








-
Liverwort (Marchantia polymorpha) was the first complete mitochondrial genome sequenced[1]. In Cucurbitaceae, sequencing of the mitochondrial genome of watermelon, zucchini[2] and cucumber[3] have been completed. Ward et al.[4] suggested that the mitochondrial genome of melon was about 2.9 Mb. In 2011, the mitochondrial genome sequence of melon was reported, mainly composed of five scaffolds and four contigs[5]. In 2020, we reported the completed mitochondrial genome of melon including a main loop and two small loops[6]. Melon mitochondrial genome can provide additional genetic information, such as cytoplasmic male sterility and mitochondrial RNA editing. Therefore, further research focusing on the mitochondrial genome of melon is expected.
Plant mitochondrial genomes have more complex structural characteristics. The size of plant mitochondrial genomes varies greatly between plants, ranging from 187 kb in liverwort[1] to 11.3 Mb in flycatcher[7], but there is no linear relationship between the size of the mitochondrial genome and the number of genes it encodes, flycatcher has 32 coding genes, but ground money has 74 coding genes. The GC content of the mitochondrial genome is about 43%−45%. The number of protein-coding genes in plant mitochondrial genomes is typically 30−50, mainly including complex I-V genes, cytochrome C biosynthetic genes, ribosomal protein genes, matR genes, mttB genes and ORFs of unknown function. The non-protein-coding genome includes rRNA and tRNA genes. rRNA includes 5s rRNA, 18s rRNA and 28s rRNA; tRNA includes three different types of tRNAs, including its own intrinsic tRNA and tRNAs transferred from the chloroplast and nuclear genomes, but with the exception of liverwort, the mitochondrial genomes of all other plants do not cover tRNA genes encoding all 21 amino acids.
RNA editing refers to a predetermined codon modification caused by nucleotide changes at the RNA level[8]. This molecular process mainly exists in chloroplasts and mitochondria of plants, where it maintains the normal biological functions of these organelles. Mitochondrial RNA editing mostly occurs along protein coding regions. In this case, RNA editing can increase the conservation of the encoded protein product, with regards to its primary structure, between different species[9]. The number of RNA editing sites can vary greatly among different species. There are only 11 RNA editing sites in the mitochondrial genome of the moss Physcomitrella patens[10], in contrast to 456 and 692 RNA editing sites identified, respectively, in the mitochondria of Arabidopsis thaliana[11] and Gossypium spp[12]. Lu et al.[13] studied the process of RNA editing in eight species from four families of gymnosperms and found a substantial difference in the number of RNA editing sites as well as their positions along the DNA of distinct families and genera of gymnosperms. Mitochondrial RNA editing can affect many important traits in plants. RNA editing of the cotton mitochondrial Ghatp1 gene, at the C1292 and C1415 loci, affects ATPase production and promotes epidermal hair and fiber elongation[12]. In tomato, a decreased RNA editing of nad3 and sdh4 genes can disrupt the biological function of mitochondria, therefore, reduce the respiratory efficiency of the fruit, which can ultimately inhibit its ripening[14]. Inadequate and deviated mitochondrial RNA editing may be associated with certain biological conditions, such as cytoplasmic male sterility[15]. The structural analysis of the mitochondrial genome will provide a theoretical basis for an in-depth study of the genetic characteristics of melon mitochondria.
-
Mitochondrial genomes were sequenced and assembled using dark-treated MR-1 yellowing seedlings as plant material[6].
Genome annotation
-
The annotation of protein-coding genes in the mitochondrial genome was carried out using BLASTN and BLASTX to search the nucleotide and protein libraries of the NCBI/GenBank database. rRNA and tRNA annotations were carried out using RNAmmer (rnammer -S euk-m lsu,ssu,tsu -xml melon.xml -gff melon.gff -hmelon.hmmreport < melon.fsa) and tRNAScan-SE (tRNAscan-SE -o tRNA.out -f rRNA.ss -m tRNA.stats /home/gjs/fasta/h.fa).
