









-
Salvia miltiorrhiza Bunge (Lamiaceae), commonly known as red sage, Chinese sage, tan shen, or danshen is one of the most commonly used herbs in traditional Chinese medicine. The major pharmacologically active ingredients of S. miltiorrhiza are diterpenoid tanshinones and phenolic acids[1], which are mainly found in the hairy roots of the plant. Tanshinones are a class of fat-soluble phenanthrene compounds derived from diterpene precursors and include, among others, cryptotanshinone, tanshinone I, and tanshinone IIA[2]. Tanshinones elicit various biological activities, including antibacterial and anti-inflammatory effects, and so far no meaningful side-effects have been observed upon long-term administration[3,4]. The phenolic acids isolated from S. miltiorrhiza roots are water-soluble and include salvianolic acid B (SAB), along with danshensu, caffeic acid, and rosmarinic acid[5]. SAB is considered as the main phenolic acid in the plant and elicited strong antioxidant activity as well as appreciable antihepatic fibrotic and anti-thrombotic effects. SAB also stimulated the proliferation and differentiation of stem cells[6−8].
Tanshinones are bio-synthetized in three steps, involving the biosynthesis of diterpene precursors, then the formation of a tanshinone skeleton, and, finally, the formation of the tanshinone through modifications of the skeleton by oxidation, methylation, decarboxylation, and/or cyclization. The production of tanshinones depends on the mevalonate (MVA) pathway in the cytosol of the hairy root cells, and the methylerythritol phosphate (MEP) pathway in the plastids[9,10]. The genes encoding for the enzymes involved in tanshinone biosynthesis include AACT, HMGR, HMGS, MK, PMK, and MDC in the MVA pathway, and DXS, DXR, MCT, CMK, HDS, and HDR in the MEP pathway.
The biosynthesis of the tanshinone precursor isopentenyl diphosphate mainly occurs in the MEP pathway[11, 12]. In addition, the protein products of the genes CYP76AK1, CYP76AH3, CYP71Ds, and 2ODD14 were recently identified to participate in the downstream biosynthesis of tanshinones[13, 14].
The SAB biosynthesis pathway has two parallel branches, one using phenylalanine as a precursor and the other using tyrosine as a precursor[15] (Fig. 1). PAL, C4H, and 4CL are the key protein-encoding genes in the phenylalanine branch of the SAB synthetic pathway, while TAT and HPPR fulfilled this function in the tyrosine branch. 4-Coumaroyl-CoA from the phenylalanine pathway and 4'-dihydroxyphenyllactic from the tyrosine pathway are coupled by the ester-forming enzyme rosmarinic acid synthase (RAS) to generate 4-coumaroyl-3',4'-dihydroxyphenyllactate. The latter compound is oxidized by CYP98A14 to generate rosmarinic acid[16,17]. Recent studies have shown that rosmarinic acid, in turn, can be oxidized to SAB by the enzyme laccase-4 (LAC4)[18].
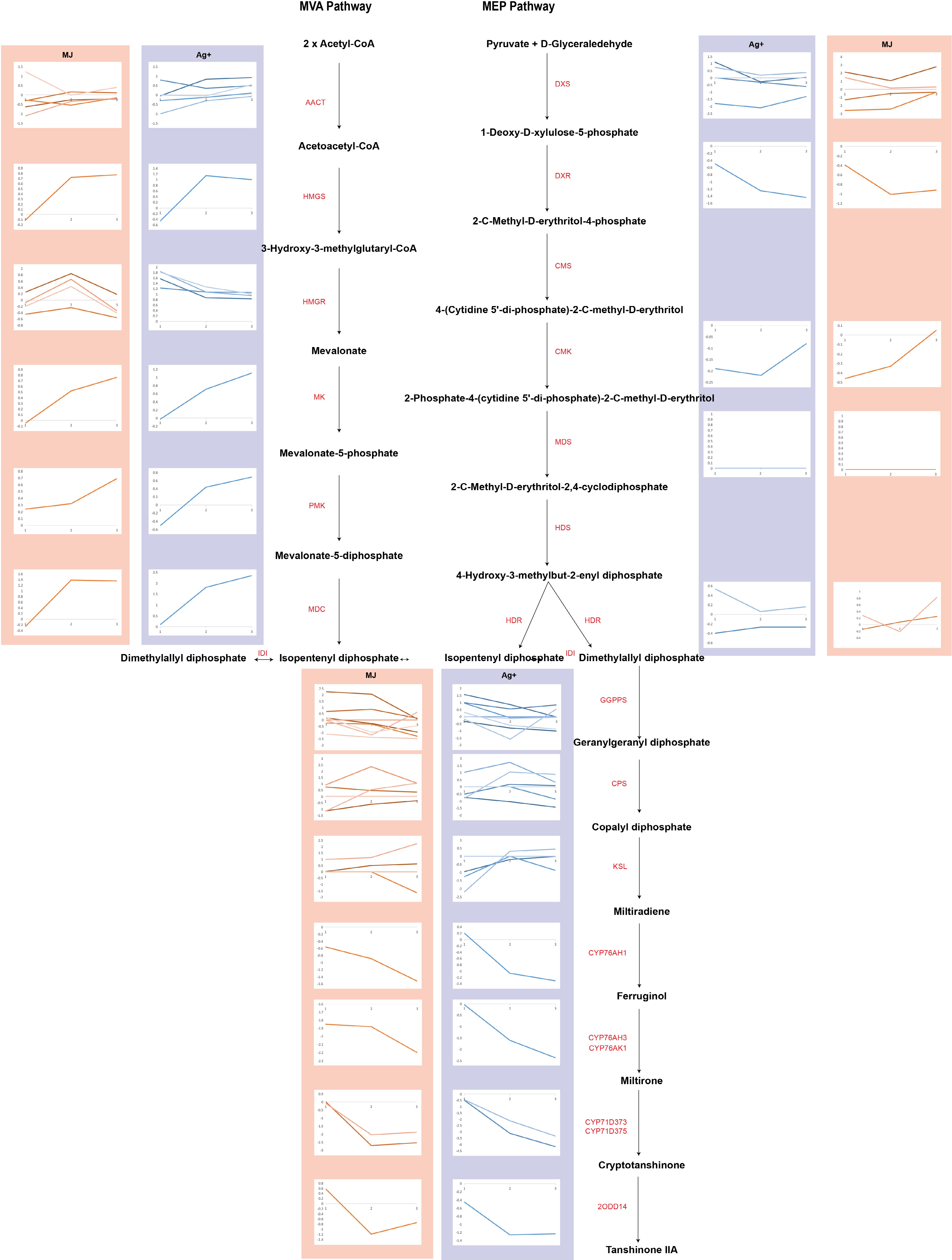
Figure 1.
The expression profiles of genes involved in tanshinone biosynthesis are shown as line charts. Line charts framed in pink represent the expression of genes under the treatment of Ag+, and line charts framed in blue represent the expression of genes under the treatment of MeJA.
