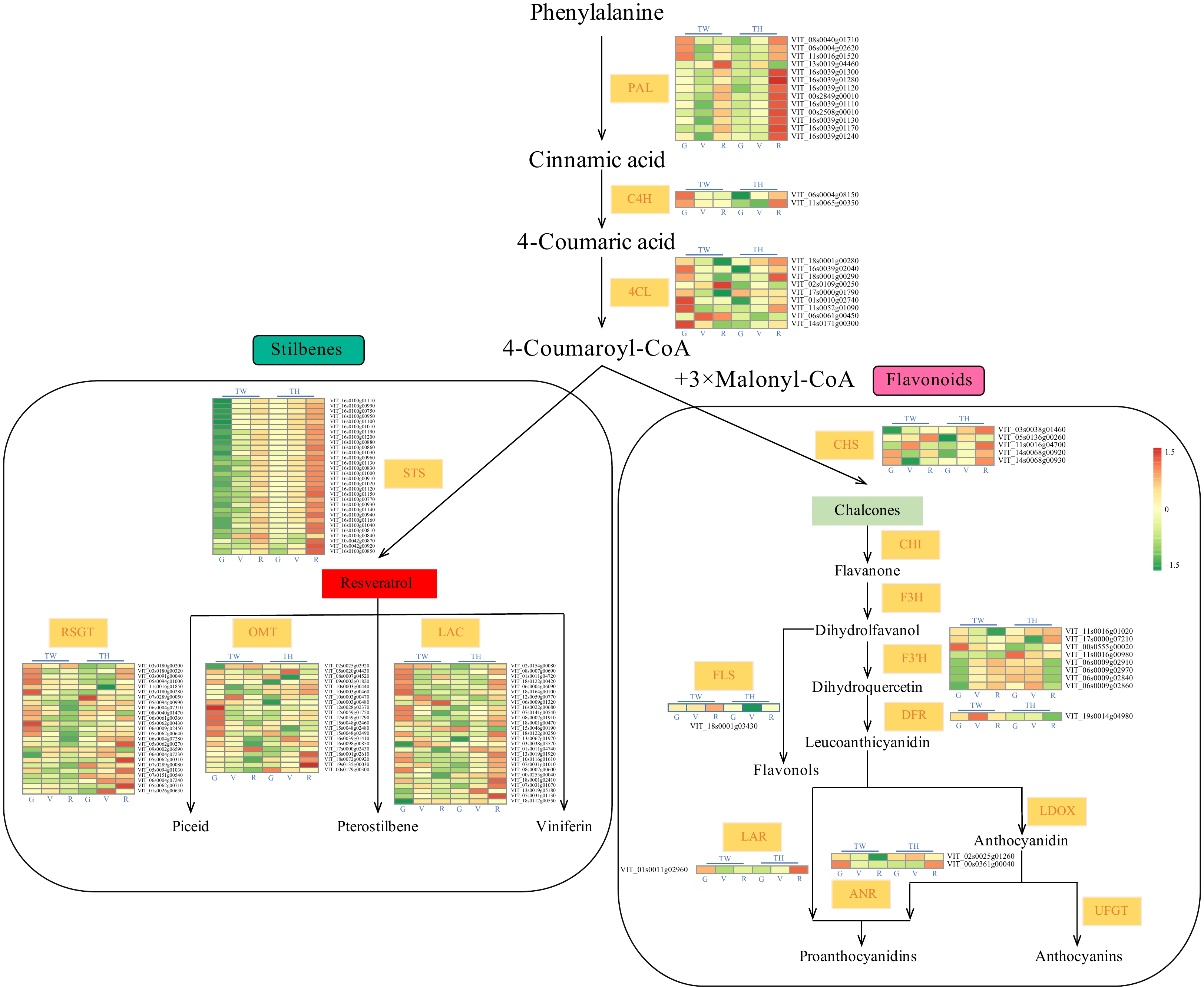
Figure 3. Expression of differentially expressed genes (DEGs) associated with phenylalanine metabolism. TW, 'Tangwei'; TH, 'Tonghua-3'. PAL, PHENYLALANINE AMMONIA LYASE; C4H, CINNAMATE 4-HYDROXYLASE; 4CL, 4-COUMARATE-COA LIGASE; STS, STILBENE SYNTHASE; RSGT, RESVERATROL GLUCOSYLTRANSFERASE; OMT, O-METHYLTRANSFERASE; LAC, LACCASE; CHS, CHALCONE SYNTHASE; CHI, CHALCONE ISOMERASE; F3H, FLAVANONE 3-HYDROXYLASE; FLS, FLAVONOL SYNTHASE; F3'H, FLAVONOID 3′-HYDROXYLASE; DFR, DIHYDROFLAVONOL 4-REDUCTASE; LAR, LEUCOANTHOCYANIDIN REDUCTASE; ANR, ANTHOCYANIDIN REDUCTASE; LDOX, LEUCOANTHOCYANIDIN DIOXYGENASE; UFGT, UDP-GLUCOSE: FLAVONOID 3-O-GLUCOSYLTRANSFERASE.
