









-
Tomato has been used as a model species for all fleshy fruits due to its excellent biological characteristics, such as short life cycle, easy self-pollination, and high growth rate[1]. Given that whether or not a respiratory burst occurs during ripening, fleshy fruits are classified into climacteric (tomato, apple, banana etc.) and non-climacteric (grape, strawberry, citrus etc) fruits[2−5]. Tomato fruit is composed of several different tissues, including pericarp (containing exocarp, mesocarp and endocarp), placenta, septum, and ovary (containing seeds and pulp)[6−8], and the pericarp accounts for 2/3 of the total fruit weight. Physical and chemical characteristics of pericarps such as texture, sugar and acid content, and lycopene content mainly determines the tomato fruit quality. The characteristics of tomato pericarp are formed during the fruit development of the fruit, which is closely related to the changes in the cell cycle[9]. In the early fruit developmental stage, active mitosis plays a dominant role in increasing the thickness of the pericarp, and cell layers dramatically increased until 5-8 d after pollination. Tomato fruit enlargement is mainly dependent on cell expansion, and this process is strongly linked to the endocycle[10,11].
The ripening of climacteric fruit mainly depends on the burst of ethylene biosynthesis[5,12,13], while the ripening of non-climacteric fruit depends on the signal transduction pathway of abscisic acid[14−16]. Fruit ripening involves changes in color, texture, flavor and aroma, the most obvious of which are changes in color due to accumulation of pigments. Tomato accumulates carotenoids, flavonoids and lycopene during ripening[17−19], while grapes accumulate anthocyanins[3,20]. Fruit softening is also one of the characteristics of fleshy fruit ripening, during which the cell wall is remodeled and the structure of all major cell wall polysaccharides changes[4,21]. The solubilization of the primary cell wall and middle lamella results in reduced intercellular adhesion, depolymerization and solubilization of hemicellulose and pectin cell wall polysaccharides, and consequently a reduction in fruit firmness[21,22]. A variety of hydrolases in the cell wall are involved in the enzymatic hydrolysis of polysaccharides, galactosidase (PG) and β-galactosidase catalyze the depolymerization of pectin in the fruit wall and middle lamella[23] Knocking out of pectin lyase (PL) makes tomato fruit firmer[24]. The pectin component homogalacturonic acid (HG) is secreted into the cell wall in a highly methyl-esterified form, followed by demethyl-esterification to balance cell wall stiffness and flexibility[25−27]. During tomato fruit ripening, the degree of methyl esterification of pectin decreased by 55%[28]. At the same time, active physiological and biochemical changes occurred in many cellular compartments including nucleus, plastid and cell wall during fruit ripening[4].
Cell size in tomato fruit pericarps does not change after the fruit development reaches the mature green stage[29]. Whether or not endocycle in tomato pericarps ceases or how it progresses after the mature green stage remains largely unknown due to the difficulties of determination of nuclear DNA ploidy distribution using a flow cytometer. Previous studies utilize unripe green fruit for the flow cytometric analysis of nuclear DNA ploidy distribution, in which nuclei with different ploidy levels could be clearly differentiated[30−32] . At the red ripe stage of tomato fruit, nuclei with ploidy levels from 2C to 32C could not be clearly differentiated from each other resulting in inaccurate calculation of nuclear DNA ploidy distribution[30]. In this study, we discovered that resuspending nuclei with buffers is critical for flow cytometric analysis of nuclear DNA ploidy distribution in red ripe tomato mesocarps, and phosphate buffered saline (PBS) buffer had the best effects. We used three buffers including 'Aru' buffer[33,34], 'Galbraith' buffer[35], 'Bourdon' buffer[36] to isolate nuclei from red ripe tomato pericarps and resuspended them with PBS buffer for flow cytometric analysis. Our results reveal that 'Aru' buffer plus PBS buffer resuspension is the best combination to retrieve nuclei from red ripe tomato pericarps for flow cytometric analysis to determine the nuclear DNA ploidy distribution.
-
Tomato plants of cultivar 'Heinz 1706' were grown in the greenhouse with the condition of 16 h light at 25 °C and 8 h dark at 20 °C. The day of performing manual pollination was determined as day 0 (Anthesis, 0 DPA).
Histological analysis of tomato pericarps
-
Pieces of fresh equatorial pericarps were fixed in FAA fixative solution containing 50% ethanol, 5% acetic acid and 5% formalin, and vacuumized for 30 minutes. Pericarp tissues were consecutively dehydrated with 50%, 70%, 85% and 95% ethanol containing 0.5% eosin, and then 100% ethanol. Tissues were cleared by 50% and 100% xylene, and embedded in wax. Embedded samples were cut into slices with 10 μm thickness using a microtome. Sections were stained with Iron vitriol-hematoxylinand observed under an optical microscope.
Nuclear DNA ploidy distribution with a flow cytometer
-
Tomato pericarp tissue was defined as reported previously[29]. Three-layer cells on the outer surface are the exocarp (Ex); the 4th to the penultimate layer of cells is the mesocarp (Me); the last layer of cells is the endocarp (En, Fig. 1b, g). About 0.5 g of fresh equatorial mesocarps were collected and chopped with a double-sided razor blade in a plastic petri dish containing 1 ml of 'Aru' buffer (10 mM MgSO4·7H2O, 50 mM KCl, 5 mM 4-Hydroxyethylpiperazineethanesulfonic acid (HEPES), 6.5 mM Dithiothreitol (DTT), 2.5% Triton X-100), or 'Galbaith' buffer (45 mM MgCl, 30 mM sodium citrate, 20 mM 4-morpho-linepropane sulfonate (MOPS), 0.1% Triton X-100), or 'Bourdon' buffer (45 mM MgCl, 30 mM sodium citrate, 20 mM MOPS, 5 mM sodium metabisulfite, 1% polyethylene glycol 10000 (PEG 10000) 0.1% Triton X-100). Chopped mesocarp samples were consecutively filtered through a 425 μm mesh sieve and a 75 μm nylon mesh sieve. The filtrate was centrifuged at 5,000 rpm for 5 min, and the pellet was resuspended with 500 μl of 1 × phosphate buffered saline (PBS, pH = 7.4) buffer. Nuclei in the resuspended solution were stained by propidium iodide (PI) at a final concentration of 50 µg/ml or 4’,6-diamidino-2-phenylindole (DAPI) at a final concentration of 1 µg/ml, which were then used for flow cytometric analysis (CytoFLEX, Backman). Endocycle index or the cycle value were calculated using the following formula[37,38]: EI = (%2C nuclei × 0) + (%4C nuclei × 1) + (%8C nuclei × 2) + (%16C nuclei × 3) + (%32C nuclei × 4) + (%64C nuclei × 5) + (%128C nuclei × 6) + (%256C nuclei × 7).
