








-
Organic acid, serving as the primary contributor of fruit acidity measured by titrable acidity and pH, influences synergistically taste and flavor quality with soluble sugars in many fleshy fruits[1,2]. Flavor quality is believed to be a vital driver of consumer preference and its formation and regulation mechanism is one of the strategic requirements for the quality optimization and variety genetic improvement of fruit[3]. Knowing the accumulation mechanism of organic acid in fruit cells is necessary to improve fruit quality.
Fruit acidity is due to the accumulation of organic acids such as malic acid, citric acid, and tartaric acid in vacuoles. The composition and content of organic acids in different fruits are significantly different, while malate is the main organic acid presented in most mature fruits[1,4]. Malate content in fleshy fruits is affected gravely by acid metabolism and transport[2,5]. In the cytoplasm, the intermediate product phosphoenolpyruvate (PEP) from glycolysis is catalyzed sequentially to malic acid by PEPC (PEP carboxylase) and NAD-cyt-MDH (NAD–dependent malate dehydrogenase)[1,6−8]. The function of NAD-cyt-MDH on increasing fruit acidity has been defined[8,9], which is believed to be the major pathway for malic acid synthesis[6,10]. Part of the organic acids accumulated in the cytoplasm are consumed as substances and energy to maintain the normal physiological activities of the cells, while the rest are transported and stored in the vacuole[2,11,12]. However, whether it can accumulate in large quantities in vacuoles is highly dependent on specific transmembrane transporters[4,12,13]. Currently, major fruit malate-regulated transporters include aluminum-activated malate transporter ALMT4 and ALMT9/Ma1[14−17] and tonoplast dicarboxylate transporter tDT[4,17,18]. Additionally, H+ pumped into the cell via proton pump protein on the vacuole membrane combine with malate2− to form protonated malate for storage in the vacuole[1,12,13,19], providing continuous driving force for malate2− to enter the cell.
The genetic mechanism of fruit acidity, a quantitative trait regulated by multiple genes, is relatively complex[2,20]. The function and regulatory mechanism analysis of malate metabolism and transport genes during fruit acidity accumulation is of great significance for scientific regulation of fruit quality. With the development of transcriptome, proteomics, and the application of gene fine-mapping technology in forward genetics, researchers have identified a series of metabolic and transport genes related to malate accumulation in apple[20,21], citrus[22,23], peach[24], watermelon[25], jujube[15], and loquat[26], and verified their regulatory functions of fruit acidity. This review focuses on the recent advances in metabolism, transport genes and upstream regulators related to malate accumulation in horticultural fruits and the future research direction of improving fruit quality by molecular biological methods are prospected.
-
The accumulation of malate in fruit cells is a complex phenomenon involving many metabolic pathways. The first step of malate synthesis in the cytoplasm is to fix CO2 to the carbon skeleton from hexose catabolism. The photocontracted products are delivered to the sink fruit over long distance transport of phloem[27], and the glucose produced by its decomposition is converted into the key substrate phosphoenolpyruvate (PEP) of malate metabolism in glycolysis. Then, PEP is converted to malate catalyzed by phosphoenolpyruvate carboxylase (PEPC) and subsequent cytoplasmic NAD-dependent malate dehydrogenase (NAD-cyMDH), and the reverse reaction is mediated by phosphoenolpyruvate carboxykinase (PEPCK)[1]. In addition, malate accumulated in the cytoplasm can be degraded to pyruvate via the NADP-dependent malic enzyme (NADP-cyME), and the remaining malate is transported and stored in the vacuole[28]. Therefore, the major enzymes involved in malate metabolism include PEPC, NAD-cyMDH, PEPCK, and NADP-cyME (Fig. 1).
