









-
The species Cucumis sativus includes a cultivated variety (var. sativus) and three wild/semi-wild varieties (var. hardwickii, var. sikkimensis, and var. xishuangbannanesis)[1,2]. Genomic structural analysis based on 119 resequencing data proposed that all cucumber germplasm can be divided into four geographic groups: a Eurasian group, an East Asian group, a Xishuangbanna (XIS) group, and an India group[3]. XIS cucumbers are semi-wild and were found in the 1980s[4], spreading in a narrow area about N20º−N23º around the Xishuangbanna prefecture of China. This cucumber group is characterized by its melon-like fruit, colorful flesh, vigorous growth, large organ size, as well as photoperiod-dependent sex expression[5,6]. One XIS assembly Cuc80 was released recently, which is generated on long-read sequencing technology[7]. There are some reports about the genetic analysis of carotenoid-rich flesh[3], fruit shape[8], and flowering time[9] by using XIS cucumbers. Generally, many characteristic traits of this group need to be determined in the future.
Higher plants can be classified into short-day (SD) plants, long-day (LD) plants, and day-neutral (DN) plants according to their photoperiodism. Short-day plants and long-day plants are found mainly in low latitude and high latitude areas, respectively. Higher plants can obtain the trait of photoperiodic flowering through simple mutation. For example, long photoperiodic barley and wheat can flower earlier at SD conditions when they have the ppd-H1 (PRR7) mutation[10]. Common bean (Phaseolus vulgaris L.) transformed from short-day plants to day-neutral plants when they migrated from southern to higher latitudes; such a flowering habit change is obtained from mutations at PHYA3 and COL2[11,12]. Cucumbers originated from the low latitude Indian subcontinent 10.1 million years ago[13] and a south-to-north adaptation must have occurred for temperature and photoperiod responses to change. During this evolutionary process, vegetative growth usually became shorter and flowering time became earlier for adaptation[14]. Nowadays, predominant cultivated cucumbers are day-neutral plants, except for XIS cucumbers and some Indian cucumbers. XIS cucumbers develop only lateral branches instead of flowers in the spring when moved from their original habitat and growers have to extend their growth into autumn for propagation purposes[15,16]. However, a recent study controversially reported that XIS and wild cucumbers are insensitive to photoperiod treatment[17].
There are already some studies focused on the flowering traits of XIS cucumbers. Three major QTLs, FT1.1, FT5.1 and FT6.2, are proposed to control late flowering under a long photoperiod in XIS cucumber, respectively[8]. Fine mapping of early flowering locus 1.1 indicates that a large deletion of the FLOWERING LOCUS T (FT) upstream should account for higher expression of FT and therefore earlier flowering during domestication[17]. A recent pan-genome analysis proposed the association of sequential variance positioned upstream of the FT coincides with flowering time in cucumber germplasm[7]. From these QTL mapping works, we can get a consensus locus that controls cucumber flowering time on the end of the long arm of chromosome 1 carrying the FT gene.
Previously, studies usually ignored the photoperiod influence and carried out the field investigation in natural field conditions, labeling the XIS cucumber as a late-flowering plant. In fact, our experiment indicates that the XIS cucumber can bloom as quickly as a cultivated cucumber when under SD conditions. In this study, we get a linear correlation between day length and flowering time, which explains why XIS cucumbers become late-flowering when moved to a higher latitude area. From QTL mapping, transcriptome analysis, and gene expression analysis, we reveal the regulation mechanism of short-day flowering in XIS cucumbers.
-
We performed photoperiod treatments to XIS49 cucumber seedlings. After the sixth leaves unfolded, the seedlings were then moved into the greenhouse and were subject to investigation of flowering traits. Only those flowers formed within the first 15 nodes were examined since cucumber seedlings with six unfolded leaves have already formed male and female flowers within 16.75 ± 1.21 nodes as observed under a microscope. Under weak light conditions, flowers were only induced by a light phase of 10 h; while under strong light conditions, plants can bear flowers at light phase of 8 h and 10 h (Fig. 1a). Weak light, under required day length (≤ 6 h), and a long day length (≥ 12 h) are unfavorable to flower formation. In addition to flowering time, XIS49 is also characterized by downward female flower (DFF), fewer female and male flowers, a lower female-to-male flower ratio, and more branches (Fig. 1b & 1c).