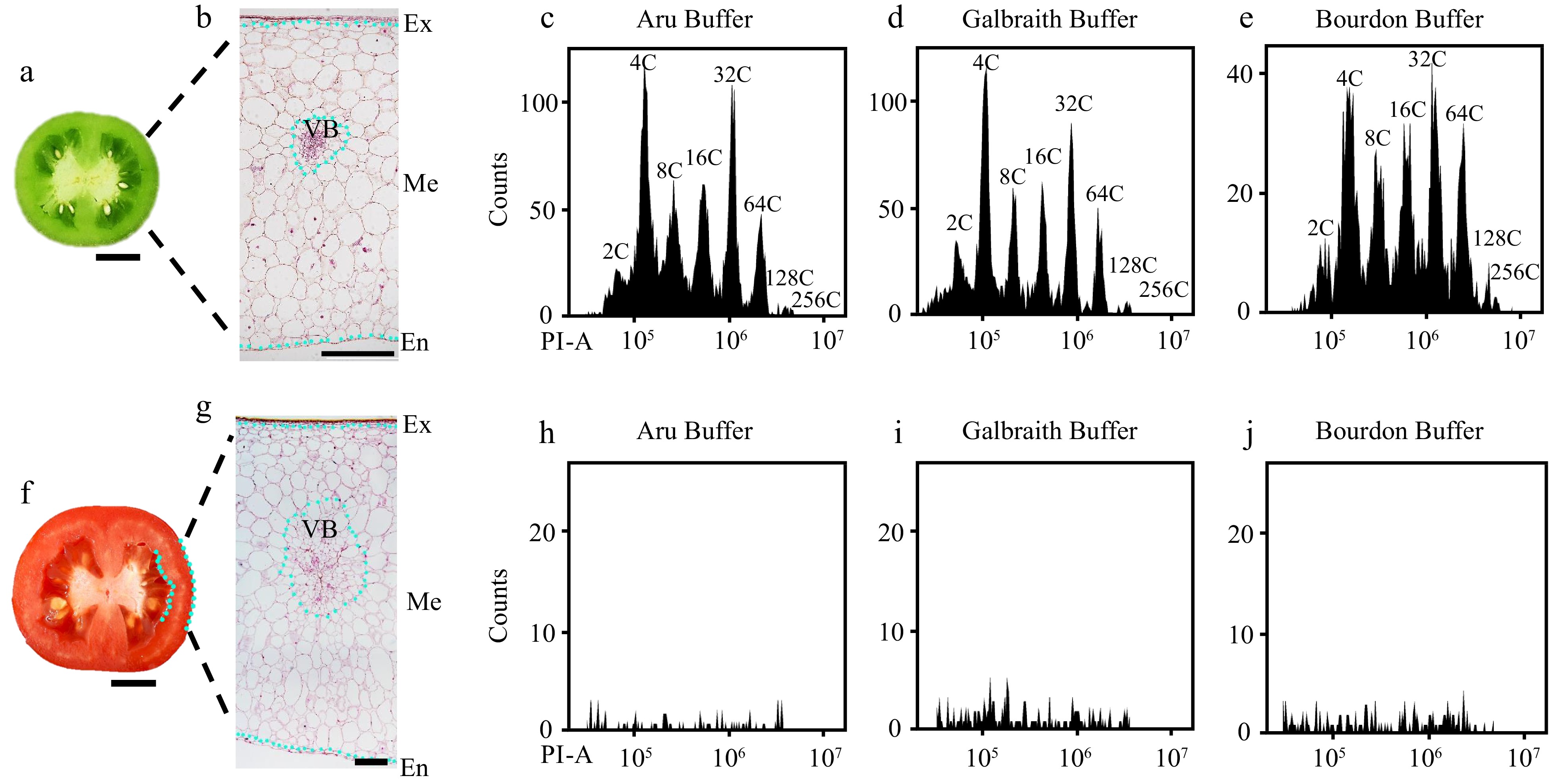
Figure 1.
Flow cytometric analysis of nuclear DNA ploidy distribution in mesocarps of green and red ripe tomato fruit. Nuclei were isolated from mesocarps of 25 d after pollination (DPA) green (c)−(e) and 55 DPA red ripe tomato fruit (h)−(j) using 'Aru', 'Galbraith' and 'Bourdon' buffer. Nuclei were stained by propidium iodide (PI) at the final concentration of 50 µg/ml for flow cytometric analysis. (a), (f) Representative picture of the equatorial plane of (a) 25 DPA and (f) 55 DPA tomato fruit. Scale bar = 1 cm. (b), (g) Paraffin section of (b) 25 DPA and (g) 55 DPA tomato pericarp. Scale bar = 500 μm. (c)−(e) Histograms of nuclear DNA ploidy distribution in 25 DPA tomato mesocarps. (h)−(j) Histograms of nuclear DNA ploidy distribution in 55 DPA tomato mesocarps. Ex, exocarp; Me, mesocarp; En, endocarp; VB, vascular bundle; PI-A, PI fluorescence pulse signal area.
Protoplast preparation
-
The method for extraction of protoplasts in tomato mesocarps was as performed in a previous study[39] with some modifications. Pieces of mesocarps were suspended in 20 ml of protoplast isolation solution containing 0.4 M mannitol, 20 mM KCl, 10 mM CaCl2, 20 mM 2-morpholinoethanesulfonic acid (MES), 2% sucrose, 1.5% 'Onozuka' R-10 cellulase (Yakult Pharmaceutical Co. Ltd., Tokyo, Japan), 0.4% macerozyme R-10 (Yakult Pharmaceutical Co. Ltd., Tokyo, Japan) and 0.05% pectinase (Yakult Pharmaceutical Co. Ltd., Tokyo, Japan), vacuumized for 30 min, and then kept in the dark at 25 °C for 12 h. An equal volume of W5 solution containing 0.15 M NaCl, 0.125 M CaCl2, 5 mM KCl, and 2 mM MES was added to the protoplast isolation solution, and the mixture was filtered through a 75 μm nylon mesh sieve. Protoplasts were collected from the filtrate by centrifugation at 500 rpm for 10 min, resuspended using 10 ml W5 solution and kept on ice for 30 min. Protoplast solution was further prepared by adding 2 ml MMg solution containing 0.4 M mannitol, 15 mM MgCl2 and 4 mM MES. For fluorescent microscopic analysis, DAPI was added to the protoplast solution at the final concentration of 1 µg/ml. For flow cytometric analysis, nuclei were isolated by adding equal volume of 'Aru' buffer to the protoplast solution, and stained by PI at the final concentration of 50 µg/ml.
Fluorescent microscopic analysis
-
Nuclei were stained by DAPI at the final concentration of 10 µg/ml and investigated by Nikon fluorescence microscope Eclipse Ni with the excitation wavelength of 328−375 nm under 4ⅹ and 10ⅹ magnification.
Measurement of pH value
-
The resuspension solution of nuclei was transferred into a 10 ml round bottom centrifugation tube, and its pH value was measured using a pH meter (PHS-3C; INESA Scientific Instrument Co., Ltd).
Data analysis
-
All data were recorded in Excel. Statistical analysis was performed using one-way ANOVA test plus Duncan's multiple range test in the data processing system (DPS) software. Different letters indicate significant difference with p < 0.05.
