









-
Passion fruit (Passiflora edulis), belonging to the Passifloraceae family and the Passiflora genus, is widely cultivated in tropical and subtropical regions[1,2]. It is renowned for its aromatic fruits, which exhibit fragrances reminiscent of various fruits, including bananas and pineapples. The fruit is not only nutritionally rich but can also be harvested year-round, contributing significantly to its economic value[3−6]. Moreover, passion fruit displays vibrant and large flowers with a distinctive secondary corona structure – a white or colorful fringe-like structure formed between the petals and pistils[2]. It also holds great significance as a horticultural plant, finding widespread use in applications like fence landscaping and small potted plants[7,8]. Furthermore, the special floral structure (sepal-petal-corona-stamen-carpel) in passion fruit also makes it an excellent and unique plant model for investigating floral organ morphology.
MADS-box gene family members are ubiquitously distributed among plants, animals, and fungi, playing crucial roles throughout plant growth and development[9−11]. MADS-box genes are instrumental in governing a spectrum of biological processes[12−14], encompassing floral transition, control of flowering time, endosperm and embryo development, seed germination, fruit development, abiotic stress responses, and notably, they assume central roles in the morphogenesis of plant floral organs[15,16]. Based on protein domain characteristics and phylogenetic tree relationships, the MADS-box gene family can be classified into two primary types: Type I and Type II[17]. Type I genes can be further divided into three subclasses: Mα, Mβ, and Mγ[18], while Type II genes, also known as MIKC-type genes in plants, consist of the MIKCC and MIKC* groups. Moreover, the MIKCc group can be further classified into 12 major subclades[19]. Notably, Type I genes have undergone more rapid evolutionary changes from birth to death compared to Type II genes[20]. Both Type I and Type II protein sequences share a conserved domain named as MADS-domain, comprising 58-60 amino acids located in the N-terminal region, which serves as an identifying feature of MADS-box proteins. Whereas, Type II MADS-box proteins in plants possess three additional domains in addition to the MADS-box domain: an intervening region with relatively low conservation, a moderately conserved Keratin-like domain, and a variable C-terminal region[21].
Among the MIKCC-type genes, specific members are known to form the well-established ABC(D)E model, providing a genetic basis for floral organ specification and development[22,23]. According to this model, the identification of floral organs in each whorl is governed by unique combinations of genes from classes A, B, C, D, and E. Sepal identification is jointly determined by A- and E-class genes, whereas petals are controlled by A-, B-, and E-class genes. The formation of stamens relies on B-, C- and E-class genes, while carpels are regulated by C- and E-class genes. Ovule specification is particularly controlled by C-, D-, and E-class genes[23−25]. In MADS-box gene family of Arabidopsis thaliana, numerous functional genes have been confirmed to be associated with the ABC(D)E model. For instance, A-class genes include APETALA1(AP1)[26]; APETALA3(AP3) and PISTILLATA(PI) belongs to class B[27,28]; AGAMOUS (AG) is a C-class gene and also involved in floral meristem development[29]; class D gene AGL11 is expressed in ovules[30]; the E class comprises SEPALLATA (SEP1, SEP2, SEP3, and SEP4), which exhibit functional redundancy and are required for specifying the identity of all floral organs[31,32]. Proteins encoded by ABC(D)E model genes form quaternary complexes that bind to CArG-box (CC[A/T]6GG) of DNA sequences, and this complex subsequently acts on downstream target genes to regulate flower development[33]. Additionally, MADS-box genes in A. thaliana perform various functions in other development processes, AGAMOUS-LIKE 20 (AGL20) serves as a key activator in integrating floral inductive pathways[34] , while FLC and SVP are involved in regulating flowering time[35]. AGL21 and AGL12 are associated with root development[36,37], and the AGL6 is involved in floral meristem and seed development[38]. FRUITFULL (FUL), SHATTERPROOF 1 and 2 (SHP1 and SHP2), collectively function in the complex regulation of fruit ripening and development[39].
The distinctive floral morphology of passion fruit makes it an excellent candidate for the study of floral organogenesis and enhances our understanding of the ABC(D)E model. While extensive research has explored MADS-box gene family members in various plants[40−43], a comprehensive investigation of these members in passion fruit has not yet been carried out. Here, we identified a total of 52 MADS-box genes in passion fruit and conducted a systematic analysis, including analysis of gene structure, motif composition, phylogenetic relationships, chromosomal localization, collinearity, and expression patterns. Notably, we also proposed an ABC(D)E model to elucidate the regulation of the unique floral structure (sepal-petal-corona-stamen-carpel) in passion fruit. Our findings could provide valuable insights for further functional investigations of passion fruit MADS-box genes and their potential applications in floral modification breeding.
-
Passion fruit genome and protein sequences were obtained from the National Genomics Data Center (NGDC) database (
https://ngdc.cncb.ac.cn/ ) under the accession number GWHAZTM00000000. We employed two distinct strategies, namely the Hidden Markov Model (HMM) search and BLAST search, to identify MADS-box genes in passion fruit. Regarding the HMM method, the MADS-box SFR family domain (PF00319) was sourced from the Pfam database (www.pfam.org ) and subsequently applied for a comprehensive search against the passion fruit protein sequences using HMMER3 (v3.3.2) software (http://hmmer.janelia.org/ )[44]. Simultaneously, plant MADS-box protein sequences were obtained from the NCBI database (www.ncbi.nlm.nih.gov ) and used as initial queries for BLASTP searches. Only homologous sequences with an E-value of less than 1e-5 were retained for subsequent analyses. The sequences identified from both the HMM search and BLAST search were consolidated and the redundant members were removed to produce a set of preliminary MADS-box candidates. These candidates then underwent rigorous verification of the MADS-box domain through the SMART (http://smart.embl-heidelberg.de )[45] and CDD (Conserved Domain Database) search (www.ncbi.nlm.nih.gov/cdd )[46] . The confirmed MADS-box genes were renamed according to their positions on the passion fruit chromosomes. Subcellular localization analysis was conducted using the WoLF PSORT tool (https://wolfpsort.hgc.jp ), while the ExPASy website tool (www.expasy.org )[47] was harnessed to predict molecular weight (MW), isoelectric point (pI), and the grand average of hydropathicity (GRAVY).Sequence alignment and phylogenetic analysis
-
Multiple sequence alignment of all 52 putative MADS-box protein sequences was performed by MUSCLE[48], and the alignment was visualized and edited by software Jalview v 2.11.2.0[49]. For phylogenetic analysis, the 105 MADS-box protein sequences of Arabidopsis were downloaded from the TAIR database (
www.arabidopsis.org )[18], those sequences were combined with putative members from passion fruit to construct the phylogenetic tree. The multiple protein sequence alignment of all 157 protein sequences was produced using software MAFFT v7.407[50]. The resulting alignments were used for exploring the phylogenetic relationship of MADS-box protein in Arabidopsis and passion fruit. Phylogenetic analysis was performed by software IQ-TREE v1.6.12[51] with maximum likelihood estimation based on the most suitable model and 1000 bootstraps. The phylogenetic tree was visualized using Evolview (https://evolgenius.info/evolview-v2/ )[52].Prediction of gene structure, conserved motif and cis-regulatory element analysis
-
To identify the conservative motifs among the putative MADS-box members of passion fruit, the full-length protein sequences were analyzed website tool MEME (
https://meme-suite.org/meme/tools/meme )[53] with parameters that the maximum number of finding motifs is 10. Exon-intron structure of MADS-box genes were distinguished using GFF files, which was acquired from the annotation information of the passion fruit genome. The exact region of conservative MADS-box domain was identified from the CDD (Conserved Domain Database) database (www.ncbi.nlm.nih.gov/cdd )[46].To explore the possible-regulator-factors of MADS-box genes from passion fruit, TBtools v1.1047 software[54] was used to extract the 2000-bp upstream region as putative promoter regions, which end with the translation initiation codon of each MADS-box genes. The cis-acting elements contained in promoter regions were marked using the PlantCARE database (
http://bioinformatics.psb.ugent.be/webtools/plantcare/html )[55], and the statistics analysis and plotting were finished using R package pheatmap (https://CRAN.R-project.org/package=pheatmap ).Chromosomal distribution and gene duplication
-
The exact location of MADS-box genes was extracted from the annotation file of the passion fruit genome, and anchored to corresponding chromosomes. Duplication events of genes occurring in the evolution of the passion fruit genome were analyzed by Multicollinearity Scanning Toolkit (MCScanX)[56] with default parameters. The genomic data of Oryza sativa (v7.0), Solanum lycopersicum (ITAG3.2), A. thaliana (TAIR10), Vitis vinifera (Genoscope.12X), Zea mays (RefGen_V4) were retrieved from the JGI Phytozome database (
https://phytozome-next.jgi.doe.gov ). A similar procedure was also applied to demonstrate the collinearity relationships and gene duplications of orthologous MADS-box genes obtained from passion fruit and these five species. The result was visualized by functions of TBtools[54]. Based on the above results of duplicated events within passion fruit, the ratios Ka/Ks of tandem duplicated and segmental duplicated gene pairs were also calculated using TBtools[54].Tertiary structures analysis of MADS-box family member
-
All MADS-box protein sequences were submitted to SWISS-MODEL (
https://swissmodel.expasy.org )[57] for predicting tertiary structure with homology modeling method, and secondary structures could be marked by the corresponding original model from PDB databases (www.rcsb.org ). The predicted models were visualized using Pymol[58].PeMADSs transcriptome analysis based on RNA-seq data
-
The fresh samples of passion fruit (P. edulis) were collected from the orchard located in the Institute of Horticulture, Guangxi Academy of Agricultural Sciences (China). The whole floral tissue was divided into bract, sepal, corona filament or corona, stamen, stigma, ovule (dividing under dissecting microscope). The identification of development stages of floral tissues was based on the horizontal width of buds with bracts (more details about distinguishing different development stages are listed in Supplemental Table S1). Samples of leaves, stems and tendril tissues were collected at 110 d after anthesis. The fruit were collected at 53 d post anthesis (DPA), DPA60, DPA100 and DPA128, respectively. The tissues samples were immediately frozen in liquid nitrogen after picking and stored at −70 °C.
The grown and healthy plants of passion fruit were subjected to abiotic stress treatments (cold and heat) with three biological replicates. The whole plants were transferred into a growth chamber with temperature of 20 °C as the cold-stress condition while the heat-stress condition had a temperature of 30 °C. Samples of floral buds were collected under different treatment times (1, 4, 12, 24 h for heat stress; 4, 24 h for cold stress). Plants cultivated under 25 °C were used as control. All samples were collected for RNA extraction.