Forward repeats in the mitochondrial genome were analyzed using Reputer software[16] with minimum repeat size set to 20 bp, hamming distance of 0, and similarity greater than 90%. Inverted repeats were analyzed using IRF (Inverted Repeats Finder) software[17] with parameters set to: match 2, mismatch 3, delta 5, match probability 80, indel probability 10, min-score 40, max-length to report 500,000, max-loop 500,000. Tandem repeat was analyzed using TRF (Tandem Repeat Finder) software with parameters set to: min. align. score 50, max. period size 500. Simple sequence repeat was analyzed using MISA software[18] with parameters set to 1 base. The parameters were set to 10 and more repetitions of 1 base, five and more repetitions of 2 bases, four and more repetitions of 3 bases, four and more repetitions of 4 bases, four and more repetitions of 5 bases, four and more repetitions of 6 bases, and only those base repetitions meeting the criteria were considered as microsatellite sequences.
Comparative analysis of mitochondrial genomes
-
Analyses were delineated using Easyfig[19], and then illustrated with MapChart 2.2[20]. Comparative analysis of sequences between organelle and nuclear genomes was performed by BLASTN and Tbtools.
Prediction of mitochondrial RNA editing sites
-
The RNA editing sites of all protein-coding gene sequences, located in the mitochondrial genomes of melon (Cucumis melo), cucumber (Cucumis sativus), watermelon (Citrullus lanatus) and zucchini (Cucurbita pepo), were predicted by PREP-Mt[21].
Phylogenetic analysis
-
Mitochondrial genomes were aligned using ClustalX (
www.clustal.org/clustal2 ). A phylogenetic tree was constructed via Neighbor Joining (NJ) using Mega7 software. -
The melon mitochondrial genome contains 4,861 pairs of forward repeats, 439 pairs of inverted repeats, 653 tandem repeats and 218 SSR sequences. The total length of these repeats is about 44.2% of those detected in the whole genome. The coding genes accounted for 1.54% of C. melo mitochondrial genome (Table 1). In fact, non-coding gene sequences accounted for 98.46% of the mitochondrial genome, but their function still require more detailed characterization.
Table 1. The basic features of the C. melo mitochondrial genome.
Feature Value Total length (bp) 2,906,673 Chromosome number 3 GC content 44.77% Gene number 88 Protein genes 40 rRNA genes 8 tRNA genes 40 Genes with introns 10 Trans-spliced genes 3 Coding sequence 1.54% Protein coding 1.23% tRNAs and rRNAs 0.31% Non-coding sequence 98.46% Repetitive content 44.2% SSRs 0.1% (218) Tandem repeats (TRs) 2.1% (653) Inverted repeats (IRs) 2.4% (439) Forward repeats (FRs) 39.4 (4,861) Chloroplast-like 2.73% Nuclear-like 48.62% Collinearity analysis of mitochondrial genomes
-
By comparing the mitochondrial genomes of four cucurbit plants, we discovered that sequences, with a consistency of no less than 80% in C. sativus, C. pepo and C. lanatus, accounted for 33%, 40% and 65% of the whole mitochondrial genome length in melon, respectively. The sequence shared by the four species accounts for about 6% of the full length of the melon mitochondrial genome. In addition, both melon and the other three crops have similar gene coding regions, but the non-coding regions are quite distinct (Fig. 1).
