









-
Nelumbo Adans. (Proteales, Nelumbonaceae), is a genus of aquatic plant species with an estimated origin of 135 million years ago (mya)[1], making it one of the earliest diverging eudicot lineages. Given the phylogenetic position in the early eudicots and the morphological similarity of extant species with fossil taxa, Nelumbo is regarded as a living fossil. Two extant species, Nelumbo lutea (Willd.) Pers. (American lotus) and Nelumbo nucifera Gaertn. (Asian lotus) are recognized in this genus[2]. The Asian lotus (also referred to as sacred lotus and 莲 'lian' in Chinese) is distributed throughout Eastern Asia and northern Oceania in freshwater habitats. The cultivation of Asian lotus is thought to have begun more than 3,000 years ago for the production of edible seeds[3]. In addition to seeds, Asian lotus is also grown for the large edible rhizome it produces, and as an ornamental in water gardens. From these different uses, Asian lotus growers and researchers categorize the different types of plants into seed, rhizome, and flower types based on morphological characteristics that best suit each of these applications[4]. In addition to the cultivated types, wild Asian lotus is common throughout east and southeast Asia in lakes and ponds.
Because Asian lotus is an important food plant, extensive molecular work has been conducted to better understand the genetic diversity and history of this species. Some examples of this work include estimating the divergence time between the two Nelumbo species from complete plastome sequences at 1.5 mya[5,6] and the discovery of an ancient whole genome duplication unique to Nelumbo using high-quality nuclear genome assemblies[7]. Population structure and genetic diversity in Asian lotus have also been extensively studied using several different markers including random amplified polymorphic DNA (RAPD)[8], inter-simple sequence repeats (ISSR)[9], amplified fragment length polymorphisms (AFLP), simple sequence repeats (SSR)[10], single nucleotide polymorphisms (SNPs)[11], and whole-genome resequencing methods[12−14]. From the above studies, higher genetic diversity was generally found among wild Asian lotus compared to cultivated Asian lotus, and among the cultivated lineages, seed and rhizome types resolved in distinct clades. However, some conflicts remained unresolved among these studies. In particular, Huang et al.[12] determined that the seed lotuses were monophyletic in respect to wild and rhizome lotuses but with low bootstrap support, while Liu et al.[14] found that seed and flower lotuses possessed higher genetic diversity, and were more often crossbred to each other than either were to rhizome lotuses. While many of the previous studies focused on patterns of genetic diversity, few have integrated geographic origin into their analyses, leaving gaps in our knowledge regarding centers of origin of the different Asian lotus types[14]. For example, wild Asian lotus from Indonesia and their relation to cultivated types has not been properly characterized in previous studies. Given issues with incomplete lineage sorting associated with whole genome duplications[15], using a pan-plastome approach can provide an improved resolution regarding questions of population structure, centers of origin, and assignment of cultivated types to well supported genetic clusters.
Chloroplasts (plastid refers to all membrane bound organelles of the same origin but serving different metabolic functions such as chloroplasts, chromoplasts, and leucoplasts) are the photosynthesis organelle in plant and algae cells, originating from cyanobacteria through an ancient endosymbiotic event and contain a distinct streamlined genome primarily made up of photosynthesis and replication related genes[16]. Compared to the nuclear genome, the plastome is uniparentally inherited and nonrecombinant, which can provide a less noisy signal for inferring relatedness especially in lineages with polyploidy, incomplete lineage sorting, and/or frequent introgression[17]. Most previous studies in phylogenomics have been focused at the species level or above and often employ genomic simplification strategies such as using only the transcriptome[18] or reduced-representation/finely-filtered genomes[19] in the final analyses. As whole-genome sequencing and assembly have gotten more accurate and complete, large intraspecies collections of genomes (often referred to as the pan-genome) are now being published that include all or nearly all major nucleotide variants found in a given lineage across the entire genome. Such pan-genomes have been produced for important agronomic plant species such as Glycine soja[20], Sorghum bicolor[21], and Brassica napus[22]. Similarly, pan-plastomes are now being generated for several plant species with the first such dataset involving 321 complete plastomes to differentiate pepper (Capsicum) cultivars and lineages[23]. As with other phylogenomic approaches pan-plastomics have several advantages over nuclear pan-genomes such as larger more complete reference sets for assembly and comparison, occurrence in higher copy number in the cell resulting in greater read depth, and the absence of large duplicate gene arrays reducing problems associated with paralogy[24]. Therefore, we employed a pan-plastome approach to address several outstanding questions regarding Nelumbo cultivation and evolution. In addition, the dataset presented here is an important comparative resource for pan-plastome studies in other species which are at present uncommon.
Here, we assembled a large plastome data set including 316 (five N. lutea and 311 N. nucifera) complete circular plastomes to: (a) construct a reliable pan-plastome map for Nelumbo, (b) identify genomic patterns in the data set, such as mutational hotspots and characterization of different nucleotide variants, and (c) resolve well supported maternal lineages within Nelumbo and relate these to the different cultivated and wild types including the sister species N. lutea to address questions regarding origin and relatedness. For convenience, the names of the different lotus types and species used in this study were simplified as follows: N. lutea (North American lotus): LA; wild Asian lotus: LW; flower lotus: LF; seed lotus: LS; and rhizome lotus: LR (all cultivated types are from N. nucifera).
-
To characterize the plastome structure and genomic organization of N. lutea and N. nucifera, comprehensive comparisons were conducted with regards to genome size, tetrad length, GC content, and gene order and function (Fig. 1, Supplemental Table S2). All plastomes assembled as part of this study retained the typical quadripartite structure found in most chloroplast genomes (comprised of a LSC and SSC separated by a pair of IRs). The plastome size across all 316 accessions varied from 163,457 to 163,672 bp (Med = 163,647 bp). Among the different plastome regions, the LSC size ranged from 91,746 bp to 91,914 bp (Med = 91,888 bp), the SSC between 19,605 and 19,639 bp (Med = 19,627 bp), and the IRs from 26,053 bp to 26,071 bp (Med = 26,066 bp). The total GC content (%) of the complete plastomes ranged from 37.95 to 38.00 (Med = 37.96) with 36.17 to 36.22 (Med = 36.19) for the LSCs, 32.22 to 32.34 (Med = 32.25) for the SSCs, and 43.18 to 43.22 (Med = 43.19) for the IRs. In all four regions and in total length the N. lutea plastomes were shorter in length than the N. nucifera plastomes.