Reference to previous common methods of RNA extraction[59], total RNA from different tissues was extracted by RNA extraction Kit (Omega Bio-Tek, Shanghai, China) with manufacturer's protocol. Following a standardized process, RNA samples were quantified and PCR library construction was performed. The analysis result was sequenced using NEB next Ultra RNA Library Prep Kit (NEB, Beverly City, MA, USA) for Illumina Biolabs. Each biological replicate used 1 µg RNA as an experiment sample. After the related processing flow of initial RNA-seq data file, TPM value of RNA-seq for each PeMADS gene were calculated, and the heatmap of the TPM value was constructed using R package (pheatmap) and chiplot (
www.chiplot.online ) with log2 (TPM + 0.0001) as the unit of measure.RNA extraction and qRT-PCR analysis
-
Based on the above standards of identifying different development stages for flowers, the tissues used for qRT-PCR analysis was br1 (bract at stage1), se1, pe1, ca1, st8, sg1 and ov5. Two-month-old healthy passion fruit plants were also subjected to ABA (100 μM) and GA (100 μM) treatments, while untreated plants served as controls. Leaf samples from phytohormone-treated plants were collected at 0, 12, 24, and 48 h post-treatment from three independent seedlings. All collected samples were immediately stored in liquid nitrogen before total RNA extraction. The Trizol reagent (Invitrogen, Carlsbad, CA, USA) was used to extract total RNA. ThermoScript RT-PCR kit (Thermo Fisher Scientific, Carlsbad, CA, USA) was used for reverse transcription. The quantitative real-time PCR (qRT-PCR) was performed by the following procedures: 95 °C for 30 s, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. The reaction volume was 20 µl, including 1 µl cDNA per sample, 10 µl 2× Taq Pro Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China) and 0.4 µl per primer, the whole system was complemented by ddH2O to 20 µl. The reaction was carried out in a Bio-Rad Real-time PCR system (Foster City, CA, USA) with three biological replicates per sample. EF1α was used as a reference gene[60]. The relative expression levels were calculated using the 2−ΔΔCᴛ method. Primers were designed using the IDT website (
https://sg.idtdna.com/pages ). Primer sequences are listed in Supplemental Table S2. -
To ensure a comprehensive screening of gene family members, the putative PeMADS genes were identified by combining the results from hidden Markov model (HMM) and the blast search. Ultimately, a total of 52 MADS-box gene family members were successfully identified from the whole genome of passion fruit (Table 1). The members were designated as PeMADS1-PeMADS51 based on their sequential positions on the chromosomes, while a single member (P_eduliaContig70023089.g) mapped to a contig was named PeMADS52. Predicted physicochemical properties of PeMADS genes coding proteins are list in Table 1. These proteins exhibit variable lengths, with the longest one being 650 aa (PeMADS7) while the shortest one is 66 aa (PeMADS29). Correspondingly, PeMADS7 possesses the heaviest molecular weight (MW) of 71,572.97 Da, while PeMADS29 has the lightest protein, only 7,697.02 Da. The protein isoelectric points (PI) range from 4.89 (PeMADS41) to 10.69 (PeMADS29). According to the Instability Index[61], only PeMADS4, PeMADS13, and PeMADS23 are considered stable proteins (value is smaller than 40). The aliphatic amino acid index (A.I.) varies from 66.05 (PeMADS4) to 100.41 (PeMADS27), indicating a significant difference in thermal stability among the PeMADS proteins. With the exception of PeMADS22, the grand average of hydropathicity score (GRAVY) of other proteins are negative, which means only PeMADS22 is a hydrophobic protein, and most proteins are hydropathicity. The prediction of subcellular localization reveals that the major action site of proteins is the nucleus, followed by chloroplasts and mitochondria. Two members, PeMADS23 and PeMADS28, are located in the cytoplasm. For detailed sequence information of all members, refer to Supplemental Table S3.
Table 1. Characteristics about 52 PeMADS proteins of passion fruit
Gene name Gene ID Chromosome Size (aa) MW (Da) PI Instability
IndexA.I. GRAVY Predicted Location PeMADS1 P_edulia010000232.g LG01 355 40,132.20 6.36 42.66 75.27 −0.63 Nucleus PeMADS2 P_edulia010000334.g LG01 190 21,875.00 9.76 48.93 76.47 −0.826 Nucleus PeMADS3 P_edulia010000557.g LG01 194 22,380.57 9.73 48.13 76.91 −0.818 Nucleus PeMADS4 P_edulia010000858.g LG01 387 42,577.71 9 35.16 66.05 −0.687 Nucleus PeMADS5 P_edulia010002143.g LG01 225 25,469.32 10.31 48.64 72.8 −0.629 Nucleus PeMADS6 P_edulia010002220.g LG01 251 29,014.90 5.84 63.89 80 −0.551 Nucleus PeMADS7 P_edulia010002256.g LG01 650 71,572.97 8.88 42.57 89.43 −0.259 Nucleus PeMADS8 P_edulia010002353.g LG01 223 25,645.29 9.51 53.22 83.54 −0.658 Nucleus PeMADS9 P_edulia010002348.g LG01 223 25,567.89 8.11 57.08 92.29 −0.209 Mitochondria PeMADS10 P_edulia010002733.g LG01 223 25,520.75 8.12 58.33 90.54 −0.242 Mitochondria PeMADS11 P_edulia010002726.g LG01 223 25,663.32 9.51 50.74 82.24 −0.671 Nucleus PeMADS12 P_edulia010003864.g LG01 227 25,943.71 9.08 53 82.91 −0.609 Nucleus PeMADS13 P_edulia010004192.g LG01 128 14,242.54 10.33 37.36 91.33 −0.087 Nucleus PeMADS14 P_edulia010004199.g LG01 166 19,203.34 9.3 45.27 93.31 −0.031 Nucleus PeMADS15 P_edulia010004299.g LG01 342 38,721.37 6.08 55.69 74.18 −0.57 Nucleus PeMADS16 P_edulia010004354.g LG01 283 32,306.41 8.22 56.25 75.51 −0.63 Nucleus PeMADS17 P_edulia010004671.g LG01 274 31,672.19 9.28 43.23 80.47 −0.444 Nucleus PeMADS18 P_edulia010004713.g LG01 212 24,765.59 9.74 44.1 84.2 −0.322 Nucleus PeMADS19 P_edulia010005413.g LG01 154 17,785.60 9.61 43.32 87.34 −0.638 Nucleus PeMADS20 P_edulia020006530.g LG02 78 9,025.34 9.69 44.24 83.72 −0.615 Nucleus PeMADS21 P_edulia020007073.g LG02 244 28,034.94 6.48 56.57 85.53 −0.696 Nucleus PeMADS22 P_edulia030008390.g LG03 141 15,691.08 6.8 40.59 89.36 0.119 Nucleus PeMADS23 P_edulia030008412.g LG03 233 27,189.87 8.39 37.53 86.57 −0.842 Cytoplasm PeMADS24 P_edulia030008784.g LG03 255 29,597.75 7.69 62.04 85.25 −0.698 Nucleus PeMADS25 P_edulia030008868.g LG03 233 27,283.18 8.32 64.57 85.75 −0.738 Mitochondria PeMADS26 P_edulia030009361.g LG03 229 25,959.02 9.2 44.3 99.21 −0.374 Nucleus PeMADS27 P_edulia040010097.g LG04 296 34,731.28 8.36 74.87 100.41 −0.349 Nucleus PeMADS28 P_edulia040010305.g LG04 226 26,236.00 9.18 45.29 75.49 −0.773 Cytoplasm PeMADS29 P_edulia040010846.g LG04 66 7,697.02 10.69 39.17 79.7 −0.239 Nucleus PeMADS30 P_edulia050011612.g LG05 67 7,764.03 9.83 43.4 93.13 −0.452 Nucleus PeMADS31 P_edulia050012002.g LG05 117 12,998.97 10.21 46.38 66.67 −0.506 Nucleus PeMADS32 P_edulia060013073.g LG06 242 27,917.83 8.72 44.44 82.64 −0.695 Nucleus PeMADS33 P_edulia060013103.g LG06 168 19,304.06 8.43 50.78 91.19 −0.642 Nucleus PeMADS34 P_edulia060013465.g LG06 88 10,152.74 9.59 76.09 82.95 −0.448 Nucleus PeMADS35 P_edulia060013580.g LG06 240 27,317.30 9.17 49.12 84.17 −0.561 Nucleus PeMADS36 P_edulia060013765.g LG06 291 31,621.31 5.33 52.13 72.51 −0.417 Chloroplast PeMADS37 P_edulia060015308.g LG06 331 37,420.77 5.28 62.06 71 −0.684 Nucleus PeMADS38 P_edulia060015605.g LG06 221 25,409.11 9.17 40.8 83.8 −0.576 Nucleus PeMADS39 P_edulia060015604.g LG06 188 21,618.59 8.81 43.18 84.04 −0.624 Nucleus PeMADS40 P_edulia060016494.g LG06 243 28,153.00 7.13 57.79 91.11 −0.658 Nucleus PeMADS41 P_edulia060016711.g LG06 175 20,210.43 4.89 54.18 69.66 −1.058 Chloroplast PeMADS42 P_edulia060016709.g LG06 179 20,635.04 5.05 58.72 73.02 −1.001 Chloroplast PeMADS43 P_edulia060016710.g LG06 198 22,057.86 5.69 58.41 81.82 −0.657 Chloroplast PeMADS44 P_edulia070017407.g LG07 181 20,894.80 8.85 71.81 70.61 −0.809 Nucleus PeMADS45 P_edulia070017456.g LG07 248 28,225.44 9.58 47.5 90.48 −0.497 Nucleus PeMADS46 P_edulia070017776.g LG07 172 19,278.41 10 50.2 88.37 −0.621 Nucleus PeMADS47 P_edulia080019101.g LG08 214 24,404.50 9.66 53.65 70.23 −0.866 Nucleus PeMADS48 P_edulia080020106.g LG08 130 14,779.15 9.38 42.35 84.69 −0.338 Nucleus PeMADS49 P_edulia090020757.g LG09 206 23,473.42 6.98 44.32 67.57 −0.681 Nucleus PeMADS50 P_edulia090021063.g LG09 204 23,738.65 9.23 44.26 86.96 −0.739 Nucleus PeMADS51 P_edulia090021542.g LG09 214 24,401.79 9.06 59.69 67.48 −0.697 Nucleus PeMADS52 P_eduliaContig70023089.g Contig7 253 28,637.17 9.46 49.64 94.47 −0.316 Nucleus Sequence alignment and 3D-structure of passion fruit MADS-domain
-
The protein sequences of 52 candidate PeMADS were subjected to alignment analysis to further elucidate the constitution and structure of MADS-domain (Supplemental Table S4). As is shown in Fig. 1a, there is a highly conserved domain in all 52 PeMADS proteins. The sequence logo was utilized to accurately depict the composition of the MADS-box region. By employing the SWISS-MODEL web server and utilizing protein homology modeling, a three-dimensional (3-D) model of MADS-box domain was constructed. This model could be divided approximately into three regions, comprising one α-helix and two β-sheets (Fig. 2b). Through analysis of amino acid distribution at each residue position within the MADS domain, 24 sites out of the 57 residues were identified as highly conserved if the percentage of a specific amino acid exceeded 80% at that particular site (Fig. 2c). Integrating the aforementioned information with the proteins' tertiary structure, the first helix contained 14 conserved residues (Arg-17, Gln-18, Val-19, Thr-20, Lys-23, Arg-24, Arg-25, Gln-27, Leu-28, Lys-30, Lys-31, Glu-34, Leu-35 and Cys-39), while the first sheet region contained one conserved residue (Phe-48), and the second region lacked any conserved residues. Among 57 residues, Arg-3, Arg-17, Arg-24, Lys-31, Glu-34 exist in all protein sequences.