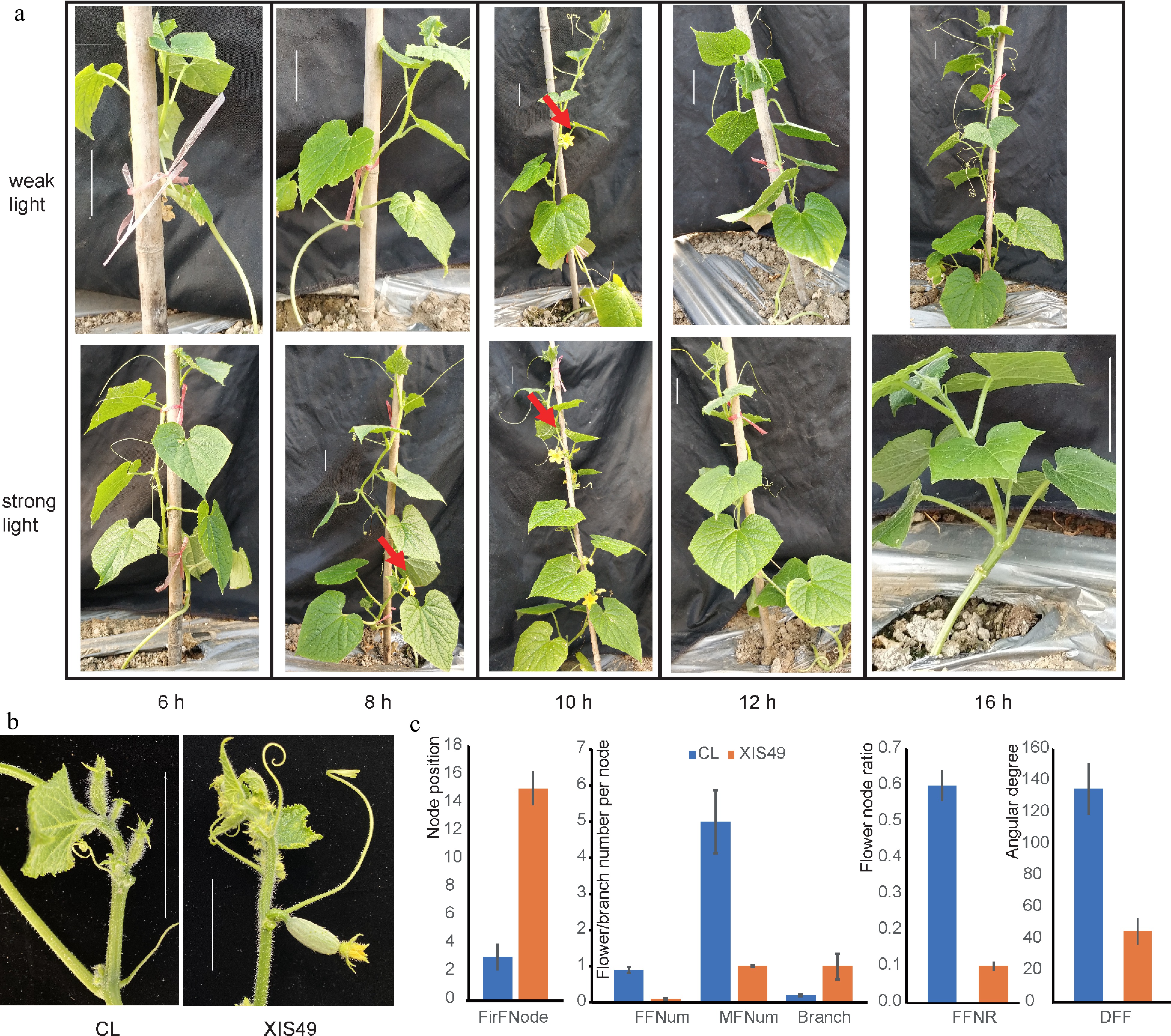
Figure 1.
Characteristic flowering-related phenotypes of XIS49. (a) Photoperiod and light intensity-dependent flowering initiation. The bars indicate a scale of 5 cm. (b) Female flower orientation. The bars indicate a scale of 5 cm. (c) XIS49 is significantly different with CL cucumber in first flower node (FirFNode), female flower number (FFNum) per node, male flower number (MFNum) per node, branch number per node, female flower node ratio (FFNR), and downward female flower (DFF) denoted by the angle at the stem.
To determine the minimum demand of a short-day condition, XIS49 seedlings were subjected to a treatment of a gradient day length change with a particularly short interval of 0.5 h between 10 and 12 h (Table 1). Here, flowers within 25 nodes were objection of field investigation. It was observed that a light phase longer than or equal to 12 h does not induce flowering within the first 15 nodes, and a 11.5 h light phase clearly meets the minimum demand of a SD condition. Interestingly, the flowering time (denoted as the first flower node) was gradually pushed back when day length increased, which means there is a negative dosage effect of light phase length on XIS49 flowering. The arrest of flowering under LD conditions is GA-independent since exogenous GA could not make amends (data not shown).
Table 1. Influence of photoperiod on flowering of XIS cucumber.
Photoperiod First flower node1 First FF node1 FF number1 8H 3.5 ± 0.2 e 8.9 ± 0.3 c 1.8 ± 0.2 a 10H 4.7 ± 0.3 d 11.3 ± 0.3 b 2.1 ± 0.3 a 10.5H 6.0 ±0.4 c 12.6 ± 0.3 ab 1.5 ± 0.4 a 11H 5.2 ± 0.2 cd 12.9 ± 0.8 a 1.6 ± 0.2 a 11.5H 10.4 ± 0.4 b − − 12H 17.6 ± 0.6 a − − 16H − − − 1 Average flower number ± SE within 25 nodes. The different letters following the numbers indicate significant difference between different photoperiod, P ≤ 0.05. FF, female flower; "−" indicates no flowers observed. Transcriptomic analysis under photoperiod treatment
-
Plants sense photoperiodic change with their leaves. To profile the global response of transcriptome to photoperiod condition change, we divided night time into trisection (N1-N3) and daytime into quinquesection (D1−D5). Leaf samples of XIS49 were collected at the midpoint of night time (N2) and daytime (D3). Then we compared transcriptome between different photoperiod conditions of 8 h (SD), 12 h (MD), and 16 h (LD). More than 6.21 Gb clean data of RNA-seq were generated for each sample by Illumina sequencing (Supplemental Table S1). The biological triplicates of each sample clustered together indicate a good reproducible (Supplemental Fig. S1). The difference of transcriptome between day and night is much bigger than the contrast between different photoperiod treatments. There were 6,205, 4,932, and 3,894 day-night differently expressed genes (DEGs) under SD, MD, and LD conditions, respectively (Supplemental Fig. S2). The numbers of the day genes (upregulated in daytime) and the night genes (upregulated in night time) are almost the same. As expected, the most significantly enriched process/pathway in Gene Ontology (GO) and KEGG enrichment analysis is about chlorophyll and photosynthesis metabolism, followed by amino acid and plant hormone-related processes and pathways.
We are more interested in those genes that show a different response to photoperiod treatments. For the daytime gene set, there are a total of 5,269 DEGs and 363 of these genes are conserved in different comparisons (Supplemental Fig. S3); for the night time gene set, there are a total of 4,976 DEGs and 293 of these genes are conserved in different comparisons (Supplemental Fig. S4). To summarize the response pattern, GO and KEGG enrichment analysis was performed on those DEGs (Supplemental Fig. S3 & S4). This was repeated with day-night DEGs and a short-long photoperiod caused DEGs involved with assimilation, amino acid/protein biosynthesis, and primary and secondary metabolites. Interestingly, ribosome genes show both circadian and photoperiodic expression mannerisms. For day-night DEGs, overwhelming predominant ribosome genes are downregulated at night under SD; in contrast, MD and LD treatment upregulate almost all the ribosome DEGs at night (Fig. 2a). On the other hand, SD enhanced the expression level of ribosome gene in the daytime, but decreased their expression in nighttime (Fig. 2b). In fact, in addition to flowering time, long photoperiod also causes serious abnormal growth, which is probably ascribed to the disturbed expression of ribosome genes.