Figure 1.
The pan-plastome of Nelumbo. The inner genes of the outer circle are transcribed counterclockwise while the outer genes are transcribed clockwise; genes with introns were marked with an asterisk (ycf3, clpP, and rps12 contain two introns all others contain one). The GC content is displayed as gray bars inside of the tetrad divisions (LSC, SSC, IRA, and IRB) SNVs, InDels, and block substitutions are represented within the GC content as orange, blue, and yellow lines, respectively. Comparisons of the complete genome length and each of the four subregions (given in median lengths) are compared for each of the different Nelumbo types N. lutea LA, LW, LF, LS, and LR.
A total of 113 unique genes were annotated and grouped into functional categories as follows: 79 protein-coding genes (PCGs), 30 transfer RNA (tRNA) genes, and four ribosomal RNA (rRNA) genes. The annotated genes were located in the following genomic regions: 83 genes (61 PCGs and 22 tRNA genes) in the LSC, 12 (11 PCGs and one tRNA gene) in the SSC, and 18 (seven PCGs, seven tRNA genes, and all four rRNA genes) duplicated in the IRs. Among the genes 18 (13 located in LSC, one in SSC and four in the IRs) including 12 PCGs (atpF, clpP, ndhA, ndhB, petB, petD, rpl2, rpl16, rpoC1, rps12, rps16, and ycf3) and six tRNA (trnA-UGC, trnG-UCC, trnI-GAU, trnK-UUU, trnL-UAA, and trnV-UAC) contained introns. Of the genes containing introns three PCGs ycf3, clpP, and trans-spliced rps12 (characterized by the first exon locating in LSC and the other two in the IRs) contained two introns (Fig. 1; Supplemental Annotation). These results indicate a highly conserved genome structure and gene content across the Nelumbo pan-plastome.
Pan-plastome polymorphisms
-
In order to assess nucleotide variants, all plastomes were aligned and scanned for variants. The pan-plastome alignment length was 164,754 bp, in which 164,058 (99.58%) sites were conserved and 696 (0.42%) sites were variable of which 577 sites were parsimony-informative and 45 were autapomorphic. Using LA001 as a reference, variants among 316 plastomes were identified as either SNVs, InDels, or block substitutions. Among these variants, SNVs were the most abundant (418, 60.06%) followed by InDels (208, 29.89%) and block substitutions (70, 10.06%). Variants were unevenly distributed throughout the pan-plastome (Table 1, Fig. 1). Most variants were located in the LSC (487, 69.97%), followed by the SSC (167, 23.99%), and the fewest in the highly conserved IRs (42, 6.03%). In regard to the location of variants to genes, intergenic spacer regions (IGS) contained the most (510, 73.28%) variants, while cds (124, 17.82%) and intronic (62, 8.91%) sequences contained far fewer (Fig. 2). A total of 499 fixed variants (294 SNVs, 151 InDels, and 54 block substitutions) were found that could distinguish American from Asian lotuses (Table 1, Supplemental Fig. S1). Nonsynonymous mutations were found most often in the PCGs accD, ccsA, cemA, matK, ndhE, ndhF, rpoC1, ycf1, and ycf2. When assessed by functional group, these nonsynonymous mutations were most abundant in the groups NADH dehydrogenase and RNA polymerase (Fig. 2, Supplemental Table S3). After excluding sites with gaps, a total of 162,648 sites were retained with 566 variable sites, among which 31 were autapomorphic and 535 were parsimony-informative including 529 bi-allelic sites and six tri-allelic sites (i.e., site 16,914 in rpl33, site 18,026 in psaJ-trnP-UGG, site 40,576 in trnL-UAA-trnT-UGU, site 40,801 in trnT-UGU-rps4, site 90,747 in psbA, and site 138,254 in ycf1).
Table 1. Number of variants among 316 Nelumbo accessions. The number of variants found only in N. nucifera are indicated in parenthesis.
Variants Total Region Location LSC SSC IRA/B CDS Intron IGS SNV 418 (294) 274 (182) 112 (89) 16 (12) 117 (91) 33 (23) 268 (180) Block substitution 70 (54) 56 (42) 12 (11) 1 (0) 3 (3) 4 (4) 63 (46) InDel 208 (151) 157 (118) 43 (27) 4 (3) 4 (2) 25 (19) 179 (130) Total 696 (499) 487 (342) 167 (127) 21 (15) 124 (96) 62 (46) 510 (356) 
Figure 2.
Variant locations categorized by genic position (CDS, Introns, and IGS) and functional group.
Population structure and genetic diversity
-
In order to elucidate matrilineal relationships among lotuses, multiple approaches were employed to resolve population structure and calculate genetic diversity as relates to the different species and cultivated types. Based on a SNV only input matrix, population structure as inferred using ADMIXTURE was determined based on the lowest CV error of 0.026 at K = 7 (Fig. 3d). All N. lutea individuals resolved into a distinct genetic cluster with no cross assignment to any of the N. nucifera genetic clusters (designated as genetic clusters I−VI, Fig. 3e). Individual assignment proportions (based on q-values) were high among nearly all Asian lotus individuals except for six accessions in genetic cluster III that had less than 35% assignment to genetic cluster I and two individuals with less than 30% assignment to genetic cluster VI as well as 15 accessions in genetic cluster in VI with less than 45% assignment to genetic cluster V. The other three methods, namely, an ML tree using a SNV only matrix, PCA using a complete plastome matrix, and a median-joining network using a gap-excluded matrix, all corroborated the population structure resolved in the ADMIXTURE analyses. For example, the close relationship between genetic clusters V and VI is resolved across methods by: (1) resolution as sister clades in the ML analyses, (2) multiple connecting nodes and few variants on branches separating V and VI in the median-joining network, (3) partial overlap between V and VI in the PCA graph, and (4) partial assignment of some individuals in VI to V in the ADMIXTURE analyses (Fig. 3a−c, 3e).