Differentially expressed TF genes
-
Among all DEGs identified in this study, 757 encoded potential TFs, and these represented 57 TF families. The most highly represented of these were the AP2/ERF, bHLH, NAC, WRKY, bZIP, HB-HD-ZIP and MYB families with a total of 76 DEGs (Fig. 4a). We found that the number of downregulated TF genes was greater than upregulated TF genes at G and V, and 48 were differentially expressed between the two cultivars at all three stages (Fig. 4b). Several members of the ERF, MYB, WRKY and bHLH families showed a strong increase in expression at the R stage (Fig. 4c). In addition, most of the TF genes showed > 2-fold higher expression in 'Tonghua-3' than in 'Tangwei' at the R stage. In particular, a few members, such as ERF11 (VIT_07s0141g00690), MYB105 (VIT_01s0026g02600), and WRKY70 (VIT_13s0067g03140), showed > 100-fold higher expression in 'Tonghua-3' (Fig. 4d).
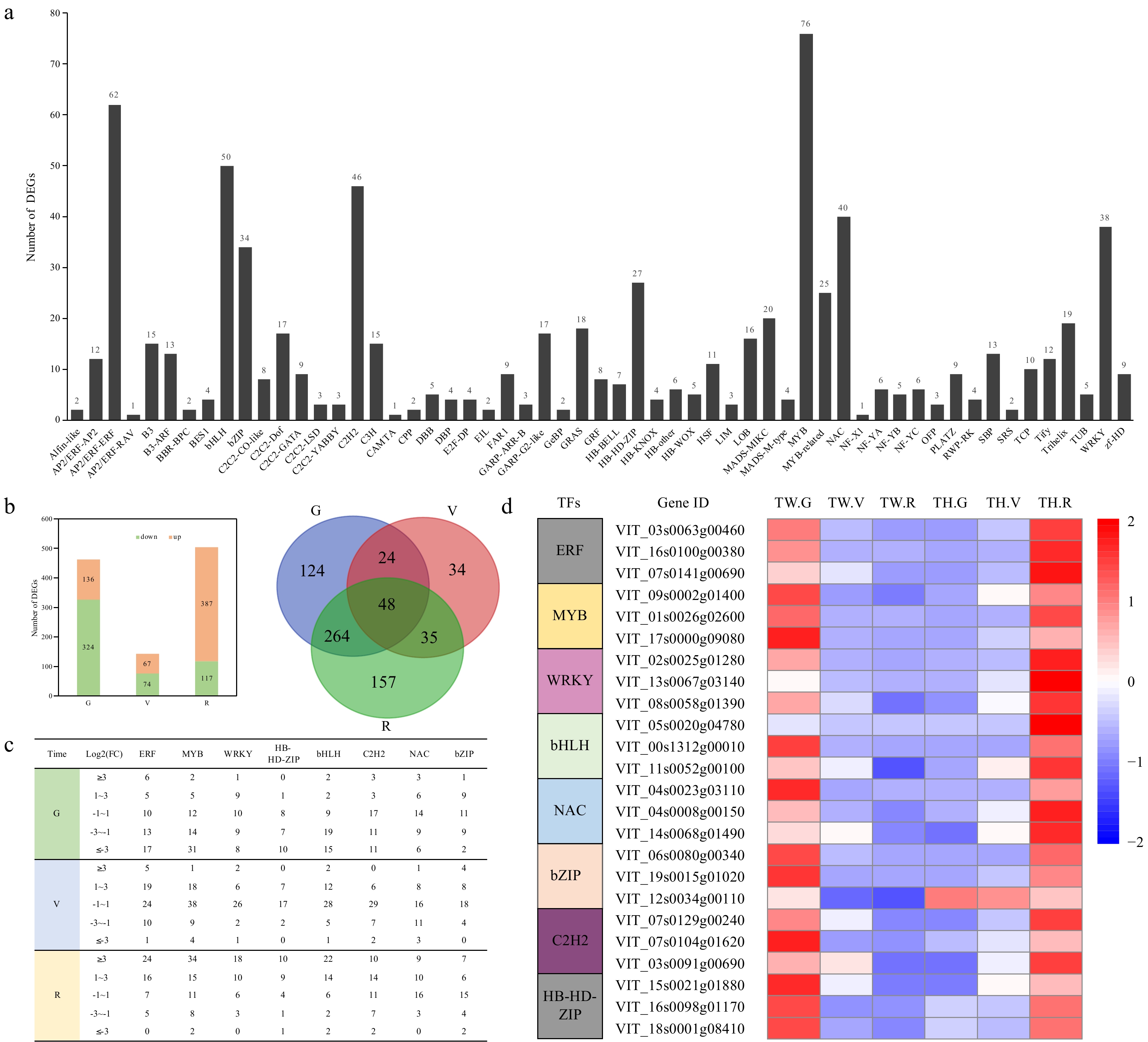
Figure 4. Differentially expressed transcription factor (TF) genes. (a) The number of differentially expressed genes (DEGs) in different TF families; (b) Number of differentially expressed TF genes, numbers of overlapping differentially expressed TF genes, and (c) categorization of expression fold change (FC) for members of eight TF families between 'Tangwei' and 'Tonghua-3' at green hard (G), véraison (V), and ripe (R) stages; (d) Heatmap expression profiles of the three most strongly differentially expressed TF genes from each of eight TF families.