-
Most studies on cell cycle in tomato fruit focus on developing stages and mature green stage, and do not extend to turning and red ripe stages[36,40,41]. In order to determine the nuclear DNA ploidy distribution in tomato red ripe fruit (55 DPA) mesocarps, we used three commonly used buffers including 'Aru' buffer, 'Galbraith' buffer and 'Bourdon' buffer to isolate nuclei for flow cytometric analysis. The sample processing follows the traditional protocol. The tomato pericarp was chopped in the lysis solution, and 50 μg/ml PI is directly added to the mixture after filtration to detection for 10 min[35,36]. Green fruits at 25 DPA had rigid pericarps with multiple layers of big cells in mesocarps, and all three buffers could successfully isolate nuclei from mesocarp cells for the determination of ploidy levels by a flow cytometer (Fig. 1a−e). However, red ripe fruits at 55 DPA had soft pericarps and cells with dramatic size differences in mesocarps, and the flow cytometer failed to determine the nuclear DNA ploidy distribution in mesocarp cells (Fig. 1f−j). To determine the reasons for poor nuclei isolation, we first isolated protoplasts from mesocarps of red ripe fruits as previously described[42]. Nuclei from 10 μl protoplast solution were stained by DAPI at the final concentration of 1 μg/ml and could be clearly detected under fluorescent microscope (Fig. 2a, b). Nuclei were extracted from 500 μl protoplast solution and stained by PI at a final concentration of 50 μg/ml for flow cytometric analysis. Flow cytometer detected nuclei with different ploidy levels, while a lot of debris affected the sensitivity and efficiency of detection resulting in 2C, 64C and 128C could not be clearly separated (Fig. 2c, d). Destruction of cellulose and pectin during the isolation of protoplasts prompted us to pretreat mesocarps with 1% cellulase and pectinase for 10 min. We isolated nuclei from pretreated mesocarps using Aru, Galbraith or Bourdon buffer, and no nuclei isolated from sole water pretreated mesocarp cells could be detected by the flow cytometer (Supplemental Fig. S1a). However, nuclei isolated in Bourdon buffer from cellulase and pectinase pretreated mesocarp cells could be detected by the flow cytometer with low efficiency (Supplemental Fig. S1b). These results suggested that pretreatment with cellulase and pectinase was not an effective way for nuclear DNA ploidy distribution analysis via the flow cytometer.
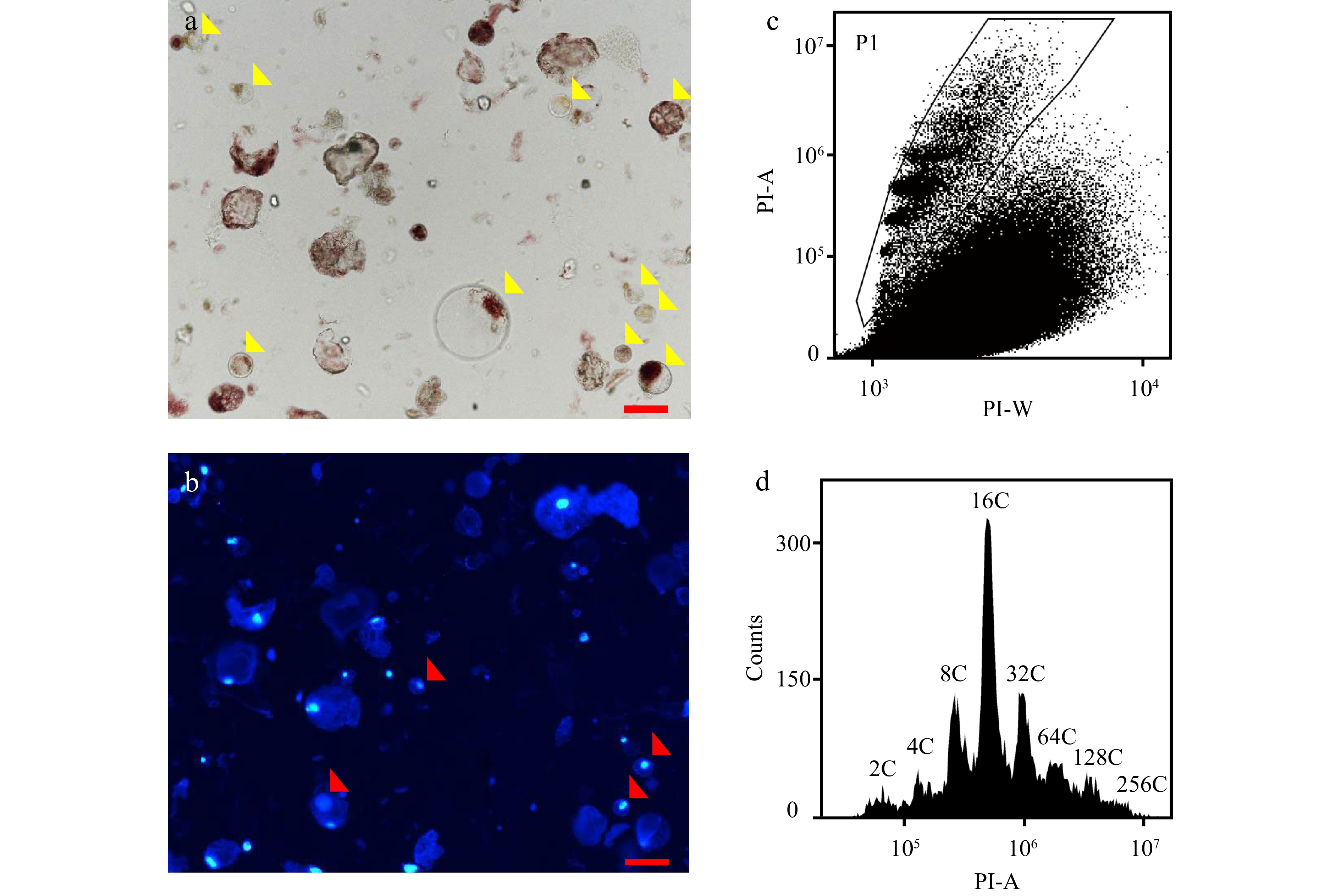
Figure 2.
Flow cytometric analysis of nuclear DNA ploidy distribution in protoplasts isolated from mesocarps of red ripe tomato fruit. (a) Microscopic examination of protoplasts isolated from 55 DPA tomato mesocarps. Yellow arrows indicate intact protoplasts. Scale bar = 100 μm. (b) Examination of nuclei in protoplasts under fluorescent microscope. Red arrows indicate nuclei. Nuclei were stained by 4’,6-diamidino-2-phenylindole (DAPI) at the final concentration of 1 µg/ml. Scale bar = 100 μm. (c), (d) Scatter plot and histogram of nuclear DNA distribution in flow cytometric analysis using nuclei isolated from protoplasts by 'Aru' buffer and stained by PI at a final concentration of 50 µg/ml . PI-A and PI-W represent PI fluorescence pulse signal area and width.
