








-
Transition from vegetative to reproductive growth is a pivotal juncture in the development of plants, orchestrated by a combination of intrinsic developmental signals and extrinsic cues[1]. Photoperiod is a major external factor, serving as a cue to trigger the transition from vegetative to floral meristem in many annual plants. In winter annuals with a life cycle spanning fall, winter, and into the next spring and summer seasons, vernalization induced by cold temperatures is a major external factor responsible for the transition[2−6]. Unlike annuals, grapevine is a perennial woody species and has unique and intricate biological processes for flower formation and development. The switch from juvenile to adult phase in grapevine is accompanied by the appearance of the first tendril along with the change of spiral to alternate leaf arrangement. Then vine's shoot apical meristem (SAM) in the main branch's apex continues producing both leaf primordia and uncommitted primordia, known as anlagen. Depending on genetic and environmental cues, an uncommitted primordium can develop into either inflorescence or tendril. Usually, the first two or three undifferentiated axillary primordia in latent buds give rise to inflorescences after they have undergone dormancy while those buds emerging in the current season's shoots are destinated to become tendrils, which serve as support structures in grapevine[7−9]. Tendrils are regarded as intermediate or modified inflorescences due to their inability to initiate floral meristem, as evidenced by their interchangeable nature and specific expression of key floral meristem identity APETALA1 (VvAP1) and LEAFY (VvLFY). Studies showed that application of cytokinins and gibberellic acid (GA) in shoots could promote anlagens to develop into inflorescences and tendrils, respectively[9−12]. Interestingly, even young tendrils treated with cytokinin can be transformed into inflorescences, whereas GA treatment effectively reverts young inflorescences back to tendrils[13,14]. This underscores the dynamic, hormone-responsive and reversable features of anlagens, tendrils and inflorescences, in a stark contrast to the terminal, irreversible nature of highly differentiated floral tissues in other woody fruit crops such as apples and peaches.
A grapevine mutant derived from a somatic mutation in the L1 cell layer of Vitis vinifera cv. Pinot Meunier manifests dominant dwarfism and insensitive to GA[15], similar to the gain-of-function GA-insensitive (gai) mutants found in Arabidopsis, rice and wheat, collectively referred to 'green revolution' dwarfism[16−18]. In-depth scrutiny revealed that the L1 dwarf mutant bears a mutation in the DELLA domain of VvDELLA1 or VvGAI1[15]. This mutation induces an amino-acid substitution in the DELLA domain of VvDELLA1 and disrupts interaction of the VvDELLA1 protein with GA signals. Consequently, VvDELLA1 proteins evade GA-mediated breakdown, resulting in highlighted accumulation of the VvDELLA1 factor and its growth-inhibitory impact on stem elongation, as evidenced by the dominant dwarf phenotype in the L1 mutant[15] and in the transgenic Arabidopsis with ectopic expression of the mutated VvDELLA1 (Vvgai1)[19]. Unexpectedly, the same mutation in the L1 dwarf mutant drastically enhanced the conversion of tendril-bound anlagens to inflorescences even in the current shoots or in juvenile seedlings[15]. This suggests that VvDELLA1 functions distinctively from its counterparts in annuals, which typically repress flowering or delay the transition from vegetative to reproductive phases[20−23].
DELLA proteins play a pivotal role as central hubs, translating hormonal stimuli, physiological cues and environmental inputs into specialized regulatory networks that govern plant growth and development. In the GA pathway characterized in Arabidopsis and other annual crops, the interactions between GA and its receptor, GIBBERELLI INSENSITIVE DWARF1 (GID1), facilitate binding to the DELLA domain of the DELLA proteins. Consequently, these interactions render DELLAs highly susceptible to polyubiquitination, marking them for subsequent degradation by the 26S proteasome[24]. This orchestrated process results in diminished DELLAs' levels, along with their growth-repressive function, leading to increased stem elongation and the promotion of flowering. Conversely, the loss of this interaction results in elevated DELLAs' levels, reinforcing their inhibitory actions, thereby yielding general outcomes of dominant dwarfism and delayed flowering[20−23].
Despite their lack of direct DNA binding capacity, DELLAs exert control over gene function by physically engaging with specific partner proteins. For example, in Arabidopsis stem tissue, TCP transcription factors interact with promoters and activate core cell-cycle genes to foster stem growth. In contrast, DELLAs directly interfere with the DNA binding activity and functions of these TCPs, ultimately suppressing stem growth and elongation[25]. In leaves, CONSTANS (CO), a key factor of flowering under long-day conditions, requires association with nuclear factor NF-YB2 to activate FLOWERING LOCUS T (FT). However, DELLAs disrupt this association, resulting in the delay of flowering[26]. In shoot apices, DELLAs have been shown to upregulate KRP3, a gene coding for a cell cycle inhibitor that curbs meristem size and inflorescence development[27]. Evidently, DELLAs enforce growth restraint across various tissues by reconfiguring distinct regulatory pathways.
A crucial inquiry revolves around the mechanism by which VvDELLA1 orchestrates and advances flowering, as well as the specific genes it targets in the grapevine. In Arabidopsis, the transition from vegetative meristem to inflorescence, followed by floral formation and development, is underpinned by an intricate network of genes. Remarkably, these genes demonstrate functional conservation across annuals, perennials, and even in woody plants like grapevine[28−38]. Plausibly, VvDELLA1 could potentially exert its influence on some of these orthologs or others that assume novel roles in shaping the developmental fate of anlagens in the grapevine. To elucidate this intricate regulation, we compared the transcriptome profiles of shoot tissues from four V. vinifera grape cultivars of Pixie, Dena, Gina and Tina containing the same gain-of-function mutated VvDELLA1 and the wild-type grape Pinot Meunier from which the gain-of-function VvDELLA1 (Vvgai1) was originally discovered[15]. Our investigation uncovered that the gain-of-function mutation in VvDELLA1 caused extensive mis-regulation of multiple genes, resulting in a collective transformation of the transcriptomic landscapes and the regulatory frameworks in the mutant vines. We identified at least two orthologs of flower regulators, VvAP1 and VvTFL1a, substantially downregulated due to the influence of the mutated VvDELLA1. This downregulation is closely linked to the significant conversion of vegetative anlagens into inflorescences. Our study examined potential interactions between identified gene candidates and VvDELLA1, as well as the plausible mechanisms underlying the regulation of flowering in grapevine.
