









-
Arbuscular mycorrhizal fungi (AM fungi) are root-associated, mutualistic fungi within the Glomeromycota phylum[1]. Over 80% of all land plants associate with AM fungi, which are thought to have coevolved with their plant partners from the beginning of terrestrial plant life[2]. However, our knowledge on AM fungal communities is still limited, particularly in many underexplored ecosystems. One understudied ecosystem is the pine barrens.
As its name suggests, the pine barrens’ distinction lies in its sandy, acidic, oligotrophic soils, and frequent fires. The New Jersey pine barrens forests have canopies dominated by pitch pine trees (Pinus rigida) and several oak species (Quercus spp.) and understories of shrubby oaks and heath plants with grasses and sedges in the open areas[3]. The pine barrens ecosystem is scattered throughout the eastern United States and to a lesser extent, worldwide, among which, the pine barrens located in southern New Jersey is the largest (1.1 million acres)[3]. Pine barrens’ soils are particularly acidic because there is little clay present, little humus, and low cation exchange capacity[3]. Little is known about the AM fungal communities that inhabit this ecosystem. Numerous novel fungal species have been recently uncovered in the pine barrens[4], demonstrating that the pine barrens is rich in fungal diversity. However, those studies focused on culturable fungi in the phylum Ascomycota. To date, only one study has looked at the microscopic abundance of AM fungi in plant roots in the pine barrens[5]. No molecular or extensive AM fungal community data has yet been collected from this ecosystem[5].
Similar to the pine barrens, hostile soil conditions, such as acidic soils, are also common in anthropogenic sites. In fact, thirty to forty percent of croplands are inhibited by acidic soils (pH < 5), making soil acidity one of the most crucial issues facing agroecosystems [6]. It has been reported that AM fungal community composition can be shaped by soil pH[7], which is considered one of the major predictors of AM fungal community change[8]. Some AM fungal species are more prominent than others in acidic soils[9], which has been explained by their adaptation to low pH[10].
In addition to soil chemistry, land management has also been shown to play a role in AM fungal community composition. A number of studies have investigated the impact of land use, such as mowing and fertilization, on AM fungal community composition. Some studies have shown that land use intensity decreases AM fungal OTU richness[11] or does not impact OTU richness at all[12]. Others have reported that intensive land use can change AM fungal community composition[13]. A more recent study by Sepp et al.[14] detected differences in AM fungal community composition between habitats with varied land use intensity. Impacts of land use on AM fungal species richness can be context dependent and can vary based on the type of fertilizer used or the particulars of the ecosystem observed[15]. Sampling from additional ecosystem types may show different trends. Gaining insight into the naturally occurring AM fungi in varying environments, including acidic soils and anthropogenic sites, may help determine which AM fungal species may be important in alleviating crop stresses.
One such important crop is switchgrass (Panicum virgatum L.), a C4 perennial warm-season grass, native to the eastern two-thirds of the USA and, most importantly, a model bioenergy feedstock species (i.e. by the Department of Energy). Switchgrass is one of the native and dominant grasses in the pine barrens and, therefore, we used AM fungi associated with switchgrass roots as a model to study AM fungal diversity. Switchgrass AM fungal communities were studied in both the pine barrens and from annually mowed switchgrass field plots (agroecosystems), as well as, an Iowa natural prairie preserve. Switchgrass was collected from an Iowa prairie in order to have a natural switchgrass site with contrasting soil properties to the other sites. The objectives of this study were to survey the diversity of AM fungal communities associated with switchgrass in pine barrens ecosystems in New Jersey and New York via Illumina sequencing, to identify novel or pine barrens-specific AM fungal clades, and to compare the AM fungal community of the pine barrens to those in a natural prairie and agroecosystems.
-
Out of 5,734,024 total raw reads (1,964 total OTUs) detected from all 33 samples, 1,822,463 reads (497 raw OTUs) belonged to the Glomeromycota phylum. This represents about 32% of our study’s raw reads (25% of raw OTUs). Several other fungal clades were observed: 542 OTUs matched to Basidiomycota, 130 OTUs to Ascomycota, 129 OTUs to Chytridiomycota, 41 OTUs to Mucoromycotina, as well as, OTUs that were identified as other Eukaryotic lineages or not assigned any taxonomy. After rarefying the data to the median number of reads, 423 AM fungal OTUs remained, consisting of 537,522 total reads. The number of AM fungal reads (rarefied) ranged from 26,451 in LIPB16 to 63,147 in CM, RP, and AF17 (Table 1). The number of AM fungal OTUs ranged from 19 in EC to 111 in IO (Table 1). Rarefaction curves show that some sites had AM fungal species saturation with the sampling effort put forth. However, other sites, specifically FAA, CM, WSF, and AF17, did not appear to reach asymptotic species saturation (Fig. 1). More sampling effort in these sites may result in additional species detection.