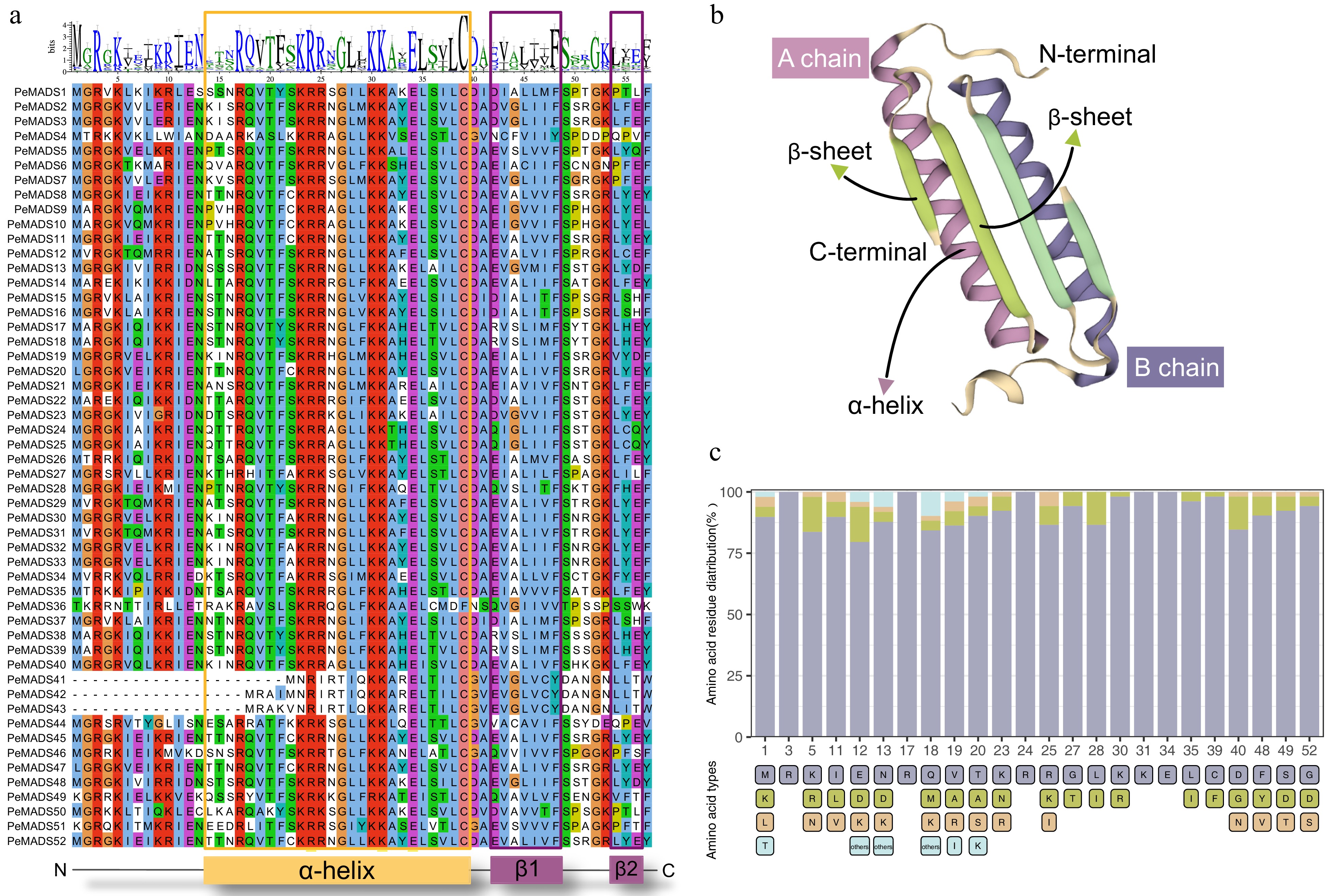
Figure 1.
Characterization of MADS-box domain. (a) Multiple sequence alignment of MADS-domain region from 52 PeMADS proteins. (b) Predicted three-dimensional structure of MADS-domain. (c) Distribution of amino acids in conserved residues of MADS-domain: number represents the positions of residues; bottom label represents species of amino acids.
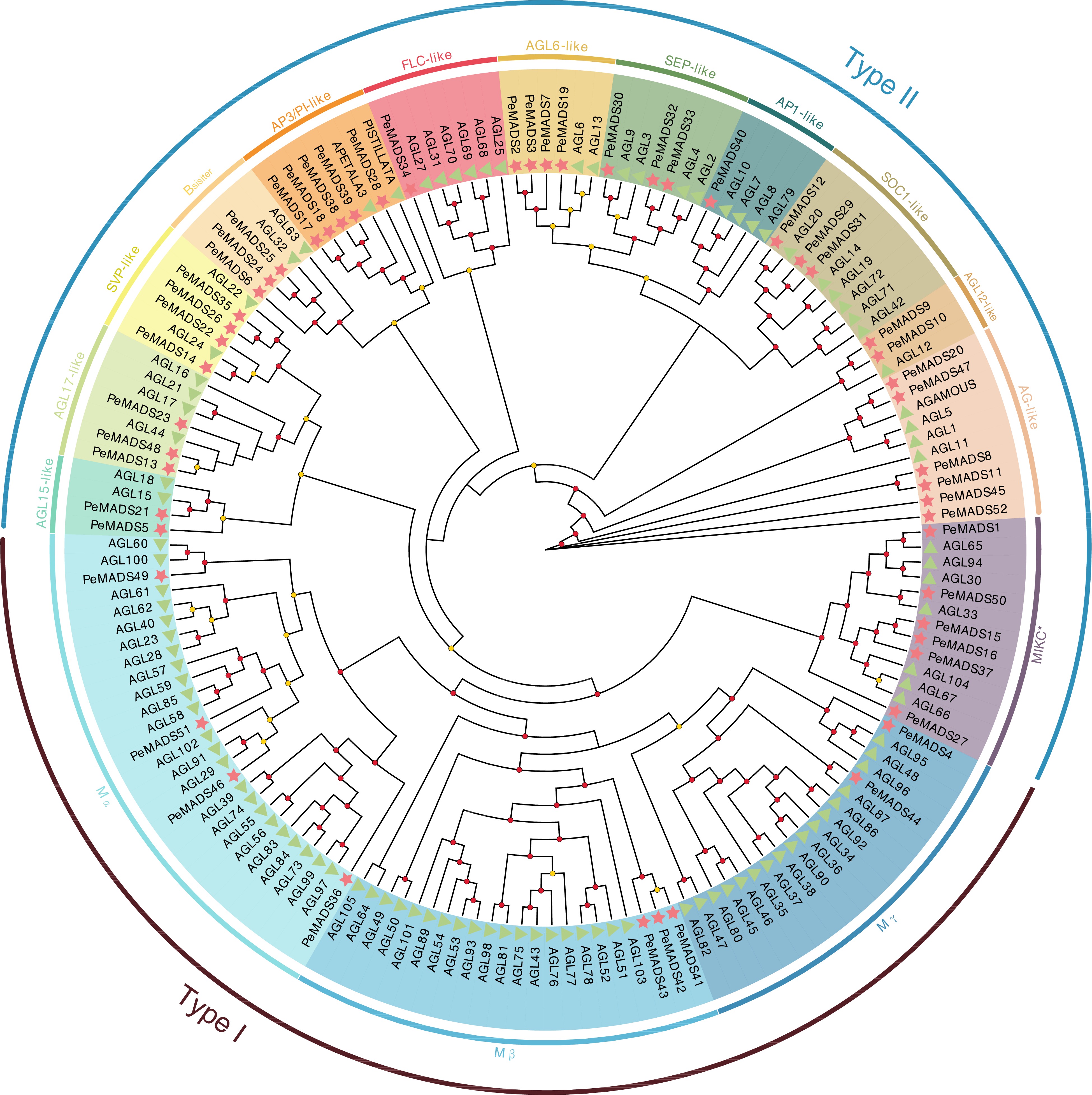
Figure 2.
Phylogenetic tree of PeMADS proteins from Passiflora edulis (Pe) and Arabidopsis thaliana (At). Red starts and green triangles indicate passion fruit and Arabidopsis, respectively. The circle placed on the tree indicated the bootstrap value (≥ 90, red circle; 60~90, yellow circle; < 60, not shown).
Classification and phylogenetic relationships of PeMADSs
-
To examine the phylogenetic relationships among MADS-box proteins from different species and ascertain the subfamily to which each PeMADS belong, a phylogenetic tree was constructed. The tree was based on the alignment result of MADS protein which is 105 sequences from Arabidopsis and 52 sequences from passion fruit (Fig. 2, Supplemental Table S5). According to previous studies and topology of the phylogenetic tree[18,62−64], the PeMADS members were classified into two groups: type I, consisting of nine members (four as Mα, three as Mβ, and two as Mγ) while 43 members were classified as type II. Within the type II group, only six PeMADS proteins belonged to MIKC* category, while the remaining members were classified as MIKCC. The MIKCC proteins were further divided into 12 clades. Sequences of both passion fruit and Arabidopsis could be identified in all clades. Most PeMADS were determined in AG-like clade (six members), while the PI-like, FLC-like and AP1-like clades each had only one member. Inside the PI-like clade, the Arabidopsis MADS-box proteins showed homology with only one other member (PISTILATA and PeMADS28).
Gene structure, conserved motifs and cis-regulatory element analysis of PeMADS members
-
The phylogenetic relationship of PeMADS proteins was depicted using a phylogenetic tree with bootstrap values. The classification obtained from the phylogenetic tree analysis aligned with the aforementioned analysis (Fig. 3a). To identify and compare the common motifs of proteins within different groups, the MEME search tool was employed to 52 PeMADS proteins, and the first ten motifs with the highest conservation were detected (motif 1−10; Fig. 3b). As illustrated in the figure, members within the same group typically exhibited similar construction of motifs, particularly noticeable in B sister, AP3-like group and AGL6-like group. PeMADS proteins displayed varying numbers and distribution of conserved motifs, with a maximum of seven motifs and a minimum of one. Except for Mβ members, motif 1 could be observed in all proteins, which could be an obvious character of MADS-box proteins. Generally, the construction of Type I members was simpler than Type II members, especially Mγ group which contained only one motif. The location of MADS-box domain sequences was mapped to the full-length proteins (Fig. 3c). All members could detect MADS domain, whereas the length of the MADS domain is relatively short in the Mβ group. Except for MIKC* group, most Type II members contained K-box domain. Type II protein lacking the K-box domain were shorter than same group members, possibly due to the loss of terminal protein sequences.
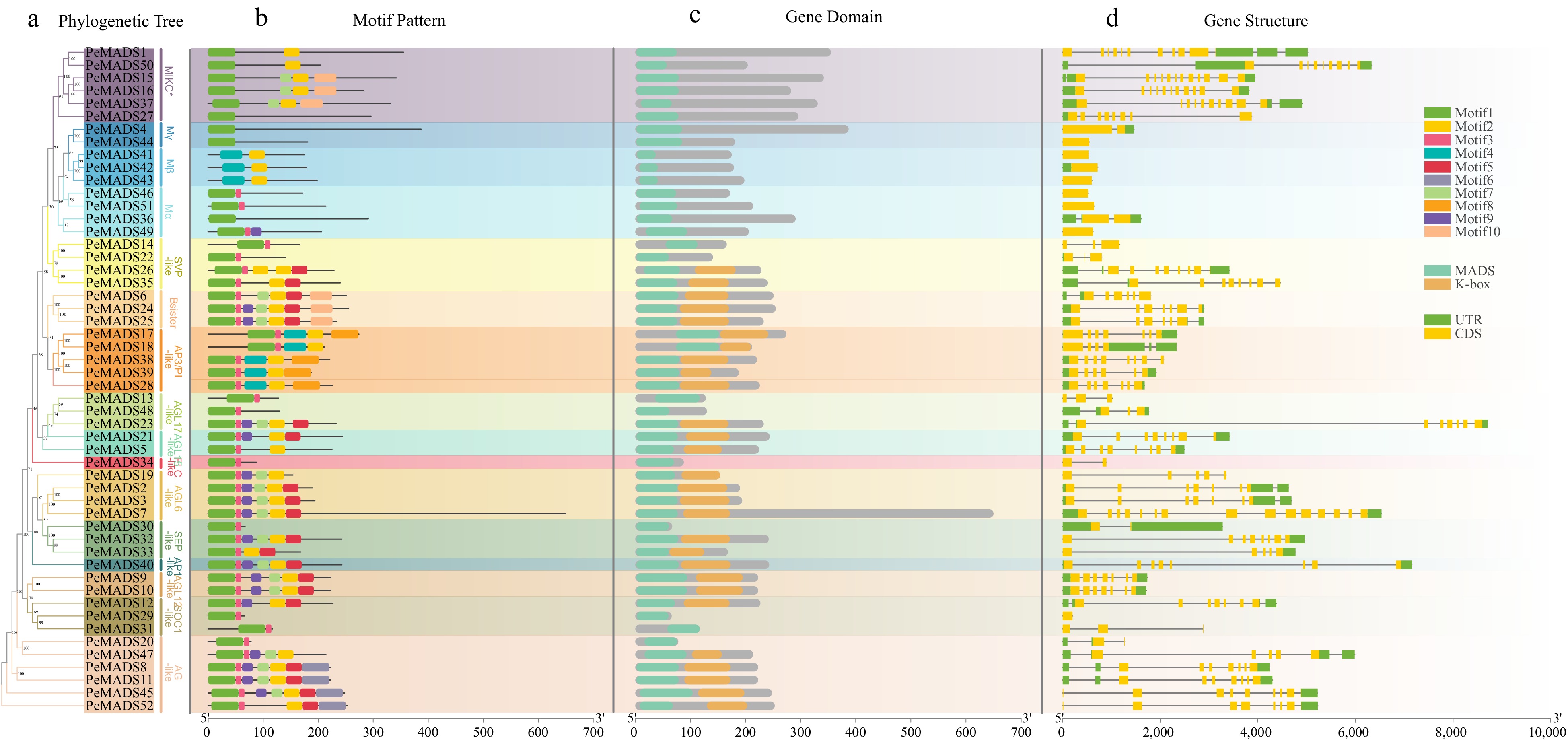
Figure 3.