The use of elicitors to influence the biosynthetic pathways of certain secondary metabolites represents an important technique to stimulate their synthesis. Both abiotic elicitors and biotic elicitors are able to promote the activity of key enzymes in metabolic processes in plants by activating and turning on the encoding genes, thus increasing the accumulation of secondary metabolites in plant cells[19]. Common abiotic elicitors are ethylene (ET), salicylic acid (SA), jasmonic acid (JA), jasmonic acid methyl ester (MeJA), and oxalic acid (OA). Xiao et al.[20] treated hairy roots of S. miltiorrhiza with silver ions (Ag+), and observed that this heavy metal elicitor significantly increased the content of SAB in S. miltiorrhiza hairy roots. PAL and TAT were key enzyme genes in the SAB biosynthesis pathway. S. miltiorrhiza hairy roots under MeJA treatment showed that elicitors significantly increased the accumulation level of SAB[21−23].
In this study, the hairy roots of S. miltiorrhiza were treated with the elicitors Ag+ or MeJA, after which transcriptome sequencing and quantitative analysis were performed in order to explore the mechanisms underlying the working of the elicitors, and screened out a set of differentially expressed genes, which were involved in the biosynthesis pathways of SAB and tanshinones. The results will contribute to the understanding of the mechanisms involved in the biosynthesis of SAB and tanshinones, providing a theoretical basis for further improving their accumulation in S. miltiorrhiza hairy roots.
-
The plants of S. miltiorrhiza were cultivated and stored in the greenhouse of Zhejiang Tech-Sci University (Hangzhou, Zhejiang, China). The growth condition of S. miltiorrhiza were retained at 26 °C and 16 h light each day for three months, and then sampled for RNA extraction and transcriptome sequencing with two biological repeats.
RNA preparation and sequencing
-
Hairy roots of S. miltiorrhiza samples were treated with Ag+ and MeJA and after 1, 3 and 6 d, duplicate samples were taken to generate RNA libraries (Supplemental Table S1 & S2). Since each sample had two independent biological repeats, nine RNA libraries were constructed. Total RNA were extracted using Trizol reagent (Invitrogen). Samples were constructed and sequenced by the Beijing Genomics Institute (BGI) (Shenzhen, Guangdong Province, China) using the Illumina HiSeq 2000 platform.
Following extraction of total RNA and treatment with DNase I, Oligo (dT) magnetic beads were used to isolate mRNA (for eukaryotes) or remove rRNAs from the total RNA (for prokaryotes). The mRNA was broken up into short fragments by mixing with fragmentation buffer, and the mRNA fragments were used as templates to synthesize cDNA. The short fragments were purified and dissolved in EB buffer for end reparation and single nucleotide A (adenine) addition, after which the short fragments were connected to adapters. Following agarose gel electrophoresis, the suitable fragments (~250 bp) were selected as templates for PCR amplification. An Agilent 2100 Bioanalyzer and an ABI StepOnePlus Real-Time PCR System were used for quantification and qualification of the sample library. Finally, all the libraries were submitted to sequences using Illumina HiSeqTM 2000.
mRNA quantitation and differential expression analysis
-
Quality control was performed on Raw pair-end sequencing data. Trimmomatic[24] (v0.39) was used for removing adapters with parameters following: 'ILLUMINACLIP:TruSeq3-PE.fa:2:30:10:8 SLIDINGWINDOW:5:15 LEADING:5 TRAILING:5 MINLEN:50'. Prinseq lite[25] (v0.20.4) was applied for removing N, and obtained clean sequencing data. Hisat2[26] (v2.1.0) mapped clean data to reference genome of S. miltiorrhiza, matching rate higher than 90% indicated the transcriptome sequencing data were reliable (Supplemental Table S1 & S2). FeatureCounts[27] (v1.6.4) was adopted to quantify total expressed transcripts with the FPKM (fragments per kilobase of exon model per million mapped reads) value. Differential analysis was programmed in R (v4.0.1) with DESeq2[28].
Co-expression network construction
-
The WGCNA[29] package of R (v4.0) was used for constructing the co-expression network, and Pearson correlation coefficients (PCC) were calculated between genes by scipy package of Python (v3.7), gene pair was considered correlated while PCC > 0.6, and p-value < 0.05 was considered to be significant.
Functional enrichment analysis
-
KEGG enrichment analysis were performed on differentially expressed gene sets under elicitors treatments by clusterProfiler package with R (v4.1). A hyper geometric distribution test was carried out to identify KEGG pathways in which differentially expressed genes are significantly enriched p-value < 0.05, comparing to total expressed transcripts.
-
Key genes in the tanshinone biosynthesis pathway were identified, and their expression after 1, 3 and 6 d treatment with the elicitors Ag+ or MeJA were calculated and visualized in Fig. 1 as line charts. The results showed that the expression of most genes involved in the MVA pathway were upregulated following Ag+ treatment. The SmMDC gene was substantially upregulated after 3 and 6 d (Supplemental Table S3), their expression having increased 2.20-fold (p-value of 8.61E-05) and 2.33-fold (p-value of 5.36E-06), respectively, when compared to that of the controls. The SmHMGR2 and SmHMGR5 genes were also statistically significantly and differentially upregulated after 3 d Ag+ treatment (Supplemental Table S3), having increased 1.31-fold and 1.54-fold, respectively, with respect to control values (p-values of 3.51E-02 and 4.56E-02, respectively). The genes in the MEP pathway were generally upregulated by the Ag+ treatment, and the downstream gene SmCPS5 was upregulated and differentially expressed after 3 d (Supplemental Table S3), its expression having increased 1.27-fold of that of the control (p-value of 3.12E-01).
In response to the MeJA treatment, the genes in the MVA pathway were generally upregulated (Supplemental Table S4 & S5). The SmGPPS2 gene, downstream in the MEP pathway, was differentially upregulated after 3 d (Supplemental Table S4), its expression having increased 1.77-fold with respect to that of the control value (p-value of 4.18E-02).
Following the Ag+ and MeJA treatments, the key genes in the MVA pathway were generally upregulated when compared to those in the MEP pathway (Fig. 1). The use of Ag+ and MeJA promoted the biosynthesis of tanshinones mainly by increasing the expression of the SmHMGR and SmMDC genes in the MVA pathway, providing evidence for a reciprocal relationship between the MVA pathway in the plastids and the MEP pathway in the cytosol.
The upstream pathway of SAB biosynthesis responded stronger to MeJA
-
The treatments with Ag+ or MeJA had positive effects on the accumulation of SAB in the hairy roots of S. miltiorrhiza. Figure 2 shows the expression of the genes in the phenylalanine and tyrosine pathway. Treatment with Ag+ led to differential up-regulation of the expression of the Sm4CL-like gene (that is involved in the phenylalanine pathway) (Supplemental Table S6 & S7), with those of Sm4CL-like1 and Sm4CL-like4 having increased 1.65-fold (p-value of 0.009) and 1.25-fold (p-value of 2.01E-02), respectively, of control values after 3 d, and that of Sm4CL-like7 having increased 1.40-fold after 6 d (p-value of 1.34E-02). In addition, the expression of SmTAT, a key gene in the tyrosine pathway, was differentially upregulated after 3 and 6 d (Supplemental Table S7), having increased 1.94-fold (p-value of 4.00E-04) and 1.73-fold (p-value of 3.01E-04), respectively, of control values. After the MeJA treatment, the Sm4CL-like7 gene involved in the phenylalanine pathway was differentially upregulated and its expression had increased (Supplemental Table S6 & Table S8), indicating that Ag+ and MeJA had stimulated the expression of Sm4CL in the phenylalanine pathway, and that, in addition, Ag+ had promoted the expression of SmTAT in the tyrosine pathway, increasing the accumulation of SAB in the hairy roots of S. miltiorrhiza.