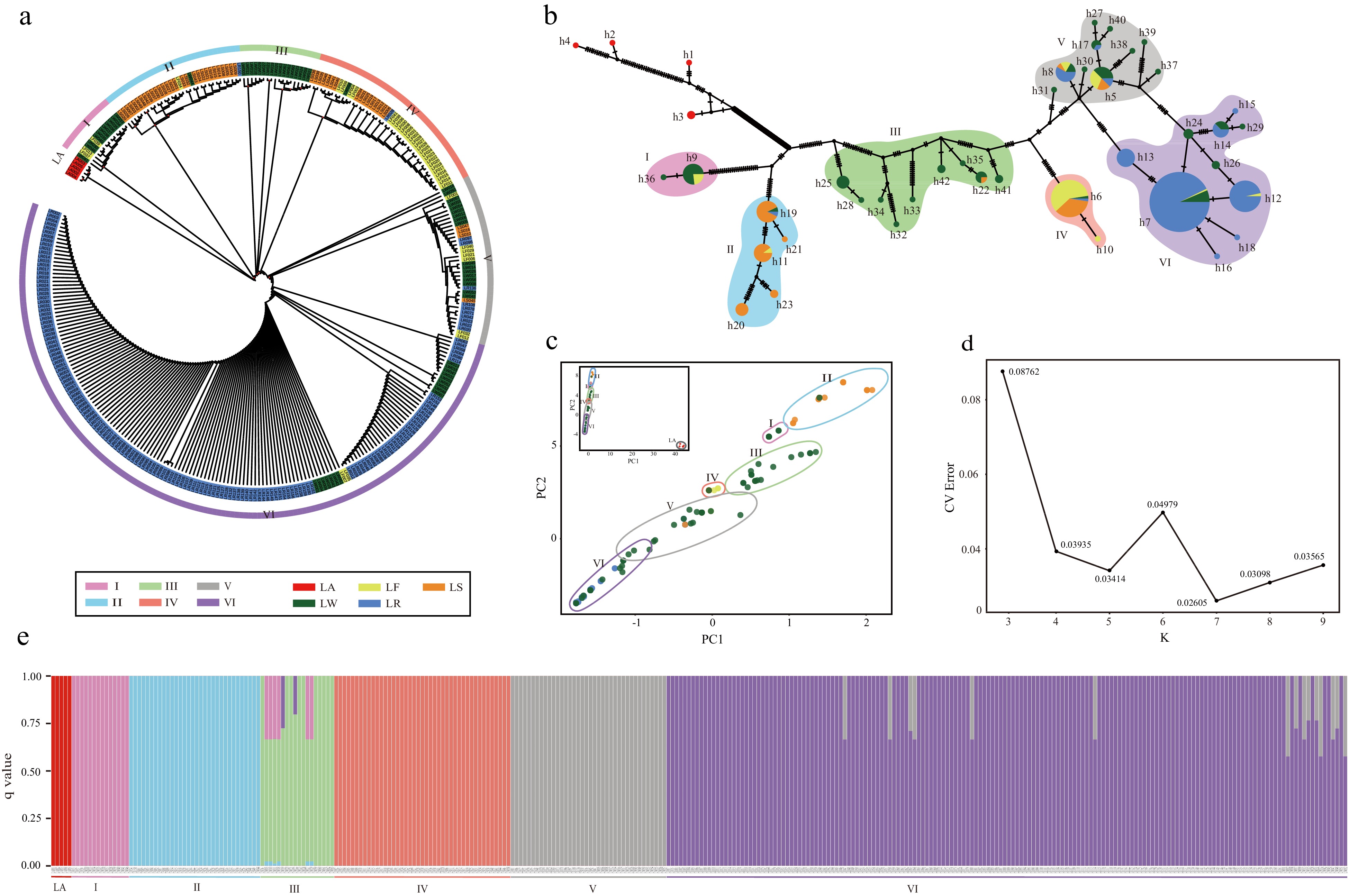
Figure 3.
Population structure of Nelumbo. All analyses resolved similar membership into six genetic clusters (I−VI). (a) ML tree of 316 accessions. (b) Median-joining network with haplotype identifiers adjacent to nodes, size of pie chart proportional to number of accessions sharing the same haplotype, colors in the pie chart represent percentage of accessions from a given Asian lotus type. (c) PCA analysis including LA (upper left inset) and excluding LA showing the first two components in both cases. (d) CV errors across a range of K values from 3−9. (e) Population structure bar-plot at K = 7.
The different Asian lotus types were unevenly represented among genetic clusters (Fig. 3a, Supplemental Fig. S2 & S3). Genetic cluster I contained three (21%) LF and 11 (79%) LW accessions; II contained 1 (3%) LF, 1 (3%) LR, 29 (91%) LS, and 1 (3%) LW; III contained 1 (3%) LS, and 34 (97%) LW; IV contained 26 (61%) LF, 1 (2%) LR, 15 (35%) LS, and 1 (2%) LW; V contained 7 (18%) LF, 4 (11%) LR, 10 (26%) LS, and 17 (45%) LW; and VI contained 2 (1%) LF, 148 (89%) LR, and 16 (10%) LW. Furthermore, rhizome lotuses were found in four of six genetic clusters with 12 haplotypes; flower lotuses found in five of six genetic clusters with nine haplotypes, and wild lotuses found in all six genetic clusters with 28 haplotypes.