-
Five V. vinifera cultivars were used in this study. Pinot Meunier is the cultivar from which the original L1 dwarf mutant was discovered. Pixie is a GA-insensitive dwarf mutant recovered via tissue regeneration from L1 layer cells of Pinot Meunier[39] and is phenotypically and genotypically identical to the L1 dwarf mutant as previously reported[15]. Pixie shows a monopodial growth, producing few, if any, lateral branches from the axillary buds (Fig. 1) and has a precocious flowering habit, producing inflorescences and bunches starting in the first year of its growth, and even in the younger/upper portion of its main branch (Fig. 1). Pixie is a hermaphrodite and was used as a pollen donor to cross with two grapevine rootstock cultivars of 187G and Freedom. Three dwarf mutant grape cultivars were selected from the F1 progenies. Two of them, named as Dena and Gina, were derived from the cross of Pixie with 187G and the third one, named as Tia, was from the cross of Pixie with Freedom (Cousins, unpublished). Dena, Gina, and Tia share the same dwarf and flowering phenotypes with their paternal parent Pixie.

Figure 1.
Pixie shoot trait characteristics. (a) Pixie shows a monopodial growth, producing few, if any, lateral branches from the axillary buds. (b) Pixie has a precocious flowering habit, producing inflorescences and bunches starting in the first year of its growth, and even in the younger/upper portion of its main branch.
All GA-insensitive dwarf mutant materials were grown in pots at the same time, and then maintained through a hydroponics system under greenhouse conditions for at least two years at the time of sampling. Nine shoots, each with 1−3 cm portion of the shoot tips that include young and unfolded leaves, were obtained from nine separately potted vines, and pooled in groups of three to make three biological replicates per genetic material. At the time of sampling, Pixie and its derived cultivars were in stages of reproductive growth, bearing inflorescences and fruit bunches along their singular main canes, but the wild-type control Pinot Meunier, the cultivar whose L1 meristem Pixie was developed from, remained in the juvenile phase. Shoot tissues of the control were likewise pooled to obtain three biological replicates. All collected/pooled tissues were taken fresh, flash frozen and stored at −80°C until further processing.
RNA-Seq library preparation and sequence reads processing
-
RNA extraction and RNA-Seq library preparation were performed as previously described[40]. RNA-Seq libraries were multiplexed for 100-bp paired-end sequencing using Illumina HiSeq 2000 at the Cornell University Biotechnology Resource Center, Ithaca, NY, USA. The RNA-seq library read qualities were assessed using FASTQC ver 0.11.9[41]. Removal of sequence adapters was done using Trimmomatic ver 0.32 (Illumina, San Diego, CA, USA). The artifact-free sequences were then individually aligned to the 12X ver 2 Vitis reference genome sequences[42] using STAR aligner ver 2.7.3[43] following a paired-end alignment protocol, with default parameters for paired-end data, which includes a standard mismatch allowance and considers canonical junctions for splice-aware alignment, as well as with an auto generation of gene counts needed for differential expression analyses. Gene expression quantification was performed using HTSeq ver 0.11.1[44] to count reads aligned to genes.
Expression analyses
-
Differential expression analyses were performed using edgeR package in R[45]. After an initial assessment, the significance threshold was set at a false discovery rate (FDR) of at least 0.05 (p < 0.001) to include all DEGs of hormone and flowering genes. Gene Ontology (GO) analysis was done using AgriGO ver 2[46], with a few modifications of the accompanying R script for visualization. Motif scan was facilitated using PlantTFDB[47]. All other visualizations such as plot and bar graph were done in R and MS excel.
Hormone and flowering genes curation
-
We evaluated the expression profiles of hormone and flowering genes. Hormone genes were retrieved from RIKEN Plant Hormone Research Network (
http://hormones.psc.riken.jp/ ). Flowering regulator genes and pathway were retrieved from flowering interactive database (FLOR-ID;www.phytosystems.ulg.ac.be/florid/ ). Grapevine homologues to the retrieved Arabidopsis genes were identified and verified from the annotation in the grape genome database using their BLAST feature[48]. -
To identify the genes that were subjected to differential regulation by the gain-of-function VvDELLA1 in the L1 dwarf mutants, we collected and conducted RNAseq profiling of shoot apices from the four mutant grape cultivars. These collected shoot apices encompass a range of structures, including the shoot apical meristem (SAM) responsible for continuous shoot growth, axially positioned primordial anlagen directed towards inflorescences or tendrils, and leaf primordia which develop into leaves. At the time of sampling, these genetic materials had been thriving in the hydroponic-fed pots for a minimum of two years, featuring both inflorescences and berries in the woody basal branches, as well as in the upper sections (Fig. 1). Simultaneously, we collected shoot apices from Pinot Meunier vines, the wild-type control (WT). WT remained in its juvenile stage at the time of sampling, indicated by their anlagens only processing into tendrils.
We conducted pairwise comparisons of Pixie, Dena, Gina, and Tia with WT, respectively. The numbers of differentially expressed genes (DEGs) at FDR ≤ 0.05 were 4,726 for Pixie, 2,744 for Dena, 2,731 for Gina and 3,210 for Tia. Our further analysis revealed that 723 DEGs were shared by at least three of the four mutants and 317 DEGs were shared by all four. Because the four grape mutants have very diverse genetic background, DEGs shown consistently across three or four of the mutants were most likely real. Among the 723 DEGs, 373 were up-regulated while 350 were down-regulated (Fig. 2; Table 1). As depicted in Fig. 3, the read abundance of these 723 DEGs spans a spectrum ranging from as low as 0.5 counts per million reads (CPM) to approximately 2,500 CPM. Remarkably, the fluctuations in expression change between the mutant and WT are considerably diverse, varying from a mere 1-fold to 550-fold difference. Notably, in line with many gene expression profiles, approximately 50% of the DEGs manifested low to moderate expression levels, usually within the range of 1−32 CPM. Within this segment, the most substantial changes in responses were observed, reaching up to a 500-fold alteration. It is intriguing to observe that the up-regulated DEGs tended to exhibit slightly more pronounced response changes, particularly those expressed at moderate to abundant levels which could soar up to 250 folds. In contrast, the changes for down-regulated genes sharing similar expression abundance fell between 30 to 60 folds. For the DEGs that were highly abundant (> 100 CPM), the alteration in their expression was relatively conservative, at around 2 folds. Genes involved in either hormone production and signal transduction, or flower formation, development, and flower regulation exhibited relatively low levels of expression or abundance. This observation underscores their vital functional significance as even minor alterations can trigger substantial physiological, metabolic, developmental, or phenotypic changes.