Table 1. The number of AM fungal operational taxonomic units (OTUs) and reads detected from each of the 11 sampled sites, obtained through Illumina sequencing. Three diversity indices (Shannon, Simpson, and Fisher's α) are compared.
Site No of Samples No of OTUs
(observed)No of OTUs
(rarefied)No of Reads
(observed)No of Reads
(rarefied)Shannon Simpson Fisher's α AF14 (M) 3 74 74 41,819 41,819 2.366 0.869 8.732 AF17 (M) 3 153 89 710,459 63,147 1.736 0.675 10.973 CM (P) 3 97 81 156,356 63,147 1.023 0.367 9.549 EC (M) 3 19 19 35,821 35,245 0.952 0.4 1.937 FAA (P) 3 83 76 169,083 54,286 2.661 0.877 8.826 IO 3 113 111 48,525 44,057 3.148 0.927 13.892 LIPB14 (P) 3 30 30 33,757 33,757 0.756 0.283 3.243 LIPB16 (P) 3 25 25 26,451 26,451 0.655 0.261 2.723 RP (P) 3 39 33 367,234 63,147 1.911 0.8 3.352 SO (M) 3 103 102 63,304 56,003 2.12 0.791 11.949 WSF (P) 3 102 84 169,654 56,463 2.089 0.811 9.428 ‘Observed’ refers to raw abundance and ‘rarefied’ refers to abundance after rarefying to the median number of reads.
Site abbreviations are: AF14 - Adelphia Research Field 2014, AF17 - Adelphia Research Field 2017, CM - Colliers Mills, EC - EARTH Center, IO - Iowa, FAA - Federal Aviation Administration, LIPB14 - Long Island Pine Barrens 2014, LIPB16 - Long Island Pine Barrens 2016, RP - Rocky Point, SO - Somerset Research Field, WSF - Wharton State Forest. (M): managed field site, (P): pine barrens site.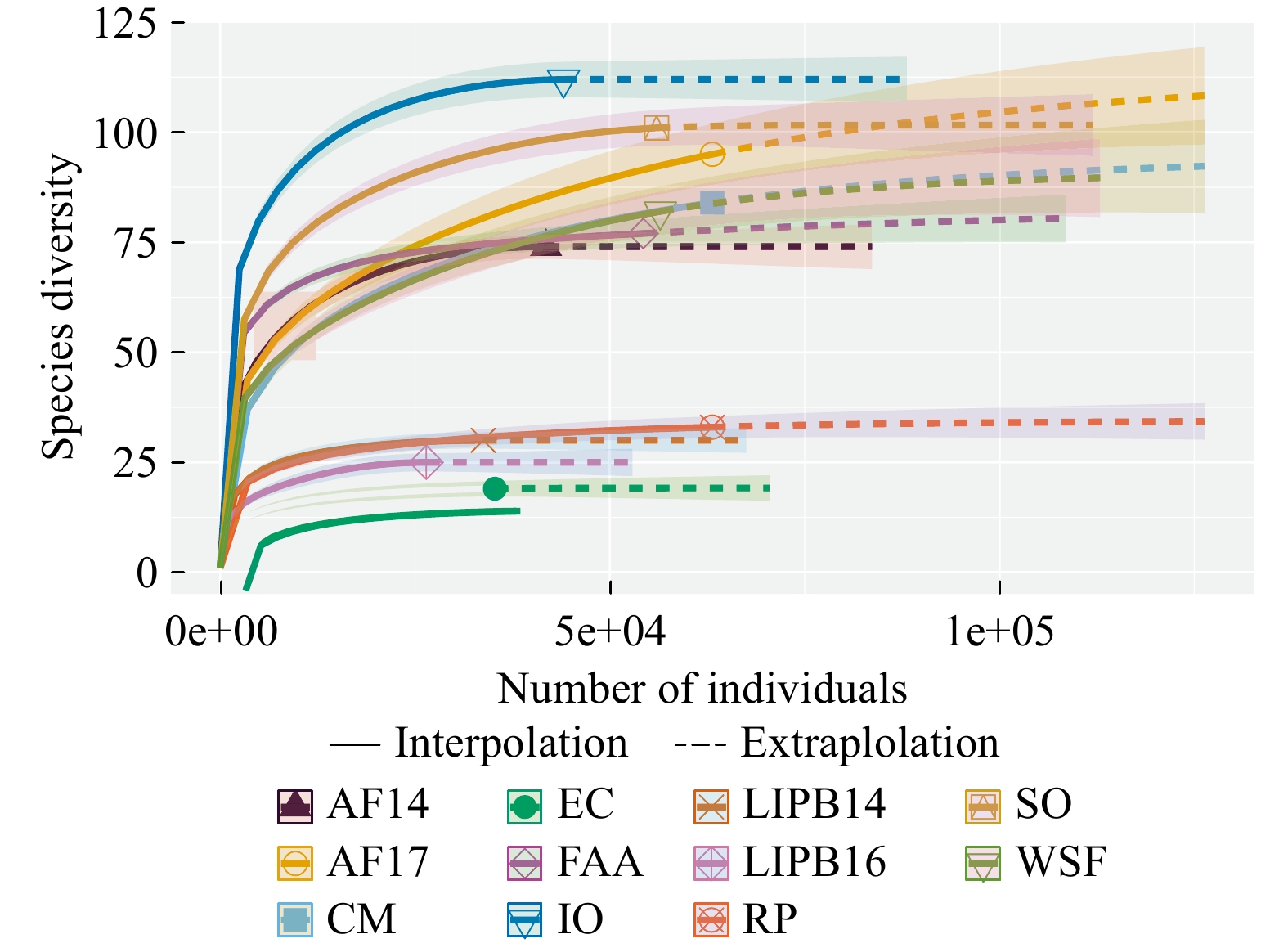
Figure 1. Rarefaction analysis of AM fungal communities from all sampled sites. AF14 - Adelphia Research Field 2014, AF17 - Adelphia Research Field 2017, CM - Colliers Mills, EC - EARTH Center, IO - Iowa, FAA - Federal Aviation Administration, LIPB14 - Long Island Pine Barrens 2014, LIPB16 - Long Island Pine Barrens 2016, RP - Rocky Point, SO - Somerset Research Field, WSF - Wharton State Forest.