The phylogenetic relationship, conserved motifs and gene structures of PeMADSs. (a) The phylogenetic tree of 52 PeMADS proteins. (b) Distribution of conserved motifs in PeMADS proteins. (c) Distribution of MADS-domain and K-domain of PeMADS proteins.(d) The gene structures of the PeMADSs, include coding sequences (CDS) and untranslated regions (UTR).
To investigate the structural diversity of PeMADS gene, the intron-exon organization of coding sequences was visualized reference to the GFF annotation file of passion fruit genome (Fig. 3c). The structure of type II group (MIKCC and MIKC*) was more complex. Nearly 33 (76.74%) type II members contained at least five CDS regions, among which the CDS regions of PeMADS7 was up to 15. Otherwise, type I groups (Mα, Mβ and Mγ) possessed less CDS regions, most members only had zero or single CDS regions, only PeMADS36 had two introns. The above discovery suggests that the transcription and splicing process of type II genes might be more complex than type I.
Cis-regulatory element analysis of PeMADS genes
-
Cis-element species and distribution could partly imply the transcription regulation and the expression pattern of associate genes. The putative promoters of PeMADS genes,spanning 2000 base pairs, were predicted using the PlantCARE server (Supplemental Table S6). A total of 39 types of cis-elements were identified and categorized into five groups: light response (16), phytohormone response (10), stress response (7) and plant growth regulation (6). Figure 4b illustrates that the majority of genes exhibited a higher proportion of light-responsive elements, followed by phytohormone-responsive elements, whereas elements associated with plant growth regulation were the least abundant. The proportions indicate that the expression of the PeMADS gene was substantially affected by light. G-box element (127), Box 4 (117), GT1-motif (70) were the most common light-responsive elements. Phytohormone-responsive elements accounted for a considerable portion of the cis-elements, with ABRE (134; abscisic acid response), AAGAA motif (106; abscisic acid response), CGTCA motif (79; MeJA response) and ERE (53; ethylene response) being the most frequently identified elements. Some types of elements centralized exist on one gene, for instance, the number of ABRE element was obviously higher in PeMADS2 and PeMADS50, implying those genes might be more possible to play a vital role in corresponding phytohormone regulation. ARE elements (135; anaerobic induction), 58 drought stress response elements (MBS and DRE core), 25 WUN-motif (mechanical injury response) and 38 LTR (low temperature response) indicated that PeMADSs could respond to abiotic stresses. ARE elements could be detected in promoter sequences of all genes, speculating anaerobic conditions was one of critical factors which induce the transcription of PeMADS gene. However, cis-elements related to plant growth such as CAT-box, CCGTCC-box, and RY-element were comparatively limited. Within the promoter region of each gene, the count of such elements did not exceed four.
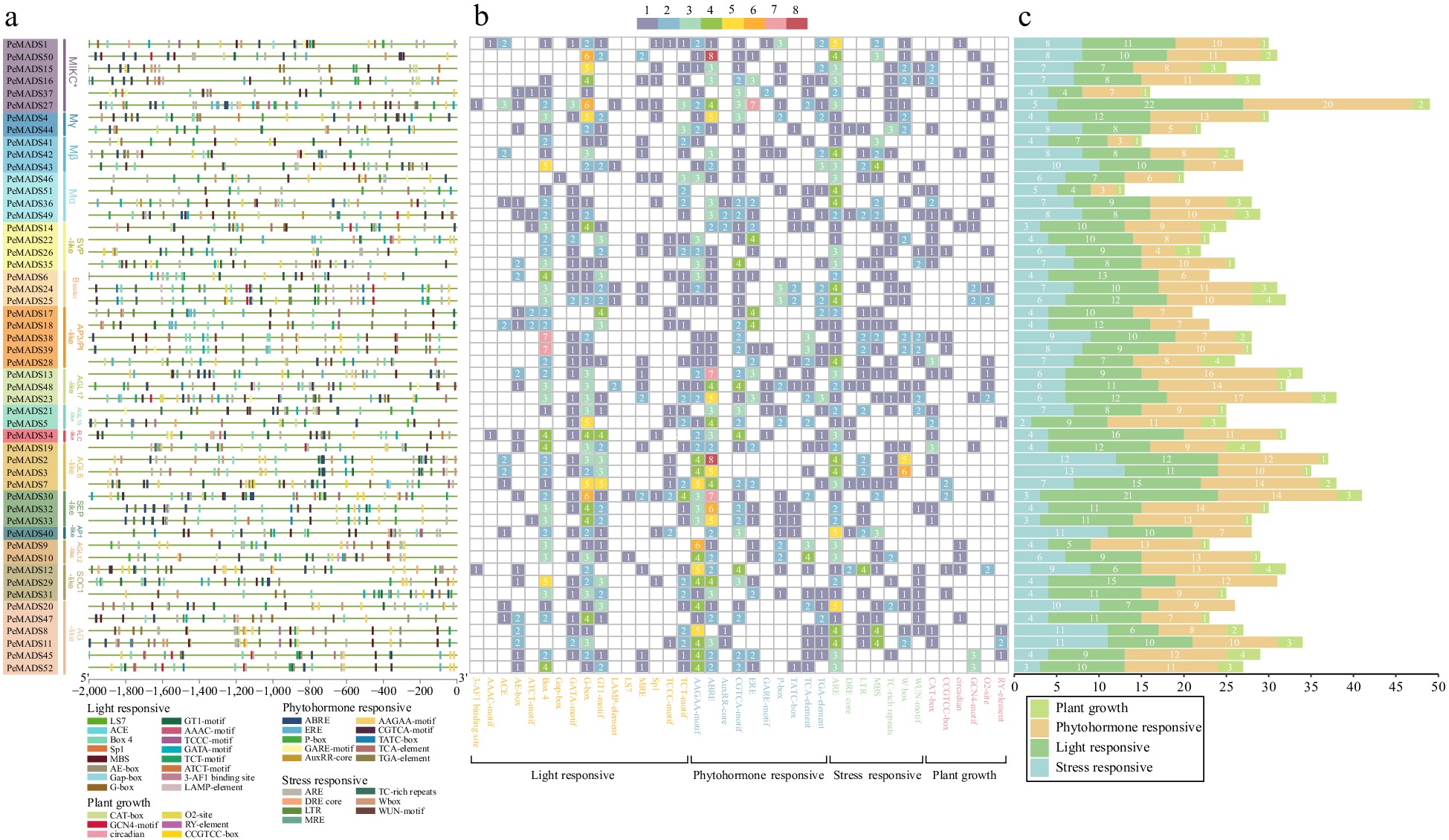
Figure 4.
(a) Analysis of cis-elements in the promoter region of PeMADS genes. (b) Heatmap of the number of cis-elements, the different color represents the number of cis-elements. (c) The sum of cis-elements in categories shown as a histogram.
Chromosomal location, synteny and evolution analysis
-
Based on the passion fruit genome's annotation files, 51 out of 52 PeMADS genes were assigned to nine linkage groups (LG01-LG09), and only one member (PeMADS52) was found on unassembled contig 7. The majority of MADS genes were concentrated on LG01 (19, 36.54%), particularly on the terminal region of LG01, encompassing approximately 33 Mb and containing 15 PeMADS genes. LG06 had the second highest number of MADS genes (12, 23.07%), followed by LG03 (5, 9.61%). The remaining linkage groups (LG02, LG04, LG05, LG07, LG08, LG09) contained only 1−3 PeMADS genes each. Numerous MADS genes were located near the proximate or the distal ends of the linkage groups. There was no apparent evidence suggesting a direct connection between the number of MADS genes and the length of linkage groups.
Based on the collinearity analysis conducted using MCScanX, only one tandem duplication gene pair, PeMADS42 and PeMADS43, was identified from LG06. Additionally, a total of 20 segmental duplication gene pairs were discovered, comprising 28 PeMADS genes located on duplicated segments across nine linkage groups (Fig. 5). Most duplicated gene pairs belong to the MIKCc group (24 pairs, accounting for 85.71% of the total). The MIKC* group and Mα have two gene pairs respectively, and those genes could be produced by whole genome duplication or the segmental genome duplication events.

Figure 5.
Distribution and collinearity of PeMADS genes in the passion fruit genome. PeMADSs marked by red has synteny with other genes. Gray lines indicate synteny blocks in passion fruit genome, brown lines indicate segmental duplicated MADS gene pairs of passion fruit.
To further investigate the evolutionary process of the passion fruit MADS-box gene family, a comparative systemic analysis was conducted between passion fruit and six representative species (Fig. 6), which cover three dicots (A. thaliana, S. lycopersicum and V. vinifera) and two monocots (O. sativa and Z. mays). The number of synteny blocks observed in A. thaliana, S. lycopersicum, V. vinifera, O. sativa and Z. mays were 996, 982, 922, 399 and 328 respectively. Among the 52 PeMADS genes, different numbers of genes exhibited syntenic relationships with the six species. A total of 33 members show colinearity with grape (33), followed by tomato (26), Arabidopsis (19) and rice (5), maize contains the least number (2). These findings suggest that the collinearity relationship between passion fruit and dicotyledons was closer than that between monocotyledons. Notably, most identified PeMADS exhibited synteny relationships with more than one species. PeMADS15 showed collinear counterparts in passion fruit and five other species, indicating its potential existence prior to ancestral divergence and its association with vital characteristics. Whereas some of the PeMADS syntenic gene pairs were exclusively found in dicotyledons or monocotyledons, suggesting their emergence after the divergence between these two groups. Syntenic PeMADS genes in five species are highlighted in Supplemental Table S7.