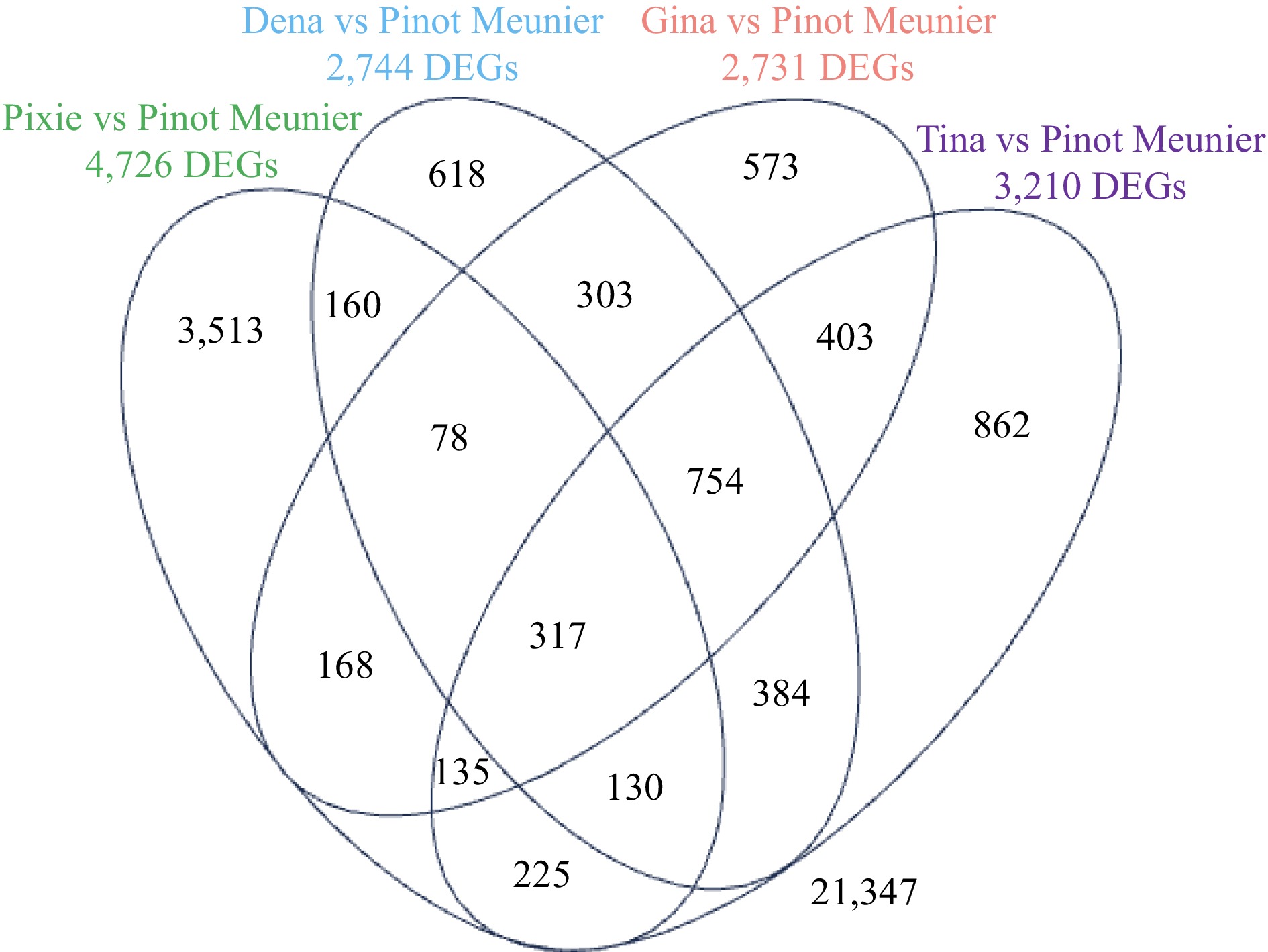
Figure 2.
A Venna diagram showing the overlaps of DEGs in 'Pixie', 'Dena', 'Gina' and 'Tia' each compared to the WT 'Pinot Meunier'. FDR ≤ 0.05.
Table 1. Numbers of DEGs that were of consistent responses in the shoots of four Pixie mutant background.
Expression change No. of DEGs1 Up-regulated 373 Down-regulated 350 Total 723 1 ≥ 1.5-fold change, FDR ≤ 0.05 in at least three of the four mutants. 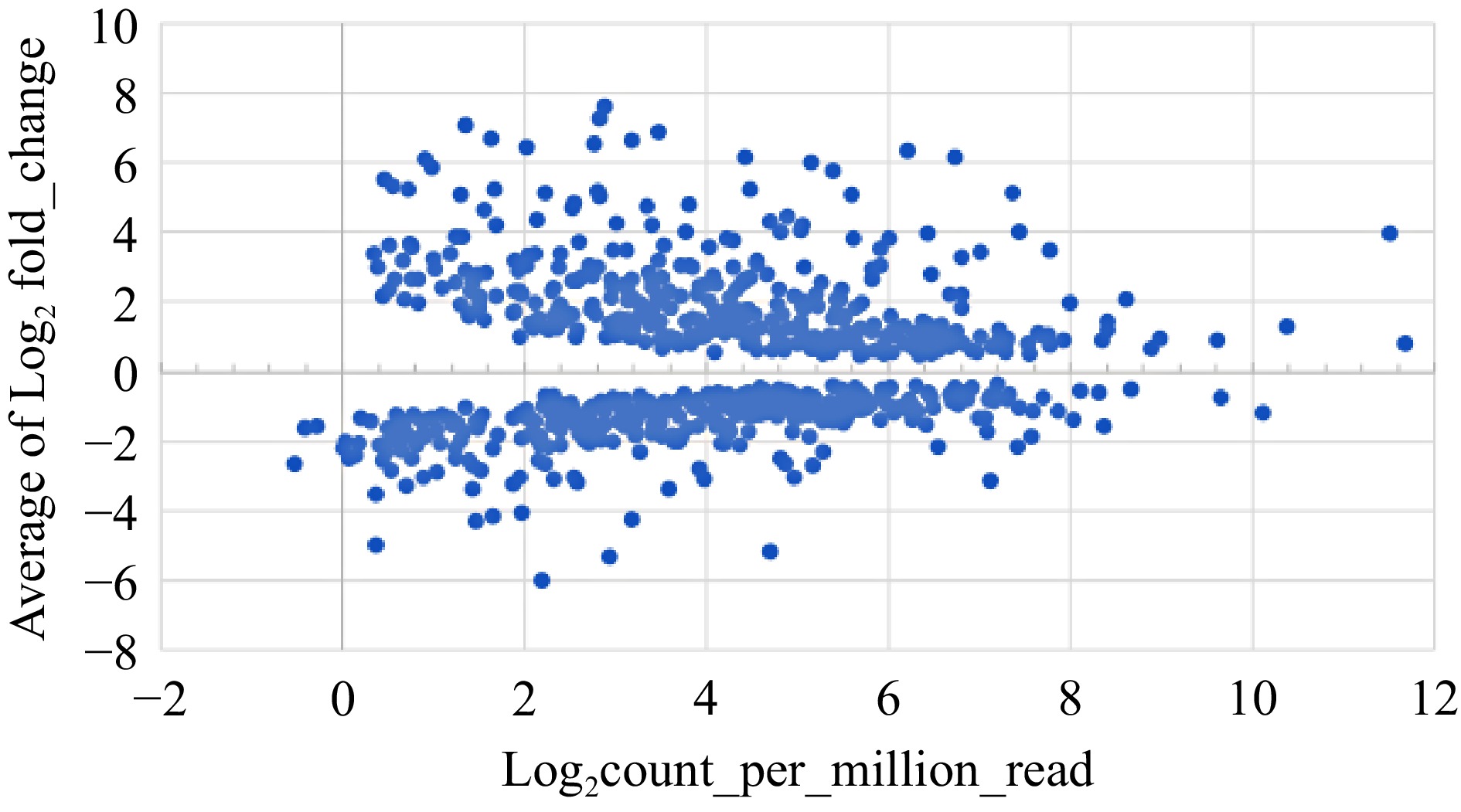
Figure 3.