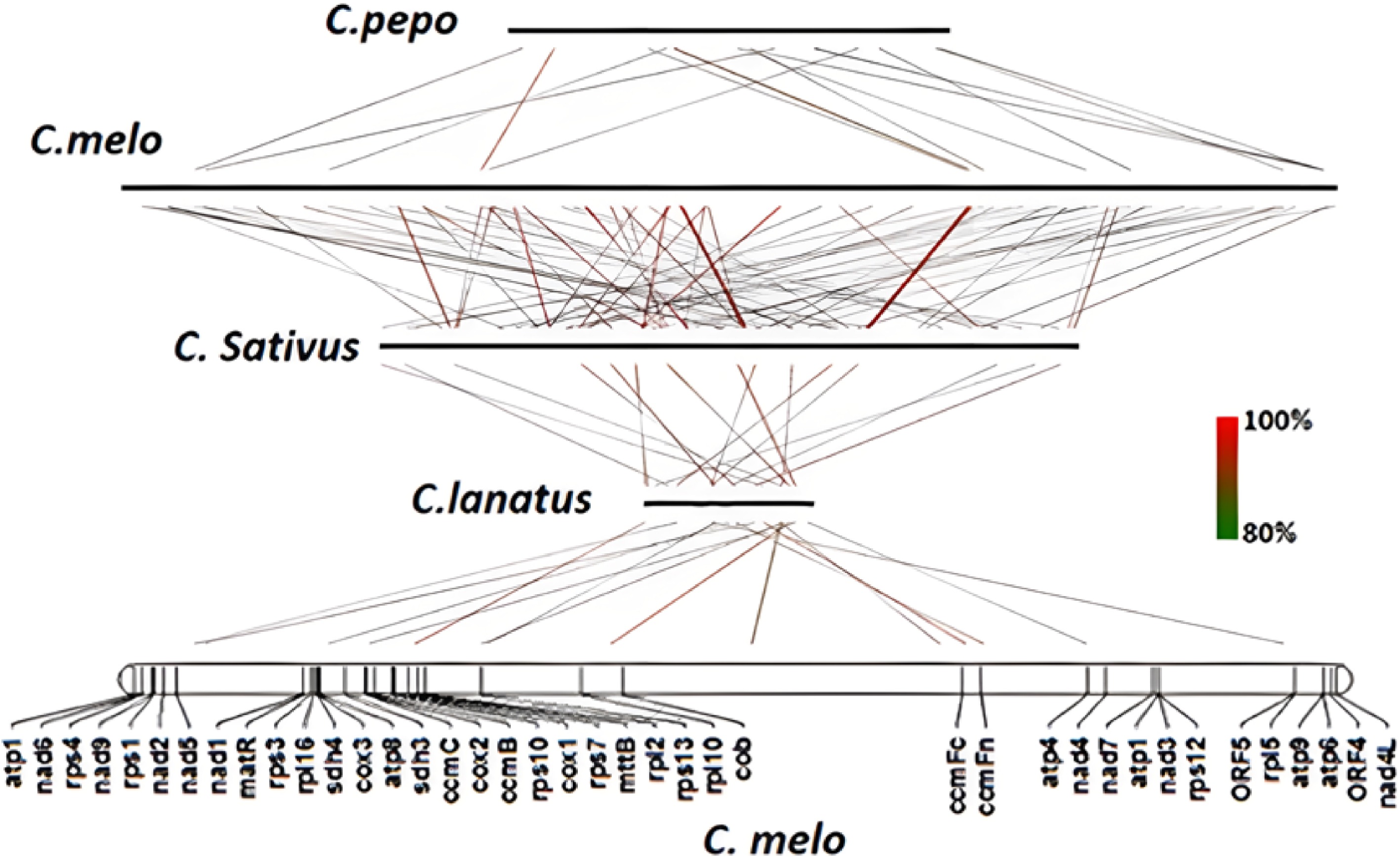
Figure 1.
Collinearity analysis of C. melo mitochondrial genome with other three Cucurbitaceae plants.
The linear relationships of mitochondrial coding genes among C. melo, C. sativus, C. pepo and C. lanatus were compared. Two or more collinear gene groups are called gene clusters, and we get 7 to 13 gene clusters with different gene numbers (Table 2). A higher number of collinear gene clusters was identified in C. melo and C. sativus, contrarily, a fewer number of collinear gene clusters was found in C. pepo and C. lanatus.
Table 2. The gene clusters of melon collinearity with three other mitochondrial genomes.
Species Amount C. melo C. lanatus 7 nad6-rps4; trnY-nad2; trnF-trnS; rps3-rpl16; sdh4-cox3-atp8; rrn5s-rrn18s; nad3-rps12 C. sativus 13 nad6-rps4; nad9-rps1; nad2-sdh3; trnF-trnS; matR-trnH; rps3-rpl16; sdh4-cox3-atp8; rrn5S-rrn18S; rpl10-trnD; ccmFc-trnW-atp4; nad3-rps12; atp9-atp6 C. pepo 8 nad6-rps4; nad9-rps1; rps3-rpl16; sdh4-cox3-atp8; rrn5S-rrn18S; nad3-rps12; atp9-atp6; trnM-trnG Gene clusters in bold are the gene clusters common to the four mitochondrial genomes. Comparison of four mitochondrial genomes
-
There were no significant differences in mitochondrial genomic GC content among C. melo, C. sativus, C. pepo and C. lanatus (between 42.8 and 45.1%, Table 3). The total number of repeat sequences in the melon mitochondrial genome was the highest among all four species, accounting for 44.2% of the total genome sequence. The number of repeat sequences in the mitochondrial genome of C. sativus, C. pepo and C. lanatus was 44.1%, 24.4% and 9.6% of the total genome, respectively.