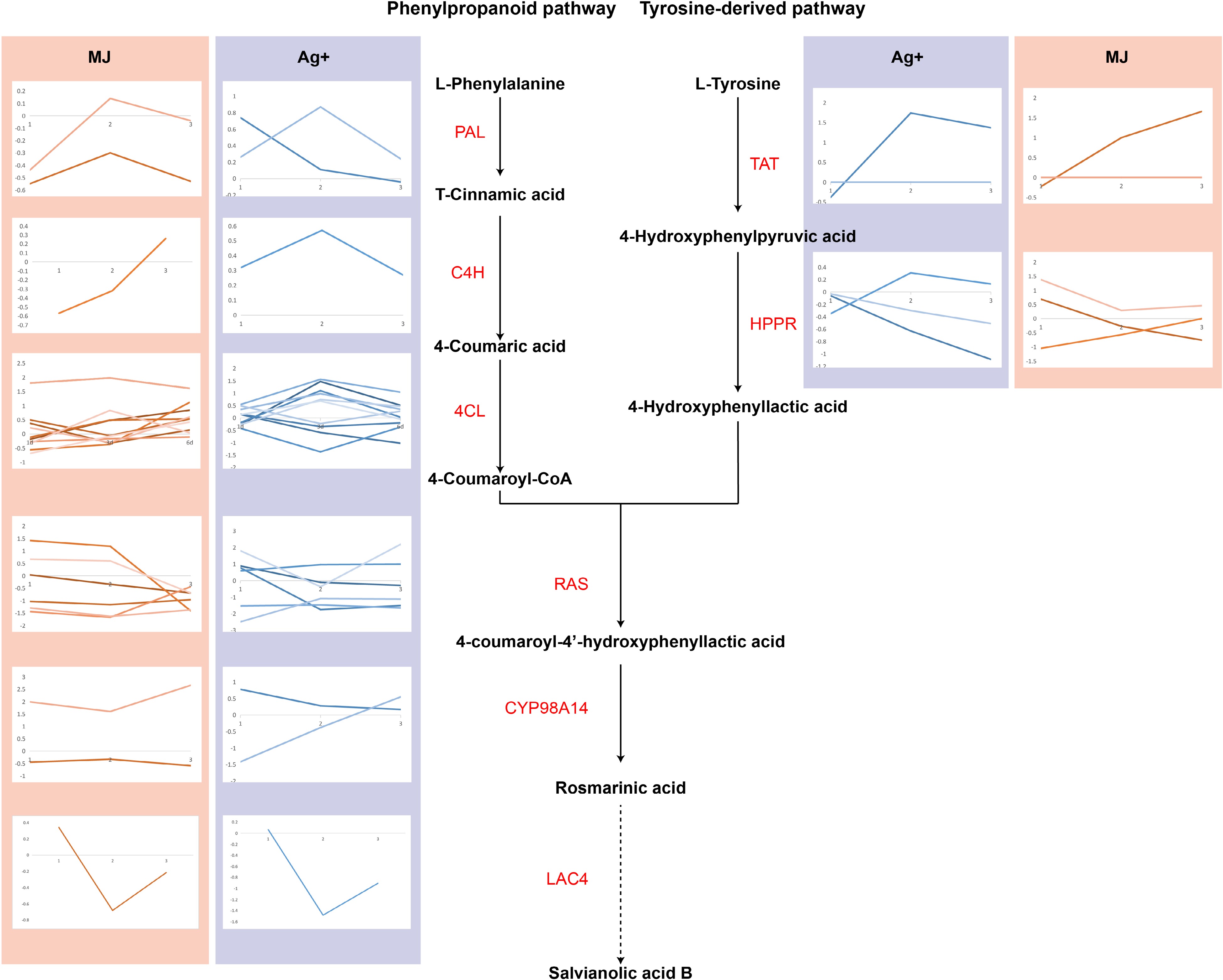
Figure 2.
The expression levels of genes involved in salvianolic acid B biosynthesis are shown using line charts. Line charts framed in pink represent the expression of genes under the treatment of Ag+, and line charts framed in blue represent the expression of genes under the treatment of MeJA.
Based on the differentially expressed genes (DEG) evaluated after Ag+ or MeJA treatment, KEGG pathway enrichment analysis was performed. This analysis showed DEG enriched in terms of the 'Phenylpropanoid pathway' (Supplemental Table S9 & S10; Supplemental Fig. S1 & S2), consisting of Sm4CL1 and Sm4CL6. At the same time, Sm4CL1 and Sm4CL6 were upregulated by the Ag+ and MeJA treatments. Moreover, SmC4H was involved in terms of the 'Phenylpropanoid pathway' after MeJA treatment, suggesting that the phenylpropanoid pathway was more sensitive to the MeJA treatment. The phenylpropanoid pathway was the upstream pathway of SAB biosynthesis, which contributed to the upregulation of SmCYP98A14.
Differentially expressed P450 highly related to the MVA pathway under Ag+ and MeJA treatment
-
Cytochrome P450 is one of the largest gene families in plants and is involved in various biochemical pathways, particularly those for the biosynthesis of secondary metabolites. Many of the downstream steps of the tanshinone pathway are catalyzed by cytochrome P450s, including CYP76AH1, CYP76AH3, and CYP76AK1 which have been established to participate in tanshinone biosynthesis. Twenty-one different candidate P450 genes were filtered from the DEG (Supplemental Fig. S3; Supplemental Table S11), including SmCYP76AH1, and SmCYP71D373.
The expression of SmCYP96A15, SmCYP716A52v2, SmCYP716A12, and SmCYP81Q32 were differentially upregulated by the Ag+ and MeJA treatments (Supplemental Fig. S3; Supplemental Table S11), suggesting that the relevant P450s may be involved in the regulation of tanshinone biosynthesis.
To verify such a function of the P450s, gene co-expression networks were constructed between candidate P450s and key genes in the biosynthetic pathways of tanshinones and SAB biosynthesis (Supplemental Fig. S4). The SmCYP96A15, SmCYP716A52v2, SmCYP716A12, and SmCYP81Q32 genes appeared to be clustered in the same gene module, suggesting a strong relationship with the SmCPS5, SmCYP76AH1, and SmCYP76AK1 genes. However, using the Pearson correlation coefficient (PCC) to determine the strength of the relationship between candidate P450s and pathway genes, a PCC of 0.96 between SmCYP716A52v2 and SmMDC, and a PCC of 0.97 between SmCYP716A12 and SmMDC were found. These findings support a meaningful contribution of Ag+ and MeJA elicitors to the MVA pathway.