Expression profiles of 723 DEGs that were consistently up-regulated or down-regulated in terms of average fold changes vs average expression levels.
Gene Ontology analysis of the shared DEGs
-
The GO enrichment analyses of the 373 up-regulated DEGs showed that most of these genes were linked to fundamental cellular activities, including responses to abiotic stimulus and regulation of cell size (Table 2 & Fig. 4). Notably, the up-regulated DEGs with substantial fold-changes played pivotal roles in biosynthesis processes, encompassing cellulose synthase, 3-ketoacyl-CoA synthase, trehalose-phosphatase/synthase, and Deoxyxylulose- 5-phosphate synthase, among others. Additionally, several significant gene families related to hormones and regulation, such as GRAS, MYB, AUX/IAA and ethylene, as well as various heat-shock proteins, 30S ribosomal, and response regulators of cytokinin, auxin response factors, and several homologs within the DOF, ERF and TCP transcription factor families were observed (data not shown). Conversely, among the 350 down-regulated DEGs, a considerable portion were also associated with fundamental cellular processes (Fig. 4). This category encompassed genes like protein kinases, along with an abundance of defense response-related genes such as lacasse and LRR-bearing genes. The down-regulated genes with notable fold changes displayed an enrichment of GO terms primarily associated with reproduction, transport, and localization processes (Table 2). These terms are exemplified by numerous carrier genes responsible for cation, potassium, auxin, and mate effluxes, as well as genes linked to floral development, including sucrose and peptide transporters. Noteworthy addition to this list were genes associated with meristem development: JAR1, a JA signaling gene; PRR7, a major gene in the temperature-sensitive circadian pathway; and KNAT1, a significant homeobox gene governing meristem cell fate determination. Evidently, these DEGs play a substantial role in influencing various pathways, culminating in a remodeling of the overall regulatory landscape, and ultimately giving rise to mutated phenotypes within the L1 mutants.
Table 2. Enriched GO terms among the 373 up-regulated and 350 down-regulated DEGs observed in the mutants.
GO terms Number UP-regulated DEGs Response to abiotic stimulus 27 Regulation of cell size 3 Down-regulated DEGs Anatomical structure development 16 Reproduction 11 Response to stimulus 36 Biological regulation 46 Transport 44 Establishment of localization 44 Localization 44 Cellular metabolic process 112 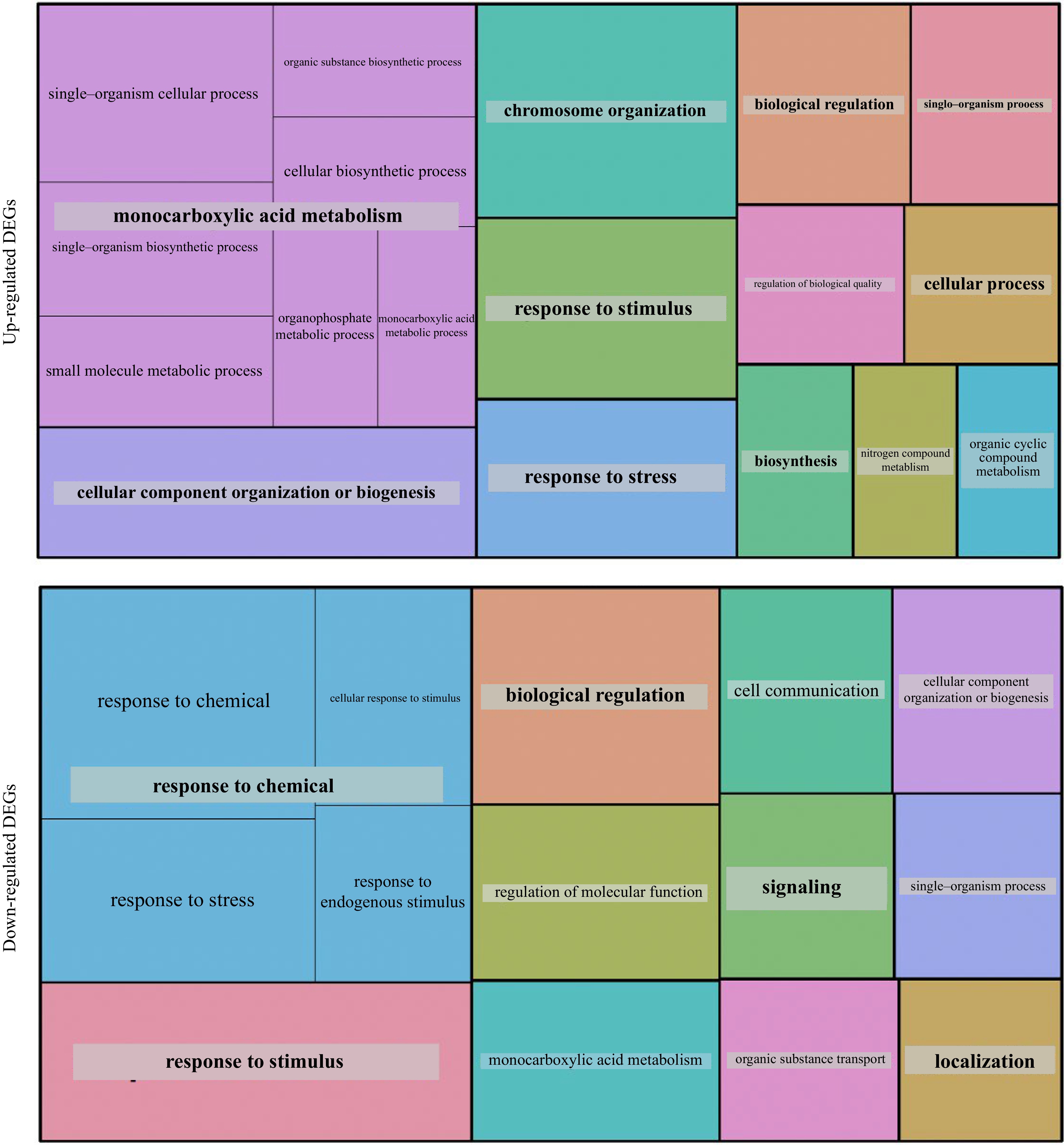
Figure 4.