Weighted gene co-expression network construction and identification of TF genes co-expressed with STSs
-
We constructed a gene co-expression network using the weighted gene co-expression network analysis (WGCNA) package, which uses a systems biology approach focused on understanding networks rather than individual genes. In the network, 17 distinct modules (hereafter referred to by color as portrayed in Fig. 5a), with module sizes ranging from 91 (antiquewhite4) to 1,917 (magenta) were identified (Supplemental Table S7). Of these, three modules (ivory, orange and blue) were significantly correlated with resveratrol content, cultivar ('Tonghua-3'), and developmental stage (R). The blue module showed the strongest correlation with resveratrol content (cor = 0.6, p-value = 0.008) (Fig. 5b). KEGG enrichment analysis was carried out to further analyze the genes in these three modules. Genes in the ivory module were significantly enriched for phenylalanine metabolism (ko00360), stilbenoid, diarylheptanoid and gingerol biosynthesis (ko00945), and flavonoid biosynthesis (ko00941), whereas the most highly enriched terms of the blue and orange modules were plant-pathogen interaction (ko04626), plant hormone signal transduction (ko04075) and circadian rhythm-plant (ko04712) (Supplemental Fig. S1). Additionally, a total of 36 genes encoding TFs including in 15 ERFs, 10 WRKYs, six bHLHs, two MYBs, one MADs-box, one HSF and one TRY were identified as co-expressed with one or more STSs in these three modules (Fig. 5c and Supplemental Table S8), suggesting that these TFs may participate in the STS regulatory network.
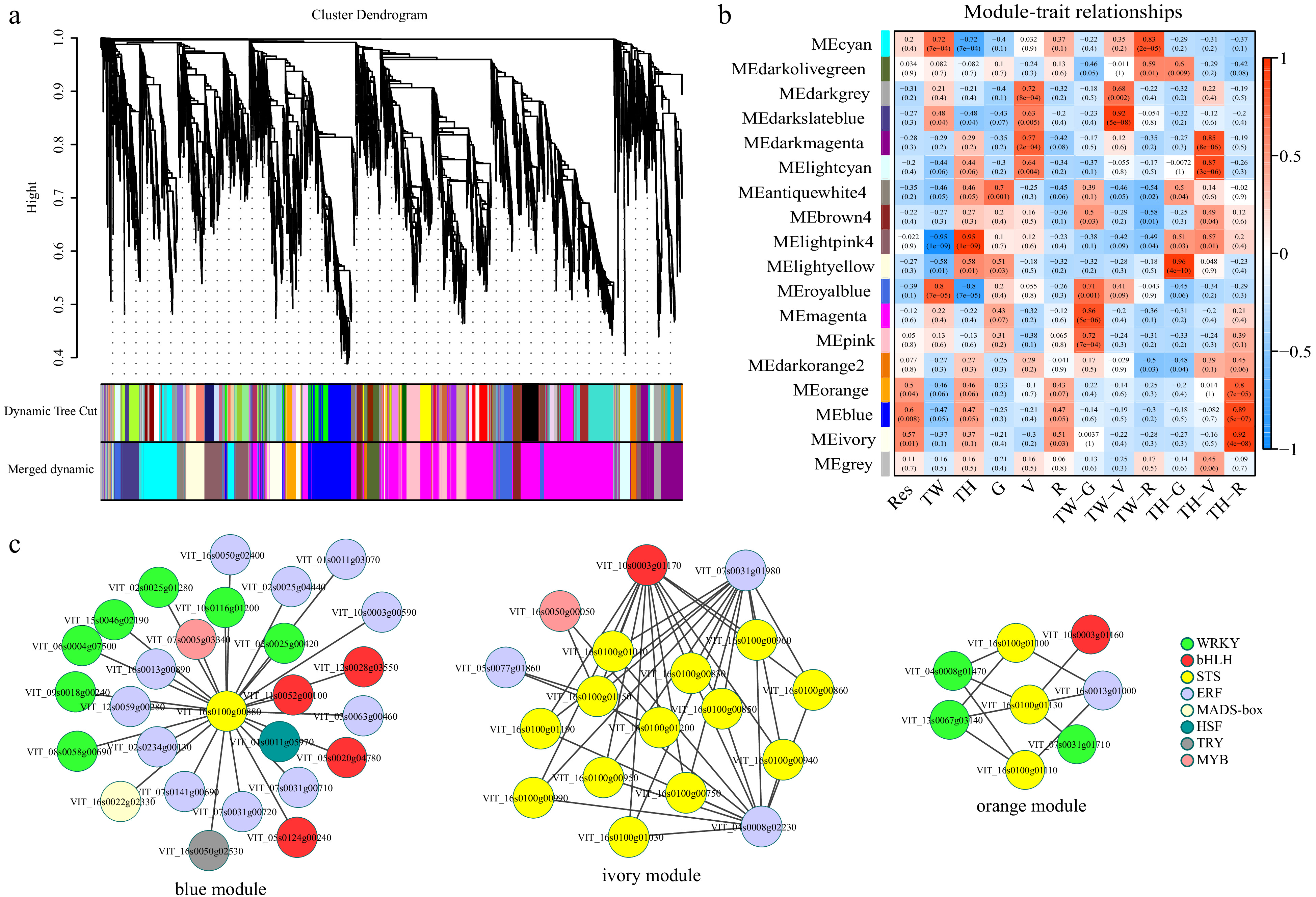