Table 3. Mitochondrial genome summary of C. melo, C. sativus, C. pepo and C. lanatus
Feature C. melo C. lanatus C. sativus C. pepo Genome Accession MG947207
MG947208
MG947209NC_014043 NC_016004
NC_016005
NC_016006NC_014050 Size in bp 2,906,673 379,236 1,644,236 982,833 Chromosome number 3 1 3 1 Topology Structure Circle Circle Circle Circle GC content (%) 44.8% 45.1% 44.3% 42.8% Gene Protein-coding genes 40 39 37 38 Protein-coding genes in bp (%) 35,613 (1.23%) 32,370 (8.5%) 32,550 (3.31%) 32,032 (3.26%) Single-copy protein genes 37 37 37 37 Single-copy protein genes in bp (%) 34,080 (1.17%) 31,986 (8.4%) 32,550 (3.31%) 31,806 (3.23%) Intron Trans-spliced 5 4 5 5 Cis-spliced 17 20 18 19 Cis-spliced introns in bp (%) 46,000 (1.6%) 32,476 (8.6%) 47,996 (2.9%) 30,557 (3.1%) tRNA genes 40 18 20 13 Native 17 3 7 3 Chloroplast-derived 23 15 13 10 Total tRNAs in bp (%) 2,999 (0.1%) 1,358 (0.4%) 1,486 (0.09%) 966 (0.1%) rRNA genes 8 3 6 3 Total rRNAs in bp (%) 5,815 (0.2%) 5,148 (1.4%) 11044 (0.67%) 5,109 (0.5%) Noncoding regions in bp (%) 2,862,246 (98.5%) 340,360 (89.7%) 1,599,156 (97.3%) 944,726 (96.1%) Repetitive content SSR (num.) 0.1% (218) 0.2% (54) 0.1% (144) 0.2% (144) TR (num.) 2.1% (653) 0.3% (14) 0.4% (120) 1.9 (287) IR (pairs) 2.4% (439) 0.4% (14) 6.3% (539) 0.2% (17) FR (pairs) 39.6% (4861) 8.7% (209) 37.3% (4974) 22.1% (1608) Maximum large repeat length (bp) 5,532 7,286 17,159 621 Number of repeats (>1 kb) 87 1 10 0 Total repeats (%) 44.2% 9.6% 44.1% 24.4% Chloroplast-like in bp (%) 79,463 (2.73%) 28,703 (7.6%) 70,702 (4.3%) 113,347(11.5%) Mitochondrial-like in bp (%) 967,450 (33.3%) 159,032 (41.9%) 907,251 (55.2%) 180,008 (18.3%) Nuclear-like in bp (%) 1,413,224 (48.62%) 24,352 (6.4%) 501,491 (30.5%) 20,638 (2.1%) A total of 40 protein-encoding genes were annotated in the mitochondrial genome of melon. The number of protein-encoding genes in the mitochondrial genome of C. sativus, C. pepo and C. lanatus was 37, 38 and 39, respectively. Melon mitochondrial genome lost rps19 gene, two copies of atp1 gene, and two more ORF genes (orf1 and orf2). The mitochondrial genome of these four plants contained three different types of ribosomal genes (rrn5S, rrn18S and rrn26S). The highest number of rRNAs were detected in melon, including six copies of rrn5S, and two single copies of rrn18S and rrn26. Six rRNA genes were present in the cucumber mitochondrial genome, and each ribosomal gene presented two repeats. Only three rRNAs were observed in watermelon and zucchini, all of which were single-copy genes (Table 4).