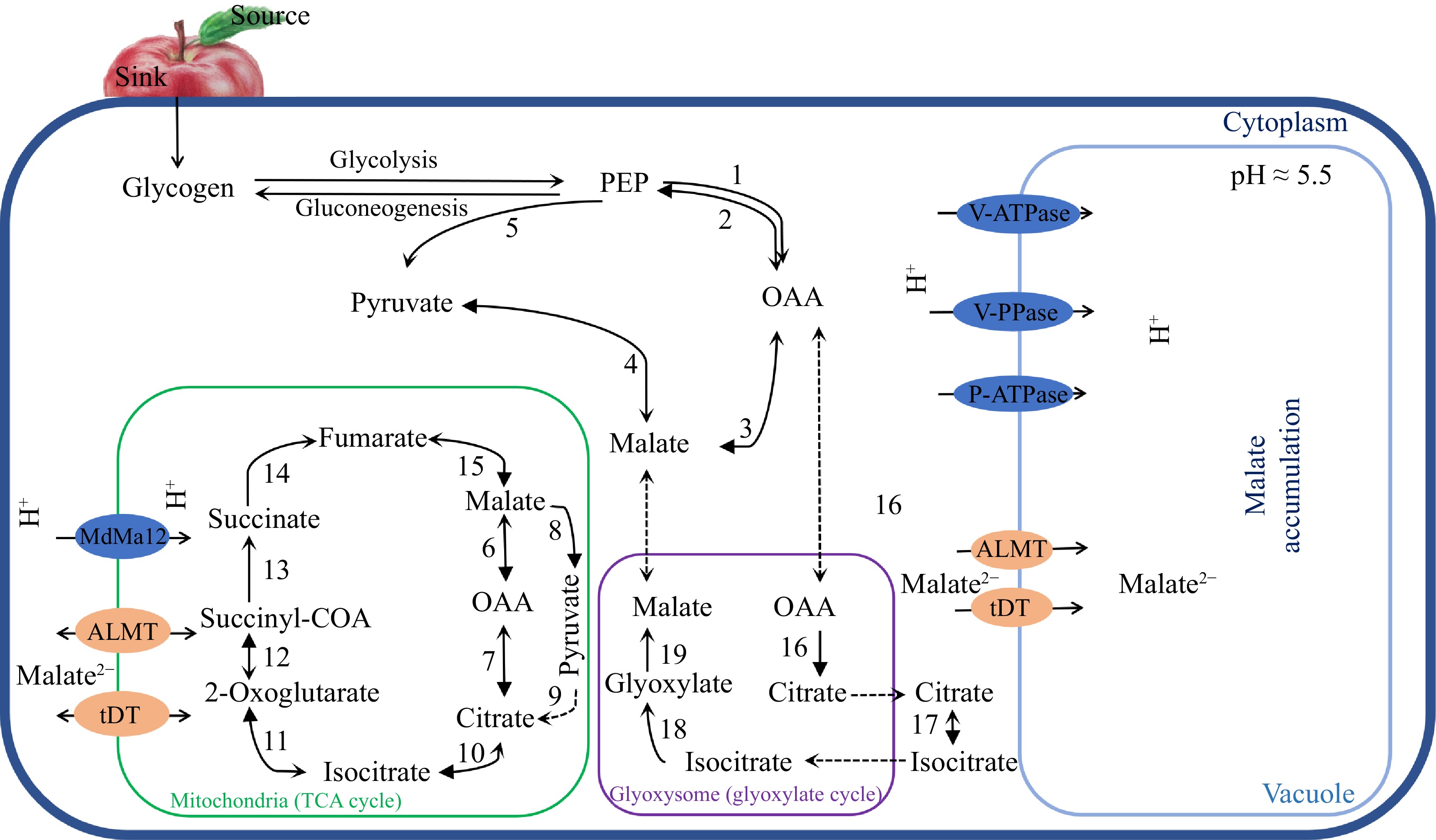
Figure 1.
A simplified model for malate metabolism, transport and accumulation in fruit. Major enzymes involved in malate metabolism in fruit: 1, phosphoenolpyruvate carboxylase (PEPC); 2, phosphoenolpyruvate carboxykinase (PEPCK); 3, NAD-cyMDH (NAD-cytoplasmic malate dehydrogenase); 4, NADP-cytoplasmic malate enzyme (NADP-cyME); 5, pyruvate kinase (PK); 6, NAD-mitochondrial malate dehydrogenase (NAD-mtMDH); 7, mitochondrial citrate synthase (mtCS); 8, NAD-mitochondrial malate enzyme (NAD-mtME); 9, pyruvate dehydrogenase (PDH); 10, mitochondrial aconitase (mtACO); 11, isocitrate dehydrogenase (ICDH); 12, α-oxoglutarate dehydrogenase (α-OGDH); 13, succinyl-coa synthase (SCS); 14, succinate dehydrogenase (SuDH); 15, fumarase; 16, cytoplasmic citrate synthase (cyCS); 17, glyoxylate aconitase (glyACO); 18, isocitrate lyase (ICL); 19, malate synthase (MS).
PEPC and NAD-cyMDH are involved in the synthesis and accumulation of malate in fruit. There was a positive correlation between the malate content and the expression levels of PEPC in apple and grape fruit during the early developmental stage[29−31]. Similarly, NAD-cyMDH has also been shown to promote malate synthesis in young grape fruits[10,32]. In apple, NAD-cyMDH1 could improve malate accumulation in apple calli and fruit, as well as increase the salt tolerance of apple plants[9,28,33,34]. Recent studies have also demonstrated the important function of another apple gene NAD-cyMDH5 in enhancing malate accumulation in fruit[35].
Physiological studies have revealed that the malate content of fleshy fruits such as apple, peach, and grape decreases significantly in the later stages of development[10,36], which is closely related to the malate decomposition genes NADP-cyME and PEPCK. Sweetman et al. pointed out that NADP-ME had low activity in the early stage of grape fruit development and was involved in the malate generating by fixing CO2[10]. As fruits gradually matured, the activity of NADP-ME increased significantly and began to catalyze the degradation of malate. Additionally, NADP-cyME is thought to be involved in the reduction of malate content during the ripening of loquat, apple, and grape fruit[6,37,38].
In the later stage of fruit development, soluble sugars accumulated in large quantities, sugar catabolic metabolism slowed down, and vacuolar malate was released into the cytoplasm as a carbon source to participate in energy metabolism and compound biosynthesis through gluconeogenesis[10]. The first step of gluconeogenesis is the production of PEP catalyzed by PEPCK or phosphopyruvate dikinase (PPDK)[39,40]. Due to the bare expression of PPDK in fruits, PEPCK is believed to be more essential in the gluconeogenesis of fruits, which has been validated in grape skins and ripe peach, blueberry, redcurrant, and raspberry fruits[41−43]. Compared with high-acid varieties, PEPCK activity in low-acid apples increased threefold, and14C-labeled malate consumption was higher, suggesting that gluconeogenesis might increase malate consumption[36]. Recent studies have suggested that the large accumulation of malate in the cytoplasm altered soluble sugar content in apple fruit, which might be caused by the up-regulating expression of MdPEPCK in gluconeogenesis[8]. In conclusion, PEPCK plays a key role in the regulation of malate accumulation in fruit.