From the median-joining network 42 haplotypes were designated among the 316 accessions examined (designated h1−h42). The four haplotypes (h1−h4) from N. lutea were denoted for possessing a large number of nucleotide variants separating the haplotypes within the species and an even larger number of variants separating these haplotypes from N. nucifera resulting in high levels of genetic and haplotypic diversity (π = 2.02e-04, Hd = 0.900) for the LA genetic cluster. Nucleotide and haplotype diversity among the six genetic clusters within Asian lotus varied widely (Fig. 4). Genetic cluster I contains 2 haplotypes with π =8.7300e-07 and Hd = 0.143; II 5 haplotypes with π = 3.7673e-05 and Hd = 0.738; III 9 haplotypes with π = 1.5533e-04 and Hd = 0.882; IV 2 haplotypes with π = 5.6860e-07 and Hd = 0.092, V 10 haplotypes π = 3.7673e-05, and Hd = 0.738; and VI 10 haplotypes with π = 8.8611e-05 and Hd = 0.533.
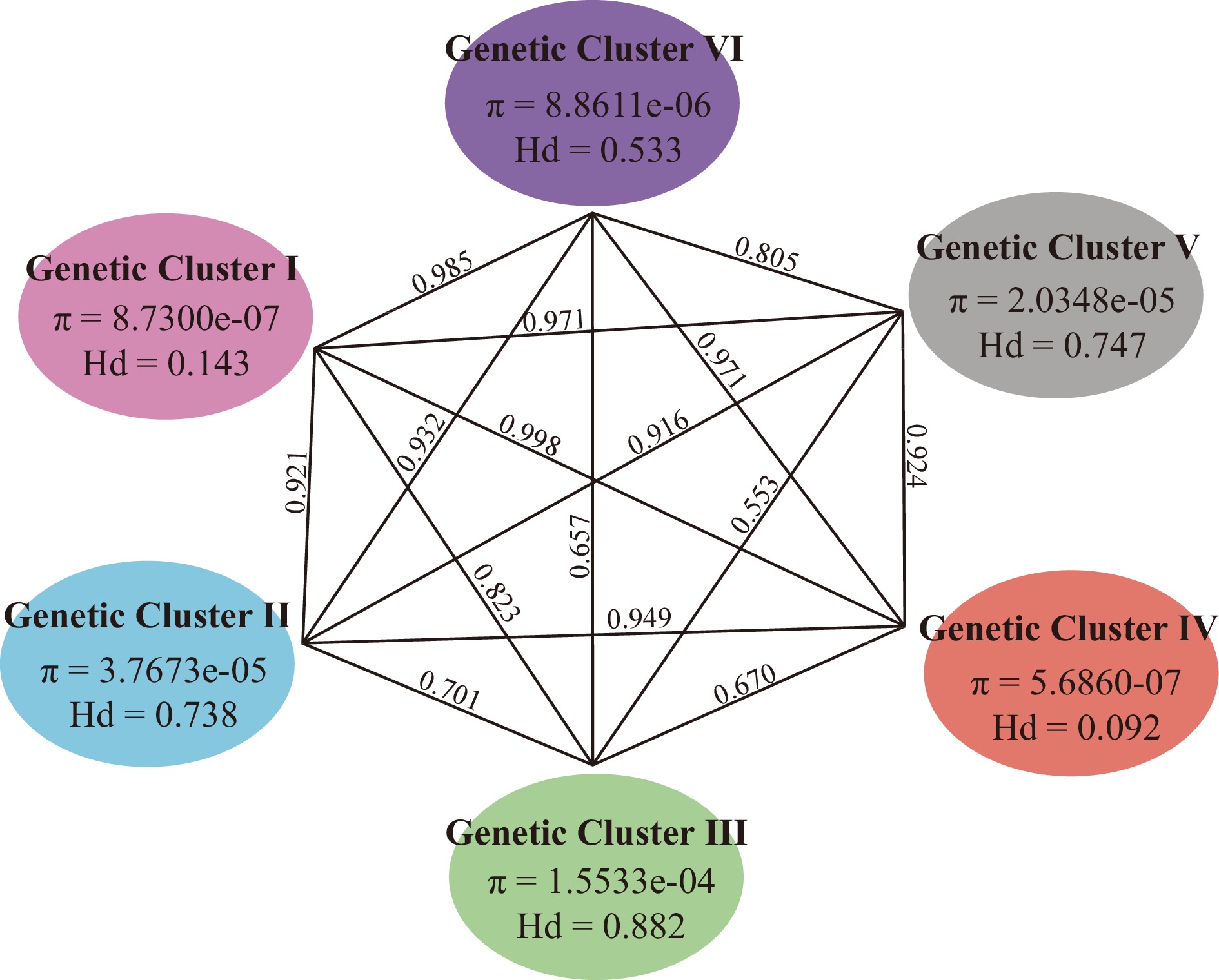
Figure 4.
Genetic diversity and differentiation of six genetic clusters of Asian lotus. Numbers above lines connecting two bubbles represent pairwise Fst calculated between respective genetic clusters.
As with genetic diversity, divergence patterns varied between genetic clusters. Genetic cluster III had the lowest level of divergence when compared to all other genetic clusters (III to I Fst = 0.823; III to II Fst = 0.701; III to IV Fst = 0.670; III to V Fst = 0.553; and III to VI Fst = 0.657). By contrast genetic cluster I had higher levels of divergence when compared to the other Asian lotus genetic clusters (I to II Fst = 0.921; I to III Fst = 0.823; I to IV Fst = 0.998; I to V Fst = 0.971; and I to VI Fst = 0.985). The typical levels of divergence between genetic clusters are more similar to that found between genetic cluster I and others, with genetic cluster III being an outlier in regard to the lower levels of divergence to all other genetic clusters.