GO analysis of the 373 up-regulated and 350 down-regulated DEGs observed in the mutants.
Expression of key GA pathway genes
-
Considering that GA represses the shift from vegetative growth to inflorescence in grapevine[13], investigating the expression patterns of genes linked to GA production and signal transduction in the L1 dwarf mutants becomes pertinent to discern whether these genes undergo feedback regulation by VvDELLA1. Among the GA signaling homologs, GID1a exhibited the most remarkable upregulation in shoot apices, with its transcript surpassing others by 100−400 folds (Fig. 5a). This suggests GID1a plays an important role in shoot apices. Nonetheless, it was not differentially expressed between WT and any of the four mutant cultivars. In terms of the two biosynthesis GA families, GA5 (GA20ox with about eight members) and GA4 (GA3ox with about three members), vital in the final stages, differential regulation was observed. Only GA20ox5 displayed consistent upregulation across all four mutants, albeit insignificantly. The remaining maintained consistent expression between WT and mutants (Fig. 5a). Similarly, VvDELLA2, one of the three DELLAs, exhibited upregulation in all mutants, yet insignificantly. Conversely, in the GA deactivation GA2ox gene family, both GA2ox1 genes and GA2ox8, particularly the latter, showed significant (FDR ≤ 0.05) downregulation across all mutants (Fig. 5b). This highlights the substantial impact of GA2ox8 downregulation, indicating that VvDELLA1 potentially targets and negatively regulates GA deactivation GA2ox family (five members), subsequently influencing GA accumulation.

Figure 5.
Notable expression of GA pathway genes in the L1 dwarf mutants. (a) Normalized transcript levels (counts per million, CPM) derived from aligned reads from three biological replicates for WT and dwarf mutants of genes involved in GA biosynthesis, including GA5 (GA20ox) with eight members and GA4 (GA3ox) with three members, and GA signal transduction, featuring GID and DELLA homologs. (b) Relative expression changes of GA deactivation genes, log2 fold change scale as calculated using edgeR at significance threshold set at FDR ≤ 0.05. The graph shows the average log2 fold change for three biological replicates between WT and the dwarf mutants. *: statistical significance at p ≤ 0.05.
VvAP1 and VvTFL1a were substantially down-regulated in the L1 dwarf mutants
-
While a myriad of genes spanning biochemical, physiological, metabolic, and regulatory pathways exhibited differential regulation in the mutants (Fig. 2; Tables 1, 2), the direct connections between these regulations and the heightened flowering phenotype in the L1 dwarf mutants remain elusive. This ambiguity arises likely due to the presence of multiple groups of meristem or primordia (e.g., SAM, anlagens, leaf primordia, and others) in the shoot tips utilized for this study, thereby complicating the analysis. Hence, we directed our attention toward genes pertinent to flowering regulation and hormone metabolism with a focus on signal transduction. Among a pool of 37 potential candidates examined, the majority of them maintained consistent expression levels between WT and the four mutants (Table 3). Only a handful of flower-positive regulators, including VvLFY, and the orthologs of FLOWERING LOCUS T (VvFT) and LATE MERISTEM IDENTITY1 (VvLMI1), were consistently upregulated in the mutants. Their transcript abundance increased by at least 2 folds on average compared to that in WT (Table 3), aligning well with their established positive flower-regulatory roles in Arabidopsis and other plants. Although these up-regulations did not reach statistical significance, even subtle changes in their expression could potentially exert significant influence on regulatory cascades and flower phenotypes. Likewise, the orthologs of Type-B ARABIDOPSIS RESPONSE REGULATOR1 (VvARR1), VvARR2b, and VvARR12, involved in cytokinin signal transduction, exhibited upregulation. This mirrors cytokinin's role in promoting the tendril-to-inflorescence transition[9−12]. As expected, the flower repressor VvTFL1a experienced a substantial and statistically significant downregulation of nearly 3 folds (p < 0.05), which implies its vulnerability to VvDELLA1 regulation. Interestingly, VvAP1, whose ortholog acts as both a floral integrator and a regulator of floral meristem identity, exhibited a significant downregulation of at least 3 folds in the L1 dwarf mutants (p < 0.05, Table 3). VvAP1 and VvTFL1a are the only two function-opposite floral regulators that were found significantly regulated in the L1 dwarf mutants, suggesting their functional importance (Table 3). Interestingly, they both were co-downregulated instead of being regulated in opposite directions. Additionally, the orthologs of another flowering integrator, SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (VvSOC1), showed moderate downregulation across all three copies: VvSOC1a, VvSOC1b and VvSOC1c. This suggests that the transformation of a positive flower regulator into a flowering-repressive factor may not be limited to VvAP1, further indicating the intricate regulatory complexity underpinning unique floral development in grapevine.
Table 3. Differential regulation of key shoot and flower regulator genes with qRT-PCR rating for selected genes.