This study uncovered a wide array of diversity, with all four AM fungal orders represented (Fig. 2, Table 2). Thirteen genera or genus-rank taxa were obtained through Illumina sequencing of switchgrass roots from all sampled sites (Table 2). OTUs that could not be identified to the genus level were labeled as ‘undescribed’ taxa, representing either undescribed clades or discrepancies in the reference database. The vast majority of this study’s total AM fungal OTUs (59%) and reads (78%) belonged to Glomus (251 OTUs, 419,908 reads) (Table 2). The second and third most diverse OTUs (clades with large amounts of OTUs) were Claroideoglomus and Acaulospora, with 42 and 38 OTUs, respectively. The top five most abundant OTUs (the OTUs with the most reads) all matched to Glomus species and these five OTUs accounted for 54% of all reads (288,355 reads). The second and third most abundant genera were Gigaspora and Clairoideoglomus, with 26,731 and 26,213 reads, respectively (Table 2).

Figure 2. Maximum-likelihood phylogenetic tree of the AM fungal diversity detected in this study. Numbers on branches indicate percent bootstrap support for 1000 replications. Bootstrap values > 70% are shown. Bold names represent sequences obtained from this study. AM fungal families are aligned vertically.
 = sequences retrieved exclusively from managed lands;
= sequences retrieved exclusively from managed lands;
 = sequences retrieved exclusively from the Iowa prairie;
= sequences retrieved exclusively from the Iowa prairie;
 = sequences retrieved exclusively from pine barrens forests.
= sequences retrieved exclusively from pine barrens forests.
Table 2. Distribution and abundance of AM fungal OTUs and reads (rarefied) among all sites. Colors represent a common order. Abundance of each taxon found under different land uses (managed, pine barrens, Iowa) is included.