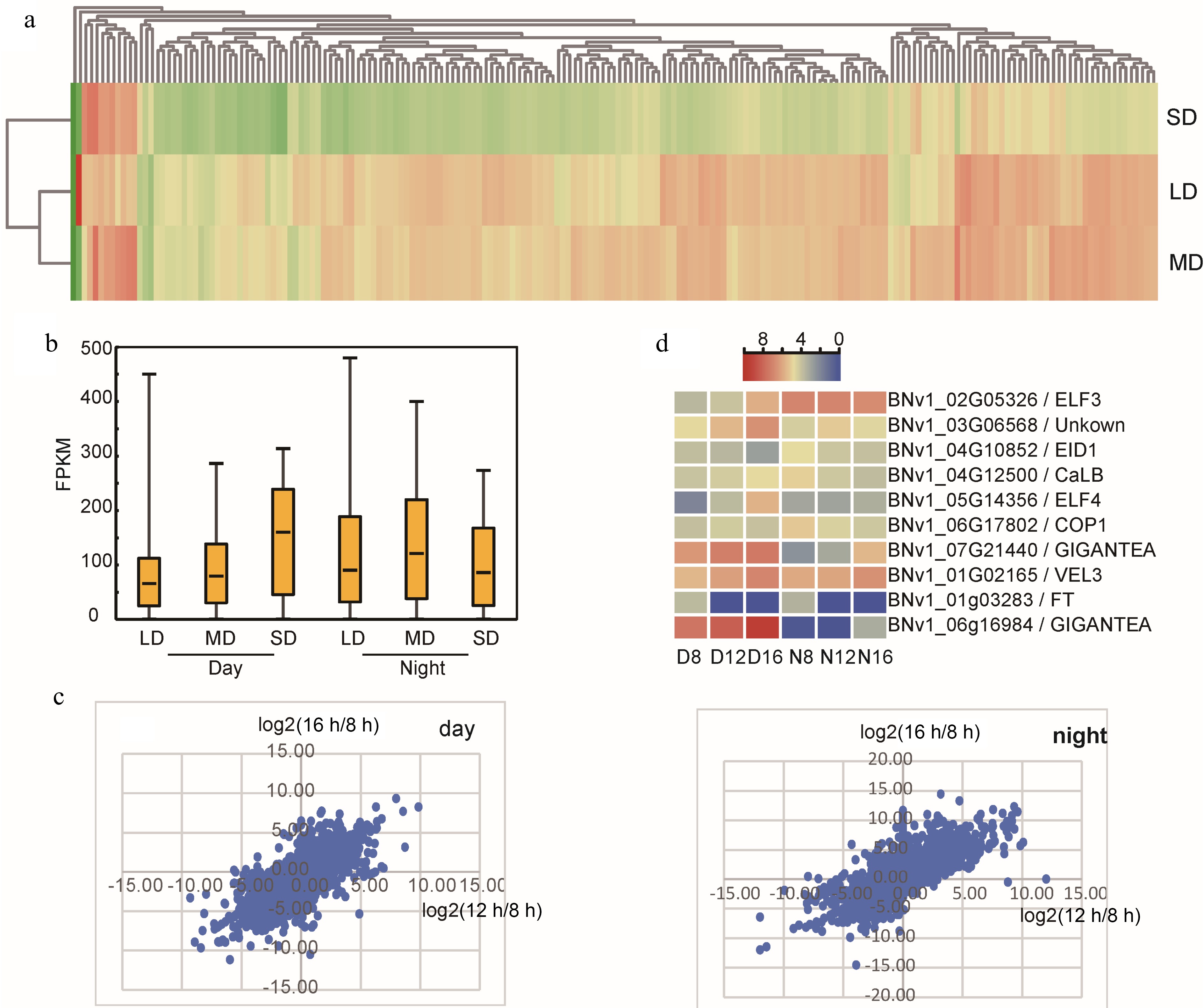
Figure 2.
Influence of photoperiod treatment on XIS49 transcriptome. (a) A great number of ribosome genes show diurnal rhythm, and these day-night ribosome DEGs (day vs night) are upregulated at night under LD and MD but downregulated at night under SD. (b) Photoperiodic DEGs (LD vs SD, MD vs SD, LD vs MD) show much higher expression level under SD in daytime but not in nighttime. (c) Plotting the value of log2(16 h-FPKM/8 h-FPKM) against log2(12 h-FPKM/8 h-FPKM) by using the photoperiodic DEG set. (d) Flowering-related photoperiodic DEGs annotated by GO term.
As there is a dosage effect of day length on flowering time (Table 1), we generated a dot plot analysis between 16-8 (16 h vs 8 h) and 12-8 (12 h vs 8 h). Although 12 h and 16 h exert the same influence on DEGs expression, there is not any expected dosage effect on global gene expression (Fig. 2c). The dosage effect on phenotype is probably a result of a single gene regulation rather than a response by the global transcriptome. Among the photoperiodic DEGs, 10 DEGs are annotated (GO annotation) to be flowering-related. FT and EID1 gene are suppressed by LD condition (Fig. 2d). For GIGANTEA (GI), EARLY FLOWERING 3 (ELF3), and EARLY FLOWERING 4 (ELF4), clearly, longer day length correlates with a higher expression level.
Response of circadian rhythm genes to photoperiod change
-
Plants sense day length change through circadian rhythm. Therefore, we profiled the expression change of genes annotated in a circadian rhythm plant (KEGG, ko04712) during the photoperiod treatment (Fig. 3a). Quantified by FPKM in RNA-seq and transcriptome analysis, there are 27 photoperiodic DEGs encoding 16 enzymes showing a significant response to photoperiod treatment in the circadian pathway. Clearly, most of these DEGs do display a circadian fluctuation in expression level as expected. For example, SPA1 is a night gene in cucumbers, and was decreased by LD; GI is a day gene and was increased by LD. The most important two genes are the FT and the CO gene. The FT gene directly regulates initiation of a decrease in shoot meristem while the CO gene is the outflow of circadian rhythm and modulates the FT gene. Impressively, the FT gene is detected by RNA-seq only under SD conditions, while the induction of CO by SD is ambiguous and unclear.