PBS buffer resuspended nuclei isolated from mesocarp cells in red ripe tomato fruit is crucial for flow cytometric analysis
-
We harvested nuclei from the filtered lysate in different nucleus isolation buffers by centrifugation (Fig. 3a). Nuclei were resuspended by water or its original nucleus isolation buffer, and water improved the detection efficiency of flow cytometer especially for nuclei isolated by 'Aru' buffer (Supplemental Fig. S2). We resuspended the pellet of nuclei with 'Aru' buffer and stained nuclei with DAPI. Fluorescence microscopic analyses revealed that many nuclei were aggregated and surrounded by debris (Fig. 3b, c). We then resuspended the pellet with five different buffers adjusted at pH 7.4 including HEPES, MOPS, Tris, phosphate (PB) and phosphate buffered saline (PBS) buffer (Fig. 4). 'Aru' buffer contains HEPES, and both 'Galbraith' and 'Bourdon' buffer contain MOPS. Interestingly, resuspended nuclei with each buffer could be effectively detected by flow cytometer. For nuclear DNA ploidy distribution analysis, HEPES buffer resuspended nuclei isolated in Aru buffer not Galbraith and Bourdon buffer, had clear separation of each peak with specific ploidy level (Fig. 4a). Either MOPS buffer or PB buffer resuspended nuclei had clear separation of peaks with high ploidy levels but not low ploidy levels such as 2C, 4C and 8C (Fig. 4b, d). Tris buffer resuspended nuclei isolated in Aru buffer or Galbraith buffer, not Bourdon buffer, had clear separation of each peak with specific ploidy level, but the proportion of low ploidy levels was less than that of high ploidy levels suggesting a nuclear aggregation (Fig. 4c). PBS buffer resuspended nuclei isolated in any of three buffers had clear separation of each peak at specific ploidy level (Fig. 4d). The proportion of nuclei at each ploidy level did not exhibit significant difference among the three buffers resulting in similar EI values (Table 1). Moreover, fluorescence microscopic analyses of nuclei resuspended by PBS buffer revealed that nuclei were separated and disassociated with debris (Fig. 3d, e) Aru buffer could retrieve 2- and 1.3-fold more nuclei than Galbraith and Bourdon buffer respectively, while the flow cytometer had the highest detection efficiency for nuclei isolated by Bourdon buffer (Table 1). Therefore, our results revealed that the utilization of PBS buffer to resuspend nuclei is effective for the flow cytometer to determine nuclear DNA ploidy distribution in mesocarp cells of red ripe tomato fruit.
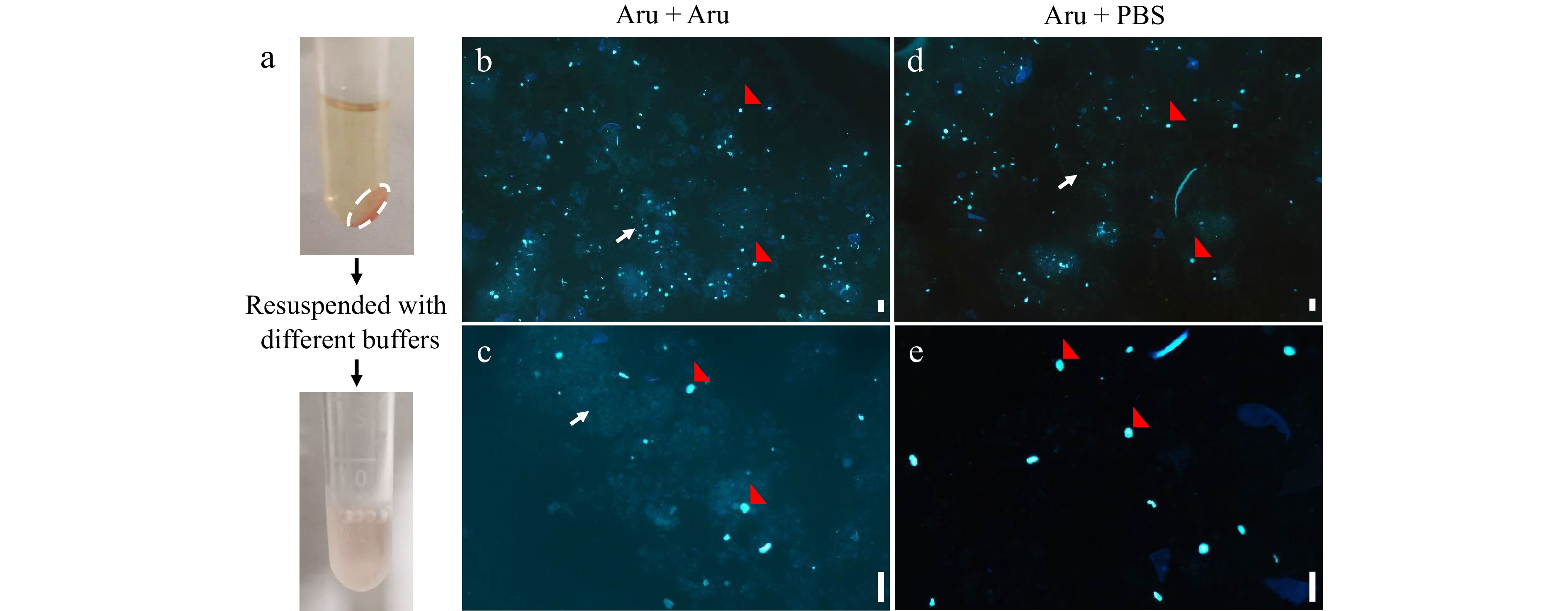
Figure 3.
Fluorescence microscopic analyses of nuclei resuspended by different buffers. Nuclei were isolated from mesocarps of 55 DPA red ripe tomato fruit using 'Aru' buffer and resuspended by 'Aru' (b), (c) or Phosphate Buffered Saline (PBS) buffer (d), (e). (a) The pellet of nuclei (white dashed circle) were collected by centrifugation and resuspended. (b)−(e) Fluorescence microscopic analyses of nuclei stained by DAPI under (b), (d) 4× and (c), (e) 10× magnification. Scale bar = 100 μm. Red arrows indicate nuclei, and white arrows indicate debris.
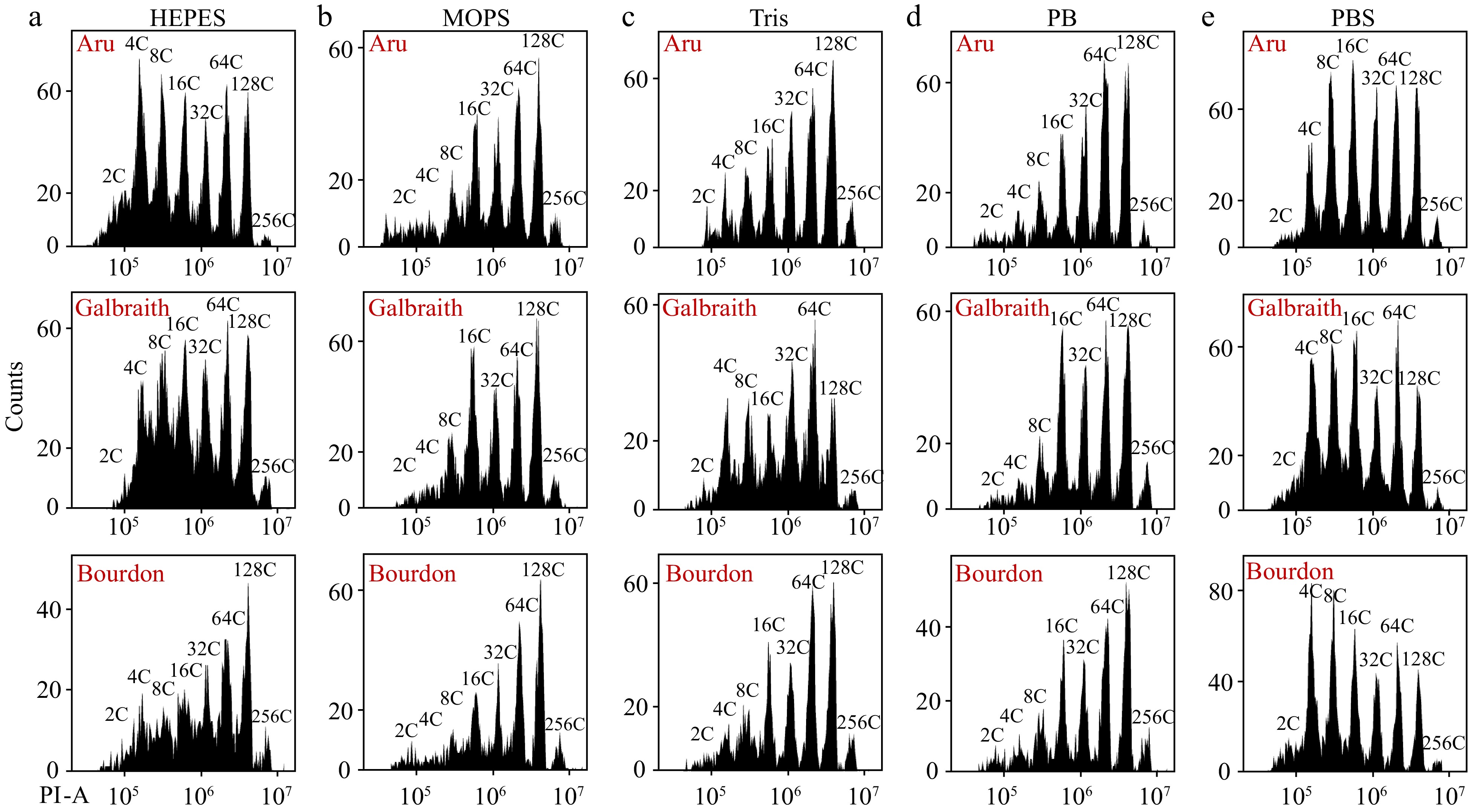
Figure 4.