Order Family Genus No of OTUs No of Reads by Land Use Managed Pine Barrens Iowa Archaeosporales Ambisporaceae Ambispora 4 0 65 0 Archaeosporaceae Archaeospora 10 301 268 0 N/A Undescribed Archaeosporales 1 0 20 0 Diversisporales Acaulosporaceae Acaulospora 38 0 23,042 643 Diversisporaceae Diversispora 3 358 87 618 Gigasporaceae Undescribed Gigasporaceae 12 1,789 12,085 0 Gigasporaceae Gigaspora 15 9,335 17,396 0 Glomerales Claroideoglomeraceae Claroideoglomus 42 14,068 7,885 4,260 Glomeraceae Glomus 251 156,470 235,695 27,743 Glomeraceae Undescribed Glomeraceae 12 742 156 1,792 N/A Undescribed Glomerales 20 12,430 266 8,993 Paraglomerales Paraglomeraceae Paraglomus 14 712 286 8 N/A N/A Undescribed Glomeromycetes 1 9 0 0 IO had the highest OTU richness. Additionally, all three diversity indices showed IO with the highest diversity index scores, indicating higher total AM fungal diversity in that location (Table 1). The second highest indices were FAA (Simpson and Shannon), and SO (Fisher’s α). The lowest diversity indices were found in LIPB16 (Simpson and Shannon) and EC (Fisher’s α) (Table 1).
Overall, Glomus was, by far, the most common genus detected from three land use categories in this study (managed, pine barrens, and Iowa). All OTUs identified in the Ambisporaceae and Acaulosporaceae families were found exclusively from the natural (non-managed) ecosystems, and almost entirely in the pine barrens sites. Only three OTUs in the Acaulosporaceae family were found in a non-pine barrens location (IO), while all Ambisporaceae OTUs were found in the pine barrens exclusively (Supplementary Table S1, Table 2). Some of these uniquely pine barrens derived OTUs had low sequence identity matches in both the NCBI and MaarjAM databases, indicating that these may represent new, undescribed AM fungal clades. These include OTUs such as CM1_1135 and FAA1_959 (Supplementary Data S1). These isolates matched Acaulospora sp. in the MaarjAM database with 94%, 92%, 92%, and 93% identity matches, respectively. Such findings may necessitate additional investigation. Additionally, there were some taxa that were more abundant in managed field sites. Diversispora spp. were more than four times more prolific in managed sites (358 reads) than in the pine barrens (87 reads) (Table 2). The most abundant Archaeospora sequence in this study (AF172_1184) was found exclusively in managed sites (Fig. 2, Table 2).
Soil and management impacts on AM fungal communities
-
Pearson’s correlation tests evidenced a positive correlation between soil pH and AM fungal diversity (Shannon, Simpson, and Fisher indices) (r = 0.53, P = 0.0017; r = 0.40, P = 0.0204; r = 0.41, P= 0.0174 respectively). ANOSIM and PERMANOVA analyses similarly showed significantly different AM fungal communities under acidic vs. non-acidic soil pH (R = 0.234, P = 0.005; pseudo-F = 3.440, P= 0.006, R2 = 0.100 respectively). ANOSIM and PERMANOVA analyses showed significantly different AM fungal communities among ‘Pine Barrens’, ‘Managed’, and ‘Iowa’ sites (R= 0.357, P = 0.001; pseudo-F = 4.286, P= 0.001, R2 = 0.222 respectively). NMDS analysis (Fig. 3) showed that pine barrens, managed, and Iowa AM fungal communities grouped separately. NMDS analysis also showed that triplicate samples grouped together, with the exception of EC.