A total of 272 P450 genes were screened after the Ag+ and MeJA treatments. Curve fitting indicated that P450 expression was statistically significantly more upregulated by Ag+ than by MeJA (Fig. 3a). A large number of P450s were downregulated after 6 d MeJA treatment. As shown in the Fig. 3b, the responses of the P450s to Ag+ and MeJA treatment were highly correlated, and this likely promoted the accumulation of tanshinones and SAB by the elicitors.
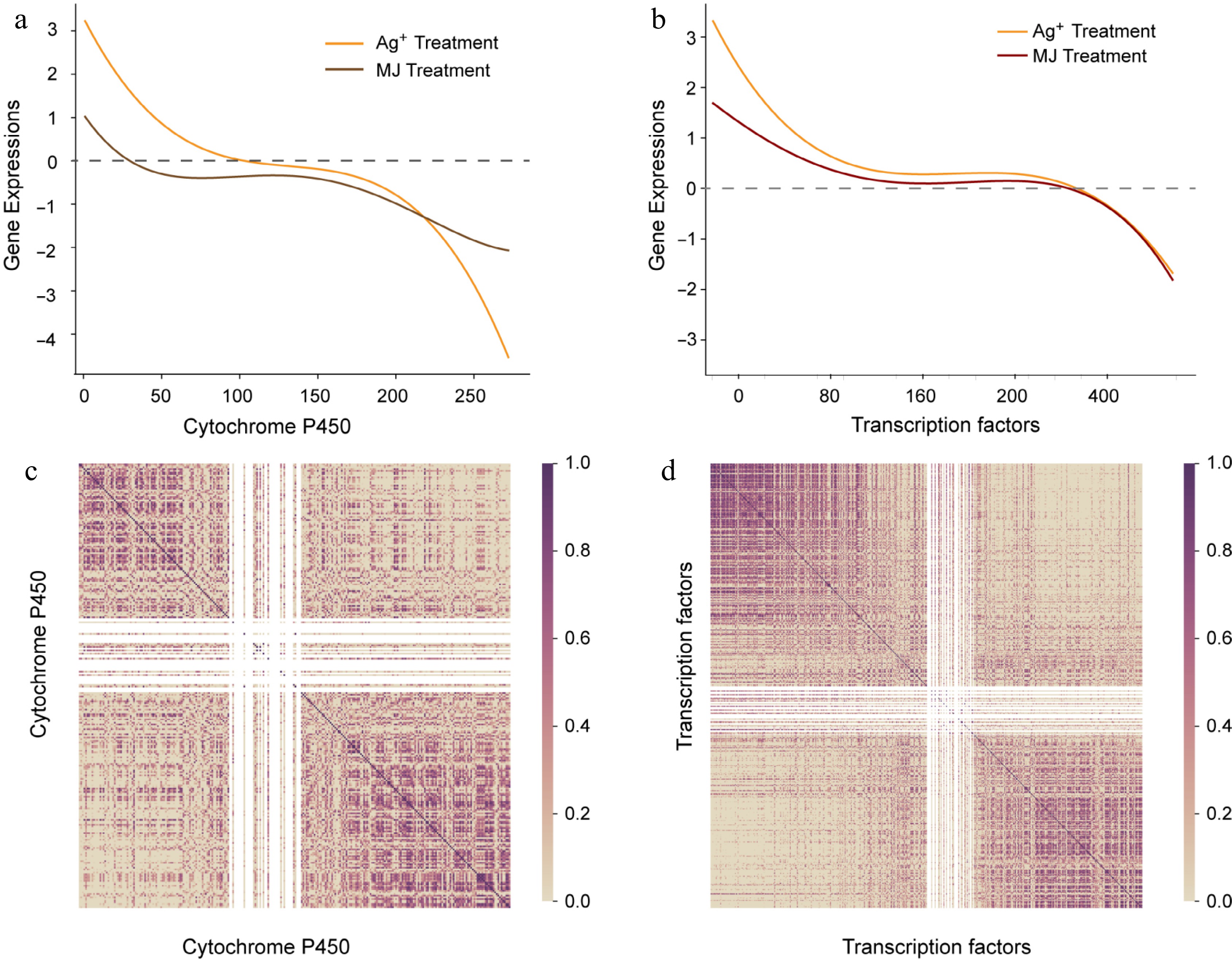
Figure 3.
The expression trend of P450 and transcript factors. (a) Expression trend of P450. X axis represents P450 ordered by descending expression levels, and the gene expression levels are shown with fitted curves. The yellow curve represents expression of P450 with Ag+ treatment, and the red curve represents expression of P450 with MeJA treatment. (b) Pearson correlation coefficient calculated with descending P450, dark purple represents high correlation. (c) The expression trend of transcript factors. (d) Pearson correlation coefficient calculated with descending transcript factors.
Regulation of tanshinone biosynthesis in response to Ag+ and MeJA by SmWRKY73 and SmbHLH25
-
In plants, families of transcription factors regulate a range of metabolic processes including responses to abiotic stresses. Many transcription factors have been observed to regulate the biosynthesis and accumulation of secondary metabolites, including those encoded by SmWRKY1, SmWRKY61, SmMYB36, SmMYB9b etc[30]. In this study, we identified a total of nine SmMYBs, ten SmWRKYs, nine SmbHLHs, and 18 SmERFs that were differentially expressed in response to Ag+ and MeJA (Supplemental Fig. S2; Supplemental Tables S12−S15).
The ERF transcription factor family is one of the most important families of transcription factors involved in the regulation of metabolic and developmental processes. The expression of SmERF112 and SmERF114 were statistically significantly and differentially upregulated by the Ag+ and MeJA treatments (Fig. 4a; Supplemental Table S12), having increased up to 6.42-fold of control values. Although SmERF112 and SmERF114 were displayed a high expression after 3 and 6 d treatment of Ag+ with only a relatively little difference between days 3 and 6, their expressions following the MeJA treatment have reached the highest level after 3 d and had decreased after 6 d. These observations suggested that the biosynthesis of tanshinones and SAB responded in a more durable fashion to Ag+ when compared to MeJA.