Malate metabolism in mitochondria and glyoxysome
-
Malate in the TCA cycle can be oxidized via two competing metabolic pathways. It is converted reversibly to OAA by NAD-mtMDH[10], or converted to pyruvate by NAD-mtME[44], which ultimately results in citrate synthesis and an altered ratio of malate and citrate. Omics analysis showed that NAD-mtMDH in mitochondria mainly catalyzed malate degradation during fruit ripening[10,32]. While NAD-mtMDH can also catalyze the malate synthesis when the conditions exist for the reversible reaction of the TCA[7,45]. In addition, the regulation of NAD-mtMDH varies among species. NAD-mtMDH appears to be regulated by the gene expression in loquat[46], while regulated at the post-transcriptional level in strawberry[47].
Malate synthase glyMS located in the glyoxysome is involved in malate synthesis in the glyoxylic acid cycle[48] (Fig. 1). Studies have shown that the glyoxylic acid cycle is activated during the ripening of banana fruit after harvest, providing substrates for gluconeogenesis[49]. Pua et al.[50] detected the expression of glyMS only in banana fruit tissues, its expression was up-regulated during the whole ripening stage, whose change was similar to the trend of malate accumulation. However, ICL proteins required for glyoxylate (MS catalytic substrates) synthesis in the glyoxylate cycle was not detected at any stage of development in raspberry, blueberry, strawberry or redcurrant fruits[41], suggesting that the malate accumulation involved in the glyoxylate cycle during fruit ripening may be species-specific.
-
Malate synthesized in the cytoplasm and mitochondria will be transported and stored in vacuoles[7,35,51]. The passage of malate into or out of the vacuole requires specific anion channels or transporters located on the vacuole membrane as carriers, which can specifically recognize and transport malate[52,53]. In the cytoplasm with near-neutral pH, most malic acid exists in anion forms, while they will combine with cations (such as K+, Na+, Ca2+), forming salt in the acidic vacuole, which maintains an electrochemical gradient between the vacuole, allowing subsequent organic acids to enter the vacuole[1,53]. The main transporters associated with malate transmembrane delivery identified in horticultural crops include malate channel protein ALMT (aluminum-activated malate transporter), malate transporter tDT (tonoplast dicarboxylate transporter), and proton pump.
Aluminum-activated malate transporter
-
ALMT encodes aluminum-activated channel protein with typical transmembrane domains that transport malate2− in different organelles[54,55]. ALMT1 was initially identified in wheat root tip tissue and its malate transport properties were confirmed by the heterologous expression of xenopus cells[56]. Subsequently, researchers identified successively a series of ALMT proteins in the model plant Arabidopsis[57,58], major crops[59,60], vegetables[61,62] and fruit trees[63,64].
The ALMT family is mainly divided into four subfamilies (ALMT I−IV) and the protein functions of each subfamily are various, mainly including aluminum tolerance, symbiotic nitrogen fixation, fertilization, ion transport, stomatal regulation, and fruit flavor[65−67]. Therein, most of the ALMTs of the subfamily I and II are mainly located on the vacuole membrane and participate in malate transportation in plant cells. For example, the ALMT subfamily I genes AtALMT1, BnALMT1/2, HvALMT1 and ZmALMT2 from respectively Arabidopsis (Arabidopsis thaliana), rape (Brassica napus), barley (Hordeum vulgare L.), and maize (Zea mays L.) are primarily expressed in root and function on the regulation of malate secretion[68−71]. The interference of LaALMT1 (Lupinus albus) led to a decrease in malate concentration in xylem sap[72], and the ALMT subfamily II genes SlALMT5 significantly increased malate and citrate contents in tomato seeds[73,74]. SlALMT11 located in leave guard cells transports malate to mediate stomatal closure in tomato[75]. AtALMT6 and AtALMT9 mediated the transcellular transport of malate or fumaric acid[57,75,71]. Moreover, it was found that AtALMT6 and AtALMT9 have strong inward rectification via membrane electrophysiology techniques, and malate transport only occurred under the condition of positive potential inside the vacuolar membrane[75,76]. As the vacuole pH decreases, the pathway by which AtALMT6 and AtALMT9 transport malate might be closed, which may be an active protection against excessive acidification of the vacuole.