Geographic distribution of Asian lotus
-
In order to resolve patterns related to origin and dispersal of Asian lotuses individuals were mapped by genetic cluster and type (Fig. 5, Supplemental Tables S1 & S4). The Asian lotus collections used in this study were made from three main areas of historical cultivation including: (1) northeastern China and North Korea; (2) east central China; and (3) southern China and several Southeast Asian countries including India, Thailand, Indonesia, and Singapore (Fig. 5). All wild lotuses in genetic cluster I were collected from Yunnan province with a single flower type collected in east central China and all remaining flower types in this cluster also collected from Yunnan. Most seed lotus accessions (28/32) in genetic cluster II were from central China with a single wild accession collected from northern Thailand, a single flower type from Japan, and a single rhizome type collected from central eastern China (Fig. 5). Accessions assigning to genetic cluster III were broadly arrayed throughout Southeast Asia in the countries of India, Indonesia, Singapore, Thailand as well as eastern China. Within genetic cluster III several haplotypes were found to have geographically narrow distributions such as h25 and h28 in China, h32 and h34 in India, h41 and h42 in Indonesia, h32 in Thailand, and h35 in Singapore. Nearly all of the accessions assigned to genetic cluster IV were collected from China (a single sample from Japan), mainly of haplotype h6 (42/43), and most of seed or flower type (26/43 flower and 15/43 seed). Accessions from genetic clusters V and VI were all collected from across eastern China except for a single accession from North Korea. Haplotypes in genetic cluster V were widespread across China except for h27, h30, h31, h37, h38, h39, and h40 which were each only collected once. All of these uncommon haplotypes in genetic cluster V were wild type lotuses. In genetic cluster VI all accessions from the northeastern most provinces of Heilongjiang and Jilin were all wild types. Most haplotypes in genetic cluster VI were geographically widespread except for h15, h16, h18, h26, and h29 which were uncommon and only collected in the provinces of Jiangsu, Guizhou, Shandong, Heilongjiang, and Anhui respectively.
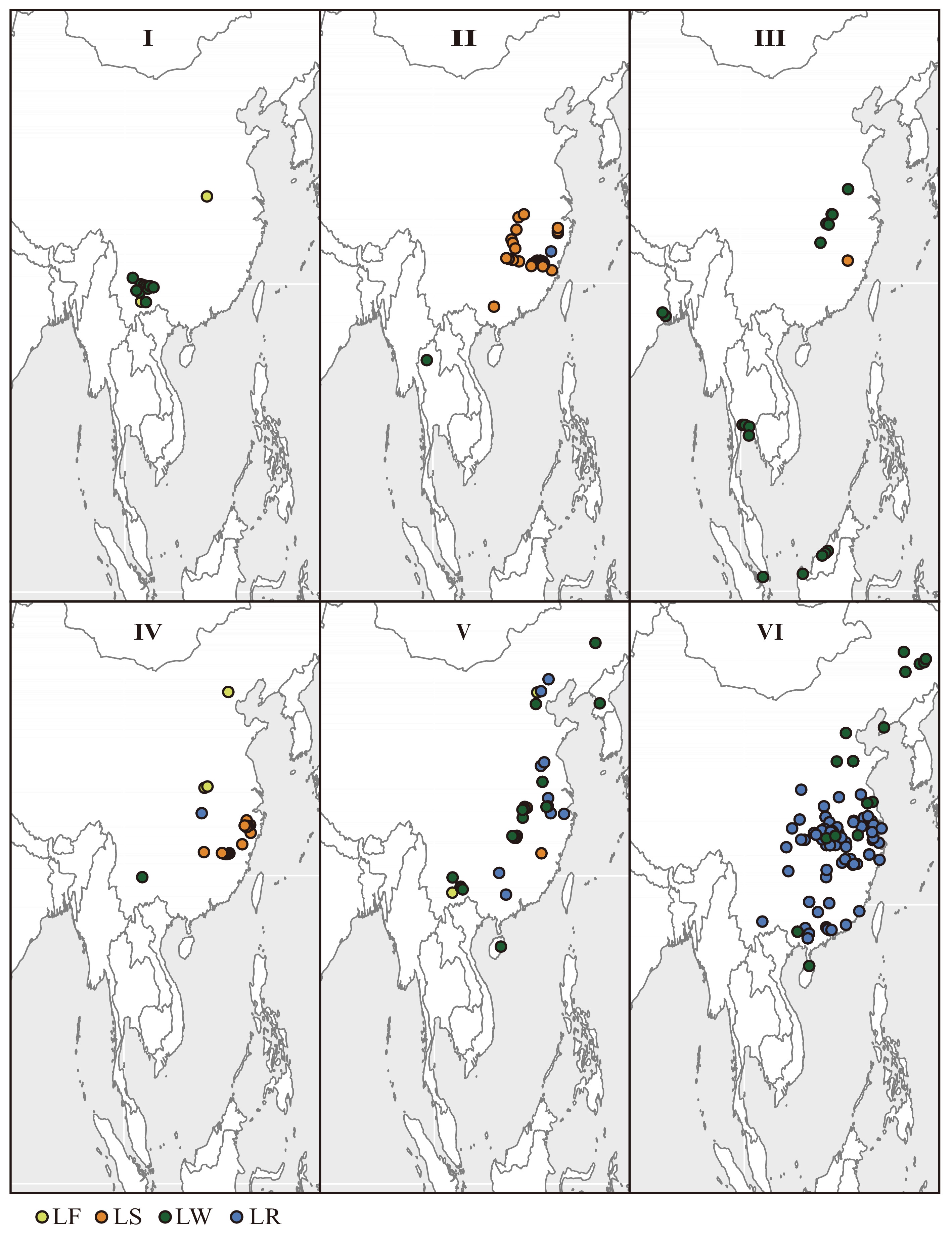
Figure 5.
Geographic distribution of Asian lotus collections used in this study separated by genetic cluster and color coded by type (see Fig. 3).