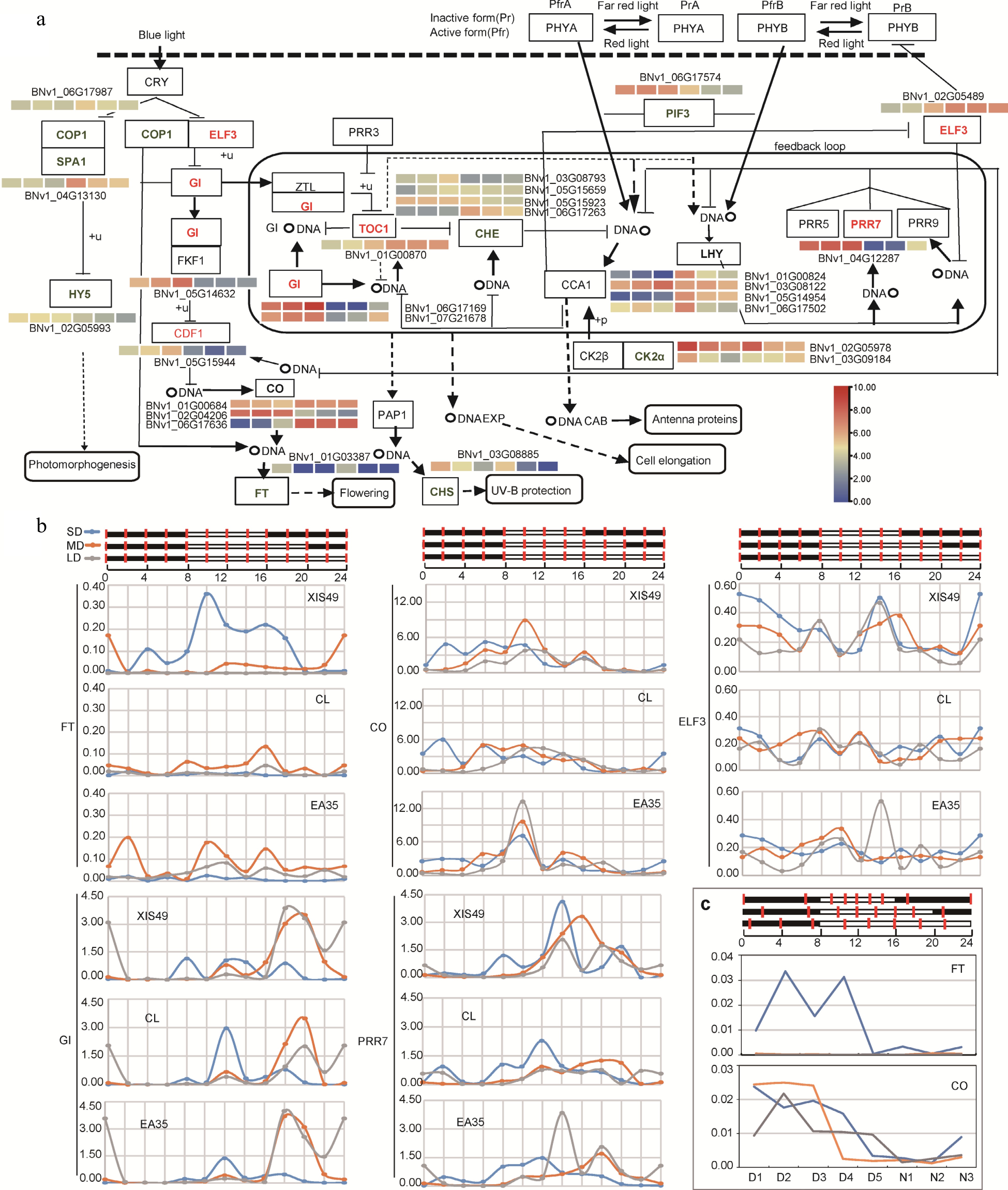
Figure 3.
Circadian expression manner of DEGs (a) Influence of photoperiod treatment on circadian pathway genes (KEGG, ko04712). Upregulated and downregulated genes by long photoperiod is denoted in red and green. A heat map is present to show transcriptional change; from left to right, 8 h-day, 12 h-day, 16 h-day, 8 h-night, 12 h-night, 16 h-night. (b) Samples were collected every two hours during a 24 h period and the expression was quantified by qRT-PCR. (c) The circadian expression of CO and FT was confirmed again by using leaf samples collected at nighttime-trisection timepoints (N1-N5) and daytime-quinquesection timepoints (D1-D5).
To precisely profile the expression response of circadian genes, leaf samples were collected every two hours in a 24 h period and gene expression was quantified by qRT-PCR (Fig. 3b). Alternatively, daytime and night time length were equally divided into five and three sections, respectively. The FT gene, the central small molecule that transmits the flowering signal from leaf to meristem, is only expressed under SD conditions from 10:00 to 18:00, and is sharply suppressed by MD conditions; under LD conditions, the FT gene is totally inactive (Fig. 3b and Supplemental Fig. S5). The CO gene 'bridges' circadian rhythm and the downstream FT gene. The CO gene is extensively expressed from 8:00 to 16:00, which coincides well with the FT genes. However, no clear substantial difference was observed for CO and those circadian clock genes between photoperiod treatments.
QTL mapping confirmed the central role of FT genes
-
We constructed an F2 population by crossing XIS49 with CL cucumber to map the QTL of first flower node (FFNode) that reflects the flowering time, as well as other flowering-related traits. All seedlings were grown under a LD condition until the six-leaf stage to ensure that the shoot apices had finished the process of sex determination within the first 15 nodes before moving them to a greenhouse. A total of 23 KASP markers screened out from 30 KASP markers were applied in the QTL mapping work (Supplemental Fig. S6). We successfully detected QTLs for all the targeted phenotypes twice in 2020 and 2021 with LOD > 5; particularly, the QTLs of the FFNode and the MFNum can explain 54.8% and 47.09% of the inheritance, respectively (Table 2). For the FFNode trait, XIS49 could not initiate flowering within the first 15 nodes, while the average value of first flower node of CL cucumber is 3.15 ± 0.55.
Table 2. QTL mapping of several flower traits by KASP.