ALMT family genes are closely related to the regulation of fruit acid quality. Therefore, the function of ALMT9 on fruit vacuole acidification in various horticultural crops is the most widely studied. The Ma1 located on chromosome 16 in apple encodes an ALMT9 homologous gene, which is considered to be the main gene controlling the acidity of apple fruit[16,63,77]. The single nucleotide polymorphism (SNP) of Ma1 at 1,455 bases causes the premature termination of its protein translation, thereby losing the capability to transport malate into the vacuole, which is closely related to the decrease of fruit acidity[77]. In general, the acidity of mature apple fruit of genotype ma1/ma1 was significantly lower than that of genotype Ma1/Ma1 or Ma1/ma1[55,78]. While vacuolar membrane-localized VvALMT9 and SlALMT9 also are the homologs of AtALMT9 in grapes and tomatoes, respectively. VvALMT9 mediates the transport of malic acid and tartaric acid in grape fruit[79]. SlALMT9, located on chromosome 6 of tomato, is a major gene leading to the variation of malate content in tomato fruit, and the deletion of 3-bp in the promoter region of SlALMT9 destroys the W-box binding site and prevents the binding of its upstream transcriptional suppressor SlWRKY42, resulting in a high accumulation of malate in fruit[62]. Recent studies have also indicated that PpALMT9 mediated the accumulation of malate in pear fruit under salt stress[14]. Additionally, overexpression of ZjALMT4 and AcALMT1 significantly promoted the increase of organic acid content in sour jujube (Ziziphus jujuba Mill.) and in kiwifruit (Actinidia spp.) fruit[15,80]. Allogeneic expression of the ALMT family gene Pbr020270.1 of pear (Pyrus bretschneideri) could increase malate accumulation in tomato fruit[64]. While, a CitALMT gene of citrus (Citrus reticulata B.) negatively affects citrate accumulation in citrus fruit[81]. These results indicate that the aluminum-activated channel protein ALMT regulates malate accumulation in fruit vacuoles.
Tonoplast dicarboxylate transporter
-
Tonoplast dicarboxylate transporters (tDT) are the first class of transmembrane transporters with malate transport properties discovered in plants[82]. In contrast to malate ion channel proteins, tDT has little rectification and plays an essential role in maintaining intracellular pH homeostasis[83]. Researchers first identified and demonstrated the acid transport function of AttDT in Arabidopsis thaliana. Overexpression of AttDT significantly increased malate content and decreased citrate accumulation in Arabidopsis leaves[84,85]. Further studies confirm that AttDT can also transport fumaric acid and succinic acid, and participate in the regulation of cytoplasmic pH homeostasis[83,85,86].
So far, AttDT homologous genes have been isolated from various fruit such as apple (Malus domestica), tomato (Solanum lycopersicum), grape (Vitis vinifera), and citrus (Citrus sinensis). During citrus maturation, the AttDT homologous gene, CsCit1, encodes a vacuolar citrate3−/H+ symporter that mediates the effluence of H+ and CitH2− in vacuole to maintain vacuolar acidic pH and citrate balance[87]. Lin et al.[88] pointed out that CitDIC, a dicarboxylate transporter, and CitCHX, a cation/H+ exchange protein, were involved in the degradation of citrate during fruit development and the reduction of citrate in fruit after harvest triggered by hot air. The content of malate in SltDT overexpressed tomato fruit was significantly increased, while the citrate accumulation was inhibited[89]. Similarly, MdtDT negatively regulates the citrate content[90] and positively participates in the accumulation of malate in cultivated apple fruit[11,91,92]. The AttDT homologous gene in grape fruit is actively transporting tartaric acid into the vacuole[93]. In addition, mitochondrial dicarboxylate transporters VvDTC2 and VvDTC3 identified in grape are likely responsible for malate transport to mitochondria in grape fruit[94]. These studies indicate that tDT positively regulates malate and negatively regulates citrate accumulation in most fruit.
Proton pump gene family
-
The transmembrane transport of malate is also affected by the vacuole pH and the electrochemical gradient (∆ψ) inside and outside the vacuole[1], while the activity and function of the proton pump greatly affects the vacuole pH and ∆ψ. Proton pump is a kind of membrane-integrated glycoprotein that can transport H+ across membranes against the concentration gradient, which mostly exists in the vacuolar membrane and plasma membrane, mainly including V-ATPase/H+-ATPase, V-PPase/H+-PPiase and P-ATPase[1,12,13,19], they pump H+ into the vacuole by hydrolyzing ATP or pyrophosphate, reducing the vacuole pH while increasing the ∆ψ on both sides of the membrane, thereby providing power for the transport of organic acids.
V-ATPase and V-PPase are widely present in a variety of horticultural crops and are involved in secondary metabolite transport, vacuole acidification, ion homeostasis, and stress tolerance[95−98]. Although V-ATPase and V-PPase are both effective in acidifying vacuoles, their activity varies in different plants and at different developmental stages of the same plant. V-ATPase is the main proton pump in the vacuoles of most horticultural plants, but V-PPase is more active than V-ATPase in some C4 plants. A large amount of highly active V-PPase is enriched in the early development stage of young tissue, hydrolyzing and removing pyrophosphate to inhibit the polymerization reactions such as RNA and starch synthesis. While, the synthesis of pyrophosphate in mature tissues is reduced and cell respiration continues to provide ATP, so V-ATPase activity dominates[4,99]. The activity analysis of the proton pump during the development of pear (Pyrus pyrifolia) fruit supported the above conclusion. V-PPase activity was highest in young fruit and decreased with the maturation of pear fruit, whereas V-ATPase activity was highest in mature fruit[100]. However, V-PPase is also the main vacuolar proton pump in grape berries whose vacuoles are strongly acidic (pH < 3)[93].