Figure 5. Results of weighted gene co-expression network analysis (WGCNA). (a) Hierarchical clustering tree indicating co-expression modules; (b) Module-trait relationship. Each row represents a module eigengene, and each column represents a trait. The corresponding correlation and p-value are indicated within each module. Res, resveratrol; TW, 'Tangwei'; TH, 'Tonghua-3'; (c) Transcription factors and stilbene synthase gene co-expression networks in the orange, blue and ivory modules.
Validation of RNA-seq data by RT-qPCR
-
To assess the reliability of the RNA-seq data, 12 genes determined to be differentially expressed by RNA-seq were randomly selected for analysis of expression via real-time quantitative PCR (RT-qPCR). This set comprised two PALs, two 4CLs, two STSs, two WRKYs, two LACs, one OMT, and MYB14. In general, these RT-qPCR results strongly confirmed the RNA-seq-derived expression patterns during fruit development in the two cultivars. The correlation coefficients between RT-qPCR and RNA-seq were > 0.6, except for LAC (VIT_02s0154g00080) (Fig. 6).
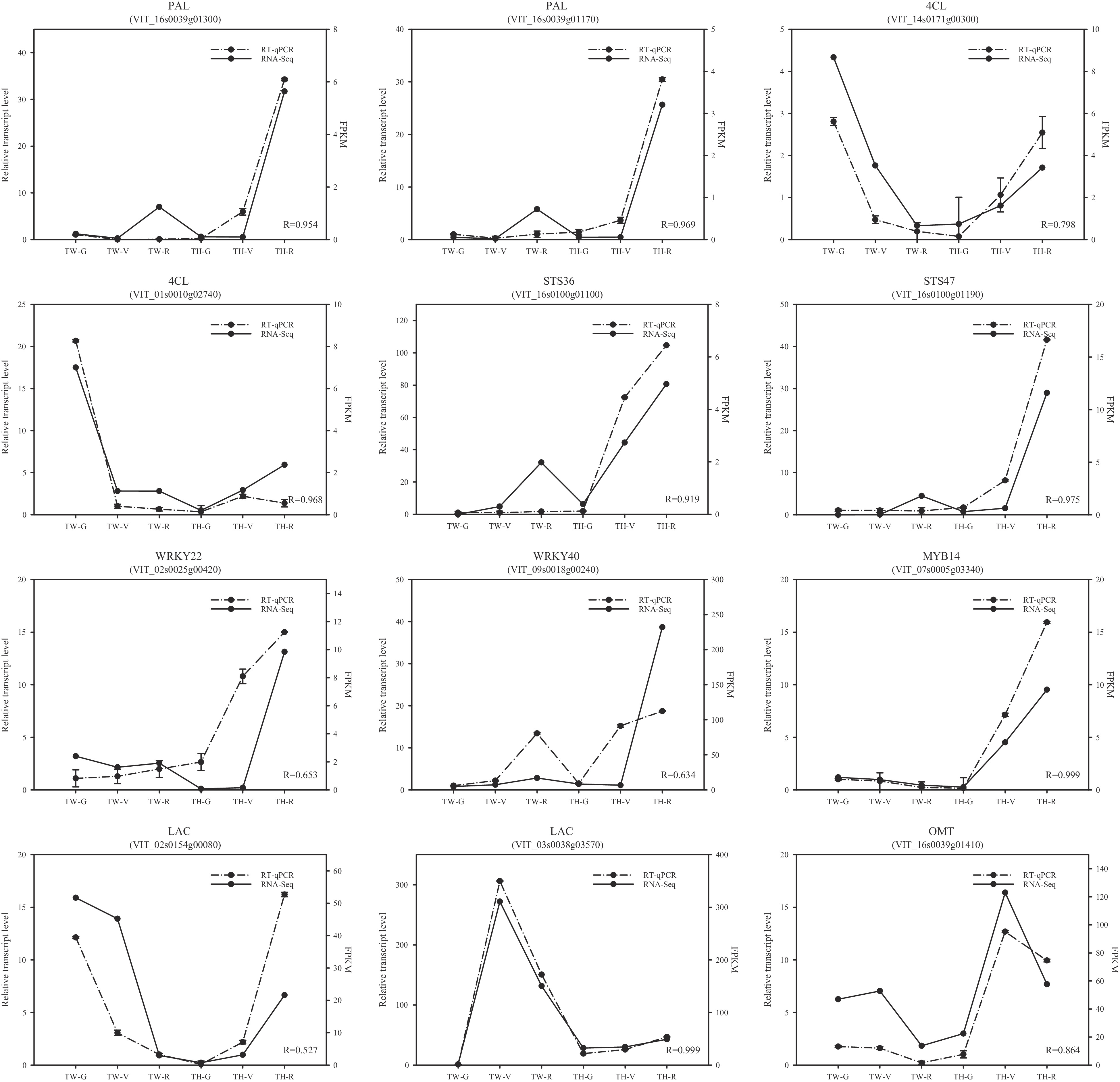
Figure 6. Comparison of the expression patterns of 12 randomly selected differentially expressed genes by RT-qPCR (real-time quantitative PCR) and RNA-seq. R-values are correlation coefficients between RT-qPCR and RNA-seq. FPKM, fragments per kilobase of transcript per million fragments mapped; TW, 'Tangwei'; TH, 'Tonghua-3'; G, green hard; V, véraison; R, ripe.
-
Grapevines are among the most important horticultural crops worldwide[26], and recently have been the focus of studies on the biosynthesis of resveratrol. Resveratrol content has previously been found to vary depending on cultivar as well as environmental stresses[27]. In a study of 120 grape germplasm cultivars during two consecutive years, the extractable amounts of resveratrol in berry