-
Plastomes are highly conserved in most land plants in terms of size, structure, and gene content, with lotuses being no exception in this regard[25]. The pan-plastome resolved here indicates that the gene content and order were highly conserved and consistent with those previously described in Xue et al.[5] and Wu et al.[6]. Despite structural and genic conservation, abundant nucleotide variants were found across the Nelumbo pan-plastome. Relatively few variants were detected in cds regions, save ycf1 which was extraordinarily rich in nucleotide variants, possible because of its position spanning the junction of IRA and SSC (Figs 1 & 2). This is similar to what has been previously reported for junction spanning genes in Passiflora trichocarpa[26]. Following ycf1 in a number of cds variants were rpoC1 and ndhF which have also been found to contain a higher number of nucleotide variants than other functional plastome genes and thus their use in phylogenetic studies for eudicot groups like Apioideae, Cactoideae, and Asteridae[27]. While mutations (especially InDels) in cds regions are expected to result in a loss of function in translated proteins, ribosomal frameshifting in plastomes has been shown to recover original functions from cds containing mutations[28]. As such mutations in some plastome cds regions may have less of an impact than predicted. Unlike the relatively limited number of cds variants the type and abundance of nucleotide variants in IGS and introns were considerably greater. For instance, block substitutions in cds regions are found only in the ycf1 and rpoA genes whereas they are relatively abundant in IGS and intronic regions (although less so in introns). Specifically the large number of variants found in the Nelumbo rpl16 intron is similar to that found in the distantly related plant families Crypteroniaceae and Poaceae where this hypervariable region was employed in phylogenetic studies[29,30]. Similarly, the presence of InDels is higher outside of cds regions. This pattern of variant abundance and type found in the Nelumbo pan-plastome follows that found in other plastomes[31]. Because frameshift mutations result in greater disruption to protein structure and function, they are often purged via selection[32]. That said, frameshift correction through translation recoding has been recently described from chloroplasts[28] which may render some cds mutations less impactful by retaining original protein function across lineages despite underlying differences in DNA. As more pan-plastome studies are completed it is becoming increasingly apparent that genomic regions previously used for higher-level systematic studies such as rbcL and matK should be supplemented with hypervariable regions found in IGS and intronic regions for improved resolution in intraspecific studies. In this study the IGS regions rps18-rpl33 and trnQ-UUG-rps16 proved especially rich with informative markers. Our pan-plastome study like those from cultivated species Brassica napus and Sorghum sp[21,22] found that SNVs are by far the most common variant type. However, one of the outstanding questions is whether variants differ in type and effect (in regard to gene function) between domesticated lineages and wild progenitors. With the completion of more pan-plastome studies from diverse cultivated taxa, patterns specific to domesticated lineages can now be resolved to try to understand the function and importance of plastids in the domestication syndrome specifically, and in plant evolutionary biology more generally.
Divergence among genetic clusters and the centers of origin for different types of Asian lotus
-
Plant population structure and genetic diversity are known to be affected through a number of different processes including genetic drift, reproductive isolation, local adaptation, demographic fluctuations, mode of reproduction, and additionally from artificial selection and human translocation associated with the domestication process[33]. Such patterns are evident in this pan-plastome study of Nelumbo wherein geographic separation between N. nucifera and N. lutea is reflected in the numerous fixed genetic differences between these species (Fig. 3). Both analyses based on nuclear[13,14] and plastomic dataset here supported the indubitable divergence between these two Nelumbo species, while some differences were also found regarding phylogeny of genetic clusters within N. nucifera mainly due to the conflicts of maternal and paternal inheritance (nucleocytoplasmic conflicts) common seen in many other species[34,35], which was also important evidence of hybridization or introgression, such as most seed lotus accessions were resolved as monophyly in the previous two researches, but into genetic clusters II and IV here. Additionally, sample differences between the two previous works, and the limited genetic information plastome carried compared to nuclear loci controlling morphologic traits used to designated lotus types could also cause these differences like seed and flower lotuses in genetic cluster IV, but not much. This was also reflected by the much lower genetic diversities of each genetic cluster compared to that in Li et al.[13], and Liu et al.[14]. Genetic clusters II and III showed much higher genetic diversity than others the same as nuclear analyses in Liu et al.[14] regarding seed and wild types, whichever genetic cluster VI (rhizome type) showed relatively lower genetic diversity.
Within N. nucifera, six well-supported genetic clusters were resolved with notable differences in the genetic and haplotypic diversity as well as the cultivated types found in each (Fig. 3). For instance, genetic cluster III is characterized by having a large number of haplotypes each separated by many genetic differences with few repeats per haplotype. In addition, the membership of genetic cluster III is made up of all wild accession except for a single seed type (LS036, h22). One possible interpretation from this pattern is that genetic cluster III represents a wild lineage from which few cultivated types have been selected. This interpretation is further confirmed by noting that the patterns resolved in genetic cluster III are similar to those found in wild N. lutea, although more sampling in N. lutea is needed to confirm this pattern. It suggested that each cultivated type — flower, rhizome, or seed lotus was not single-originated (cultivated from the single wild population) because no cultivated type was found solely within single genetic cluster, implying potential multiple origins for all types and/or maternal introgression into cultivated types from different origins, as like the instances where it was clearly known when the cultivated rice was initially selected from certain cultivated lineages[36,37]. Types can be further resolved by haplotype wherein several types are sometimes found within a single haplotype. For instance, the largest haplotype h7 (genetic cluster VI) with 109 accessions is made up of 1% flower, 93% rhizome, and 6% wild types. Furthermore, the relatively narrow distributions of wild lotus in genetic clusters I and III, while cultivated types in genetic clusters V and VI further expanded their range, indicating the domestication and cultivation history of lotus has gradually expanded under the action of human activity. Genetic diversity also showed a decreasing trend from wild to cultivated types (genetic clusters III to V to VI), which may also be a signal of human domestication. It showed that cultivated types were selected to be cultivated from multiple origins or if it has maternal introgression, both of which could result in a polyphyletic pattern among the cultivated types, for instance, rhizome lotuses were found in four of six genetic clusters with 12 haplotypes; flower lotuses found in five of six genetic clusters with nine haplotypes, and wild lotuses found in all six genetic clusters with 28 haplotypes (Fig. 3a, b, & Fig. 5). Given this pattern among our plastomic data a monophyletic origin for seed lotuses is not supported[12], however because seed lotuses were found in four out of six genetic clusters with eight unique haplotypes, claims regarding diversity are supported by our data. Based on this wild type, lotuses contain the highest level of genetic diversity with cultivated types also exhibiting high levels of plastomic diversity. It should also be noted that the classification of cultivated types was based on their primary use, and some types also have traits that make them usable for other purposes, which might cause some tenuous designations to bring out conflicting results in determining monophyly. An important step in understanding the evolution of cultivated lotuses would be to analyze the nuclear genes involved with lotus domestication[13,38] in concert with the pan-plastome data to better understand how plastome divergence is concordant with patterns of artificial selection detected in the nuclear genome. Such findings may help to elucidate patterns of introgression in the domestication of lotus and how plastomes might have been involved in controlling the directionality of crosses through cytonuclear-incompatibility.