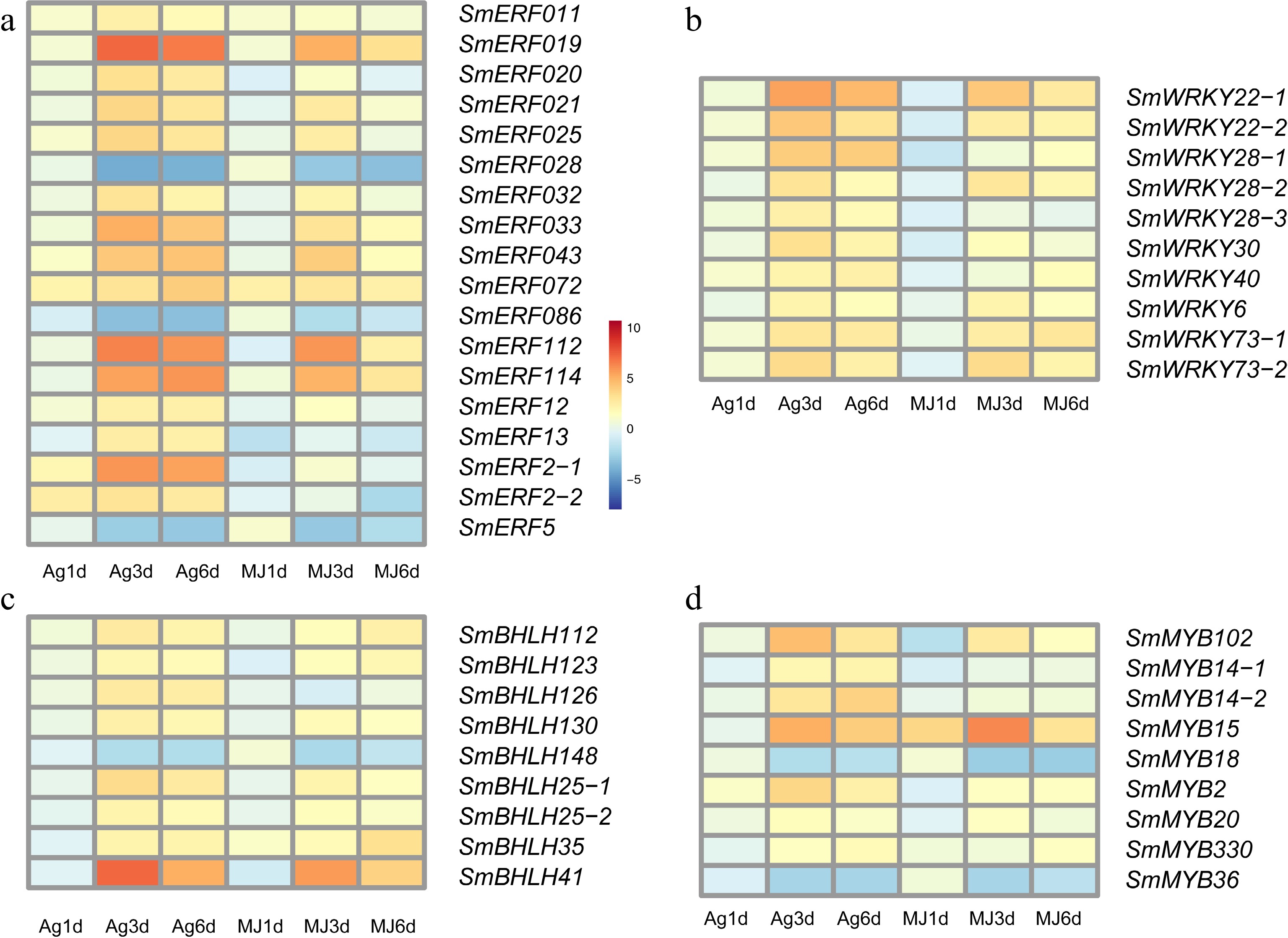
Figure 4.
The expression heatmap of transcript factors differentially expressed under the treatment of Ag+ and MeJA. (a) ERF family, (b) WRKY family, (c) bHLH family and (d) MYB family.
Of ten candidate WRKYs, two WRKY22, three WRKY28, and two WRKY73 were statistically significantly and differentially upregulated by the Ag+ and MeJA treatments (Fig. 4b; Supplemental Table S13). SmWRKY22-1 reached its highest expression after 3 d following Ag+ (5.57-fold of the control value; p-value of 1.11E-21), and after 6 d MeJA (2.62-fold of the control value; p-value of 6.83E-05). At the same time, the expression of SmWRKY73-1 after 3 d was 3.39-fold upregulated with respect to that of the control. SmWRKY22-1 and SmWRKY73-1 responded stronger to the MeJA treatment when compared to that with Ag+. Similarly, SmbHLH25, SmbHLH41, and SmbHLH112 in the candidate bHLHs gene set (Fig. 4c, Supplemental Table S14) were statistically significantly and differentially upregulated in response to Ag+ and MeJA.
Candidate MYBs were generally upregulated following the treatment with Ag+ or MeJA, with the expression of SmMYB14, SmMYB15, and SmMYB102 differentially upregulated after 3 and 6 d treatment with the elicitors (Fig. 4d; Supplemental Table S15). Notably, the expression of SmMYB15 had increased 5.08-fold (p-value of 6.12E-04) of the control value after 3 d Ag+, and 6.23-fold (p-value of 4.01E-04) of the control value following the MeJA treatment. Thus, SmMYB15 may be involved in inhibiting the biosynthesis of tanshinones.
Based on the clustering results of gene modules, SmWRKY22, SmWRKY28, SmWRKY73, SmERF112, SmERF1, SmbHLH41, and SmbHLH25 clustered in ME1 module and showed a high correlation with SmCPS5, SmHGMS1, SmMDC, SmMK, and SmPMK, while SmWRKY73 and SmbHLH25 showed a high correlation with SmKSL1 (PCCs of 0.66 and 0.70). Therefore, SmWRKY73 and SmbHLH25 may coordinately regulate the biosynthesis of tanshinones in response to Ag+ and MeJA in S. miltiorrhiza hairy roots.
A total of 494 transcription factors were identified from the S. miltiorrhiza genome, involving WRKY TFs, MYB TFs, and bHLH TFs. TF expressions were sorted in descending order following Ag+ treatment, and plotted as a scatter graph with best fits following MeJA treatment (Fig. 3c). The results showed that the TFs responded to both Ag+ and MeJA, but showed a more pronounced increase in expression following the Ag+ treatment when compared to the MeJA treatment. On the basis of descending order, correlation coefficients were calculated within total TFs, and presented in a heatmap in purple (Fig. 3d). The heatmap showed highly correlating TFs concentrated on the diagonal line. Furthermore, correlation coefficient in the upper left suggested the upregulated expression of TFs by the Ag+ and MeJA treatments were highly correlated. Thus, these TFs are likely to promote the accumulation of tanshinones and SAB jointly in S. miltiorrhiza hairy roots.
Promotion of tanshinone and SAB accumulation by genes in the pathway for hormone signaling
-
Plant hormones play an important role in regulating plant growth. Jasmonic acid (JA), gibberellin acid (GA), and abscisic acid (ABA) have been reported to promote the accumulation of secondary metabolites in the hairy roots of S. miltiorrhiza[31, 32]. The genes involved in the biosynthetic pathways of the secondary metabolites were identified and expressions were evaluated. SmGA2ox-8 gene expression was statistically significantly and differentially upregulated by the Ag+ and MeJA treatments, having increased 5.17-fold (p-value of 3.47E-12) and 5.51-fold (p-value of 2.62E-14), respectively, of control values after 3 d, (Fig. 5a) (Supplemental Table S16). And that of the SmGA20x-9 gene was also statistically significantly and differentially upregulated after 3 d Ag+, which was 2-fold higher than the control value (p-value of 4.31E-04).