Table 4. Comparison of the gene content among C. melo, C. sativus, C. pepo and C. lanatus mitochondrial genome.
Gene C. melo C. lanatus C. sativus C. pepo Complex I nad1,2,3,4,4L,5,6,7,9 + + + + Complex II sdh3 + 2 + + sdh4 + + + + Complex III cob + + + + Complex IV cox1,2,3 + + + + Complex V atp1 2 + + + atp4,6,8,9 + + + + Cytochrome c biogenesis ccmB, C, Fc, Fn + + + + Ribosomal RNAs rrn5S 6 + 2 + rrn18S + + 2 + rrn26S + + 2 + Ribosomal proteins rpl2,5,16 + + + + rpl10 + − + − rps1,3,4,7,10,12,13 + + + + rps19 − 2 − 2 Other ORFs matR, mttB + + + + orf1,2 + − − − Total number 48 42 43 41 +: indicates the presence and uniqueness of this gene; −: represents the absence of this gene, and the number represents the copy number of this gene. We compared the tRNA use of C. melo, C. sativus, C. pepo and C. lanatus (Table 5). The results showed that the mitochondrial genome sequences of all four species could not encode a complete set of tRNA that could recognize all codons or transport a complete set of 20 amino acids.
Table 5. Comparison of the tRNA genes.
tRNA C. melo C. lanatus C. sativus C. pepo trnC-GCA M MM M M trnD-GUC CCMM − C − trnE-UUC M M MM M trnF-GAA CM M CCC M trnfM-CAU − M M M trnG-GCC CM MM M M trnH-GUG CCM C CC C trnI-CAU − M MM M trnK-UUU − M − M trnL-CAA CM − − − trnM-CAU CCCMMM C M C trnN-GUU CCMM C − C trnP-UGG M M M M trnQ-UUG MM MM M M trnR-ACG M − M − trnS-GCU M M M − trnS-UGA C M − − trnV-GAC C − − − trnW-CCA CCCMMMM − C − trnY-GUA M M M M Choloroplast-derived 17 3 7 3 Mitochondrial-derived 23 15 13 10 Total (type) 40 (18) 18 (15) 20 (15) 13 (13) Prediction of mitochondrial RNA editing sites
-
Our results show that C. lanatus had the largest number of RNA editing sites along its mitochondrial genome (509 sites). C. melo and C. sativus contained 507 and 498 RNA editing sites, respectively. C. pepo presented the lowest number of RNA editing sites (total of 486). Nad4, ccmB and ccmFn were the three genes with the highest RNA editing sites (Table 6).
Table 6. Number of RNA editing sites in the four mitochondrial genomes.
Order Genes C. melo C. lanatus C. sativus C. pepo 1 Atp1 4 5 5 5 2 Atp4 11 13 12 13 3 Atp6 23 22 22 22 4 Atp8 2 4 2 2 5 Atp9 5 6 5 6 6 ccmB 34 34 33 30 7 ccmC 28 27 27 26 8 ccmFc 17 17 18 17 9 ccmFn 36 36 36 35 10 Cob 16 14 16 14 11 Cox1 17 17 18 19 12 Cox2 13 13 13 13 13 Cox3 8 9 8 7 14 matR 12 12 12 12 15 mttB 27 24 23 24 16 Nad1 21 21 21 20 17 Nad2 25 25 25 24 18 Nad3 12 12 12 10 19 Nad4 37 38 36 33 20 Nad4L 13 13 13 13 21 Nad5 28 27 28 23 22 Nad6 15 10 10 10 23 Nad7 25 27 27 26 24 Nad9 7 7 7 7 25 Rpl2 5 3 3 2 26 Rpl5 10 10 9 8 27 Rpl16 5 5 5 5 28 Rps1 3 4 3 4 29 Rps3 7 9 7 8 30 Rps4 17 17 17 18 31 Rps7 2 2 2 2 32 Rps10 6 5 6 5 33 Rps12 5 7 7 7 34 Rps13 4 3 3 4 35 Rps19 pseudo 3 pseudo 4 36 Sdh3 3 4 3 5 37 Sdh4 4 4 4 3 Total − 507 509 498 486 Note: pseudo indicates that the gene is a pseudogene. Phylogenetic analysis
-
In this study, the phylogenetic relationship of C. melo, C. sativus, C. pepo and C. lanatus were analyzed based on 10 conserved coding genes (atp6, nad6, cox3, rps12, atp1, nad4, nad9, nad7, nad4L) that are present in all 14 species. Evolutionary relationships were analyzed (Fig. 2) and it was found that grapes and Cucurbitaceae are closely related, and Cucurbitaceae can cluster well in a clade, but the genetic distance of C. pepo in the phylogenetic tree is closer to C. melo and C. sativus compared to C. lanatus.