Different resuspension buffers of nuclei have distinct effects on flow cytometric analysis. Nuclei were isolated from mesocarps of 55 DPA red ripe tomato fruit using 'Aru', 'Galbraith' and 'Bourdon' buffer. Nuclei were harvested by centrifugation, resuspended by (a) HEPES, (b) MOPS, (c) Tris, (d) Phosphate or (e) PBS buffer, and stained by PI at a final concentration of 50 µg/ml. Shown are histograms of flow cytometric analyses. PI-A, PI fluorescence pulse signal area.
Table 1. Impacts of different isolation buffers with PBS buffer resuspension of nuclei on the nuclear DNA ploidy distribution in the mesocarp of red ripe tomato fruit.
Treatment Nuclear DNA ploidy distribution EI Value Number
of nucleiEfficiency value 2C 4C 8C 16C 32C 64C 128C 256C Aru + PBS 4.25 ± 0.40%b 11.05 ± 0.38%b 12.57 ± 0.40%a 11.63 ± 0.76%b 17.86 ± 1.17%a 25.48 ± 0.92%a 14.93 ± 0.24%a 2.22 ± 0.40%a 3.75 ± 0.02a 4312 ± 254a 0.39 ± 0.02%b Gal + PBS 3.68 ± 0.46%b 13.39 ± 0.93%a 13.81 ± 0.92%a 12.76 ± 0.12%b 19.38 ± 0.83%a 21.72 ± 0.66%b 13.01 ± 1.74%a 2.24 ± 0.68%a 3.59 ± 0.10a 2133 ± 118c 0.16 ± 0.01%c Bou + PBS 5.35 ± 0.21%a 12.14 ± 0.71%ab 12.18 ± 0.57%a 14.14 ± 0.61%a 19.12 ± 0.21%a 21.24 ± 1.23%b 13.24 ± 0.24%a 2.55 ± 0.23%a 3.59 ± 0.05a 3442 ± 130b 0.58 ± 0.05%a Different letters indicate significant difference with p < 0.05 determined by one-way ANOVA plus Duncan's multiple range test. PBS buffer resuspension of nuclei does not alter the nuclear DNA ploidy distribution
-
To investigate whether PBS buffer resuspension of nuclei affects the nuclear DNA ploidy distribution, we isolated nuclei from green fruit (25 DPA) mesocarp cells using three different buffers for flow cytometric analysis with or without PBS buffer resuspension. Nuclei isolated in Aru, Galbraith and Bourdon buffer with or without PBS buffer resuspension were clearly separated at different ploidy levels in flow cytometric analysis, and Aru or Bourdon buffer isolated nuclei after PBS resuspension were much cleaner and less affected by debris (Fig. 5). The flow cytometer had enhanced detection of nuclei with high ploidy levels especially 128C and 256C after PBS resuspension, and had better detection efficiency for nuclei isolated in either Aru or Bourdon buffer (Table 2). The nuclear DNA ploidy distribution in nuclei isolated by either Aru or Bourdon buffer was not altered after PBS buffer resuspension resulting in similar EI values, while PBS resuspension increased the proportion of 128C and 256C in nuclei isolated by Galbraith buffer resulting in a slight increase of EI value. Again, Aru buffer retrieved significantly more nuclei than Galbraith and Bourdon buffer. Taken together, our results suggest that the nuclear DNA ploidy distribution in mesocarp cells of tomato fruit at all developmental stages can be accurately determined by a flow cytometer using nuclei isolated by Aru buffer and resuspended by PBS buffer.
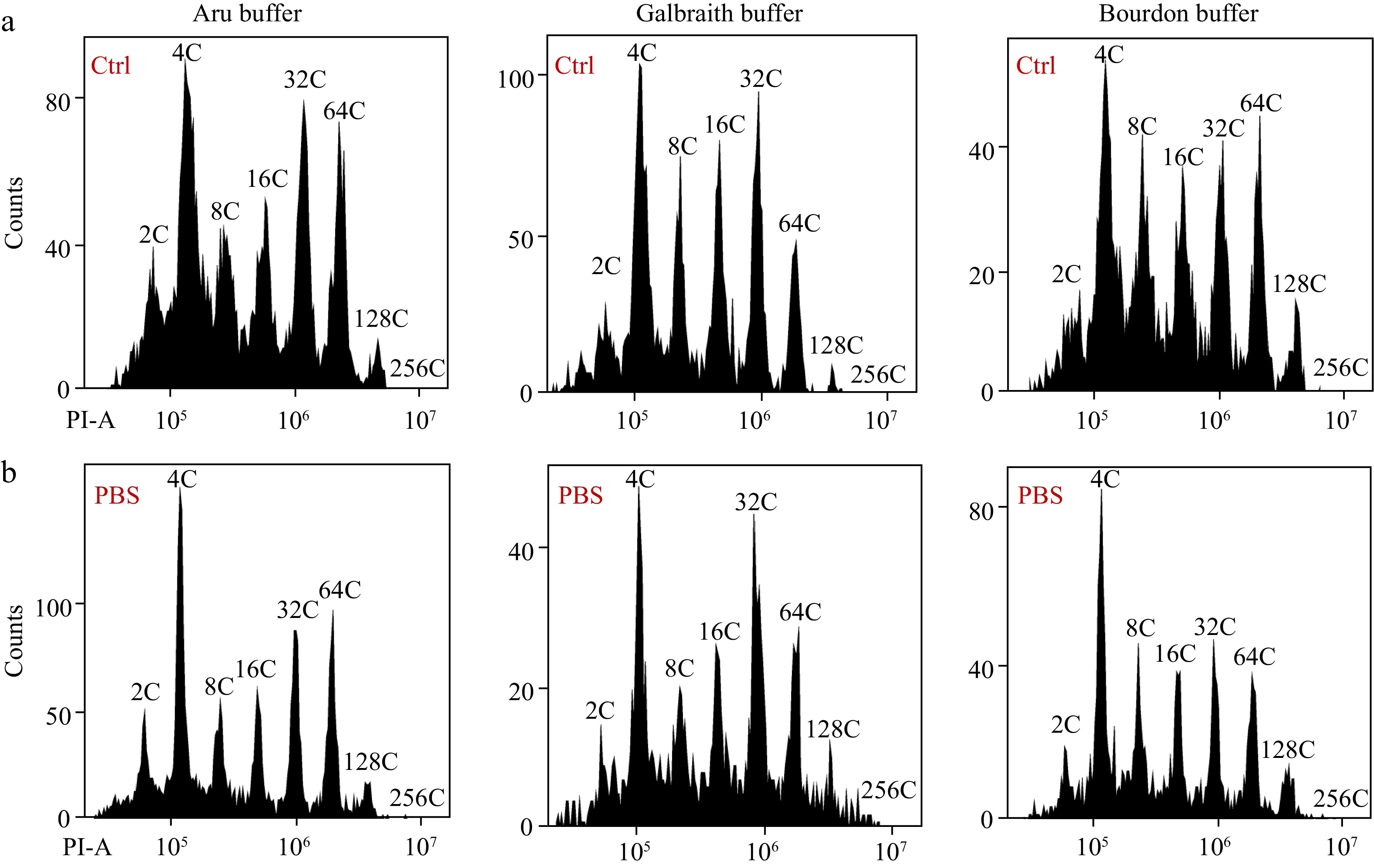
Figure 5.