skin were significantly higher in seeded cultivars than in seedless ones, and were higher in both berry skin and seeds in wine grapes relative to table grapes[28]. Moreover, it was reported that total resveratrol content constantly increased from véraison to complete maturity, and ultraviolet-C (UV-C) irradiation significantly stimulated the accumulation of resveratrol of berry during six different development stages in 'Beihong' (V. vinifera × V. amurensis)[9]. Intriguingly, a recent study reported that bud sport could lead to earlier accumulation of trans-resveratrol in the grape berries of 'Summer Black' and its bud sport 'Nantaihutezao' from the véraison to ripe stages[29]. In the present study, resveratrol concentrations in seven accessions were determined by high performance liquid chromatography (HPLC) in the seed, pulp and skin at three developmental stages (G, V and R). Resveratrol content was higher in berry skins than in pulp or seeds, and were higher in the wild Chinese accessions compared with the domesticated cultivars. The highest resveratrol content (2.99 µg g−1 FW) was found in berry skins of 'Tonghua-3' at the R stage (Table 1). This is consistent with a recent study of 50 wild Chinese accessions and 45 cultivars, which reported that resveratrol was significantly higher in berry skins than in leaves[30]. However, we did not detect trans-resveratrol in the skins of 'Tangwei' during the G or V stages (Table 1). To explore the reason for the difference in resveratrol content between 'Tangwei' and 'Tonghua-3', as well as the regulation mechanism of resveratrol synthesis and accumulation during berry development, we used transcriptional profiling to compare gene expression between these two accessions at the G, V, and R stages.