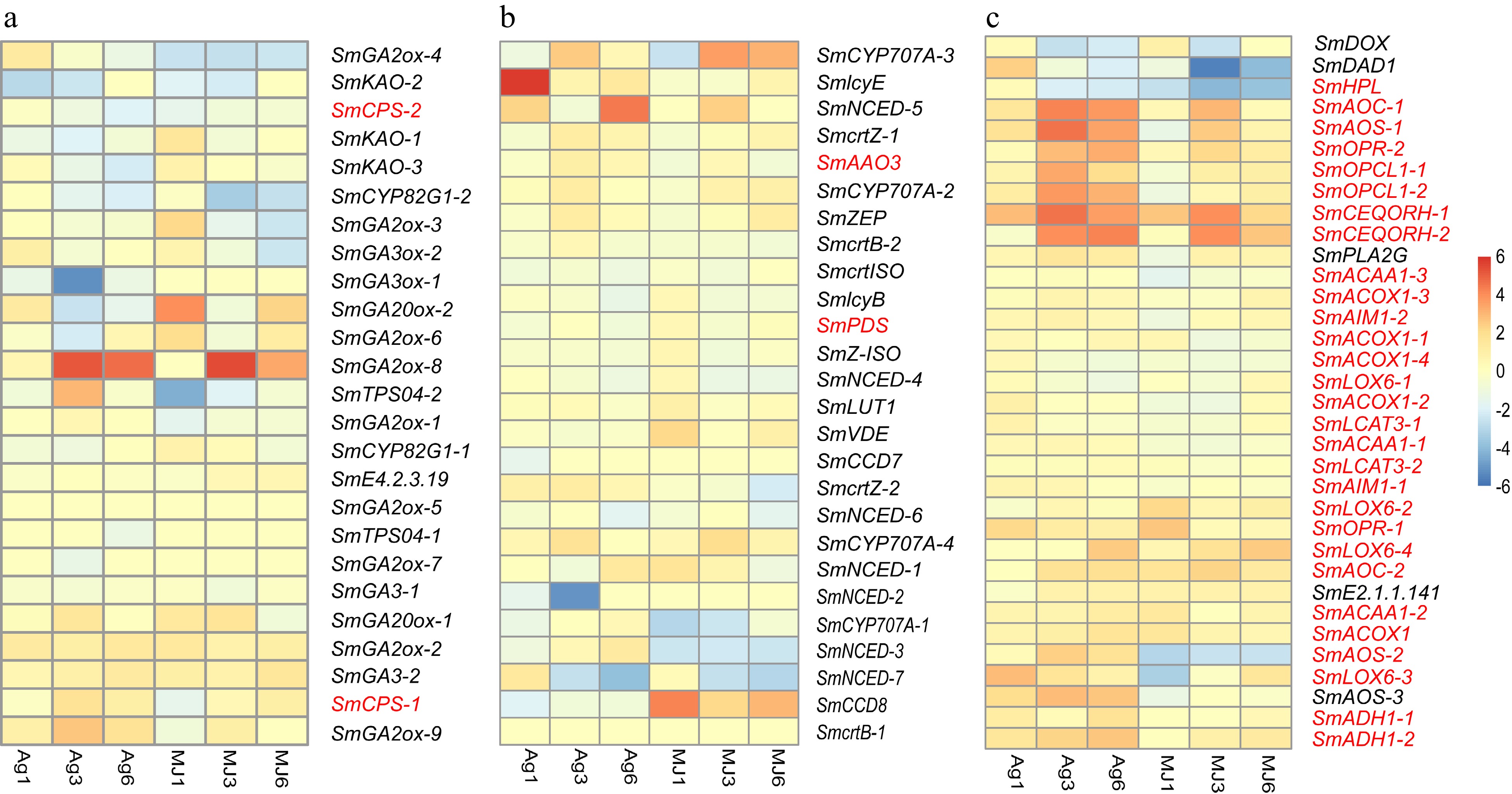
Figure 5.
The expression heatmap of the signaling pathways differentially expressed under the treatment of Ag+ and MeJA. (a) Gibberellin signaling pathway. (b) Abscisic acid signaling pathway. (c) Jasmonic acid signaling pathway.
Lipoxygenase (LOX), Allen oxidase (AOS), and Allen oxidative cycle enzyme (AOC) are involved in the biosynthesis of JA[33]. SmOPCL1, SmOPR, SmAOS were statistically significantly and differentially upregulated in response to Ag+ (Supplemental Table S17). Although there were no statistically significant differences with respect to control values, examination of the heat maps suggested that the expression of most of the genes involved in the JA signaling pathway were upregulated following the Ag+ treatment (Fig. 5b). In contrast, the expression of these genes did not seem to be affected by the treatment with MeJA. These observations suggest that the JA signaling pathway was more sensitive to Ag+ when compared to MeJA.
Activation of the ABA signaling pathway effectively stimulated the production of tanshinones. According to heat map analysis (Fig. 5c), the expression of the SmlycE and SmNCED-5 genes were relatively high and responsive to Ag+ and MeJA treatment (Supplemental Table S18).
KEGG enrichment results of DEGs showed a set of genes involved in terpenoid biosynthesis, including SmDXS, SmGGPPS, SmGA20ox1, and SmGA30ox1 due to Ag+ and MeJA treatment, and SmCYP707A1, SmNCED due to MeJA treatment (Supplemental Table S9 & S10). Based on the calculated PCCs between candidate genes and pathway genes, SmCYP707A-1 showed a high correlation with SmAACT1 in the upstream pathway (PCC of 0.78) while SmNCED highly correlated with downstream SmCYP76AK1 and SmDXR (PCCs of 0.96 and 0.95, respectively). Both SmCYP707A-1 and SmNCED participated in the biosynthesis of ABA, which indicated that ABA regulated both upstream and downstream biosynthesis of tanshinones following MeJA treatment. In addition, SmGA2ox1 and SmGA3ox1 showed a high correlation with SmCYP76AH1 (PCCs of 0.69 and 0.70, respectively). This suggested that the upregulated expression of SmGA2ox1 and SmGA3ox1 led to an increase in the accumulation of tanshinones by regulating the biosynthesis of GA induced by Ag+ or MeJA.