Effects of PBS buffer resuspension on the nuclear DNA ploidy distribution in mesocarps of green tomato fruits. (a) Nuclei were isolated from mesocarps of 25 DPA green tomato fruit using 'Aru', 'Galbraith' and 'Bourdon' buffer and directly used for flow cytometric analysis. (b) Isolated nuclei in (a) were harvested by centrifugation and resuspended in PBS buffer. All nuclei were stained by PI at a final concentration of 50 µg/ml. Shown are histograms of flow cytometric analyses. PI-A, PI fluorescence pulse signal area.
Table 2. Impacts of different isolation buffers with or without PBS buffer resuspension of nuclei on the nuclear DNA ploidy distribution in the mesocarp of green tomato fruit.
Treatment Nuclear DNA ploidy distribution EI Value Number
of nucleiEfficiency value 2C 4C 8C 16C 32C 64C 128C 256C Aru 7.88 ± 0.58%bc 27.11 ± 0.30%a 13.70 ± 1.57%bc 14.76 ± 1.86%ab 20.98 ± 1.28%a 13.56 ± 2.71%a 2.02 ± 0.26%bc 0c 2.63 ± 0.06bc 3450 ± 102a 0.81 ± 0.05%b Aru + PBS 10.37 ± 0.23%a 27.35 ± 0.57%a 12.47 ± 1.26%c 13.45 ± 1.28%b 19.01 ± 0.98%ab 14.07 ± 2.01%a 3.11 ± 1.03%abc 0.17 ± 0.14%bc 2.59 ± 0.07bc 2968 ± 449a 1.01 ± 0.15%b Gal 9.39 ± 1.48%ab 27.57 ± 1.47%a 13.82 ± 0.48%bc 16.40 ± 1.48%a 20.48 ± 0.97%a 10.78 ± 0.59%a 1.56 ± 0.12%c 0c 2.50 ± 0.09c 2979 ± 125a 1.38 ± 0.11%a Gal + PBS 7.56 ± 0.53%c 23.23 ± 1.60%b 13.03 ± 0.81%c 15.19 ± 0.83ab 21.14 ± 1.14%a 13.82 ± 1.07%a 5.19 ± 0.49%a 0.84 ± 0.32%a 2.86 ± 0.05a 1676 ± 225b 0.94 ± 0.06%b Bou 7.82 ± 1.00%bc 24.65 ± 0.66%b 16.57 ± 1.23%a 16.27 ± 0.33%a 16.17 ± 1.91%bc 14.00 ± 0.67%a 4.42 ± 1.70%ab 0.10 ± 0.11%c 2.69 ± 0.08b 1922 ± 37b 0.81 ± 0.09%b Bou + PBS 7.45 ± 0.57%c 27.87 ± 0.56%a 15.70 ± 0.96%ab 14.71 ± 0.26%ab 15.62 ± 2.35%c 13.05 ± 1.09%a 4.97 ± 1.99%a 0.64 ± 0.46%ab 2.65 ± 0.07b 2064 ± 214b 1.56 ± 0.20%a Different letters indicate significant difference with p < 0.05 determined by one-way ANOVA plus Duncan's multiple range test. High pH value of PBS buffer to resuspend nuclei facilitates the detection of the flow cytometer
-
To determine whether pH value in nuclei resuspension buffer is important for flow cytometric analysis, we adjusted the pH value of PBS buffer to 5.4, 7.4 and 9.0 to resuspend nucleus solutions for flow cytometric analysis. We first chopped the mesocarp of red ripe tomato fruit in 'Aru' buffer with a pH value of 6.0 and filtrated lysates with sieves. We measured the pH value of the filtrate which was about 4.0. We collected nuclei from the filtrate by centrifugation and then resuspended them with PBS buffers at different pH values, and observed that nuclei in PBS buffer at either pH value could be clearly separated and detected at each ploidy level by a flow cytometer. However, PBS buffer of pH 5.4 retrieved slightly less nuclei with higher ploidy levels than that of pH 7.4 and 9.0 resulting in significant reduction of EI values (Fig. 6a−c, Table 3). Overall, PBS buffer with high pH value retrieved more nuclei than low pH value (Table 3). A pH value of 7.4 and 9.0 did not affect the detection efficiency of flow cytometry and nuclear ploidy distribution (Table 3). The combined results suggest that PBS buffer with a high pH value may facilitate the separation of nuclei from debris for accurate flow cytometric analysis.
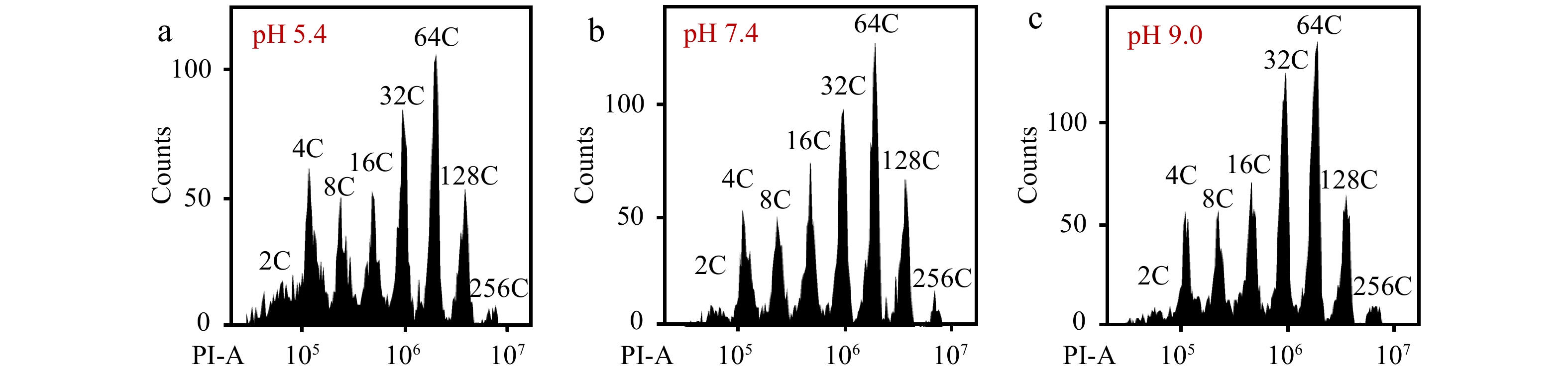
Figure 6.