After sequence read alignment and transcript assembly, 23,649 and 23,557 unigenes were documented in 'Tangwei' and 'Tonghua-3', respectively. As anticipated, due to the small number of structures sampled, this was less than that (26,346) annotated in the V. vinifera reference genome[18]. Depending on the sample, 80.47%−88.86% of sequence reads aligned to a single genomic location (Supplemental Table S1); this is similar to the alignment rate of 85% observed in a previous study of berry development in Vitis vinifera[19]. Additionally, 1751 novel transcripts were excavated (Supplemental Table S2) after being compared with the V. vinifera reference genome annotation information[18, 31]. A similar result was also reported in a previous study when transcriptome analysis was performed to explore the underlying mechanism of cold stress between Chinese wild Vitis amurensis and Vitis vinifera[32]. We speculate that these novel transcripts are potentially attributable to unfinished V. vinifera reference genome sequence (For example: quality and depth of sequencing) or species-specific difference between Vitis vinifera and other Vitis. In our study, the distribution of genes based on expression level revealed an inverse trend from G, V to R between 'Tangwei' and 'Tonghua-3' (Fig. 1). Furthermore, analysis of DEGs suggested that various cellular processes including metabolic process and catalytic activity were altered between the two cultivars at all three stages (Fig. 2 and Supplemental Table S3−S5). These results are consistent with a previous report that a large number of DEGs and 100 functional subcategories were identified in 'Tonghua-3' grape berries after exposure to UV-C radiation[8].
Resveratrol biosynthesis in grapevine is dependent on the function of STSs, which compete with the flavonoid branch in the phenylalanine metabolic pathway. Among the DEGs detected in this investigation, genes directly involved in the resveratrol synthesis pathway, STSs, C4Hs and 4CLs, were expressed to significantly higher levels in 'Tonghua-3' than in 'Tangwei' during V and R. On the other hand, DEGs representing the flavonoid biosynthesis pathway were upregulated in 'Tangwei', but downregulated in 'Tonghua-3' (Fig. 3 and Supplemental Table S6). These expression differences may contribute to the difference in resveratrol content between the two cultivars at these stages. We note that 'Tangwei' and 'Tonghua-3' are from two highly diverged species with different genetic backgrounds. There might be some unknown genetic differences between the two genomes, resulting in more than 60 functional subcategories being enriched (Fig. 2d) and the expression levels of genes with putative roles in resveratrol biosynthesis being significantly higher in 'Tonghua-3' than in 'Tangwei' during V and R (Fig. 3). A previous proteomic study also reported that the expression profiles of several enzymes in the phenylalanine metabolism pathway showed significant differences between V. quinquangularis accession 'Danfeng-2' and V. vinifera cv. 'Cabernet Sauvignon' at the véraison and ripening stages[33]. In addition, genes such as RSGT, OMT and LAC involved in the production of derivatized products of resveratrol were mostly present at the G and V stages of 'Tangwei', potentially resulting in limited resveratrol accumulation. However, we found that most of these also revealed relatively high expression at R in 'Tonghua-3' (Fig. 3). Despite this situation, which does not seem to be conducive for the accumulation of resveratrol, it still showed the highest content (Table 1).
It has been reported that overexpression of two grapevine peroxidase VlPRX21 and VlPRX35 genes from Vitis labruscana in Arabidopsis may be involved in regulating stilbene synthesis[34], and a VqBGH40a belonging to β-glycoside hydrolase family 1 in Chinese wild Vitis quinquangularis can hydrolyze trans-piceid to enhance trans-resveratrol content[35]. However, most studies mainly focus on several TFs that participate in regulation of STS gene expression, including ERFs, MYBs and WRKYs[13, 15, 17]. For example, VvWRKY18 activated the transcription of VvSTS1 and VvSTS2 by directly binding the W-box elements within the specific promoters and resulting in the enhancement of stilbene phytoalexin biosynthesis[36]. VqWRKY53 promotes expression of VqSTS32 and VqSTS41 through participation in a transcriptional regulatory complex with the R2R3-MYB TFs VqMYB14 and VqMYB15[37]. VqMYB154 can activate VqSTS9/32/42 expression by directly binding to the L5-box and AC-box motifs in their promoters to improve the accumulation of stilbenes[38]. In this study, we found a total of 757 TF-encoding genes among the DEGs, including representatives of the MYB, AP2/ERF, bHLH, NAC, WRKY, bZIP and HB-HD-ZIP families. The most populous family was MYB, representing 76 DEGs at G, V and R between 'Tangwei' and 'Tonghua-3' (Fig. 4). A recent report indicated that MYB14, MYB15 and MYB13, a third uncharacterized member of Subgroup 2 (S2), could bind to 30 out of 47 STS family genes. Moreover, all three MYBs could also bind to several PAL, C4H and 4CL genes, in addition to shikimate pathway genes, the WRKY03 stilbenoid co-regulator and resveratrol-modifying gene[39]. VqbZIP1 from Vitis quinquangularis has been shown to promote the expression of VqSTS6, VqSTS16 and VqSTS20 by interacting with VqSnRK2.4 and VqSnRK2.6[40]. In the present study, we found that a gene encoding a bZIP-type TF (VIT_12s0034g00110) was down-regulated in 'Tangwei', but up-regulated in 'Tonghua-3', at G, V and R (Fig. 4). We also identified 36 TFs that were co-expressed with 17 STSs using WGCNA analysis, suggesting that these TFs may regulate STS gene expression (Fig. 5 and Supplemental Table S8). Among these, a STS (VIT_16s0100g00880) was together co-expressed with MYB14 (VIT_07s0005g03340) and WRKY24 (VIT_06s0004g07500) that had been identified as regulators of STS gene expression[13, 15]. A previous report also indicated that a bHLH TF (VIT_11s0016g02070) had a high level of co-expression with STSs and MYB14/15[15]. In the current study, six bHLH TFs were identified as being co-expressed with one or more STSs and MYB14 (Fig. 5 and Supplemental Table S8). However, further work needs to be done to determine the potential role of these TFs that could directly target STS genes or indirectly regulate stilbene biosynthesis by formation protein complexes with MYB or others. Taken together, these results identify a small group of TFs that may play important roles in resveratrol biosynthesis in grapevine.