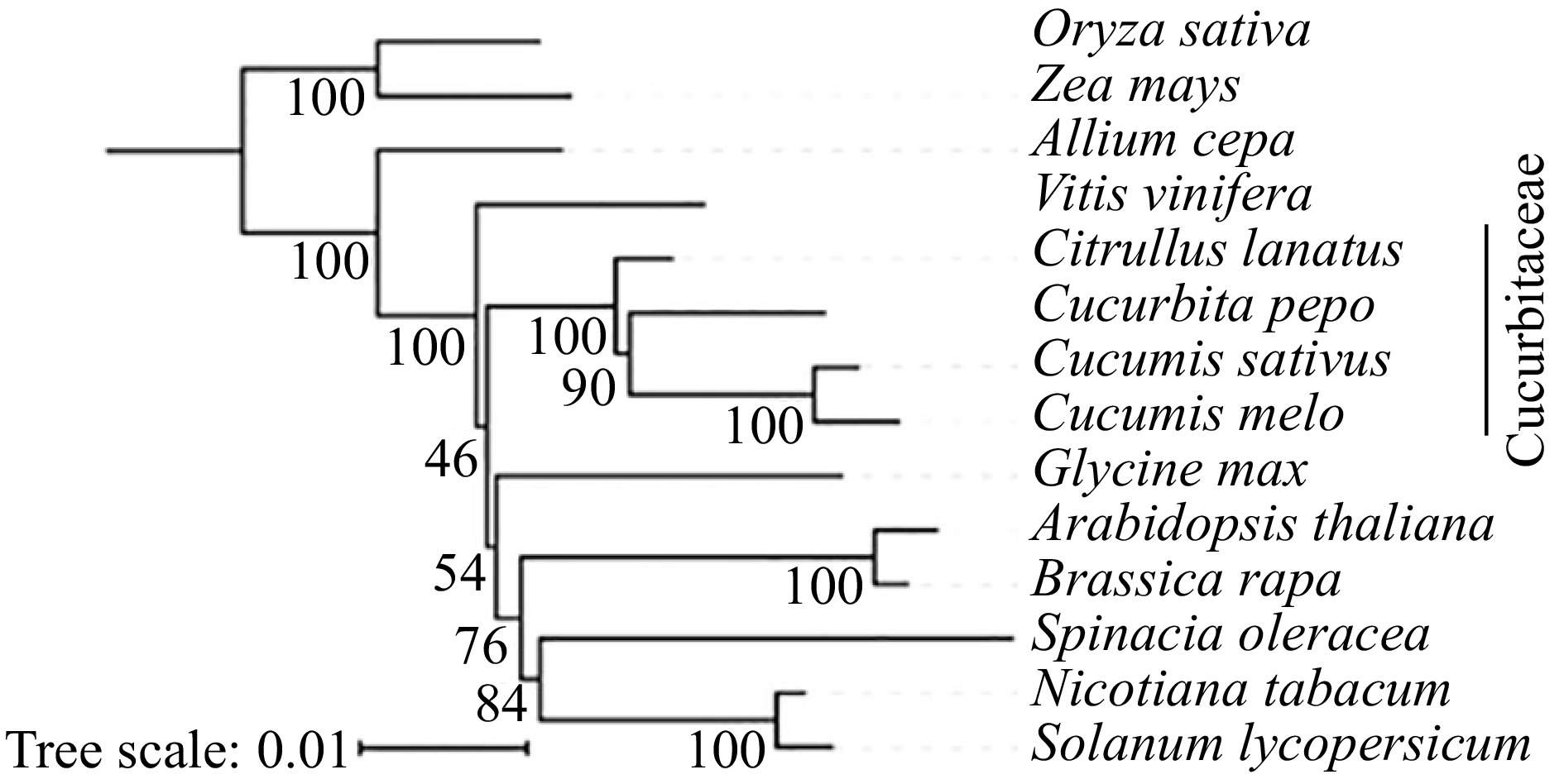
Figure 2.