The expression patterns of V-ATPase and V-PPase were similar in high-acid and low-acid loquat varieties, but their expression levels were higher in low-acid varieties[26]. Etienne et al.[101] found that the expression of V-ATPase and V-PPase in the fruit of different peach varieties were positively correlated with organic acid accumulation, indicating that V-ATPase and V-PPase were involved in the regulation of organic acid accumulation in fruit. Overexpression of V-ATPase proton pump MdVHP1 in apple calli increased the accumulation of malate and soluble sugar in vacuoles[91]. Further studies confirmed the active function of MdVHA-A3, MdVHA-D2, MdVHA-B1, MdVHA-B2, MdVHA-E2, CitAHA10, and CitVHA-c4 in fruit vacuole acidification[92,102−104]. Moreover, a mitochondria-targeted PPase gene, Ma12, was identified in apple and its overexpression increased malate accumulation in apple calli and tomato fruit by up-regulating the expression of mitochondrial malate dehydrogenase mMDH12[7]. These results fully demonstrated the indispensable functions of V-ATPase and V-PPase in fruit vacuole acidification.
P-ATPase is a new class of proton pump genes with proton transport and vacuolar acidification properties, which are divided into five subfamilies (P1−P5), among which the P3 subfamily ATPase is involved in the transport of organic acids. P3A-ATPase proton pump PhPH5, which is localized in the vacuolar membrane in petunias interacts with the P3B-ATPase proton pump PhPH1 to form a complex, which affects petal color by acidifying the vacuole[105,106]. Interestingly, the vast majority of P3A subfamily members are located in the plasma membrane, while only PhPH5 belongs to the vacuole membrane localization gene, exhibiting a strong ability to transport protons across membranes. It is unclear how PhPH5 acquired this unique cellular localization during evolution. The pH1−pH5 complex can reduce the stoichiometric value of H+/ATP from 1.0 to 0.5 for super acidifying vacuoles[107].
The function of PH1−PH5 complex highly acidifying vacuoles exists only in a few angiosperms, and PH1 homologs are lacking in most plants[106]. How the independent loss of PH1 homologs occurs in multiple plants are unclear. Studies have shown that PH1 and PH5 can be expressed ectopically in plants where certain tissues do not express them, resulting in a decrease of vacuole pH in the corresponding tissues[108]. At present, the researchers have begun to explore whether the pH1−pH5 complex has the function of acidifying fruit vacuoles. The hyper-acidification of citrus fruit is regulated by the P-ATPase complex CitPH1-CitPH5[109]. Similarly, the PhPH5 homolog Ma10 and the PhPH1 homolog Ma13 in apple were reported to regulate the malate accumulation in apple calli and tomato fruit[12,110], however, it is not clear whether there is an interactive relationship between the Ma10 and Ma13. The interference of VvWRKY26 and VvMYB5 in grape leaves significantly decreased the transcriptional expression of VvPH5 and VvPH1, causing increased vacuole pH[111]. In the same year, researchers identified and proved that the expression level of CsPH8, a homologous gene of PhpH5, was highly consistent with the changing trend of citrate content in various citrus fruits at different developmental stages, and overexpression of CsPH8 significantly increased the citrate accumulation in strawberry fruit[22]. Similarly, overexpression of the P3A-ATPase proton pump gene PbPH5 also significantly increased the malate accumulation in pear fruit[112]. It can be seen that P-ATPase alone or in the complex form, both play an important roles in vacuole acidifying.
-
Many transcription factors play essential roles in the regulation of malate content by influencing the expression of genes related to malate metabolism and transport (Table 1). MdcyMDH1 is identified as a major gene associated with malate accumulation via MapQTL in 'Honeycrisp' × 'Qinguan' F1 hybrids, and after MdcyMDH1 is overexpressed, the malate concentration of fruit is enhanced[8,9,28]. MdbHLH3 and MdWRKY126 directly activated the transcriptional expression of MdcyMDH1, and also increased the expression of malate transport-related genes such as MdtDT, increasing malate content in fruit[34,35]. The indel of a repeat sequence in MdcyMDH1 (MA7) promoter region in 'Gala' (MA7/MA7) and 'Fuji' (ma7/ma7) apple varieties were named respectively 'MA7' and 'ma7', and the upstream regulator, MdbHLH74, could enhance the expression of MdcyMDH1 in apple with MA7/MA7 genotype, but not with ma7/ma7 genotype, affecting the malate content in different varieties[9]. Additionally, TRXL1 up-regulates NADP-cyMDH activity, increases malate accumulation, and inhibits superoxide radical formation in response to high-temperature stress, and the expression of TRXL1 is positively regulated by CPN60A and negatively regulated by CLPC1[113]. Based on current research, the main object of transcriptional regulation of malate metabolism is cytoplasmic malate dehydrogenase, and the regulatory network of other malate metabolism-related genes need to be further studied.
Table 1. The crucial genes and their upstream regulatory factors of fruit acidity regulation.