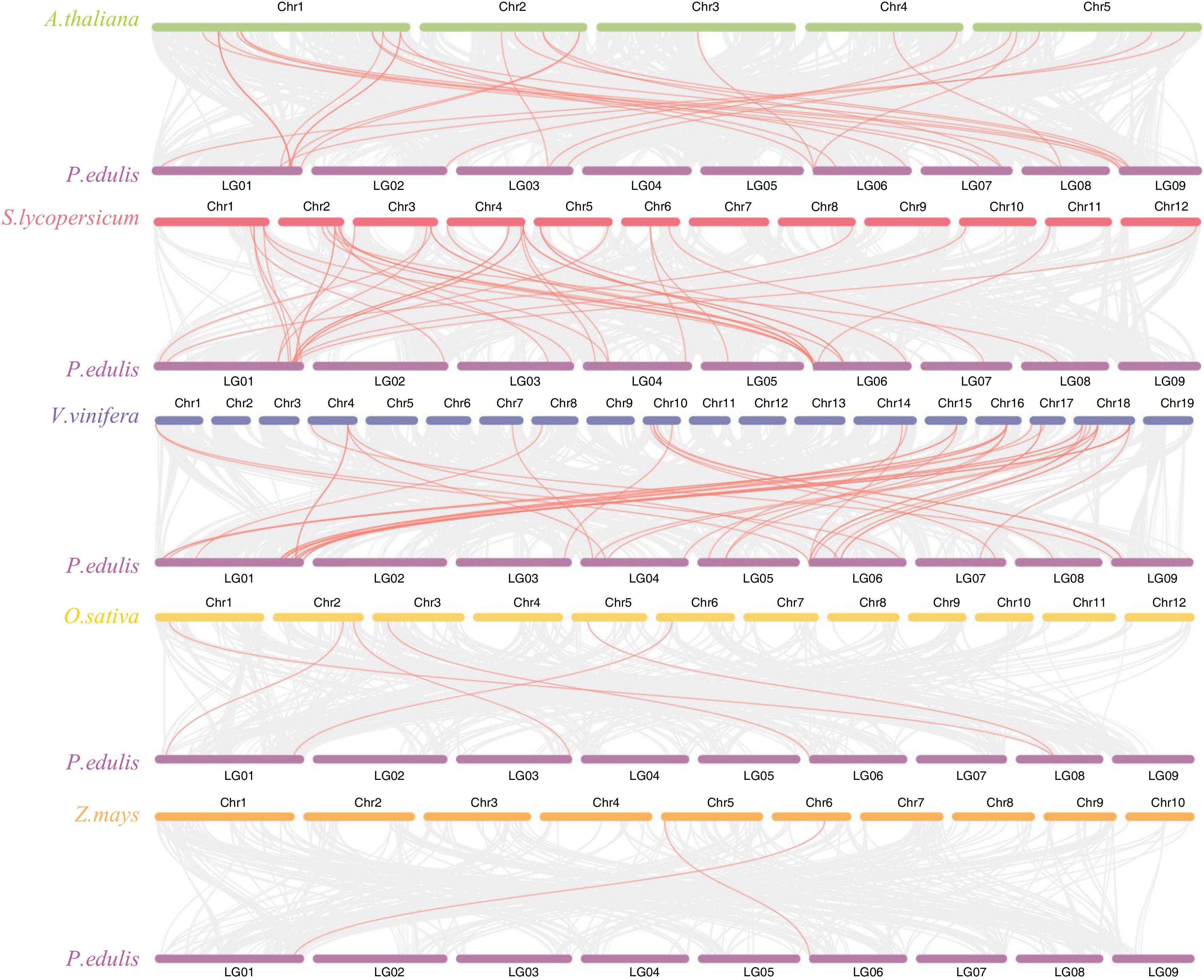
Figure 6.
Synteny analysis of PeMADS genes and five representative plants species. Gray lines in the background indicate the collinear blocks between passion fruit and other plant genomes, while red lines highlight the systemic gene pairs of PeMADSs between passion fruit and other plant genomes.
To gain insights into the evolutionary constraints affecting the passion fruit MADS-box gene family, we calculated the Ka/Ks (non-synonymous substitution/synonymous substitution) ratios for both segmental duplicated gene pairs and tandem duplicated gene pairs using the NG method. As shown in Table 2, it can be observed that all segmental duplicated gene pairs have Ka/Ks values below 1, indicating a strong purifying selective pressure acting on these duplicated gene pairs throughout evolution. In contrast, the only tandem duplicated gene pairs (PeMADS42 and PeMADS43) exhibited Ka/Ks > 1, suggesting that positive selection has influenced these gene pairs[65].
Table 2. The Ka/Ks ratios of the duplicated PeMADS gene pairs.
Duplicated gene pairs Ka Ks Ka/Ks Group Duplicated type PeMADS2 & PeMADS3 0 0.01 0 MIKCC/MIKCC Segmental PeMADS3 & PeMADS7 0.13 0.57 0.24 MIKCC/MIKCC Segmental PeMADS3 & PeMADS19 0.26 1.38 0.19 MIKCC/MIKCC Segmental PeMADS8 & PeMADS11 0.01 0.02 0.28 MIKCC/MIKCC Segmental PeMADS8 & PeMADS45 0.04 0.54 0.08 MIKCC/MIKCC Segmental PeMADS9 & PeMADS10 0.01 0.01 0.43 MIKCC/MIKCC Segmental PeMADS15 & PeMADS16 0 0.02 0.29 MIKC*/MIKC* Segmental PeMADS17 & PeMADS18 0.01 0.01 0.42 MIKCC/MIKCC Segmental PeMADS17 & PeMADS28 0.29 1.29 0.22 MIKCC/MIKCC Segmental PeMADS17 & PeMADS38 0.05 0.67 0.08 MIKCC/MIKCC Segmental PeMADS18 & PeMADS28 0.22 1.15 0.19 MIKCC/MIKCC Segmental PeMADS18 & PeMADS38 0.07 0.55 0.12 MIKCC/MIKCC Segmental PeMADS20 & PeMADS47 0.03 0.51 0.07 MIKCC/MIKCC Segmental PeMADS24 & PeMADS25 0 0.01 0.3 MIKCC/MIKCC Segmental PeMADS26 & PeMADS35 0.22 0.71 0.31 MIKCC/MIKCC Segmental PeMADS28 & PeMADS38 0.32 1.52 0.21 MIKCC/MIKCC Segmental PeMADS29 & PeMADS31 0.01 0.29 0.05 MIKCC/MIKCC Segmental PeMADS32 & PeMADS33 0.01 0.02 0.28 MIKCC/MIKCC Segmental PeMADS46 & PeMADS51 0.55 3.98 0.14 Mα/Mα Segmental PeMADS52 & PeMADS45 0.16 0.22 0.71 MIKCC/MIKCC Segmental PeMADS42 & PeMADS43 0.14 0.06 2.42 Mβ/Mβ tandem Homology modeling of PeMADSs tertiary structures
-
The protein structure homology modeling was performed using the SWISS-MODEL database, utilizing homologous templates retrieved from the PDB database. The three-dimensional models for all PeMADS protein sequences were generated and are presented in Supplemental Table S8. The predicted structures with highest GMQE and QMEAN scores for each subfamily were visualized using PyMOL and displayed in Fig. 7. Based on previous research on MADS transcription factors, the highly conserved MADS domain structure could be broadly categorized into one α-helix and two β-sheets. The flexible intervening domain was predominantly α-helical, while the K domain consisted of three α-helices. C-terminal domain generally exhibits variable structures[66,67]. Analogously, except for PeMADS7, which has the longest protein chain and a more complex structure, the remaining members displayed comparable structures, consisting of one to three α-helices, two β-sheets. Notably, nearly all members exhibited αββ structures in the N-terminal region, resembling the MDAS domain structure identified in human and yeast. However, the structure of other regions did not correspond to the expected characteristics of the I-domain and K-domain. This discrepancy may be attributed to the availability of suitable homologous models for the majority of PeMADS protein sequences. The coverage of homology modeling was approximately 50%, primarily focused on the N-terminal region, suggesting that successful modeling of certain structures towards the C-terminal region may have been limited.
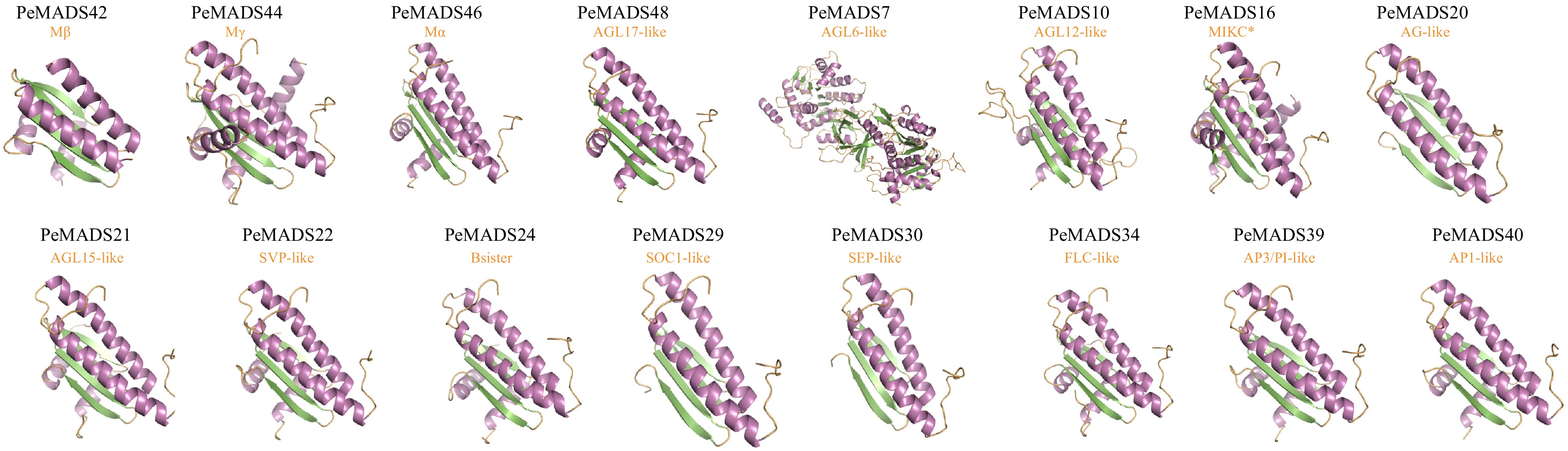
Figure 7.
Predicted three-dimensional structures of the passion fruit MADS-box protein sequences.
Expression patterns of PeMADSs in different floral tissues of passion fruit
-
Based on the conclusions drawn from previous studies, the 'ABC(D)E model' which proposes a hypothesis for the formation and identity of floral organs, was closely relevant to the MADS-box gene family[68]. In order to gain insights into the expression pattern of MADS-box genes, the flower (dividing into six species of floral structures) and the fruit were selected as sampling parts. The expression levels of 52 PeMADS genes were detected from these tissues during different development stages (Supplemental Table S9). Only the genes with the highest TPM value exceeding 6 are shown in Fig. 8.
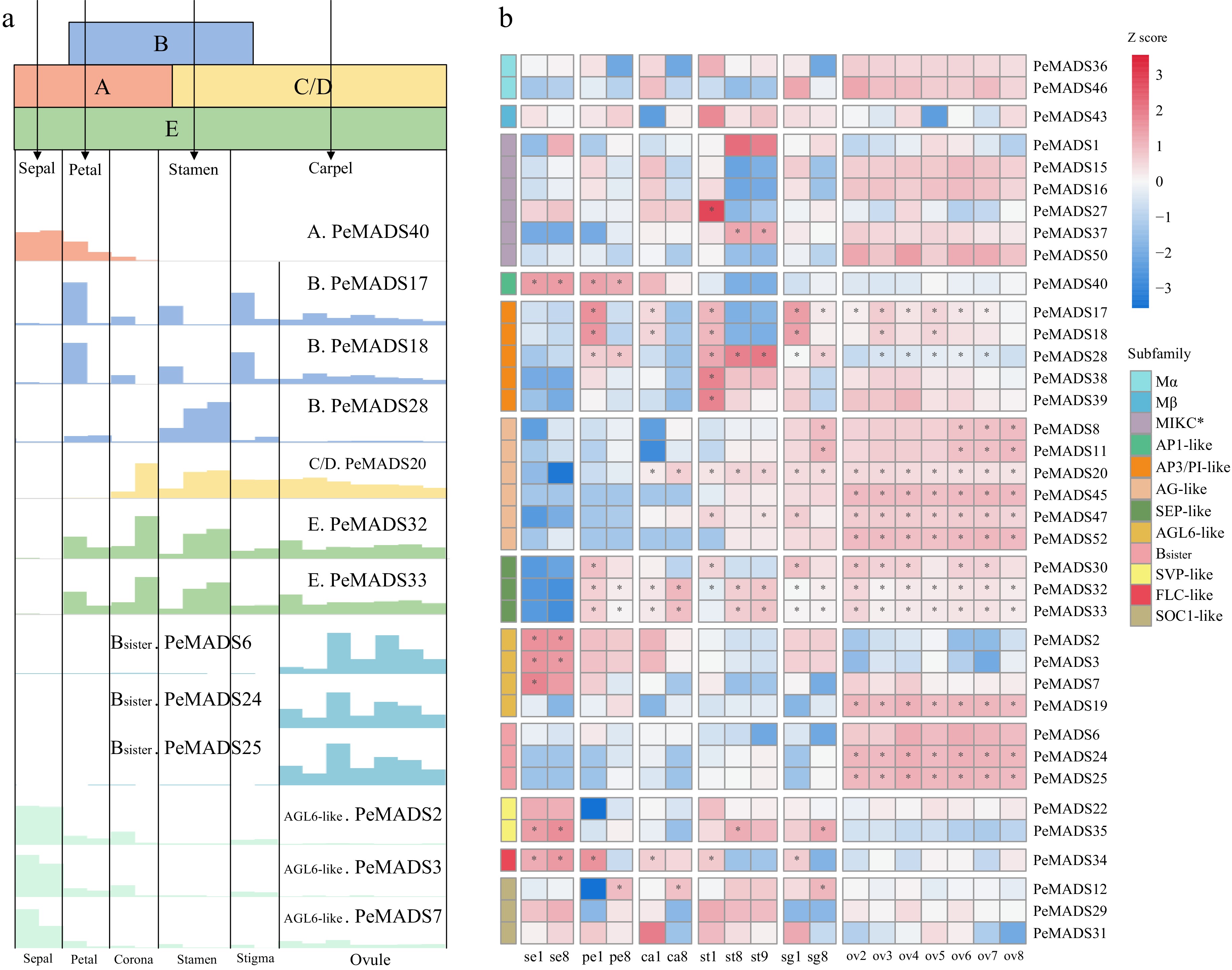
Figure 8.