The construction of phylogenetic tree among 14 species based on mitochondrial conserved genes.
-
Our study corroborates with previous reports that utilized renaturation kinetics and restriction endonuclease technology to analyze the mitochondrial genome analysis of Cucurbitaceae[4]. The structure of both C. melo and C. sativus mitochondrial genomes is polycyclic. Likewise, this genome structure has been observed in other plants, including wheat[22] and rape[23].
The protein-coding genes in melon mitochondrial genome are like those in the three mitochondrial genomes. Similar conclusions have been reached in studies of other higher plants, where the coding regions of plant mitochondrial genomes are more conserved than the non-coding regions[24]. Melon mitochondria contain two more ORF genes than the other three reference genomes, and ORFs may encode proteins with important functions. In fact, some mitochondrial ORFs have been associated with cytoplasmic male sterility in many plants.
With the expectation of T-urf13, atp6 of radish and orf256 of wheat, it has been verified that most of the transcripts of protein-coding genes are edited in the mitochondria of higher plants, but the editing degree of transcripts from different genes is variable[25, 26]. The male sterility of plants may be caused by some genes in the mitochondrial genome recombining with ORF to form chimeric genes or inadequate RNA editing[27]. In the present study, a slightly distinct number of RNA editing sites was detected in the mitochondrial genomes of four Cucurbitaceae crops. The elucidation of these RNA editing sites may provide data support for the research of RNA editing in cucurbits.
The mitochondrial genomes of both melon and three other kinds of Cucurbitaceae plants were linearly analyzed. C. melo and C. sativus showed more collinearity between mitochondrial gene clusters, which further shows that C. melo has a closer relationship with C. sativus. In the four kinds of Cucurbitaceae plants some gene cluster, rps3-rpl16 is widespread in the plant mitochondrial genome typical gene cluster[28].
-
This study upon in-depth comparative analysis of the mitochondrial gene structure of C. melo, C. sativus, C. pepo and C. lanatus, revealed that the large number of repetitive and nuclear genome sequences were the potential reasons for the increasing scale and variation of the melon mitochondrial genome. These results provide the basis for the genetic variation of the mitochondrial genome in Cucurbitaceae plants.
This study was funded by Scientific Research Foundation of Hebei Normal University of Science and Technology (2022YB004) and the National Nature Science Foundation of China (U21A20229 and 31960607).
-
The authors declare that they have no conflict of interest.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Cui H, Ding Z, Zhu Q, Gao P. 2022. Structural characteristics of the melon mitochondria genome. Vegetable Research 2:20 doi: 10.48130/VR-2022-0020 

Structural characteristics of the melon mitochondria genome
- Received: 24 May 2022
- Accepted: 21 October 2022
- Published online: 14 December 2022
Abstract: The mitochondrial genome can provide important genetic information of melon. We reported the mitochondrial genome sequence of melon in a previous study, the structural characteristics of the melon mitochondrial genome were further analyzed in the present study. The mitochondrial genome of melon is comprised of three circular DNA molecules, with a total length of about 2.9 Mb, contains 4,861 pairs of homologous repeats, 439 pairs of inverted repeats, 653 tandem repeats and 218 SSR sequences. The coding genes accounted for 1.54% and non-coding gene sequences accounted for 98.46% of the melon mitochondrial genome. The total repetitive sequence of mitochondrial genome of melon was the highest among Cucumis melo, Cucumis sativus, Cucurbita pepo and Citrullus lanatus. The large number of repeated sequences and nuclear genome sequences were the main reason for the increasing size and variation of melon mitochondrial genome. Melon mitochondrial genome has the highest GC content and tRNA quantity. These regions were the main source of mitochondrial genome differences among all species here analyzed.
-
Key words:
- Cucumis melo /
- Mitochondria genome /
- Coding genes /
- RNA editing