With regard to the geographic origins of cultivated lotuses, several inferences can be made. Genetic cluster I has a probable origin in Yunnan province as all wild accessions were collected there and this genetic cluster has been the matrilineal source for a very small number of flower type cultivars (two flower types from genetic cluster I collected in this study). Of any of the geographic patterns genetic cluster I is the most restricted and least selected from in generating lotus cultivars. The only wild accession in genetic cluster II was from Chiang Mai, Thailand suggesting that this may be the origin of the many seed types collected from this genetic cluster in central eastern China (Fig. 5). However alternative inferences include matrilineal gene flow into wild Thai populations or the Thai accession is the result of an escaped cultivar[39]. Given that higher genetic diversity is present in China within genetic cluster II, the alternative inferences cannot be ruled out as centers of origin can also be centers of genetic diversity. Genetic cluster III like I is made up primarily of wild type accessions but unlike genetic cluster I, III is geographically distributed throughout Southeast Asia and eastern China. Additionally, haplotypes within genetic cluster III are restricted to a given geographic location. As such, genetic cluster III may represent a lineage that broadly dispersed in the distant past and thereafter through adaptation and drift have produced localized haplotypes. The geography of island and peninsula formation in the Sunda Shelf over the last 50 million years may have helped drive this pattern[40]. Genetic clusters IV, V, and VI all appear to originate in eastern China as no wild accession were found outside this geographic area. In genetic cluster IV, a single wild accession from Yunnan shares the h6 haplotype with 98% of the mostly flower and seed type accessions in this genetic cluster. The low nucleotide and haplotype diversity of matrilineal genetic cluster IV is counter to findings found among seed and flower lotuses where high levels of admixture have been noted in these lotus types using nuclear data. That said it is possible to have had a matrilineal bottleneck induced from cytoplasmic incompatibility within a lineage while maintaining a highly diverse and admixed nuclear genome over time[41]. That said, flower and seed types are found in nearly all of the genetic clusters, albeit only two out of 166 are flower type and no seed types in genetic cluster VI, suggesting high levels of maternal introgression among flower and seed types across genetic clusters. Genetic cluster VI is clearly the source of most rhizome type lotuses and because the plant part selected for is unrelated to sexual reproduction, a few very common haplotypes (resulting from asexual reproduction via rhizome cuttings to plant fields) account for nearly all rhizome types in this genetic cluster. Despite most accessions in genetic cluster VI having only a few haplotypes, numerous wild haplotypes were also assigned to this cluster with some having restricted geographic distribution (Supplemental Fig. S4). This suggests that a good deal of wild diversity remains throughout eastern China and especially in the northeastern region.
The domestication of aquatic plants for human consumption is unsurpassed in diversity and extent outside of the eastern coastal plain of China. Lotus, because of the many parts of the plant that can be used for human consumption and the health benefits from eating these parts, has been and will continue to be an important food crop for humans. As with any crop, genetic diversity is essential to maintain high levels of nutrition, disease resistance, yields, and improving or developing traits of interest[42]. Wild populations of Asian lotus are known to be threatened by human development and environmental pollution[13] making the characterization and mapping of genetic diversity all the more important in prioritizing conservation efforts. Our study has shown that cultivated and wild Asian lotus are divided into at least six maternal lineages with geographic distribution and selection of lotus types differing between genetic clusters. From these results, several regions in China (namely Yunnan and the northeast) as well as regions in southeast Asia should be explored further to more properly characterize the unique genetic diversity of lotuses from these areas. In addition, these wild haplotypes should be assessed for their potential use in developing new lotus cultivars. The experimental breeding of diverse lotuses may also provide useful insights into cytonuclear incompatibility and further our understanding of genomic evolution in this living fossil lineage. In summary, the pan-plastome resources presented here for lotus will provide new insights into the natural and domestication history of this lineage as well as prove useful in applied studies such as marker-assisted breeding or the development of transplastomic lines for improved yield or disease resistance.
-
The de novo assembly of all plastomes to complete circular molecules was conducted rather than SNV calling to a reference genome because de novo assembly allows for the assessment and removal of intergenomic (e.g., horizontal gene transfer from chloroplast to nuclear genome) transfer sequences[43]. From the total 365 accessions provided in Li et al.[13] and Liu et al.[14], some accessions (one LA, 17 LW, 11 LF, four LS, and 16 LR) had to be discarded because complete plastomes could not be assembled due to lower sequencing quality in these samples. In the final set, a total of 316 plastomes (five LA, 63 LW, 39 LF, 49 LS, and 160 LR) were successfully assembled for use in downstream analyses. Except for N. lutea sampled from North America, the N. nucifera accessions were broadly collected across China and Southeast Asia (Supplemental Table S1). Based on Illumina next-generation sequencing (NGS) reads from whole-genome sequencing (WGS), de novo assembly of all plastomes was completed using SPAdes 3.14[44] from which a graph of major contigs was used to generate a circular molecule in Bandage 0.8.1[45]. The clean raw WGS reads were first randomly extracted using the 'sample' function in SeqKit 0.13.1[46] to generate approximate 6–8 Gb datasets, and were then aligned against two published congeneric plastomes (NC_015605, and NC_025339) to filter out plastomic reads using BWA-MEM algorithm in bwa 0.7.17[47] with default settings[47]. Filtered reads were used for de novo assembly in SPAdes 3.15.2 with five k-mers (51, 71, 91, 101, and 121) and further combined automatically in SPAdes 3.15.2 following settings provided in He et al.[37]. Bandage 0.8.1 was used to obtain the final circular molecule for each accession. Average assembly depth of each accession surpassed 100×. All plastome sequences were manually adjusted to start with the first base of the LSC region using Blastn 2.9.0[48] against the sequence itself.