Gene family Gene name Protein name Activation (+)/
inhibition (−)Module control Species MYB transcription factor MdMa1 MdMYB73 + MdBT2-MdCIbHLH1
-MdMYB73Malus domestica MdMa1/MdMa11 MdMYB123 + — Malus domestica MdMa1 MdMYB44 − MdbHLH49-MdMYB44 Malus domestica MdMa1 MdMYB21 − — Malus domestica MdVHA-A3/D2 Ma10 MdMYB44 − WD40-MdbHLH49
-MdMYB44Malus domestica MdVHA-B1/E MdVHP1
MdtDTMYB1/10 + MdTTG1-MdbHLH3
-MdMYB1/10Malus domestica MdVHA-A MdVHP1 MdMYB73 + MdBT2-
WD40-MdbHLH1
-MdMYB73Malus domestica CitPH5 CitPH4 + CitTRL-CitPH4 Citrus reticulata WRKY transcription factor SlALMT9 SlWRKY42 − — Solanum lycopersicum PpALMT9 PpWRKY44 + PpABF3-PpWRKY44 Pyrus spp. ZjALMT4 ZjWRKY7 + — Ziziphus jujuba MdMa1 MdWRKY31 + — Malus domestica MdMDH1 MdWRKY126 + — Malus domestica bHLH transcription factor MdMDH1 MdbHLH3 + — Malus domestica NAC transcription factor AcALMT1 AcNAC1 + — Actinidia spp. ERF transcription factor MdMa1 MdERF72 − MdWRKY31-MdERF72 Malus domestica CitVHA-C4 CiERF13 + — Citrus reticulata PP2C family MdVHA-A3/B2/D2 Ma10 MdPP2CH − SAUR37-MdPP2CH Malus domestica Transcriptional regulation of malate transportation
-
Jia et al.[20] identified three major genes associated with malate transport (MdPP2CH, MdMYB44, and MdSAUR37) via MapQTL and BSA-seq and verified their functions. MdPP2CH reduced the malate accumulation by phosphorylating the proton pump gene in apple calli, while MdSAUR37 could inhibit the phosphorylation activity of MdPP2CH and positively regulate malate content. Another acid accumulation major gene, MdMYB44, negatively regulated proton pump gene Ma10, MdVHA-A3, and MdVHA-D2 and malate transporter Ma1 to inhibit fruit vacuole acidification. A further study indicated that the presence of SNP (A/T) in the MdMYB44 promoter affected the ability of its upstream transcription factor MdbHLH49 to regulate the activity of the MdMYB44 promoter and malate accumulation of fruit[104]. Additionally, other MYB transcription factors also play an essential role in proton pump regulation. Apple MdMYB1/10 directly binds and activates the expression of proton pump genes MdVHA-B1, MdVHA-B2, MdVHA-E2, and MdVHP1, accelerating the malate accumulation of vacuoles[92]. The MdCIbHLH1-MdMYB73 module regulates downstream proton pump genes MdVHA-A, and MdVHP1 for the acidification of fruit vacuole[11], while the MdBT2 response to nitrate treatment could ubiquititatively degrade MdCIbHLH1, and malate content in MDBT2-silenced apple calli is significantly upregulated[114].
Studies on the transcriptional regulation of malate transporters focus on tDT and ALMT proteins. tDT positively regulates the malate content and negatively participates in the accumulation of citrate in tomato and apple fruit[89,90], which was regulated by transcription factor MdMYB1[92], MdMYB73[11] and MdbHLH3[34]. The latest study revealed that AP2 domain-containing transcription factor MdESE3 activate their expression of MdtDT, MdMa11, and MdMDH12 to increase malate accumulation in apple[18]. While, ALMT9, the major contributor of fruit malate accumulation, has constant attention from reseachers, and an increasing number of transcriptional regulatory mechanisms regarding the influence of ALMT9 on fruit acidity have emerged. The ALMT9 homologous genes have been identified as a crucial gene functioning in enhancing malate transport, and vacuole acidification in various horticultural crops such as apple[77], tomato[62], grape[79], and pear[14], where expression is regulated respectively by the transcription factors MdWRKY31-MdERF72[115], MdMYB73[11,114], MdMYB21[116], MdMYB123[117], MdMYB44[104], SlWRKY42[62], and PpABF3-PpWRKY44[14] (Fig. 2). Interestingly, the malate content in stable Ma1-overexpressed apple fruits was significantly reduced. Further studies showed that alternative splicing generates two Ma1 isoforms (higher expression of MA1α and lower expression of Ma1β). Ma1β is only able to form polymers with MA1α protein for strong malate transport function and the absence or reduced transport activity of MA1α/Ma1β polymers in Ma1 transgenic fruits decreased malate accumulation, which was regulated by MdMYB73[16].