Gene name Pathway Grapevine gene
ID ver 2Grapevine gene
ID ver 3Arabidopsis gene ID Average folds of changes between mutants and WT Average expression across all libraries VvFT Meristem identity VIT_00s0203g00080 − AT1G65480 2.71 0.26 ± 0.18 VvTFL1 VIT_06s0080g00290 − AT2G27550 (−2.81) * 1.38 ± 0.88 VvLFY VIT_08s0007g04200 − AT5G61850 2.22 18.18 ± 8.99 VvTFL1B FT gene family VIT_08s0007g03450 − AT5G03840 1.77 1.05 ± 0.54 VvTFL1C VIT_16s0100g00700 − 1.16 0.1 ± 0.18 VvMFT VIT_17s0000g02630 − AT1G18100 (−2.46) 0.08 ± 0.06 VvAP1 VIT_01s0011g00100 − AT1G69120 (−3.03) * 5.9 ± 4.45 VvCALa VIT_01s0010g03890 Vitvi01g01673 AT1G26310 (−1.47) 42.91 ± 18.08 VvCALb VIT_17s0000g04990 Vitvi17g00470 1.09 3.2 ± 1.23 VvFUL VIT_14s0083g01030 Vitvi14g01341 AT5G60910 (−1.56) 6.73 ± 3.53 VvLMI1 VIT_08s0007g04200 − AT5G03790 2.22 18.18 ± 8.99 VvWUS VIT_04s0023g03310 − AT2G17950 3.2 0.17 ± 0.19 VvFDa Vernalization VIT_00s0349g00050 − AT4G35900 2.45 2.89 ± 1.69 VvFDb VIT_18s0001g14890 Vitvi18g01165 (−1.13) 13.85 ± 1.95 VvFLC VIT_15s0048g01270 Vitvi15g00776 AT5G10140 1.14 3.16 ± 3.44 VvAGL24 Agamous / MADS MIKC gene family VIT_18s0001g07460 Vitvi18g00517 AT4G24540 1.34 38.16 ± 10.25 VvSVPa VIT_00s0313g00070 Vitvi07g01441 AT2G22540 (−1.01) 32.78 ± 8.94 VvSVPb VIT_03s0167g00070 − (−1.47) 28.24 ± 11 VvSVPc VIT_15s0107g00120 Vitvi15g00225 1.25 18.23 ± 4.9 VvSVPd VIT_18s0001g07460 Vitvi18g00517 1.34 38.16 ± 10.25 VvSOC1.1 VIT_15s0048g01250 − AT2G45660 (−1.38) 32.46 ± 9.71 VvSOC1.2 VIT_16s0022g02380 − AT2G45660 (−1.51) 8.7 ± 2.8 VvSOC1.3 VIT_15s0048g01240 − (−1.67) 74.18 ± 26.71 VvSPL3a SPL/ Ageing pathway VIT_04s0210g00170 Vitvi04g01556 AT2G33810 (−1.03) 60.11 ± 53.82 VvSPL3b VIT_10s0003g00050 Vitvi10g00481 (−1.14) 30.95 ± 7.85 VvSPL9 VIT_08s0007g06270 Vitvi08g01720 AT2G42200 1.01 136.75 ± 49.19 VvSPL4 VIT_12s0028g03350 Vitvi12g00280 (−1.11) 74.42 ± 31.95 VvSPL13 VIT_01s0010g03910 Vitvi01g01678 (−1.54) 48.51 ± 15.96 Vvlog5 Cytokinin VIT_06s0004g02680 − (−1.73) 22.17 ± 17.55 VvRR VIT_05s0077g01480 − 1.53 83.62 ± 20.74 VvARR12 VIT_11s0206g00060 − 1.26 21.69 ± 4.58 VvARR11 VIT_01s0010g02230 − 1.41 2.09 ± 1.71 VvARR2 VIT_02s0012g00570 − (−1.13) 152.22 ± 31.65 VvARR2b VIT_01s0011g05830 − 1.41 69.94 ± 13.7 VvARR12 VIT_04s0008g05900 − 1.37 23.28 ± 6.45 Vvyabby VIT_15s0048g00550 − 1.39 186.31 ± 79.49 * Significant at p ≤ 0.05. -
Grapevine has evolved a unique flower developmental programming, as characterized by specialized but versatile anlagen primordia that can give rise to tendrils in current shoots or inflorescences when emerging from latent buds. However, the fate of these anlagen, whether vegetative or reproductive, is susceptible to hormonal fluctuations influenced by vine age and seasonal changes[7,8,37]. This intricate regulation, while complex, is completely disrupted in the L1 dwarf mutants due to a gain-of-function mutation in VvDELLA1. This mutation leads to a pronounced de-repression of the transition from anlagens to inflorescence, a phenotype indicating the positive role of the mutated VvDELLA1 in promoting this transition[15]. Strikingly, this contradicts the repressive functions of its counterparts in annual plants[20−23]. The specific mechanism by which VvDELLA1 diverges and facilitates anlagen-to-inflorescence development in grapevine remains unclear. This study delves into transcriptome profiles of both WT and four L1 dwarf mutants to shed light on this phenomenon. Our analyses underscores VvDELLA1 as a central regulatory node, and its dominant mutation leads to the mis-regulation of several hundred genes. This comprehensive shift significantly alters the transcriptome and regulatory landscapes in the L1 dwarf mutants (Fig. 2; Tables 1 & 2).
Although various genes associated with biological processes, metabolism pathways, and regulatory circuits exhibit mis-regulation, their direct roles in governing anlagen-to-inflorescence development remain elusive. This is further complicated by the presence of multiple meristematic cells and tissues (e.g. SAMs, leaf primordia, and anlagens) in the shoot apices used for RNA-seq analysis in this study. Through a meticulous investigation focused on genes involved in the regulation of vegetative-to-floral meristem transition, floral meristem identity, and cytokinin signal transduction, we have identified VvTFL1a and VvAP1 as potential pivotal flower regulators. These findings suggest potential regulatory modules orchestrating the developmental trajectory of anlagens in grapevine.
The mis-regulation of flower-regulators and cytokinin signal genes in the L1 dwarf vines may be responsible for the remarkable augmentation of tendrils transitioning into inflorescences. At least seven genes, including VvLMI1, VvFT and VvLFY, which are known to positively regulate flowering in Arabidopsis, and VvARR1, VvARR2b and VvARR12, which are associated with cytokinin signaling, showed moderate upregulation in the L1 mutants (Table 3). These genes are functionally conserved across plant species, including grapevine[29], and cytokinin has been documented to induce the tendril-to-inflorescence transition in grapevine[9−11]. Thus, even a slight increase in expression of these genes could have a profound regulatory impact and lead to a noteworthy phenotypic response. In contrast, VvTFL1a, a known flower repressor, was one of two flower regulators that underwent significant downregulation in the L1 dwarf mutants (Table 3). This observation is consistent with the fact that TFL1 in apple has been demonstrated to repress the juvenile-to-adult transition and flowering[49], and ectopic expression of one of the three VvTFL1s in Arabidopsis results in delayed flowering[50]. Consequently, VvTFL1a likely assumes a similar repressive role in impeding the anlagen-to-inflorescence development or promoting the anlagen-to-tendril transition. This notion is further supported by its specific expression during the initial stages of tendril development[29].