Chr. Start End LOD PVE% Size (Mb) SF1.1 1 29,795,488 29,931,423 32.09 46.90 0.14 SF6.1 6 12,580,505 21,116,696 6.65 7.90 8.54 FirFFNode1.1 1 190,063 10,053,157 8.97 15.72 9.86 FFNum1.1 1 190,063 10,053,157 6.03 8.70 9.86 FFNum2.1 2 12,405,113 22,000,804 5.81 11.55 9.60 MFNum1.1 1 29,369,853 29,795,488 18.10 14.11 0.43 MFNum6.1 6 2,507,906 12,580,505 14.14 16.15 10.07 MFNum6.2 6 12,580,505 21,116,696 14.85 12.18 8.54 FFNR1.1 1 190,063 10,053,157 7.89 13.20 9.86 DFF3.1 3 135,870 11,923,221 6.76 11.00 11.79 branch1.1 1 29,795,488 29,931,423 5.19 7.17 0.14 Short-day flowering (SF) was mapped based on the phenotype of first flower node; FirFFNode, first female flower node; FFNum, female flower number; MFNum, male flower number; FFNR, female flower node ratio; DFF, downward female flower; branch, branch number per node. To shorten the QTL region, nine more markers positioned from 28,294,532 to 30,080,987 were applied. As a result, the QTLs were detected twice around 29,795,488 and the final QTL was positioned between 29.08 and 29.62 Mb on chromosome 1 (Fig. 4a; Table 2). At this locus, there are two big polymorphic fragments between the parent plants: a sequence variant (SV) cluster and the big insertion in XIS49 (Fig. 4b). The FT gene is positioned between the SV cluster and the big insertion. Since there is no sequential difference of the FT genes in term of both gene body and promoters between parents, we focused on the two big polymorphic fragments. The SV cluster region consists of serious SVs within a segment of 29.1MB-29.4Mb. These SVs caused polymorphic promoters of 16 genes, which contribute all the SV genes in the QTL region (Table 3). The big insertion (with a length of 39.88kb) was formed during the evolutionary period when cucumbers separated from melons and is found in wild cucumbers (WI22) and semi-wild cucumbers (XIS49), but later was lost in cultivated cucumbers (Supplemental Fig. S7). A polygalacturonase (PG) gene and four TIR transposon with high quality (length > 1,000; score > 2,000) were positioned in the big insertion. The closest ITR TE was 976 bp upstream of coding start site of PG gene. Interestingly, PG genes in the big insertion also showed a photoperiod-dependent expression response, which is the same as the FT gene.
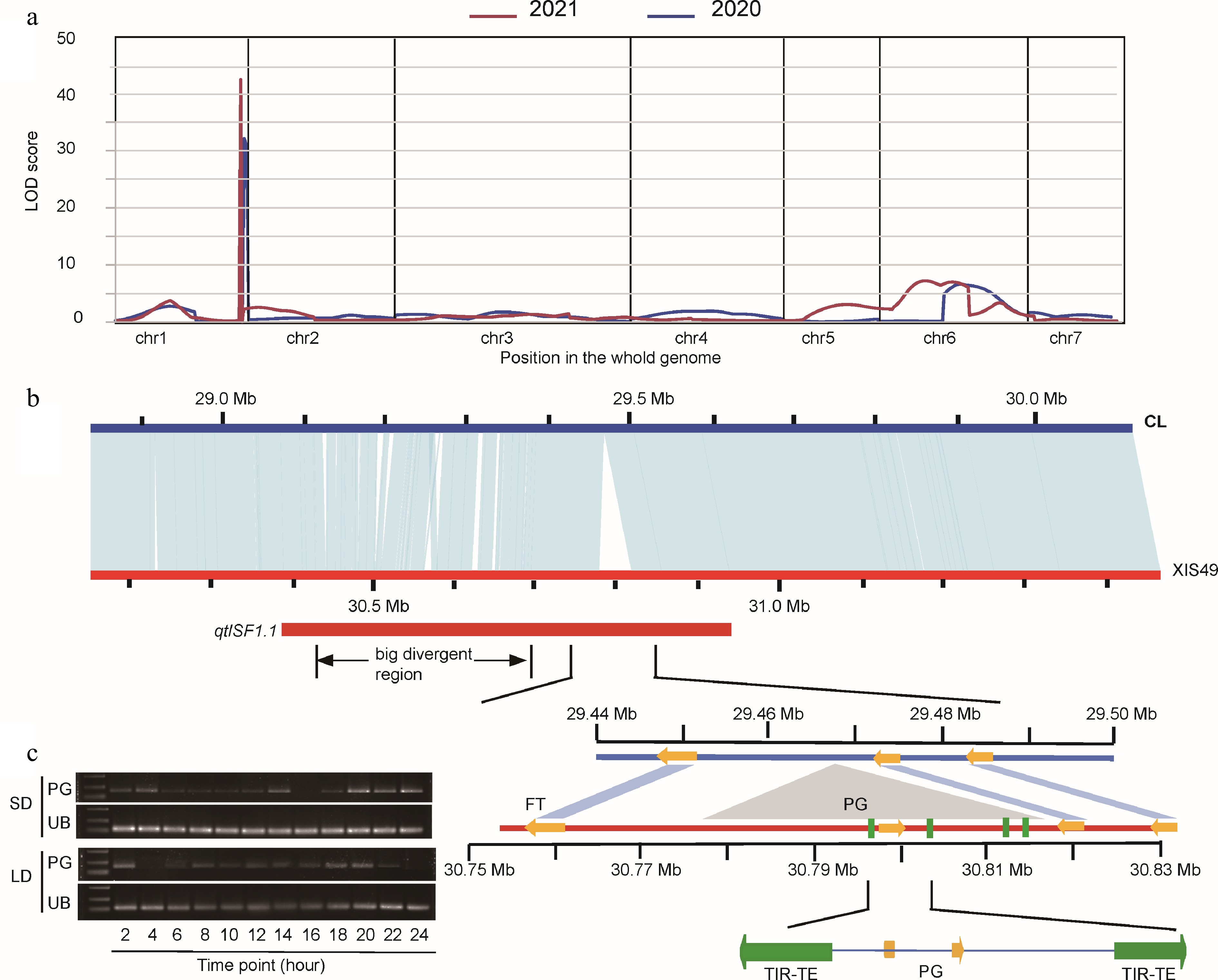
Figure 4.