The pH value of PBS buffer affects the detection of flow cytometer. Nuclei from mesocarp cells of red ripe tomato fruits were isolated in 'Aru' buffer, harvested by centrifugation and resuspended by PBS buffer with a pH value of (a) 5.4, (b) 7.4 and (c) 9.0. All nuclei were stained by PI at a final concentration of 50 µg/ml. Shown are histograms of flow cytometric analyses. PI-A, PI fluorescence pulse signal area.
Table 3. Impacts of different pH values of PBS buffer on the nuclear DNA ploidy distribution in the mesocarp of red ripe tomato fruit.
Treatment Nuclear DNA ploidy distribution EI Value Number
of nucleiEfficiency value 2C 4C 8C 16C 32C 64C 128C 256C PBS (pH 5.4) 5.22 ± 0.79a 14.72 ± 0.24a 11.93 ± 0.36a 12.75 ± 0.77b 19.95 ± 0.42b 22.56 ± 0.78b 11.63 ± 1.18a 1.22 ± 0.27b 3.48 ± 0.01b 3058 ± 104c 0.43 ± 0.01a PBS (pH 7.4) 3.11 ± 0.14b 11.10 ± 0.45b 11.27 ± 0.25ab 13.65 ± 1.11a 21.69 ± 1.05a 24.75 ± 1.03a 12.40 ± 0.56a 2.02 ± 0.19a 3.74 ± 0.03a 3339 ± 14b 0.37 ± 0.01b PBS (pH 9.0) 2.60 ± 0.14b 10.44 ± 0.22b 10.76 ± 0.32b 14.25 ± 0.12a 22.57 ± 0.48a 25.63 ± 0.27a 11.77 ± 0.16a 1.96 ± 0.05a 3.78 ± 0.02a 3598 ± 36a 0.38 ± 0.01b Different letters indicate significant difference with p < 0.05 determined by one-way ANOVA plus Duncan's multiple range test. -
Tomato fruit development is a consequence of active cell cycle progression including mitotic cell cycle and endocycle, which controls cell division and cell expansion, respectively. Previous studies on the connection between cell cycle regulation and tomato fruit growth and development mainly focus on stages before or at the mature green. The endocycle plays a dominant role in tomato fruit expansion, while its progression and roles in tomato fruit after the mature green stage remains largely unknown. The traditional method to analyze the nuclear DNA ploidy distribution in vegetative tissues or green tomato fruit using a flow cytometer is less effective and unstable for fruits after mature green stages. Here we establish a protocol to effectively and accurately analyze the nuclear DNA ploidy distribution in red ripe fruit mesocarp cells using a flow cytometer. Resuspending nuclei with PBS buffer is critical for the separation between nuclei and debris, resulting in an effective detection of the flow cytometer. Our results can also facilitate cell cycle related studies or ploidy determination in organs of other crops with difficulties via flow cytometric analysis.
At the end of the cell expansion phase, the single cell volume of the mesocarp increased 30,000 times compared to the initial cell volume[10], which is closely related to the endocycle. However, there is no evidence linking the endocycle to fruit ripening possibly due to the technical limitation of endocycle analysis using a flow cytometer. Fruit ripening is associated with the changes of cell wall structure and accumulation of secondary metabolites such as cellulose, saccharide and pectin, which may hinder the isolation of nuclei for flow cytometric analysis[22,26,28]. In order to solve this problem, we first adopted the method of extracting protoplasts, in which cell walls and pectin are destroyed and cells are released. As expected, the flow cytometer detects nuclei at different ploidy levels especially 8C, 16C, 32C with substantial enrichment suggesting that cellulose, pectin and other substances are barriers for the separation of nuclei. However, protoplast isolation causes loss of cells and nuclei from broken cells, which results in the alteration of nuclear DNA ploidy distribution. The preparation of protoplast is also time- and labor-consuming, and therefore more costly. To remove barriers influencing nucleus isolation, we filter mesocarp lysates twice through a 425 μm steel mesh sieve and a 75 μm nylon mesh sieve, and harvest nuclei by centrifugation. We observed a large number of nuclei surrounded by debris after DAPI staining under fluorescent microscope, which prompts us to use different buffers to resuspend the nucleus pellet and release nuclei. This strategy drastically improves the detection of the flow cytometer, and resuspension of nucleus pellet with PBS buffer has better effects than other buffers on flow cytometric analysis of nuclear DNA ploidy distribution. PBS buffer is usually used to resuspend animal cells prior to flow cytometric analysis[43,44]. In our study, we confirm that PBS buffer resuspension of nuclei does not alter the nuclear DNA ploidy distribution in green fruit mesocarp cells during flow cytometric analysis, suggesting that this method is accurate and suitable for all developmental stages of tomato fruit cells. PBS disperses the nuclei encased in debris, and the detection efficiency of the flow cytometer increases with the rising pH value. Considering that high pH value may cause damage to the tube line of the flow cytometer, we recommend the neutral buffer of PBS (pH 7.4) for long-term analysis.
To summarize our method, we produced a workflow for the detection of nuclear DNA ploidy distribution in mesocarp cells of red ripe tomato fruits using a flow cytometer (Fig. 7). Firstly, 0.5 g or so equatorial mesocarp tissues are placed in a plastic petri dish containing 1 ml of 'Aru' buffer, and then chopped into fine pieces with a razor blade. Secondly, the lysate of mesocarps is initially filtered through a 425 μm steel mesh sieve, and then filtered through a 75 μm nylon mesh sieve. The filtrate is centrifuged at 5000 rpm for 5 min to collect pellets and discard the supernatant. The pellet containing nuclei is resuspended with 500 μl of PBS buffer (pH 7.4). Finally, the resuspension solution is stained with PI at the final concentration of 50 μg/ml and analyzed by a flow cytometer.