The expression profile of MADS-box genes in floral tissues. (a) The ABC(D)E model in Eudicots, the bottom illustration indicates the gene expression values (bar heights) of ABC(D)E members in passion fruit. (b) Gene expression patterns of MADS-box gene family from floral tissues of passion fruit.
As shown in Fig. 8b, the expression level of Type I genes (PeMADS36/46/43) were generally lower compared to most Type II genes. As to Type II genes, PeMADS27 and PeMADS37 (MIKC* group) were detected in the early development stage and mid-and-late stage of stigmas, respectively, which might indicate genes from the MIKC* group influence stigmas development. Moreover, the expression profile within MIKCc group exhibited greater variability, with many genes showing high expression in multiple tissues. For example, PeMADS8/PeMADS11 were highly expressed in the last stage of stigmas and the last three stage of ovules. PeMADS32/33 were primarily expressed in any sampled tissues except sepals and the early stage of stamens. PeMADS17/18 were abundant in the early stages of petals, coronas, stamens, stigma and nearly all stages of ovules. Similarly, PeMADS28, also from the same subfamily, exhibited preferential expression in stamens, with slightly higher expression levels in petals and stigmas compared to other tissues. PeMADS40 was highly expressed in sepals and petals. PeMADS20 was mainly expressed in the late stage of coronas and stamens, nearly all stage of stigmas and ovules, and it was similar to the expression profile of PeMADS47, which exhibited relatively low expression in the corona, part stages of stamens and stigmas. PeMADS2/3/7 were abundant in sepals, petals and coronas. On the contrary, the expression patterns of many PeMADS genes were highly tissue-specific. For instance, PeMADS6/24/25/45/52 were abundant in all stages of ovules. PeMADS38/39 were only highly expressed in the first stage of stamen. Additionally, the expression levels of certain genes changed during the development process of specific tissue. For example, during the development of sepals and stigmas, the expression level of PeMADS35 gradually increased, and PeMADS28 displayed a similar ascending trend in stamens.
Based on the expression profiles described above, PeMADS genes that match the expression patterns of homologues genes within the same subfamily were identified as potential members of the ABC(D)E model of passion fruit. Ultimately, PeMADS40 was categorized as an A group member, while PeMADS17, PeMADS18 and PeMADS28 belong to the B group, PeMADS20 was identified as a member of the C group, PeMADS32/PeMADS33 were classified as members of the E group. Additionally, three genes, namely PeMADS6, PeMADS24, and PeMADS25, belong to the B sister group, while PeMADS2, PeMADS3, and PeMADS7 belong to the AGL6-like group. The details of the expression trends are shown in Fig. 8a.
To validate the reliability of predicted ABC(D)E model for passion fruit. Six PeMADS genes from different groups were selected as representatives for qRT-PCR analysis (Fig. 9b). PeMADS40, as an A group member, exhibited high expression levels in sepals, petals, and corona. B group member, PeMADS28 highly expressed in petals, stamens and sigmas. PeMADS20 (C/D group member) mainly expressed in the inner four whorls (ovule, stigma stamen and corona). PeMADS33 (E group gene) expressed in nearly all tissues excepting sepal. PeMADS6 (B sister) specifically expressed in ovules. PeMADS2 from AGL6-like group presented preferential expression in the first three whorls (sepal, petal and corona). Overall, the qRT-PCR analysis results were consistent with the RNA-seq data.
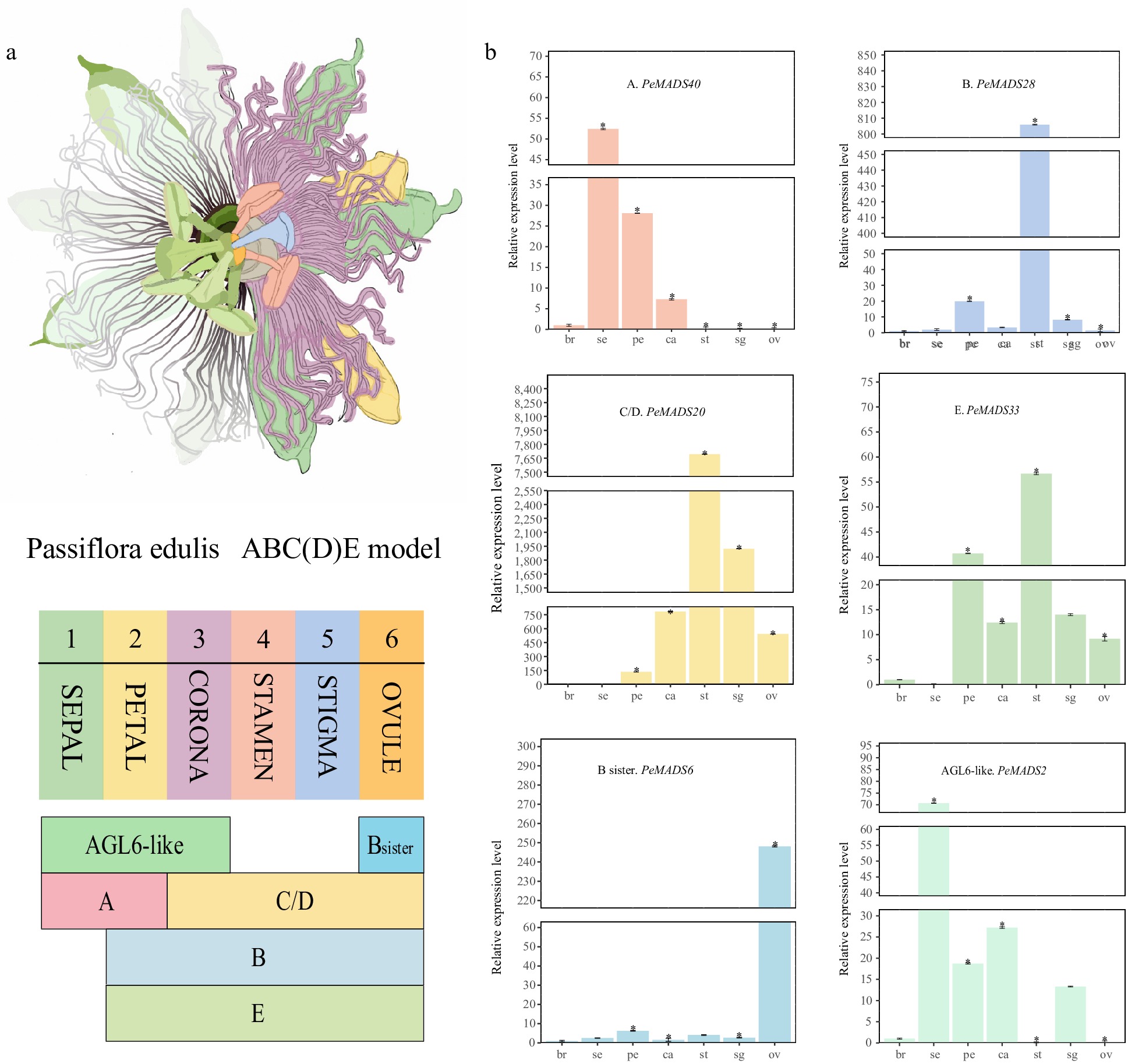
Figure 9.
qRT-PCR analysis of part members from the ABC(D)E model. (a) Illustration of passion fruit floral structure and summarization of the ABC(D)E model in passion fruit. (b) qRT-PCR results of six representative members from the ABC(D)E model, all experiments were performed independently at least three times. Error bars represent the standard deviation. Asterisks indicate significant differences in transcript levels compared with the early development stage of bract (br1). (* p < 0.05).
Expression profiles of PeMADSs genes in non-floral organs and under temperature and phytohormone treatment
-
Except for the core functions in floral meristem, MADS-box genes have been reported to be involved in various growth regulatory processes, like the maturation of fruit[39]. To further investigate the tissue-specific expression of PeMADS genes, the RNA-seq data of vegetative organs (tendril, stem, leaf) and fruits with different stages of maturation were extracted for analysis. Excluding the MADS-box members with rather lower expression level (highest TPM value is less than 5), 31 PeMADSs were retained (Fig. 10a). The expression levels of partial genes were higher in fruit tissue, like PeMADS2/11/32/33/34/47/45. Notably, PeMADS34 showed significant induction during the early development stage of fruits, with its expression decreasing as the fruit matured. Most genes expressed during the fruit maturation belong to the AG-like and SEP-like subfamilies. Conversely, certain PeMADS genes were primarily detected in vegetative organs. PeMADS9/12/40 highly expressed in tendril and stem. PeMADS22/35 mainly expressed in tendril, stem and leaf. The expression level of PeMADS1/29/31 were higher in stem and leaf. It should be noticed that the members from ABCD(E) model were also associated with the development of these non-floral organs, which might indicate the diversified functions of MADS-box members.