QTL mapping of short-day flowering in XIS49. (a) LOD profiles of QTL for FFNode (first flower node). Plants were pretreated by LD condition. (b) A syntenic analysis of 1 Mb segment that carries the QTL between XIS49 and CL cucumber. The qtlSF1.1 carry two major sequence variants nearing the FT gene, 'the bid divergent region' and 'the big insertion'. The orange arrows indicate protein coding genes. The four green rectangles indicate TIR-TE insertion. (c) The expression profile in a 24-h period of the inserted polygalacturonase (PG) gene shows photoperiodic manner. UB, ubiquitin.
Table 3. Genes at the locus LF1.1 with sequential divergent between CL and XIS49.
Gene ID Position/Mb SVs in promoter SNP Gene function CDS Promoter2 CsaV3_1G043880 29.04 HDR Unknown CsaV3_1G043890 29.04 DEL; DEL T(-31)A; C(-30)G; A(-5)C Unknown CsaV3_1G043980 29.14 INS; INS T18I; D214N; I304Y Choline monooxygenase CsaV3_1G043990 29.15 HDR A51T A(-26)C Unknown CsaV3_1G044000 29.15 INS Y158S Phosphatidylglycerol transfer protein CsaV3_1G044010 29.17 DEL Unknown CsaV3_1G044020 29.18 CPL DnaJ protein CsaV3_1G044060 29.23 HDR; INS L23H; K127R; A130S; S202L T(-43)A Cytochrome P450 CsaV3_1G044110 29.33 HDR YTH domain, RNA binding CsaV3_1G044160 29.38 HDR Thioredoxin F-type CsaV3_1G044440 29.78 CPG; INS A27T Protein TIC 20-II CsaV3_1G043900 29.05 DEL Unknown CsaV3_1G043920 29.06 DEL T(-43)G; A(-22)T DnaJ protein CsaV3_1G043930 29.08 DEL A572T Serine/threonine-protein phosphatase CsaV3_1G043960 29.12 DEL Glycosyl transferase CsaV3_1G044070 29.25 DEL S locus-related glycoprotein 1 binding pollen coat protein CsaV3_1G044130 29.36 INS Glycoside hydrolase CsaV3_1G044170 29.39 DEL Unknown CsaV3_1G044180 29.43 DEL Activator of Hsp90 ATPase CsaV3_1G043940 29.10 F281I; Q332R; I441T; I574L Unknown CsaV3_1G043950 29.11 E13D; K34E; P26L; Y29F; L67S Unknown CsaV3_1G043970 29.13 Protein IQ-DOMAIN CsaV3_1G044030 29.19 D47N; C81R; V113A; A119G; H145Y; Q160K; F207L; Y211N 23 kDa jasmonate-induced protein CsaV3_1G044040 29.20 F208L; G381D; I424M; S753F; G790D beta-galactosidase 7 CsaV3_1G044050 29.22 G431R; L569S Microtubule-associated protein CsaV3_1G044080 29.28 S20A Pentatricopeptide repeat-containing protein tubulin-folding cofactor E CsaV3_1G044090 29.28 CsaV3_1G044100 29.32 Heavy metal-associated domain CsaV3_1G044120 29.35 S90A Unknown CsaV3_1G044140 29.37 D317E Aspartic proteinase CsaV3_1G044150 29.37 Unknown CsaV3_1G044210 29.45 G82R FT CsaV3_1G044450 29.80 R64G Phosphoglycolate phosphatase 1 Sequential/Structural variance (SV) including (HDR), (CPL), (CPG), and insertion (INS) and deletion (DEL); Regular INS and DEL, SV length > 50 bp; italic INS and DEL, SV length < 50. 2 T(-31)A, the nucleotide with a distance of upstream-31 bp to the transcription start site is T in CL, and A in XIS49. -
XIS cucumbers were reported as late-flowering plants and as having no different response between SD and LD conditions[18]. However, flowering time of the XIS cucumber is deeply influenced by photoperiod condition in many other studies[15,16]. Moreover, CG9192 (an XIS cucumber) was reported to be about 80 days later than '404' (a cultivated cucumber) in terms of flowering time, regardless of photoperiod condition[18]. Our study indicates that XIS49 can form blooms as quickly as a cultivated cucumber when grown under SD conditions. The negative dosage effect and the linear correlation of day length and FFNode explains why these cucumbers perform as late-flowering plants when introduced to a middle latitude. In summary, XIS cucumbers are short-day plants, not late-flowering. In addition to day length, light intensity also affects FFNode; weak light and day length that is too short (≤ 6 h) both suppress flower initiation even under SD conditions, which must be due to deficient vegetative growth (Fig. 1). Indeed, there is no difference in flowering time between SD and LD treatment when light is inadequate.
There have been a few reports about QTL mapping of a late-flowering trait in XIS cucumbers[8]. In a recent QTL mapping work, an XIS cucumber CG9192 and an East Asian cucumber '404' were used as a donor parent and a recruit parent to construct an NIL population[18]. All these studies identified the same locus that carry the FT gene. These studies did field investigation under natural photoperiod conditions and focused on "flowering time". Here, flowering traits were all investigated under a photoperiod treatment of 16h. Interestingly, we detected the same FT-QTL on chromosome 1 but with a much higher LOD value (30−40) than previous reports. In XIS49, the circadian expression of the FT gene during 10:00−18:00 (SD conditions) was totally suppressed by LD conditions. Such an influence of photoperiod on FT expression is not observed in CL cucumber. These results confirm that the FT gene contributes to short-day flowering in XIS cucumber.
Despite of the success of QTL mapping and the consensus of the FT gene, the detailed mechanism remains unknown because there is no sequence and gene structure diversity of the FT gene. A recent pan-genome analysis summarized that the big insertion around the FT gene associates with late flowering[7]. The big insertion which carries a PG gene and four TEs was also detected in our study (Fig. 4). The PG gene shows a similar photoperiodic-dependent expression patten with the FT gene, which may imply cis-regulation from the big insertion. However, since the big insertion is too far from the FT gene (>15 kb), a detailed regulation mechanism needs to be clarified in the future. It is still too early to exclude the possible regulation from the SV cluster region as well as the SV genes (Fig. 4; Table 3).