Contribution of gene clustering to the biosynthesis of S. miltiorrhiza
-
The genome of S. miltiorrhiza and transcriptomic data were uploaded to plantiSMASH[34], an analysis platform for the identification of gene clusters, and a total of 32 gene clusters were identified (Supplemental Table S19). Multiple CYP71Ds and TFs were observed in the gene clusters. Based on the correlation coefficients of key genes in the gene clusters calculated by plantiSMASH, it was hypothesized that tanshinone biosynthesis was determined by the joined contribution of gene clusters. Combined with the tanshinone biosynthesis pathway, the sequential chain of cluster18-cluster26-cluster16-cluster3-cluster13-cluster23-cluster11-cluster7-cluster5 was screened, key genes in the downstream pathway of tanshinone biosynthesis distributed in these clusters (Fig. 6). Two SmHDR were found on cluster18, one SmGPPS3 on cluster16, two SmCYP76AH1 and one SmCYP71D55 on cluster3, and one SmCYP71D373 on cluster7, all of which had positive effects on tanshinone biosynthesis[13]. In addition, SmWRKY22-1 was found on cluster18, and SmWRKY22-1 showed significantly increased expression levels after 3 and 6 d Ag+ or MeJA treatment, reaching 6.04- and 4.53-fold, respectively, higher levels than those of the controls after 3 d. These findings suggest that SmWRKY22-1 might regulate SmHDR in a gene cluster and were co-involved in the biosynthesis of tanshinones in S. miltiorrhiza roots. Notably, four Sm2OGD were found in cluster 5, and the expression level of these Sm2OGD increased after 1 d of treatment with MeJA, which was 1.60 - 3.91 times higher than that of the controls. According to our results, Sm2OGD on cluster 5 was located downstream of SmCYP71D373, therefore, Sm2OGD was likely involved in the biosynthesis of 16-hydroxymiltiron to tanshinone.
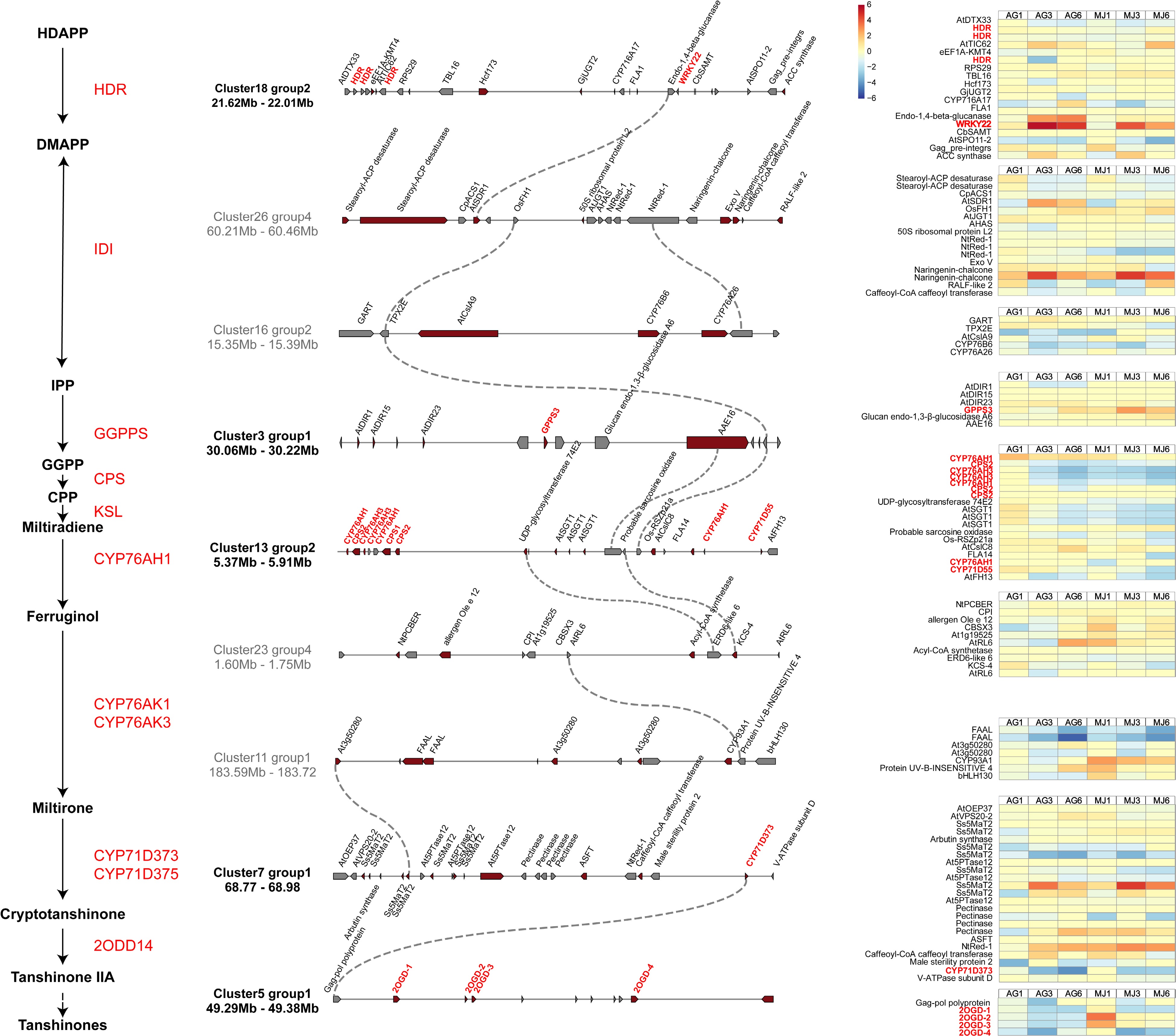
Figure 6.
Gene clusters of S. miltiorrhiza. Left is the downstream pathway of tanshinone biosynthesis. Gene clusters involved in tanshinone biosynthesis were identified by plantiSMASH. Pathway genes and candidate genes are labeled in red, and dotted lines represent co-expressed relationship between genes. The expression of genes in clusters under the treatment of Ag+ and MeJA are on the right of the figure.
-
Tanshinones and phenolic acids are the main active compounds of S. miltiorrhiza hairy roots, and the pathways for their biosynthesis have been widely been. The elicitors Ag+ and MeJA can induce the accumulation of tanshinones and phenolic acids. Tanshinones are mainly biosynthesized through the MEP pathway. Our results showed that the genes involved in the MVA pathway were more sensitive to Ag+ and MeJA elicitors when compared to the MEP pathway. A set of candidate genes was screened, consisting of P450, transcription factors, and genes in hormone signaling pathway. The statistically significant differences in the expression of the genes involved in the MVA pathway may be related to the crosstalk between the two pathways[35].
Ag+ and MeJA have been reported to promote the accumulation of secondary metabolites in S. miltiorrhiza hairy roots, including tanshinones and SAB. In the current study, the genes encoding for the enzymes in the MVA pathway correlated more with Ag+ and MeJA than the MEP pathway, and their expression was generally up-regulated by treatment with Ag+ or MeJA. Whereas tanshinones were mainly biosynthesized by the MEP pathway in plastids, the response of the enzymes in the MVA pathway to elicitors may be attributed to crosstalk between both pathways[35]. Ag+ and MeJA had a facilitating effect on SAB accumulation, and SmTAT was differentially upregulated in the tyrosine-derived pathway, consistent with a previous report[36]. The expression of Sm4CL was differentially upregulated by Ag+ and MeJA in the phenylpropanoid biosynthetic pathway. In addition, the expression of SmCYP98A14 was upregulated in response to MeJA treatment, implying that the SAB biosynthetic pathway was more responsive to MeJA when compared to Ag+.