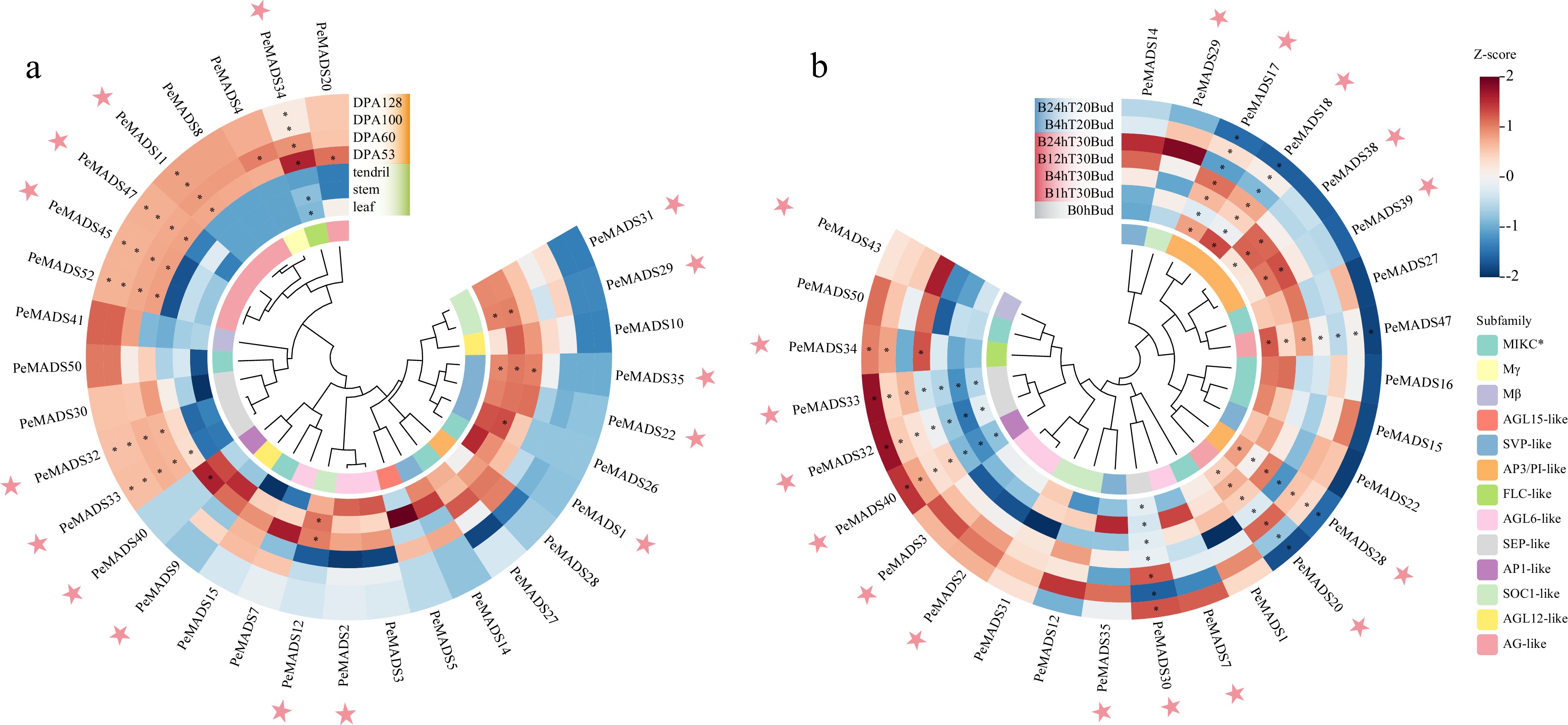
Figure 10.
Expression profiles of PeMADS genes in (a) non-floral organs and (b) under temperature stresses, DPA indicates days post anthesis; '1, 4, 12, 24 h' represents the time of stress treatment. Temperature of 20 °C (T20) is regarded as cold stress, while a temperature of 30 °C is regarded as heat stress (T30). The pink stars highlight the members with specific expression characteristics during fruit development or under temperature treatment.
The differentiation of flower buds and the development of floral organs in passion fruit are highly temperature-sensitive. As observed by Chang & Cheng, high temperatures (30/25 °C) could make flower buds more susceptible to abortion, while low temperatures (20/15 °C) result in the inhibition of flower bud formation[69,70]. To explore the response of PeMADS genes under low-temperature (20 °C) and high-temperature stress (30 °C), the RNA-seq data was acquired from examined buds with a series of processing-time gradient. After excluding members with low expression levels, significant differences in expression profiles were observed among the remaining PeMADS genes under temperature stresses (Fig. 10b). Some members were induced under cold conditions, including PeMADS2/3/30/32/33/34/40. Among them, the expression of PeMADS30/32/33/40 were significantly increased with longer processing time. The change in expression were more complex under heat conditions, the expression level of most genes increased in different degrees. PeMADS38/39, were up-regulated at 1 and 4 h of processing time, while down-regulated at 12 and 24 h. As for PeMADS7/35/20/28/29/30/43, their expression levels noticeably increased at specific processing times, while the expression of PeMADS17/18/47 were repressed under heat stress. Presumably, the E-class genes (PeMADS32/33) and the A-class genes (PeMADS40) might respond to cold stress, while the C/D-class gene (PeMADS20) and the B-class gene (PeMADS28) were mainly induced under heat stress. The expression levels of the aforementioned genes are listed in Supplemental Table S10.
To elucidate the response patterns of PeMADS genes to phytohormone treatments, qRT-PCR was employed to analyze the relative expression profiles of six selected PeMADS genes in plant leaves subjected to different phytohormones (ABA and GA) at various time points (0, 12, 24, and 48 h) (Fig. 11, Supplemental Table S11). Under ABA treatment, the expression of PeMADS2, PeMADS28, and PeMADS38 was significantly downregulated at different time points. The expression of PeMADS17 and PeMADS34 were initially induced and gradually upregulated at 12 and 24 h post treatment, followed by downregulation at 48 h. PeMADS19 exhibited a significant induction of high expression within a short period (12 h), but as the treatment time extended, the expression gradually decreased, with notable suppression observed at 48 h. In the case of GA treatment, PeMADS2 showed a gradual downregulation with the prolongation of treatment time, while the relative expression of PeMADS17 was initially suppressed at 12 h post-treatment, followed by induced upregulation, reaching its highest expression level at 24 h post-treatment. The relative expression of PeMADS19 was induced and upregulated after GA treatment, with the highest expression level observed at 48 h. Additionally, the expression of PeMADS28, PeMADS34, and PeMADS38 was all inhibited, with significant differences observed at 12 h. These findings suggested that PeMADS genes generally play roles in responding to phytohormones (Supplemental Table S11).
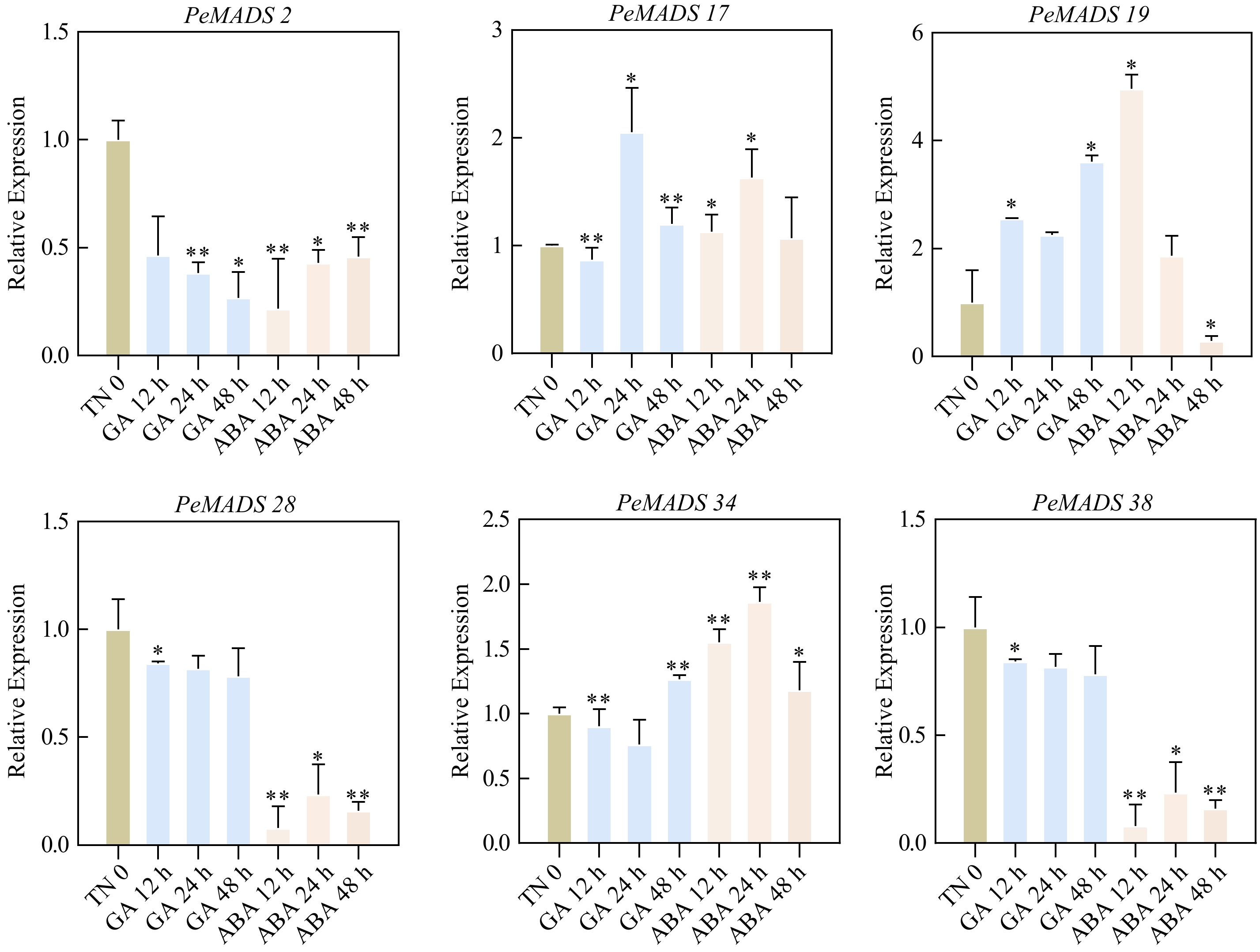
Figure 11.
Expression patterns of PeMADS genes in response to phytohormones. Leaf samples were collected at 0, 12, 24 and 48 h after GA and ABA treatments. Significant differences were analyzed by the Student's t-test (* p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001, and **** p-value < 0.0001).
-
The MADS-box gene family plays crucial roles in the morphology development of diverse plant organs, particularly in the floral development of angiosperms[71]. Since the completion of the genome sequences of numerous significant plant species, MADS-box genes have been systematically identified and examined at the genome level in various plants. In the present work, we have successfully identified 52 MADS-box genes in the genome of the passion fruit, and conducted analyses on their phylogenic relationships, gene structure, gene synteny and prediction of cis-elements in all members. However, it should be noted that the gene number is relatively low compared to other flowering plants (angiosperms). Several common crops, such as Arabidopsis, wheat, and tomato, possess over 100 members in the MADS-box gene family. It is well-known that whole genome duplication events play a significant role in the expansion of gene numbers and species diversification. Whole-genome duplication events exert a significant influence on gene proliferation and species diversification, prominently shaping the evolutionary trajectory of flowering plants through subsequent gene losses[72]. In the case of passion fruit, the reduction of type I genes resulted in a reduction in the size of the MADS-box gene family. Moreover, on a genome scale we reveal that specific subfamilies of PeMADS primarily evolved via segmental duplication rather than tandem amplification. Furthermore, the higher number of exons in type II (MIKC) genes (ranging from 5 to 15) compared to type I (1−2) is consistent with observations in other species, including sesame, rice, and soybeans. This findings align with the more intricate and versatile functions attributed to type II (MIKC) genes in contrast to type I (M-type) genes[73,74].
Expression of the ABC(D)E model
-
In this study, we provide a summary of the ABC(D)E model in passion fruit based on tissue-specific expression profiles. The six parts of floral organs in passion fruit were regulated by different combinations of ABC(D)E-class genes. Specifically, the carpel (including ovules and stigmas), stamens and coronas were controlled by the same pattern, B-, C(D)- and E-class genes. Petals are regulated by A-, B- and E-class genes. A-class genes is the only type functions in sepals (Fig. 9a).
Referring to the well-studied A. thaliana MADS gene family, the classic ABC(D)E model of floral organ identities can be described as follows: AP1 belongs to A group (performing functions for sepals and petals), AP3 and PI are classified as B group (performing functions for petals and stamens), AG is identified as C group (performing functions for stamen and carpel), while SEP genes belongs to E group (performing functions in all floral whorls)[75,76]. In our study, the expression profiles of passion fruit ABC(D)E homologues partly agree with the putative model derived from A. thaliana. The regulation of petals and stamens in passion fruit is similar to that of A. thaliana, but differs in terms of carpels and sepals. With the exception of previous detected C- and E-genes, we found that B-class genes are found expressed in carpels and corona, indicating an expanded regulatory role of B-class in passion fruit. Conversely, the types of genes that regulate sepals are reduced, with only A-class genes associated with sepal development. Notably, the mutation of the B-class gene DoLL1, in Physalis floridana leads to abnormal development of ovules and stigmas, suggesting the regulatory functions of B-class genes in carpels[77]. Previous studies have speculated that the wider expression patterns could be associated with subfunctionalization and neofunctionalization of ABC(D)E genes[78]. Hence, we suggest that the increased number of whorls regulated by B-class gene in passion fruit might be a result of adaption to its floral development.