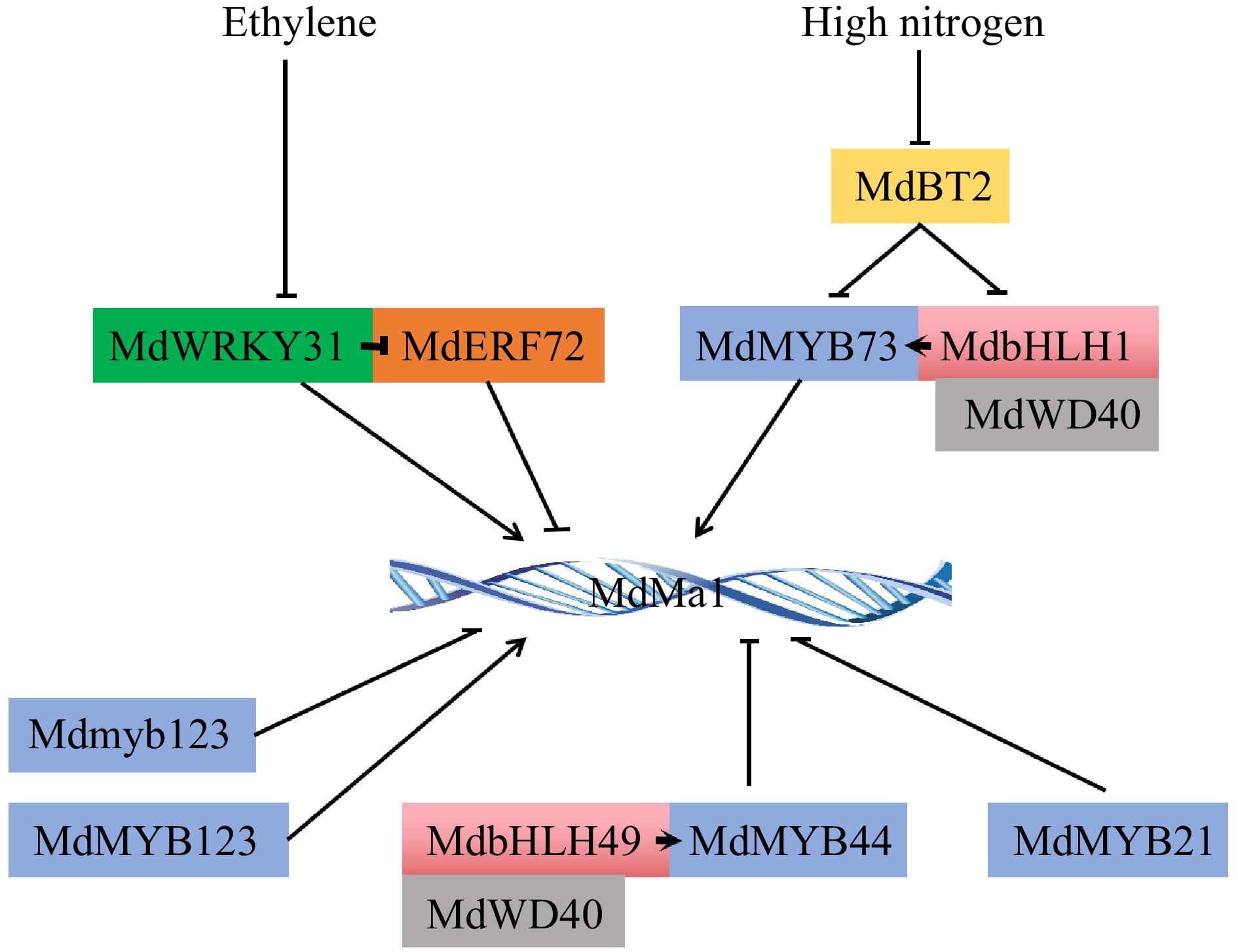
Figure 2.
Upstream regulators of major gene Ma1 regulating apple fruit acidity. The arrows represent positive regulation and the rest represent negative regulation.
-
Protein post-translational modification is a kind of chemical modification existing in the late stage of protein biosynthesis that affects the protein stability and activity by changing different biochemical functional groups on amino acid residues of proteins, including protein phosphorylation, ubiquitination, acetylation and methylation. MdPP2CH decreased the malate content via phosphorylating the proton pump MdVHA-A3, MdVHA-B2, MdVHA-D2 and ALMT transporter MdALMTII, and its dephosphatase activity was inhibited by MdSAUR37 in apple[20]. Some enzymes involved in malate metabolism, such as malate dehydrogenase (MDH2) and phosphoenolpyruvate carboxykinase (PEPCK1), are also regulated by either the glucose-induced degradation-deficient pathway or the vacuole import and degradation pathway[118], but the specific protein modification mechanisms remain to be investigated.
Posttranslational modification except for the above directly modified functional proteins associated with malate accumulation, can also modify upstream regulatory proteins of functional genes to affect fruit acidity. The ubiquitin E3 ligase MdCOP1 degrades MdMYB1 in the dark through a ubiquitin-dependent pathway to regulate anthocyanins and malate accumulation[92,119]. Similar, high glucose-inhibited U-box-type E3 ubiquitin ligase MdPUB29 and glucose sensor MdHXK1 ubiquitinates and phosphorylates MdbHLH3, respectively, affecting malate concentration[120,121]. It was found that the apple transcription factor MdCIbHLH1 acidified fruit vacuole by enhancing the activity of MdMYB73, which promoted the up-regulated expression of MdVHA-A, MdVHP1 and MdALMT9. While BTB-BACK-TAZ domain protein MdBT2 degrades ubiquititatively MdCIbHLH1 and MdMYB73 via ubiquitin/26S proteasome pathway to regulate the malate content of vacuole in apple plants under nitrate stress[114,122]. These studies provide groundbreaking insights into the direct posttranslational modification of organic acid-related functional proteins and their upstream regulatory proteins, which is helpful to cultivate high-quality horticultural crop varieties from the perspective of post-translational modification.
-
To date, researchers have performed some basic research on the accumulation of malate in horticultural crops. Members of the acid metabolism and transport families have been screened and identified at the genome-wide level in most horticultural crops, and their function on vacuole acidification has been demonstrated in apple, tomato, pear, and Arabidopsis. However, the systematic regulatory network of malate accumulation and the cross-regulation between sugar and acid metabolism and transport remain to be further explored. Therefore, the following suggestions are put forward for the future research direction of malate accumulation regulation in horticultural crops.