Length, together with GC content of LSC, SSC, and IR for each plastome was detected using Perl script, which were processed in IBM SPSS Statistics 22 (SPSS Inc., Chicago, USA). Accessions LA001 (N. lutea) and LF001 (N. nucifera, flower lotus) were annotated as exemplars from each of the two Nelumbo species respectively using GB2sequin[49] with published plastomes of N. lutea (NC_015605) and N. nucifera (NC_025339) from NCBI used as comparative references to check the accuracy of the de novo assemblies.
Plastome polymorphisms
-
Assembled plastome sequences were aligned in MAFFT 7[50] using the default settings. All aligned plastomes were manually scanned with variants detected by DnaSP 6[51] using LA001 used as the reference. Nucleotide variants were classified into SNVs, Block substitutions (consecutive nucleotide substitutions greater than 1 nucleotide in length which in some cases includes gaps), and InDels (insertions or deletions one nucleotide in length). The position of each variant was mapped to the reference to characterize the location in the genome as cds, intronic, or intergenic. The stacked graph was plotted in package ggplot2 in R v 4.0[52].
Population and phylogenetic analyses
-
Based on the complete aligned plastome sequences, the median-joining network was resolved in Popart 1.7[53] to resolve haplotype diversity. The principal component analysis (PCA) of the two datasets (including or excluding LA) was performed in TASSEL 5.2[54], using the highest (in regard to percent explanation) two eigenvectors for plotting in two dimensions. Using a SNVs only matrix extracted from the aligned plastomes (alignment file of SNVs from 316 accessions was available at Figshare,
https://doi.org/10.6084/m9.figshare.17694764.v2 ), IQ-tree 2.1[55] was used to reconstruct a maximum likelihood (ML) tree using a TVMe+ASC+R2 nucleotide substitution model chosen by the Bayesian information criterion (BIC) and 1,000 bootstrap replicates to assess branch support. Population structure analysis was performed using ADMIXTURE 1.3[56] using default settings for haploid data with runs on different K values from 3−9. The optimal K was chosen from the lowest cross-validation (CV) error value compared across all values of K. Nucleotide diversity (π), haplotype diversity (Hd), and genetic differentiation (Fst) were calculated in DnaSP 6 to assess the genetic diversity and divergence within and among different genetic clusters. Source information of each accession was collected from previous publications and plotted using ggplot and map packages in R v 4.0. This work was supported by the National Natural Science Foundation of China (Grant No. 31970244), also co-funded by the Training of Excellent Science and Technology Innovation talents in Shenzhen - Basic Research on Outstanding Youth (STIC: RCYX20200714114538196) and the Zhejiang Provincial Natural Science Foundation of China [LY21C160001].
-
The authors declare that they have no conflict of interest.
-
# These authors contributed equally: Jie Wang, Xuezhu Liao, Cuihua Gu
- Supplemental Table S1 Sampling Information of 316 lotus accessions.
- Supplemental Table S2 Plastome size and GC content of different types of Asian lotus.
- Supplemental Table S3 Functional groups of CDS with variant events.
- Supplemental Table S4 Genetic clusters and haplotypes of lotus accessions.
- Supplemental Fig. S1 An example of some species-specific variants among the pan-plastome.
- Supplemental Fig. S2 Liner ML tree of 316 accessions.
- Supplemental Fig. S3 Liner BI tree of 316 accessions. Genetic clusters were colored as ML.
- Supplemental Fig. S4 Geographical distribution of wild Asian lotuses from different haplotypes. Blue, red, and green dotted boxes represented three distributive regions, namely, blue: northeastern China and North Korea; red: east central China; green: southern China and several Southeast Asian countries including India, Thailand, Indonesia, and Singapore.
- Supplemental Annotation.txt Annotation of plastome sequence of LA001 (Nelumbo lutea) in this study.
- Copyright: © 2022 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang J, Liao X, Gu C, Xiang K, Wang J, et al. 2022. The Asian lotus (Nelumbo nucifera) pan-plastome: diversity and divergence in a living fossil grown for seed, rhizome, and aesthetics. Ornamental Plant Research 2: 2 doi: 10.48130/OPR-2022-0002 

The Asian lotus (Nelumbo nucifera) pan-plastome: diversity and divergence in a living fossil grown for seed, rhizome, and aesthetics
- Received: 15 November 2021
- Accepted: 04 January 2022
- Published online: 20 January 2022
Abstract: The Asian lotus (Nelumbo nucifera) has a history of cultivation in Asia dating back over 3,000 years where it has been an important food crop producing edible rhizomes and seeds as well as flowers of great aesthetic and cultural value. Here, we de novo assembled the plastomes of 316 lotus accessions including five North American lotus (N. lutea) and 311 Asian lotus (N. nucifera) to construct a pan-plastome genome map, and investigate the phylogeography and genetic diversity among the only two extant species within this living fossil lineage. A total of 113 unique genes were annotated and plastome sizes varied between 163,457 and 163,672 bp with only minor differences in each of the four major genomic units. The most abundant nucleotide differences among plastomes were single nucleotide variants followed by insertions/deletions and block substitutions mainly found in intergenic spacer regions of the large single copy portion of the plastome. Seven well-supported genetic clusters were resolved using multiple different population structure analyses. The different lotus types (flower, seed, rhizome, or wild) were disproportionally assigned to multiple different genetic clusters. This pattern indicates that the domestication of Asian lotus involved multiple genetic origins and possible matrilineal introgression. Geographic mapping of accessions also revealed that genetic diversity is unevenly distributed with eastern China possessing the highest genetic diversity and regions such as Yunnan, Indonesian, and Thailand possessing unique haplotypes. These results provide an important maternal history of Nelumbo and necessary groundwork for future studies on intergenomic gene transfer, cytonuclear incompatibility, and conservation genetics.
-
Key words:
- Phylogeography /
- Sacred lotus /
- North American lotus /
- Centers of origin /
- Aquatic plants /
- Domestication