An unexpected revelation emerges as VvAP1, akin to VvTFL1a, showed significant downregulation in the L1 dwarf mutants, a departure from the roles its counterparts play in annual plants. In a parallel manner, the three VvSOC1s genes were also moderately downregulated in these mutants. In model species such as Arabidopsis, both AP1 and SOC1 collaborate with FT and LFY to consolidate signals arising from photoperiod fluctuations, temperature changes, vernalization, and hormonal balance. This concerted effort aims to initiate the transition from vegetative meristem to inflorescence meristem transition and the subsequent formation of floral meristems. In this intricate process, FT is initially synthesized in leaves and subsequently migrates to the shoot apex, where it engages with FLOWERING LOCUS D (FD). This interaction subsequently triggers the upregulation of AP1, CAULIFLOWER (CAL), and FRUITFUL (FUL), collectively driving the transformation of the shoot apical meristem into the inflorescence meristem (IM)[28,30,33,34,38]. IM sustains its indeterminate inflorescence growth and eventually gives rise to flower meristem. The dynamics of these two developmental events are delicately managed through an intricate interplay involving TFL1, LFY and AP1. At the heart of IM, TFL1 is expressed and migrates from the core to the outer layer, where it exerts transcriptional repression over LFY and AP1. This repression is pivotal in preserving indeterminate growth[31,32]. Simultaneously, AP1 and LFY proteins generated in floral meristem suppress the transcription of TFL1, ensuring the progression of floral formation and development[35,36]. This reciprocal repression elegantly synchronizes the two spatiotemporally intertwined developmental events in Arabidopsis. However, co-downregulation of VvAP1 with VvTFL1a in the L1 dwarf mutant apparently seems to challenge this reciprocal repression relationship and the flower-positive regulatory role attributed to AP1 in Arabidopsis.
The divergent regulation of VvAP1 and VvLFY in the L1 dwarf mutants point to distinct functional roles, where the former fosters the transition of anlagen to tendrils, while the latter enhances anlagen's development into inflorescences. This interpretation is further supported by their specific expression patterns within tissues[51]. In grapevine, tendrils and inflorescences represent vegetative and reproductive growth states, respectively, yet they are homologous structures/organs as indicated by their reciprocal homeotic transformations, shared meristematic origin, co-existence in intermediate structure, and the activation of floral meristem regulator genes VvFT, VvLFY, VvAP1, and VvFUL [7,9,51−53]. This evidence strongly suggests that tendril is, in essence, a modified inflorescence lacking floral meristems. Consequently, the primary distinction between these tissues lies in their capacity to generate floral meristems, a process primarily regulated by AP1 and LFY in Arabidopsis[35,36]. As anticipated, the upregulation or downregulation of VvAP1 or VvLFY, or both, exerts direct control over the formation of floral meristems and developmental fate of anlagens.
Supporting this prediction, both VvLFY and VvAP1 are actively transcribed in inflorescences across five Vitaceae species examined[51]. However, VvAP1's expression is only limited to tendrils[51]. These unforeseen tissue-specific expression patterns challenge the notion that both VvAP1 and VvLFY play, like their Arabidopsis counterparts, similar roles in grapevine. Instead, they suggest that these two genes have functionally diverged and play contrasting roles in regulating floral meristems in tendrils and inflorescences: VvAP1 likely promotes and maintains tendrils by inhibiting floral meristem formation, while VvLFY enhances inflorescence development by promoting the floral meristem. However, the relative balance or abundance of these factors ultimately governs whether tendrils or inflorescences are formed. This is reinforced by the observation that the extensive conversion of the tendril-bound anlagens into inflorescences in the L1 dwarf mutant is correlated with the downregulation of VvAP1 (Table 3). Consequently, VvAP1 seems to have adopted a novel but negative regulatory role in relation to floral meristems. Conceivably, genes coregulated with VvAP1 (VvSOC1a, VvSOC1b and VvSOC1c) or with VvLFY (VvLMI1, VvFT) may functionally interact with VvAP1 and VvLFY, respectively. Taken together, our findings, combined with previously documented tissue-specific expression data, uncover a novel role for VvAP1 and elucidate a potential mechanism underlying the regulation of floral meristems, tendrils, and inflorescence development in grapevine, as illustrated in Fig. 6.
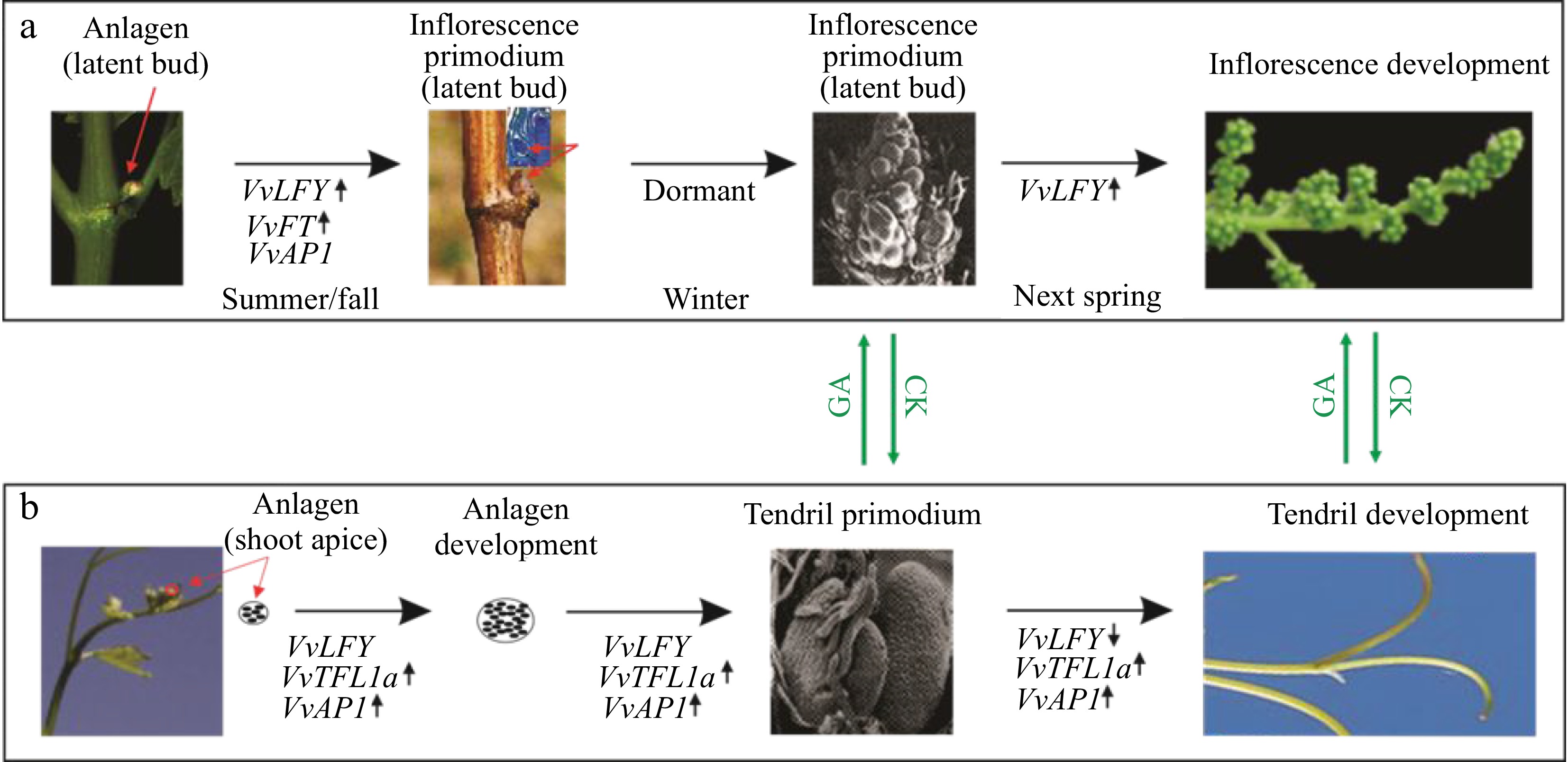
Figure 6.