-
In summary, we documented the trans-resveratrol content of seven grapevine accessions by HPLC and performed transcriptional analysis of the grape berry in two accessions with distinct patterns of resveratrol accumulation during berry development. We found that the expression levels of genes with putative roles in resveratrol biosynthesis were significantly higher in 'Tonghua-3' than in 'Tangwei' during V and R, consistent with the difference in resveratrol accumulation between these accessions. Moreover, several genes encoding TFs including MYBs, WRKYs, ERFs, bHLHs and bZIPs were implicated as regulators of resveratrol biosynthesis. The results from this study provide insights into the mechanism of different resveratrol accumulation in various grapevine accessions.
-
V. davidii 'Tangwei', V. amurensis × V. Vinifera 'Beibinghong'; V. amurensis 'Tonghua-3' and 'Shuangyou'; V. vinifera × V. labrusca 'Jumeigui'; V. vinifera 'Red Globe' and 'Thompson Seedless' were maintained in the grapevine germplasm resource at Northwest A&F University, Yangling, Shaanxi, China (34°20' N, 108°24' E). Fruit was collected at the G, V, and R stages, as judged by skin and seed color and soluble solid content. Each biological replicate comprised three fruit clusters randomly chosen from three plants at each stage. About 40−50 representative berries were separated into skin, pulp, and seed, and immediately frozen in liquid nitrogen and stored at −80 °C.
Extraction and analysis of resveratrol content
-
Resveratrol extraction was carried out as previously reported[8]. Quantitative analysis of resveratrol content was done using a Waters 600E-2487 HPLC system (Waters Corporation, Milford, MA, USA) equipped with an Agilent ZORBAX SB-C18 column (5 µm, 4.6 × 250 mm). Resveratrol was identified by co-elution with a resveratrol standard, and quantified using a standard curve. Each sample was performed with three biological replicates.
RNA extraction, transcriptome library construction and sequencing
-
Three biological replicates of each stage (G, V and R) from whole berries of 'Tangwei' and 'Tonghua-3' were used for all RNA-Seq experiments. Total RNA was extracted from 18 samples using the E.Z.N.A. Plant RNA Kit (Omega Bio-tek, Norcross, GA, USA). For each sample, sequencing libraries were constructed from 1 μg RNA using the NEBNext UltraTM RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA). The library preparations were sequenced on an Illumina HiSeq2500 platform (Illumina, San Diego, CA, USA) at Biomarker Technologies Co., Ltd. (Beijing, China).
RNA-seq data analysis
-
Raw sequence reads were filtered to remove low-quality reads, and then mapped to the V. vinifera 12X reference genome[18, 31] using TopHat v.2.1.0[41]. The mapped reads were assembled into transcript models using Stringtie v2.0.4[42]. Transcript abundance and gene expression levels were estimated as FPKM[43]. The formula is as follows:
$ \text{FPKM =}\frac{\text{cDNA Fragments}}{\text{Mapped Fragments}\left(\text{Millions}\right)\text{× Transcript Length (kb)}} $ Biological replicates were evaluated using Pearson's Correlation Coefficient[44] and principal component analysis. DEGs were identified using the DEGSeq R package v1.12.0[45]. A false discovery rate (FDR) threshold was used to adjust the raw P values for multiple testing[46]. Genes with a fold change of ≥ 2 and FDR < 0.05 were assigned as DEGs. GO and KEGG enrichment analyses of DEGs were performed using GOseq R packages v1.24.0[47] and KOBAS v2.0.12[48], respectively. Co-expression networks were constructed based on FPKM values ≥ 1 and coefficient of variation ≥ 0.5 using the WGCNA R package v1.47[49]. The adjacency matrix was generated with a soft thresholding power of 16. Then, a topological overlap matrix (TOM) was constructed using the adjacency matrix, and the dissimilarity TOM was used to construct the hierarchy dendrogram. Modules containing at least 30 genes were detected and merged using the Dynamic Tree Cut algorithm with a cutoff value of 0.25[50]. The co-expression networks were visualized using Cytoscape v3.7.2[51].