In addition to the classic ABC(D)E model, two other types of homologues, B sister and AGL6-like genes, also exhibit tissue-specific characteristics in passion fruit flowers. The phylogenetic relationship indicates that AGL6-like genes are sister to SEP-like gene(E-class)[79,80]. Correlational research suggests that AGL6-like in rice could regulate the development of four whorls of floral organs, which is similar to the function of E-class genes[81]. The expression of AGL6 homologous gene has been detected in the first and second whorl of flowers in Hyacinthus orientalis[82]and Petunia hybrida[83]. AGL-6 like genes may be associated with inner perianth formation in angiosperms[84]. In passion fruit, E-class gene are detected in all flower whorls except for sepals. While the expression level of AGL6-like genes is extremely high in sepal. We propose that AGL6 homologues partly assume the function of E-class genes in the evolution of passion fruit and assist A-class genes in sepal identification. Similarly, B sister genes are primarily transcribed in the female reproductive organs of plants[85], and their aberrant expression leads to ovule development failure in wheat[86]. The same expression pattern is observed in passion fruit, where B sister genes are specifically expressed in ovule tissues, suggesting their role in ovule development. In addition to the above results, three members of MIKC* gene family exhibit prominent expression in stamens, aligning with previous speculations regarding the conserved function of MIKC* gene in male gametophytes of angiosperms[87]. Therefore, we propose the MIKC* genes may serve a similar function in male gametophytes of passion fruit.
Corona filaments are a distinctive structure in passion fruit and a significant attraction of its floral organs. However, the exact regulatory pathway underlying corona filament formation remains unknown. In this study, we found that corona filaments are mainly controlled by B-, C/D- and E- class genes, with C/D-class exhibiting the highest expression levels among all genes. Previous researches has also detected the expression of B- and C/D-class genes in coronas and the speculated that the tissue's origin is from stamens based on the species of regulated genes and its developmental characteristics[88]. Nevertheless, stronger evidence is still needed to elucidate the origin of the corona tissue.
Expansion of MADS-box genes in passion fruit
-
Arabidopsis has reported a total of 108 MADS-box genes[18], whereas passion fruit has only identified 52 MADS-box members. However, there is no significant difference in the number of Type II genes between two species (46 in Arabidopsis, 43 in passion fruit). Thus, the disparity mainly lies in the number of Type I genes (59 in Arabidopsis, nine in passion fruit), and duplication events of Type I genes are frequent in Arabidopsis[20]. These duplication events of Type I genes may explain the imbalanced distribution of MADS-box gene members between Arabidopsis and passion fruit.
Based on the phylogenetic relationship, the 37 MIKCc genes were unevenly classified into 12 gene clades (Fig. 2). Interestingly, compared to their homologous members in Arabidopsis within the same clade, there is an expansion of corresponding members in passion fruit, particularly in the AG-like, AGL6-like, AP3/PI-like and SVP-like clades. This expansion is especially prominent in the AG-like (C-class homologous of ABCE model) and AP3/PI-like (B-class homologous of ABCE model homologous) clades. In our study, most genes in the AP3/PI-like clade and all genes in the AG-like clade were generated through segmental duplications, and nearly all members showed high expression levels in various tissues. This broader regulation scope of the B- and C-class genes, compared to the classic ABC(D)E model, may explain the increase in these two clades. Moreover, there is a decrease in the number of gene members in certain clades, including SOC1-like, AP1-like and FLC-like. SOC1-like genes mediate the control of flowering through vernalization, photoperiod and gibberellin-dependent pathways[89]. The FLC gene serves as a central factor in Arabidopsis vernalization[64,90]. The loss of genes from these two clades in passion fruit could potentially affect the flowering machinery of the plant.
Involvement of PeMADS genes in development of non-floral organs and stress response
-
Beside the function in flower development, MADS-box genes also play a crucial role in multiple physiological processes of plants, including abiotic stress response and development of non-floral organs[91]. AGL4 encodes a MADS and are involved in weakening cell walls during dehiscence, abscission, and cellular expansion[92]. In passion fruit, its homologue (PeMADS32/33) exhibits expression in various tissues, with a significant up-regulation observed under cold stress conditions. Based on these findings, we propose that these two genes could play a role in cell wall remodeling during cold stress. Besides, hormone-responsive elements were detected in the putative promoter regions of many PeMADSs including PeMADS2/19/34, and expression analysis using qRT-PCR also revealed that these genes were widely involved in phytohormone responses. The diverse cis-regulatory elements in the promoter regions may contribute to the functioning of PeMADS genes in different processes. Our results shown that the expression profile of AP1 homologue (PeMADS40) exhibits exceptionally high expression in tendrils, which is consistent with previous studies[93]. It provides some evidence supporting the origin of tendrils. In banana and tomato, members of the AG-like and SEP-like subfamily are considered key regulators of fruit development and ripening processes[40,94,95]. Similarly, among the PeMADS genes that highly expressed in fruit of passion fruit, a significant portion belong to the AG-like and SEP-like subfamilies.
-
Passion fruit is an economic valuable tropical fruit crop characterized with nutritious juice, aromatic smell and bright flowers with distinct coronal filaments. Here, we present a comprehensive analysis of MADS-box genes in passion fruit, covering gene identification, phylogenetic relationships, gene structure, motif composition, chromosomal mapping, gene duplication and synteny, cis-element predictions, and tissue-specific expression profiles. A total of 52 PeMADS genes were identified and classified into two types with five subgroups (type I: Mα, Mβ, Mγ; type II: MIKCC, MIKC*) based on phylogenetic analysis. The relatively limited abundance of this gene family in passion fruit can be attributed to the reduction in type I genes, while certain subfamilies undergo amplification of PeMADSs predominantly through segmental duplications. Structural analysis including exon-intron organization, motif composition and homologous protein modeling reveals the relatively conserved features of PeMADSs within the same subfamily. Furthermore, we systematically discussed MADS-box genes involved in the ABC(D)E model of flower organ identity. Our findings indicate that the regulation of petals and stamens in passion fruit is similar to that of Arabidopsis but diverges concerning carpels and sepals. Notably, the distinctive floral organ in passion fruit, the corona, is primarily controlled by B-, C(D)-, and E-class genes, supporting the speculation of its stamen origin. In addition to the classic ABC(D)E model, B sister and AGL6-like genes exhibit tissue-specific characteristics in passion fruit flowers. AGL6 homologues might be involved in sepal identification, while B sister genes are specifically expressed in ovule tissues, suggesting their role in ovule development. Besides, many of PeMADSs tend to express in both reproductive and vegetative organs, and some members are also induced under temperature stresses. These results give an insight into the relationship between the structure and function of MADS-box genes in passion fruit. Ultimately, this study lays a foundation for further investigations into the functions of MADS-box genes in passion fruit tissue development, particularly in flower organ identity.
-
The authors confirm contribution to the paper as follows: study conception and design: Qin Y, Zheng P; data collection: Lu L, Chen S, Shen M; analysis and interpretation of results: Cai X, Liu R, Wang X; draft manuscript preparation: An C, Liao J. All authors reviewed the results and approved the final version of the manuscript.
-
The data presented in this study are available in the article, Supplementary materials and online repositories. The passion fruit genome data and transcriptome data used in this work were deposited in the National Genome Data Center (NGDC) (
https://ngdc.cncb.ac.cn ) database under accession number GWHAZTM00000000 and CNP0002747, respectively. This work was supported by Science and Technology Innovation Project of Pingtan Science and Technology Research Institute (PT2021007, PT2021003), General Project of Guangxi Natural Science Foundation (2022GXNSFAA035535), Guangxi Academy of Agricultural Sciences basic Research Project (Gui Nong Ke 2021YT046). We thank Zhenjiang Zheng from Fujian Lianmi Ecological Agriculture Development Co., LTD for his assistance during sample collection.
-
The authors declare that they have no conflict of interest.
-
accompanies this paper at (https://www.maxapress.com/article/doi/10.48130/tp-0024-0004)
-
Received 3 November 2023; Accepted 23 January 2024; Published online 20 February 2024
-
52 MADS-box genes were identified in passion fruit genome, and the reduction of type I genes contributed to the reduction in the size of the MADS-box gene family.
A putative flower regulation ABC(D)E model of passion fruit were proposed, and the unique floral structure--coronas, similar to stamens, were predominately controlled by B-, C(D)- and E-class genes.
Many PeMADS genes were also involved in the development regulation of non-floral tissues and/or in phytohormone and stress responses.
-
# Authors contributed equally: Chang An, Jingyi Liao, Lin Lu
- Supplemental Table S1 Transcriptome sample information for organ development of passion fruit flower.
- Supplemental Table S2 Specific primers for PeMADS genes for qRT-PCR.
- Supplemental Table S3 List of CDS and protein sequences of 52 PeMADS genes.
- Supplemental Table S4 Multiple sequence alignment results of MADS-domain in PeMADS proteins.
- Supplemental Table S5 Protein sequences of MADS gene families used in phylogenetic analysis.
- Supplemental Table S6
- Supplemental Table S6 Cis-acting elements in promoter region of PeMADS genes.
- Supplemental Table S7 Results of intraspecific collinearity analysis(intraspecific and interspecific).
- Supplemental Table S8 PeMADS protein homology modeling results.
- Supplemental Table S9 Expression matrix of PeMADS genes in different development stages of floral organs.
- Supplemental Table S10 Expression matrix of PeMADS genes in vegetative organs.
- Supplemental Table S11 Expression patterns of PeMADS genes in response to phytohormones.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
An C, Liao J, Lu L, Cai X, Liu R, et al. 2024. From gene expression to flower patterns: genome-wide characterization of the MADS-box gene family in passion fruit (Passiflora edulis). Tropical Plants 3: e004 doi: 10.48130/tp-0024-0004 

From gene expression to flower patterns: genome-wide characterization of the MADS-box gene family in passion fruit (Passiflora edulis)
- Received: 03 November 2023
- Revised: 15 January 2024
- Accepted: 23 January 2024
- Published online: 20 February 2024
Abstract: Passion fruit (Passiflora edulis) is an economically valuable tropical fruit crop renowned for its nutritious juice, aromatic fragrance, and vibrant flowers with distinct floral structure, known as corona. These unique floral features make passion fruit a good candidate for the study of floral organogenesis. MADS-box genes play essential functions in various aspects of plant growth and development, especially for floral morphogenesis. However, a comprehensive investigation of MADS-box gene family members in passion fruit has not yet been conducted. Here, 52 MADS-box genes were identified in the passion fruit genome and classified into two types with five subgroups (type I: Mα, Mβ, Mγ; type II: MIKCC, MIKC*) based on phylogeny. The notable reduction in the abundance of type I MADS-box genes within the passion fruit genome, in contrast to Arabidopsis, is thought to be a contributing factor to the diminished gene number within this gene family. The structural analysis illustrated that PeMADS proteins within the same subfamily are relatively conserved. Moreover, we proposed the putative flower regulation ABC(D)E model of passion fruit and explored the regulatory genes of the characteristic structure corona. We found that the regulation of petals and stamens in passion fruit is similar to that of Arabidopsis, but differs in terms of carpels and sepals. The distinct coronas were predominately controlled by B-, C(D)- and E-class genes, supporting the speculation that the corona might have originated from stamens. Except for the core functions in the floral meristem, many PeMADS genes were also involved in the development regulation of non-floral tissues and/or in phytohormone and stress responses. The co-presence of diverse cis-regulatory elements associated with growth/developmental regulation, phytohormone and stress responses in the promoter regions of PeMADSs might be closely related to their diverse regulatory roles. The results of this study provide valuable insights into the MADS-box genes in passion fruit and their involvement in the development of floral structure. These discoveries lay the foundation for the cultivation of exceptional ornamental varieties of passion fruit.
-
Key words:
- Passion fruit /
- MADS-box gene /
- Floral organ identity /
- ABC(D)E model.