A schematic illustration of the possible opposing roles of VvAP1 and VvLFY in regulating the developmental fate of anlagens in grapevine. (a) At the anlagen-to-inflorescence transition, the lateral primordial meristem gives rise to the latent bud in the leaf axil, which contains the shoot apical meristem (SAM), leaf primordium, and uncommitted anlagen. Expression or upregulation of VvFT, VvAP1, and VvLFY is observed in this stage. The anlagen is then differentiated into an inflorescence primordium in late seasons and eventually develops into a mature inflorescence in the following spring. (b) The anlagen-to- tendril developmental course is associated with increased expression of VvAP1 and VvTFL1a. The shoot apex produces lateral anlagens with the expression or activation of VvAP1, VvLFY, and VvTFL1a. These anlagens are then destined to become tendril primordia, followed by the development of tendrils in current growing shoots. The ratio between VvLFY and VvAP1 likely controls the developmental route of the anlagens to either inflorescences or tendrils. It is noted that anlagens at any stage or derived primordia are sensitive to hormone regulation, with cytokine (CK) promoting inflorescences and GA favoring tendrils. The red arrows indicate the anlagen in either shoot apex or latent bud, or inflorescence primordium in the latent bud. Up or down arrows indicate the upregulation and downregulation, respectively.
The noteworthy observation that VvTFL1a and VvAP1 among flower regulator genes were significantly modulated in the L1 dwarf mutant implies their central roles in grapevine regulation. However, their simultaneous downregulation challenges the conventional view, indicating that they actually exert negative control over flowering, which appears to contradict their reciprocal repression observed in their Arabidopsis orthologs[35,36]. Plausibly, internal physiological cues such as vine age, growth state, and hormone equilibrium, or external signals including temperature, photoperiod, and chilling, could directly or indirectly target VvTFL1a, VvAP1, or both, possibly through the intermediary of the VvDELLA1 factor. This orchestration determines when and where anlagen evolve into tendrils or inflorescences. Given that GA-mediated degradation of the VvDELLA1 factor results in the de-repression of both VvTFL1 and VvAP1, it becomes evident why GAs repress flowering in grapevine. The fact that GA2ox1 and GA2ox8, constituents of the GA2ox family responsible for GA deactivation, experienced substantial downregulation in the L1 mutants (Fig. 5b), suggests that VvDELLA1 is a negative regulator of these two genes. This regulation likely leads to an accumulation of more GAs, which, in turn, triggers the degradation of DELLA1. This intricate regulatory interplay among GA2ox1 and GA2ox8, GAs and the VvDELLA1 could perpetually maintains VvTFL1a and VvAP1 in repression, thus steering the developmental trajectory of anlagen toward tendril formation in growing shoots and juvenile vines.
-
The authors confirm contribution to the paper as follows: study conception and supervision: Zhong GY; data collection: Arro J; analysis and interpretation of results: Arro J, Liu Z, Zhong GY; RNAseq library preparation: Yang Y; development of the genetic materials: Cousins P; manuscript revision and discussion: Song G; draft manuscript preparation: Arro J, Liu Z; finalization of the manuscript: Liu Z, Zhong GY. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article.
We wish to thank Debra Johnston of USDA-ARS Grape Genetics Research Unit for providing her assistance in maintaining the L1 DELLA mutant vines in hydroponic tanks in the greenhouse. Jie Arro is a participant of the ORISE-ORAU Education and Training Program.
-
The authors declare that they have no conflict of interest. Guo-qing Song is the Editorial Board member of Fruit Research who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and the research groups.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Arro J, Yang Y, Song GQ, Cousins P, Liu Z, et al. 2024. Transcriptome analysis unveils a potential novel role of VvAP1 in regulating the developmental fate of primordia in grapevine. Fruit Research 4: e011 doi: 10.48130/frures-0024-0004 

Transcriptome analysis unveils a potential novel role of VvAP1 in regulating the developmental fate of primordia in grapevine
- Received: 27 September 2023
- Revised: 02 December 2023
- Accepted: 26 December 2023
- Published online: 04 March 2024
Abstract: The grapevine shoot meristem contains undifferentiated primordia known as anlagen, which can develop into either inflorescences or tendrils depending on vine age, growth status, hormone balance, and other factors. Interestingly, a gain-of-function mutation in the DELLA domain of VvDELLA1 in the dwarf mutant grape, Vitis vinifera L. cv. Pixie, virtually disrupts the normal developmental course of anlagen and reroutes tendril-bounded anlagen toward inflorescence development even at the juvenile stage. To understand the underlying mechanism(s), we compared the transcriptome profiles of V. vinifera cv. Pinot Meunier (from which Pixie was derived), Pixie, and three other V. vinifera grape cultivars (Dena, Gina, and Tia) which were derived from crosses involving Pixie and carry the same DELLA mutation. Our findings revealed significant mis-regulation of hundreds of genes, profoundly reshaping both transcriptome landscapes and regulatory pathways in the mutant grapes. Interestingly, VvAP1, a central positive flower regulator in annuals, was unexpectedly co-downregulated with VvTFL1a, a flowering repressor. We also found several other key flower regulators which were either upregulated (e.g., VvFT, VvLFY) or downregulated (e.g., VvSOC1s) in all mutant grapes, although the overall effect was moderate. These findings, along with the previous identification of tendril-specific expression of VvAP1 and inflorescence-specific expression of VvLFY, support that VvAP1 promotes anlagens to develop tendrils, whereas VvLFY favors inflorescences formation. The balance between these factors, particularly the abundance of VvAP1 transcripts, ultimately dictates whether anlagens develop into tendrils or inflorescences.
-
Key words:
- Vitis /
- Grapevine /
- Anlagen /
- Primordia /
- VvAP1 /
- Inflorescences /
- Tendrils /
- Transcriptomes