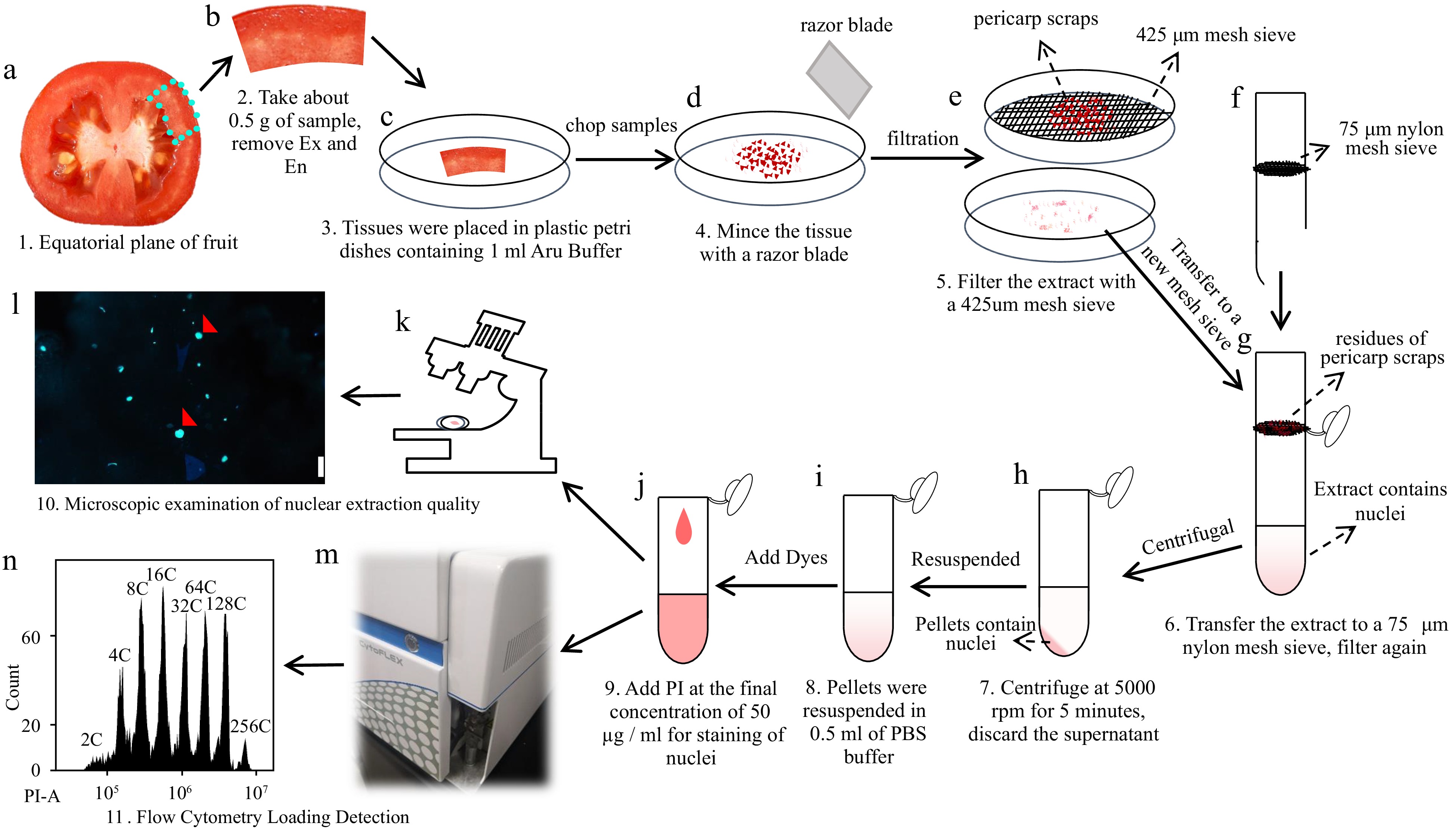
Figure 7.
Workflow diagram illustrating the detection of nuclear DNA ploidy distribution in tomato mesocarp using a flow cytometer. (a)-(d) About 0.5 g of the equatorial mesocarp is taken and chopped into pieces with a razor blade in a plastic petri dish containing 1 ml of 'Aru' buffer. (e)-(g) Lysates were filtered through a 425 μm steel mesh sieve and a 75 μm nylon mesh sieve sequentially. (h), (i) The filtrate is centrifuged at 5000 rpm for 5 min and the supernatant is discarded. The pellet of the nuclei is resuspended using 500 μl PBS buffer (pH 7.4). (j)-(n) PI is added to the resuspended solution at a final concentration of 50 µg/ml for flow cytometric analysis.
Recently single-cell or single-nucleus sorting techniques are increasingly used to decipher functions of cells with different ploidy levels or in different tissues of various plant species[45−50]. In tomato, single-nuclear sorting technology and in situ hybridization are applied to illustrate the developmental regulation and metabolite accumulation in cells with different ploidy levels at the mature green stage[36,40,41]. The technical breakthrough in our method not only facilitates the detection of nuclear DNA ploidy distribution in cells of other fleshy fruits (eg, grapes, strawberries, etc), but also the sorting of nuclei with different ploidy levels in ripe fruits to study the relationship between endocycle and intracellular events.
This research was supported by National Natural Science Foundation of China (31872951 to Zhilong Bao, 31872952 to Fangfang Ma), and Taishan Scholar Foundation of Shandong Province (tsqn201812034) for Zhilong Bao. We thank Tomato Genetics Resource Center (TGRC) for providing tomato seeds. We thank plant growth facility members in the State Key Laboratory of Crop Biology at Shandong Agricultural University (China).
-
The authors declare that they have no conflict of interest.
- Supplemental Fig. S1 Effect of cellulase and pectinase on nucleus isolation. Mesocarps of 55 DPA red ripe tomato fruit were pretreated in water without (a) or with (b) cellulase and pectinase for 10 minutes. Nuclei were isolated from pretreated mesocarps using “Aru”, “Galbraith” and “Bourdon” buffer for flow cytometric analysis. Shown are histograms of flow cytometric analyses. PI-A, PI fluorescence pulse signal area.
- Supplemental Fig. S2 Effects of nucleus resuspension on flow cytometric analysis. Nuclei were isolated from mesocarps of 55 DPA red ripe tomato fruit using “Aru”, “Galbraith” and “Bourdon” buffer, harvested by centrifugation and resuspended by water (a) or original nucleus isolation buffers (b) for flow cytometric analysis. Shown are histograms of flow cytometric analyses. PI-A, PI fluorescence pulse signal area.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang J, Zhang P, Wang G, Chen C, Wang X, et al. 2023. Determination of nuclear DNA ploidy distribution in the mesocarp of tomato red ripe fruit using a flow cytometer. Vegetable Research 3:8 doi: 10.48130/VR-2023-0008 

Determination of nuclear DNA ploidy distribution in the mesocarp of tomato red ripe fruit using a flow cytometer
- Received: 29 November 2022
- Accepted: 04 January 2023
- Published online: 24 February 2023
Abstract: Metabolic changes at the ripening stage are critical for the final formation of tomato fruit quality, which may have a close relationship with nuclear DNA ploidy level. The lack of efficient isolation of nuclei from tomato red ripe fruit mesocarp cells hinders the studies of relationships between nuclear DNA ploidy level and fruit quality. We established a protocol to determine the nuclear DNA ploidy level in tomato red ripe fruit mesocarps using a flow cytometer. Tomato mesocarps were chopped in nucleus isolation buffer and filtered with a 425 μm steel mesh sieve and 75 μm nylon mesh sieve sequentially, and nuclei were collected from the filtrate by centrifugation. Three nucleus isolation buffers including Aru, Galbraith and Boudon buffer could isolate nuclei from green fruit mesocarp cells but not red ripe fruit for flow cytometric analysis. We noticed that resuspending nuclei isolated from red ripe fruit mesocarp cells with buffers was a critical step for flow cytometric analysis, and phosphate buffered saline (PBS) buffer had better effects than HEPES, MOPS, Tris and phosphate buffer. PBS buffer resuspension did not change the nuclear DNA ploidy distribution in green fruit pericarps during flow cytometric analysis. Aru buffer retrieved more nuclei with PBS resuspension for flow cytometric analysis than Galbraith and Boudon buffer, and the high pH value of PBS buffer improved the detection efficiency. Our protocol could effectively isolate nuclei from tomato red ripe fruit pericarps for flow cytometric analysis, which may facilitate the nuclear sorting with different ploidy levels in the future.
-
Key words:
- Tomato /
- endocycle /
- nucleus isolation /
- red ripe /
- flow cytometry.