Since photoperiodic flowering is determined by the length of light phase, the circadian pathway plays a critical role in flowering regulation. Nowadays, the circadian clock has been experimentally determined and well-reviewed by many researchers[19,20]. Taking Arabidopsis for example, a complex CO-FT module is suggested to control its long photoperiod flowering[21−23]. One mechanism is that the SPAs-COP1 complex degrades the CO protein in the dark phase, while longer nighttime in SD conditions covers the critical evening expression of the CO in LD conditions, which subsequently results in suppression of FT. One other mechanism relies on GI-FKF1 activator and CDF repressor of the FT gene. LD conditions upregulated many circadian pathway genes like morning gene CDF1, evening genes ELF3 and GI, afternoon gene PRR7, and night gene TOC1. Night genes SPA1, COP1, HY5, and CHE, are downregulated (Fig. 2). Clearly some of the circadian clock is disturbed as with the GI gene. However, these changes might not account for short-day flowering since these disturbances seem to appear in both the XIS cucumber and cultivated cucumbers.
-
As demonstrated in this study, XIS49 is a restrictive short-day plant, a distinct divergence, as it's been previously reported as late-flowering. The flowering time was gradually decreased with the increase of day length and the minimum light phase demand for flowering is 11.5 h. LD conditions can completely suppress the flowering process in XIS49. Our study indicates that the cis-regulation of the FT gene accounts for short light phase-dependent flowering and the photoperiodic cis-regulation probably comes from a 30 kb insertion. Outflow of circadian clock disturbance caused by photoperiod change cannot explain the acquisition of short-day flowering and there is likely not any trans-regulation mechanism. This conclusion is based on gene expression analysis during a diurnal cycle and QTL mapping work. The mapped locus FFNode1.1 can explain 46.9% of the flowering time trait with an LOD value of 32.09. We also mapped QTLs controlling other flowering-related phenotypes at the same time.
-
The homozygous cucumber XIS used in this study was collected by Sichuan Agricultural University from Wenshan prefecture, Yunnan Province (China) and was assigned ID number XIS49 after being purified for six generations. Chinese long cucumber, also known as '9930', is the sequencing material. EA35 is a pure line of the East Asian group, the same type as '9930'. XIS49 and EA35 were sequenced by nanopore sequencing technology and the assemblies and annotations were downloaded from Gene Bank with the project ID of PRJNA956325.
Photoperiod treatment and sample collection
-
Photoperiod treatment was started at seed-sowing and once six leaves were unfolded, the seedlings were then moved to a greenhouse in April. Photoperiod conditions were controlled by a plant incubator with long-day length (LD, 16 h/8 h, day/night), medium-day length (MD, 12 h/12 h, day/night), and short-day length (SD, 8 h/16 h, day/night). The photoperiod treatment was carried out under moderate temperatures (30 °C/25 °C, day/night). We also studied the effect of light density inanition in regards to day length, with strong light (1,700 μmol·m2·s−1) and weak light (200 μmol·m2·s−1). The entire third unfolded leaf samples were collected for total RNA extraction for RNA-seq and qRT-PCR. Leaves from three seedling individuals were homogenate as one repeat and there were triplicates for each sample. Leaf samples were collected every 2 h over a 24 h period at the stage of four unfolded leaves. While, night time and daytime were divided into trisection (N1−N3) and quinquesection (D1−D5), respectively. Samples collected at N2 and D3 were used for RNA-seq and transcriptome analysis.
RNA-seq and transcriptome analysis
-
Total RNA was extracted using the Trizol method as previously described[24]. A total amount of 1 μg RNA per sample was used. Purity of the RNA sample was examined by NanoDrop, while Agilent 2100 (Agilent Technologies, De Novo Santa Clara, CA, USA) was used to assay RNA integrity. Qualified RNA was processed for library construction and illumine sequencing by Biomarker (Beijing, China).
Sequencing bases quality score was calculated to evaluate the probability of an incorrect base[25]. After trimming adapter contaminations and removing low quality-score nucleotides, clean data were obtained. Clean reads were mapped to the XIS49 genome by using an alignment tool HISAT2[26]. StringTie was used to assemble the mapped reads[27]. The mapped reads were compared with original annotation; novel transcripts were determined and annotated by searching against databases[28]. The expression level of a gene or transcript was quantified by StringTie in a form of FPKM (Fragments Per Kilobase of transcript per Million fragments mapped)[27]. Reproducibility of biological replicates was determined by Pearson correlation coefficient R (Pearson's Correlation Coefficient)[29]. DESeq2 was used in differential expression analysis and the criteria for differentially expressed genes was set as Fold Change (FC) ≥ 2 and FDR < 0.01[30]. Functional annotation and enrichment analysis were done by using a database of GO (Gene ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes).
Gene expression was quantified by qRT-PCR
-
Relative gene expression was quantified by qRT-PCR. Total RNA was extracted using the MolPure TRIeasy Plus Total RNA Kit (Yeasen Biotechnology, Shanghai, China). Reverse transcription was carried out by Hifair V one-step RT-gDNA digestion SuperMix for qPCR (Yeasen Biotechnology, Shanghai, China). For qRT-PCR, the PCR mix regent is SYBRPRIME qPCR Kit(Fast HS)(BIOGROUND Biotechnology, ChongQing, China), and the apparatus was Bio-Rad CFX96 (BIO-RAD, USA). The inner reference gene is ubiquitin (CsaV3_7G003730). The primer sequence and annealing temperature is listed in Supplemental Table S2.
Phenotyping of important trait in the F2 population
-
An F2 population was constructed by crossing XIS49 and CL. All the F2 seedlings were treated with LD conditions before being removed to a greenhouse in April. The parental genotypes and a segregating population were grown for two years (2020 and 2021) in Chengdu (China) N30.6°. For each time period, the population size was 250 individuals. Six important traits were investigated. (1) First flower node (FFNode), the first node bearing a flower, regardless of whether it is a male or a female flower. (2) Female flower number per node (FFNum) within 15 nodes. (3) Male flower number per node (MFNum) within 15 nodes. (4) Female flower node ratio (FFNR), the ratio of nodes (< 15 nodes) that bear any female flowers. (5) Female flower angle (FFA), the angle between the female flower and the stem. There are four values assigned to describe the angles during investigation: 0−45º, 1; 45º−90º, 2; 90º−135º, 3; 135º−180º, 4. Branch number (BN) per node (< 15 nodes). For the above traits, we investigated flowers and branches within 15 nodes.