Validation of RNA-Seq data by RT-qPCR
-
RT-qPCR was carried out using the SYBR Green Kit (Takara Biotechnology, Beijing, China) and the Step OnePlus Real-Time PCR System (Applied Biosystems, Foster, CA, USA). Gene-specific primers were designed using Primer Premier 5.0 software (PREMIER Biosoft International, Palo Alto, CA, USA). Cycling parameters were 95 °C for 30 s, 42 cycles of 95 °C for 5 s, and 60 °C for 30 s. The grapevine ACTIN1 (GenBank Accession no. AY680701) gene was used as an internal control. Each reaction was performed in triplicate for each of the three biological replicates. Relative expression levels of the selected genes were calculated using the 2−ΔΔCᴛ method[52]. Primer sequences are listed in Supplemental Table S9.
- This research was supported by the National Key Research and Development Program of China (2019YFD1001401) and the National Natural Science Foundation of China (31872071 and U1903107).
- The authors declare that they have no conflict of interest.
- Supplemental Table S1 Number of quality-filtered reads and mapping rate for individual sequencing libraries.
- Supplemental Table S2 The sequences of 1751 novel transcripts.
- Supplemental Table S3 KEGG pathway enrichment analysis of DEGs at the G stage between ‘Tangwei’ and ‘Tonghua-3’ (p < 0.05).
- Supplemental Table S4 KEGG pathway enrichment analysis of DEGs at the V stage between ‘Tangwei’ and ‘Tonghua-3’ (p < 0.05).
- Supplemental Table S5 KEGG pathway enrichment analysis of DEGs at the R stage between ‘Tangwei’ and ‘Tonghua-3’ (p < 0.05).
- Supplemental Table S6 DEGs involved in the phenylalanine metabolism pathway at the G, V and R stages between ‘Tangwei’ and ‘Tonghua-3.
- Supplemental Table S7 Number of genes included in each module.
- Supplemental Table S8 Genes encoding transcription factors co-expressed with STS genes.
- Supplemental Table S9 Genes and oligonucleotide primer pairs used for RT-qPCR.
- Supplemental Fig. S1 Significantly enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways for genes in the orange, blue and ivory modules using the weighted gene co-expression network analysis (WGCNA) analysis. The corrected p-values are marked to the right of each bar.
- Copyright: © 2022 by the author(s). Exclusive Licensee Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang X, Wu Y, Zhang Y, Yin X, Nocker S, et al. 2022. Identification of potential key genes in resveratrol biosynthesis via transcriptional analyses of berry development in grapevine (Vitis spp.) genotypes varying in trans-resveratrol content. Fruit Research 2: 6 doi: 10.48130/FruRes-2022-0006

Identification of potential key genes in resveratrol biosynthesis via transcriptional analyses of berry development in grapevine (Vitis spp.) genotypes varying in trans-resveratrol content
- Received: 21 November 2021
- Accepted: 28 March 2022
- Published online: 16 May 2022
Abstract: Resveratrol is an important secondary metabolite not only owing to its function as a phytoalexin, but also its potential benefits to human health. In this study, the content of trans-resveratrol was documented in seven accessions of grapevine, in the seed, pulp and skin of berries, and at three developmental stages. The highest amount (2.99 µg g−1 FW) was found in the skin of berries at the ripe stage from V. amurensis 'Tonghua-3'. Resveratrol was not detected in several samples, including skin of berries at the green hard or véraison stage from V. davidii 'Tangwei'. We carried out transcriptional profiling of developing 'Tonghua-3' and 'Tangwei' berries to identify gene expression patterns that may be linked with the difference in resveratrol content between these accessions. The expression levels of several differentially expressed genes (DEGs) with presumed function in resveratrol biosynthesis, including STILBENE SYNTHASEs (STSs), CINNAMATE 4-HYDROXYLASEs (C4Hs) and 4-COUMARATE-COA LIGASEs (4CLs), were significantly higher in 'Tonghua-3', than in 'Tangwei' during the véraison and ripe stages. Gene ontology and Kyoto Encyclopedia of Genes and Genomes analyses suggested that these DEGs were enriched for multiple biological processes at the three stages of fruit development. Additionally, we identified a total of 36 transcription factors, including MYBs, WRKYs, ERFs, bHLHs and bZIPs, that were co-expressed with 17 STSs via a weighted gene co-expression network analysis, suggesting roles as regulators of resveratrol biosynthesis. Overall, these findings provide insight into genotypic differences in resveratrol biosynthesis in grapevine, as well as the molecular genetics of its regulation.
-
Key words:
- Grapevine /
- Resveratrol /
- RNA-seq /
- Transcription factor