SmCPS2, SmCYP76AH1, and SmCYP76AH3 have previously been reported to be clustered in a gene cluster for tanshinone production[13, 37]. Our study re-identified the gene clusters and calculated the correlation between gene clusters in the genome of S. miltiorrhiza (Supplemental Fig. S5). Also, the gene cluster of SmCPS2, SmCYP76AH1, and SmCYP76AH3 was observed to consist of two SmCYP76AH3, three SmCYP76AH1, and two SmCPS, while there were three SmCYP71D55. The latter gene has been reported to be a sesquiterpene synthase involved in the metabolism of terpenoids. This is in accordance with the current results, implicating SmCYP71D55 in the production of tanshinones in S. miltiorrhiza.
Gene expression analysis constructed a co-expression network of gene clusters, and identified genes involved in tanshinone biosynthesis, implying that gene clustering contributed to the production of these compounds. SmWRKY22 was located on the same gene cluster with three SmHDRs, indicating that SmWRKY22 was likely involved in regulating upstream elements of tanshinone biosynthesis. In addition, we identified four Sm2OGD in cluster 5, which have been reported to catalyze the hydroxylation of sugiol at the C-15 and C-16 positions to produce the diterpenoids hypargenin B and crossogumerin C, respectively[38]. And all Sm2OGD were significantly upregulated by MeJA treatment, while Ag+ had almost no effect on them.
-
Our study reported key genes related to tanshinone and SAB biosynthesis following Ag+ and MeJA treatment of S. miltiorrhiza hairy roots, comprising genes in the biosynthesis pathways of P450, TFs, and phytohormone signaling. Regulatory networks of tanshinones and SAB biosynthesis treated with abiotic elicitors were constructed. Gene cluster analysis and gene expression analysis further explored the role of gene clustering on tanshinone production. The results from this study contribute to understanding the accumulation of tanshinones and SAB in S. miltiorrhiza hairy roots and the mechanism involved in these processes.
This work was financially supported by Zhejiang Provincial Natural Science Foundation of China (LR21H280002); Key Scientific and Technological Grant of Zhejiang for Breeding New Agricultural Varieties (2021C02074); National Natural Science Foundation of China for State Key Laboratory (81973415, 31800255) and Key project of the Central Government: Capacity Building of Sustainable Utilization of Traditional Chinese Medicine Resources (2060302) .
-
The authors declare that they have no conflict of interest.
-
# These authors contributed equally: Ying Cheng, Xinyu Hong
- Supplemental Table S1 RNA-Seq Mapping Rate Statistics of CK group and Ag+ group.
- Supplemental Table S2 RNA-Seq Mapping Rate Statistics of MeJA group.
- Supplemental Table S3 Differential genes of tanshinone biosynthesis pathway under the treatment of Ag+.
- Supplemental Table S4 Differentially expressed genes on tanshinone pathway under the treatment of MeJA.
- Supplemental Table S5 Genes expression in tanshinone pathway under the treatment of Ag+ and MeJA.
- Supplemental Table S6 Genes esxpression on SAB pathway under the treatment of Ag+ and MeJA.
- Supplemental Table S7 Differential genes of SAB pathway under the treatment of Ag+.
- Supplemental Table S8 Differential genes of SAB pathway under the treatment of Ag+.
- Supplemental Table S9 The KEGG results of Ag+ treatment.
- Supplemental Table S10 The KEGG results of MeJA treatment.
- Supplemental Table S11 Differentially expressed P450 under the treatment of elicitors.
- Supplemental Table S12 Differentially expressed ERF under the treatment of elicitors.
- Supplemental Table S13 Differentially expressed WRKY under the treatment of elicitors.
- Supplemental Table S14 Differentially expressed bHLH under the treatment of elicitors.
- Supplemental Table S15 Differentially expressed MYB under the treatment of elicitors.
- Supplemental Table S16 Differentially expressed genes on GA signal pathway.
- Supplemental Table S17 Differentially expressed genes on JA signal pathway.
- Supplemental Table S18 Differentially expressed genes on ABA signal pathway.
- Supplemental Table S19 The information of Gene clusters involved in tanshinone pathway.
- Supplemental Fig. S1 The KEGG enrichment results of genes differentially expressed under the treatment of Ag+.
- Supplemental Fig. S2 The KEGG enrichment results of genes differentially expressed under the treatment of MeJA.
- Supplemental Fig. S3 The expression heatmap of P450 differentially expressed under the treatment of Ag+ and MeJA.
- Supplemental Fig. S4 Co-expressed heatmap of gene modules and salvianolic acid B biosynthesis pathway genes.
- Supplemental Fig. S5 Co-expressed heatmap of gene modules and tanshinone biosynthesis pathway genes.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Cheng Y, Hong X, Zhang L, Yang W, Zeng Y, et al. 2023. Transcriptomic analysis provides insight into the regulation mechanism of silver ions (Ag+) and jasmonic acid methyl ester (MeJA) on secondary metabolism in the hairy roots of Salvia miltiorrhiza Bunge (Lamiaceae). Medicinal Plant Biology 2:3 doi: 10.48130/MPB-2023-0003 

Transcriptomic analysis provides insight into the regulation mechanism of silver ions (Ag+) and jasmonic acid methyl ester (MeJA) on secondary metabolism in the hairy roots of Salvia miltiorrhiza Bunge (Lamiaceae)
- Received: 15 February 2023
- Accepted: 03 April 2023
- Published online: 26 April 2023
Abstract: Salvia miltiorrhiza Bunge (Lamiaceae) is a traditional medicinal plant. Its main active components are tanshinones and salvianolic acid B (SAB). Abiotic elicitors such as silver ions (Ag+) and jasmonic acid methyl ester (MeJA) are reported to promote the accumulation of secondary metabolites in the hairy roots of S. miltiorrhiza. However, the mechanisms underlying the working of the elicitors remains unclear. For this reason, transcriptome quantitative analysis was conducted to describe the overall expression of elicitors-treated hairy roots, and key protein-encoding genes were identified in the MVA pathway which had a positive effect on tanshinone accumulation. Upstream genes in the biosynthetic pathway of SAB were more responsive to MeJA. Transcription factors (SmWRKY73, SmbHLH25), genes in the GA signaling pathway (SmGA2ox, SmGA3ox) and ABA signaling pathway (SmNCED, SmCYP707A) may be involved in regulating tanshinone and SAB biosynthesis. Upregulated transcription factors were highly correlated in response to Ag+ treatment and MeJA treatment, and Ag+ treatment had a better induction effect on transcription factors than MeJA treatment. Gene cluster mining and co-expression network construction further revealed the contribution of gene clusters to tanshinone biosynthesis, and Sm2OGD was likely involved in tanshinone biosynthesis. In conclusion, the current study screened the key genes for transcription factors and hormone signaling pathways that may promote the accumulation of tanshinones and SAB. The results improved the regulatory network of Ag+ and MeJA-mediated secondary metabolite biosynthesis in S. miltiorrhiza hairy roots, and improved our insight into the regulatory mechanisms involved in the biosynthesis of tanshinones and SAB.
-
Key words:
- Salvia miltiorrhiza /
- Elicitors /
- Ag+ /
- MeJA /
- Secondary metabolites /
- Tanshinones /
- Salvianolic acid B