Epigenetic regulation
-
Epigenetics is a kind of 'post-genetics' that can achieve genetic heritability under the premise that the nuclear DNA sequence remains unchanged through the methylation modification or histones acetylation, phosphorylation, and ubiquitination of gene promoter DNA. This regulatory mechanism of epigenetic modification has been elucidated to some extent in fruit soluble sugar and anthocyanin accumulation[123−125], indicating epigenetic modification plays a crucial role in fruit quality formation. In pummelo (Citrus maxima LCA) and lemon (Citrus limon (L.) Burm f.), DNA methylation changes in promoters of key genes involved in citrate synthesis and accumulation directly affect the citrate content in the flesh[23,126], revealing a previously unexplored link between epigenetic regulation and organic acid accumulation of horticultural fruits. A recent study identified a CgAN1, BHLH-type regulator coupling citrate and anthocyanin, from citrus varieties with high citrate, anthocyanin, and low citrate, anthocyanin, and confirmed that the reduction of the methylation level of the gene promoter can enhance the citrate accumulation of fruit[23]. However, the epigenetic mechanism related to fruit acidity, especially malate, is relatively limited, the urgent task is to uncover the complex DNA methylation mechanisms controlling key genes in malate synthesis, and accumulation pathways in horticultural crops.
Cross-regulation on the accumulation of soluble sugars and organic acids
-
The differences in the composition and content of soluble sugars and organic acids play a decisive role in fruit quality, and flavor[127]. Physiological studies have revealed that during the ripening process of fleshy fruit such as apple, peach, and grape, organic acid content decreases, and soluble sugar accumulation increases[10,28,36,128]. The variousness in malate content caused by the overexpression of MDH or ME genes could lead to a change in the redox state of the plastid thus affecting the accumulation of starch and sugar in tomato fruit cells[129,130]. Yao et al.[28,91] pointed out that the overexpression of MdVHP1 and MdcyMDH1 in apple calli both increased malate and soluble sugar contents, and the effect of MdcyMDH1 on malate and soluble sugar accumulation was regulated by transcription factor MdbHLH3[34]. While high glucose-inhibited U-box-type E3 ubiquitin ligase MdPUB29 and glucose sensor MdHXK1 ubiquitinates and phosphorylates MdbHLH3, respectively, affecting the expression of its downstream genes[120,121]. FaMYB44.2 could inhibit the expression of FaSPS, reducing both the sucrose and malate content in banana fruit[131]. Similarly, the increased accumulation of malate, citrate, glucose, and fructose was observed in SlAREB1 overexpressed red ripe peel compared to antisense-inhibited lines[132]. A recent study has also indicated that the accumulation of malate in the cytoplasm mediated by MdcyMDH1 increased the sucrose content in apple fruit by up-regulating the expression of MdSPS, which is likely to be achieved via starch cleavage or gluconeogenesis[8]. These above studies indicate that there is an interactive relationship between carbohydrate and organic acid accumulation in fruit. However, the spatiotemporal crosstalk between sugars and acids during fruit development remains unclear. Therefore, elucidation of the potential cross-regulatory mechanisms of sugars and acids in fruit is important to optimize the ratio of sugars to acids in fruit and improve fruit quality.
The exploration of a clear and thorough regulatory network of malate accumulation and regulation in horticultural crops relies on the functional identification of major genes in malate metabolism and transport in fruit and the in-depth study of the above directions, which lays an important foundation for the improvement of fruit quality via molecular-assisted breeding.
-
The authors confirm contribution to the paper as follows: study conception and design: Zhang LH; data collection: Li MJ, Zhang LH, Zhang AN, Xu Y; analysis and interpretation of results: Zhang LH, Zhang AN, Xu Y, Zhu LC, Ma BQ; draft manuscript preparation: Zhang LH, Zhang AN, Xv Y, Zhu LC, Ma BQ. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article.
This work was supported by the Outstanding Youth Science Foundation of Heilongjiang Province (YQ2023C006), the China Postdoctoral Science Foundation (2023MD744175), the Talent Introduction Program of Northeast Agricultural University of China, and Modern Agricultural Industrial Technology Collaborative Innovation and Promotion System of Heilongjiang Province. The authors would like to thank Mr. Li Dalong from Northeast Agricultural University for providing the instruments.
-
The authors declare that they have no conflict of interest.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang LH, Zhang AN, Xu Y, Zhu LC, Ma BQ, et al. 2024. Accumulation and regulation of malate in fruit cells. Fruit Research 4: e031 doi: 10.48130/frures-0024-0025 

Accumulation and regulation of malate in fruit cells
- Received: 01 May 2024
- Revised: 02 July 2024
- Accepted: 13 July 2024
- Published online: 02 September 2024
Abstract: Fruit acidity is an important component of flavor quality in fleshy fruit. The accumulation of malate, the dominant organic acid in the acidity formation of most mature fruit, is highly regulated by metabolism and transportation during fruit development. The knowledge on the mechanism of fruit acidification, as well as the major genes and substances is however still limited. In the present paper, the research advances on the relevance between malate accumulation and the genes associated with malate metabolism and transportation, as well as the transcriptional regulation of malate in fruit was reviewed. Furthermore, positive future research could provide a theoretical reference for optimizing fruit quality and genetic improvement.
-
Key words:
- Malate /
- Fruit /
- Metabolism /
- Transporter /
- Regulation