QTL mapping
-
Comparative genomic analysis between XIS49 and CL cucumber genome has been carried out. We used BWA software to align clean reads of XIS49 to the reference genome (CL cucumber), and only uniquely mapped reads were used for the following analysis. SNPs were called using the Samtools software[31], and low quality SNPs were filtered using Coval scripts[32]. Finally, polymorphic SNPs between XIS49 and CL were identified after sequence alignment and variation calling.
To design SNP-KASP markers, we retrieved the upstream and downstream sequences of selected SNPs and used BLASTN to determine their specificity in genomic sequences. Newly designed KASP markers were validated for polymorphisms and genotyping quality by application on the parents. Only those KASP-SNP markers having correct and clear genotyping results were used to genotype the entire mapping population. PCR reaction reagents were purchased from GENTIDES Biotech Co. Ltd. (Wuhan, China) and the SNP genotyping technique is penta-primer amplification refractory mutation system (PARMS). In this detection system, the report fluorophores were FAM and HEX KASP, while ROX was used to normalize the fluorescent reporter signal. PCR amplification and fluorescence detection was performed by Bio-Rad CFX96 TouchTM system (Bio-Rad Laboratories, Inc., Irvine, USA). Soft QTL lciMapping4.2 was used to estimate the genetic distance, LOD values, and finally QTLs[33].
Data availability
-
RNA-seq data of LD-D3, MD-D3, SD-D3, LD-N2, MD-N2, SD-N2 were deposited in the NCBI sequence read archive (SRA) under the accession number PRJNA951446.
This work was supported by the National Natural Science Foundation of China (32072599). We appreciate Prof. Xixiang Li from the Institute of Vegetable and Flowers, Chinese Academy of Agricultural Sciences (CAAS), for valuable suggestions on this study.
-
The authors declare that they have no conflict of interest.
-
# These authors contributed equally: Shan-Shan Song, Qian Hao, Li-Hong Su
- Supplemental Fig. S1 Evaluation of sequencing quality by (a) Principal Component Analysis (PCA) and (b) Correlation heat map between samples.
- Supplemental Fig. S2 Day-night DEGs. (a) A Venn diagram of day-night DEGs under SD, MD, and LD. (b) GO enrichment analysis of day-night DEGs. (c) KEGG enrichment analysis of day-night DEGs.
- Supplemental Fig. S3 Photoperiodic DEGs in daytime. (a) A Venn diagram of day-night DEGs under SD, MD, and LD. (b) GO enrichment analysis of photoperiodic DEGs. (c) KEGG enrichment analysis of photoperiodic DEGs.
- Supplemental Fig. S4 Photoperiodic DEGs in nighttime. (a) A Venn diagram of day-night DEGs under SD, MD, and LD. (b) GO enrichment analysis of photoperiodic DEGs. (c) KEGG enrichment analysis of photoperiodic DEGs.
- Supplemental Fig. S5 Daytime and night were equally divided into 5 and 3 sections, respectively. SD, short daylength. MD, medium daylength. LD, long daylength. D1-D5, the 5 sections of daytime. N1-N3, the 3 sections of night time. Relative expression level was quantified by qRT-PCR and modified by inner ubiquitin gene.
- Supplemental Fig. S6 SNP-KASP markers and the linkage map used in the QTL mapping.
- Supplemental Fig. S7 Syntenic analysis of the QTL region. (a) Wild cucumber WI22 and CL cucumber. (b) Cucumis hystrix and CL cucumber. (c) Melon (DHL92) and CL cucumber. The red arrow indicates the FT gene.
- Supplemental Table S1 Sequencing data statistics.
- Supplemental Table S2 Primers used in this study.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Song SS, Hao Q, Su LH, Xia SW, Zhang RJ, et al. 2023. FLOWERING LOCUS T (FT) gene regulates short-day flowering in low latitude Xishuangbanna cucumber (Cucumis sativus var. xishuangbannanesis). Vegetable Research 3:15 doi: 10.48130/VR-2023-0015 

FLOWERING LOCUS T (FT) gene regulates short-day flowering in low latitude Xishuangbanna cucumber (Cucumis sativus var. xishuangbannanesis)
- Received: 17 December 2022
- Accepted: 17 March 2023
- Published online: 04 May 2023
Abstract: Xishuangbanna (XIS) cucumber is a semi-wild cucumber originating from a low latitude. XIS cucumbers are strictly short-day plants, while cultivated cucumbers are day-neutral plants. The length of the light phase has a dosage effect on flowering time and the day length requirement for flowering is 8−11.5 h. Out-of-range photoperiod conditions and weak light conditions are unfavorable for blooming. Transcriptomic and gene expression analysis indicate circadian pathway genes as well as CONSTANS (CO) did not show a differential response to photoperiod treatment between XIS and cultivated cucumbers. The FLOWERING LOCUS T (FT) gene is activated from 10:00 to 18:00, and long- (16 h) and medium- (12 h) day length suppressed this diurnal rhythm expression. We designed Kompetitive allele specific PCR (KASP) markers based on genomic SNPs between the mapping parents (XIS49 and CL) to genetically map the short-day flowering gene. Field investigation was performed after long-day photoperiod (16 h) treatment. Finally, we detected a strong quantitative trait locus (QTL) signal at a 540-kb segment (chr1: 29.08−29.62 Mb) that carries the FT gene. We found a 30-kb TE-rich insertion with a distance of 15 kb to the FT gene in XIS49, which may contribute to the gain of the short-day flowering trait in XIS49. The 30-kb insertion endowed the FT gene and the inserted polygalacturonase (PG) gene with a photoperiod-dependent expression manner. Our study indicates that the FT gene, but not its upstream circadian clock genes, regulate short-day flowering in the XIS cucumber, and the cis-regulation of the FT gene is probably due to a TE insertion.
-
Key words:
- Circadian clock /
- Short-day cucumber /
- Flowering time /
- CONSTANS /
- Photoperiod treatment