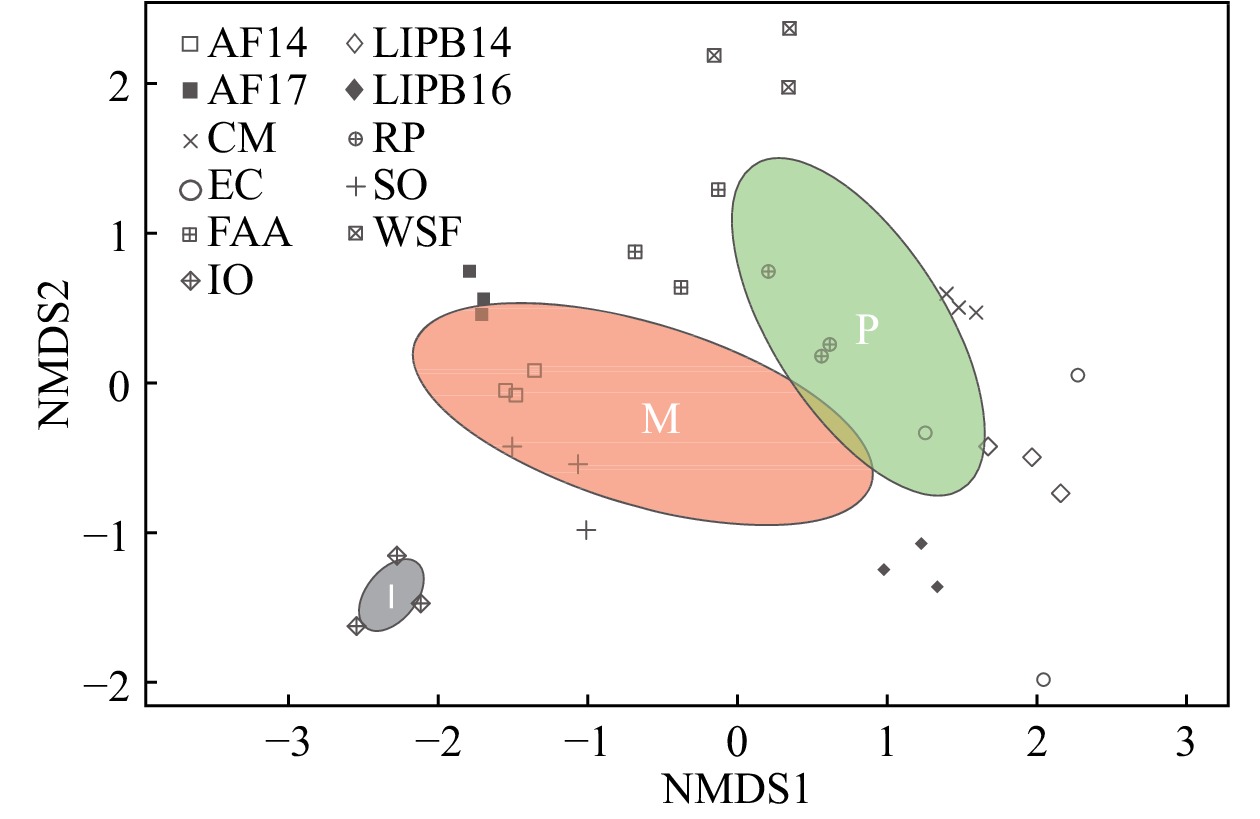
Figure 3. Nonmetric multidimensional scaling (NMDS) plot of AM fungal communities from 33 switchgrass samples from 11 sites. This analysis was based on Bray–Curtis dissimilarity measurements between samples (non-pooled) (stress = 0.147). A green ellipse with a ‘P’ indicates pine barrens AM fungal diversity, a red ellipse with an ‘M’ indicates managed AM fungal diversity, and a grey ellipse with an ‘I’ indicates Iowa AM fungal diversity. Pine barrens sites included CM, FAA, LIPB14, LIPB16, RP, and WSF. Managed sites included AF14, AF17, EC, and SO. The Iowa site was exclusively IO. Different shapes represent the sampling sites.
-
Our study found that switchgrass populations surveyed in this study are inhabited by a large diversity of AM fungi, representing all four known AM fungal orders. OTUs detected from the Ambisporaceae and Acaulosporaceae families were derived almost entirely from pine barrens sites; some of these may represent new AM fungal lineages. Additionally, different land uses and soil conditions harbored different communities of AM fungi. We found that managed switchgrass sites had a significantly different AM fungal community compared to natural sites. Soil acidity was also correlated with AM fungal diversity. However, this study was not a controlled experiment and, therefore, conclusions on the drivers of diversity in these sites cannot be made on these data alone.
Our finding that soil pH was correlated with AM community diversity is in line with several other studies[7,16]. However, some meta-analyses have shown little impact of soil pH on regional differences in AM fungal communities[17] and, instead, found biogeographic history and AM fungal dispersal (or lack thereof) to play a larger role[18]. It was surprising to find that the New Jersey pine barrens forests had, in general, more AM fungal diversity compared to the Long Island pine barrens forests (Table 1) because we expected there to be a common trend between all pine barrens sites. The biogeographic history of Long Island and climatic differences due to its being a thin island strip may, in this case, be playing a larger role than plant community and soil properties. The extent to which these fungi are specifically adapted to certain soils is an ongoing debate[16,19]. Nevertheless, environmental features are likely the underlying ‘proxy indicators’ of AM fungal communities[14,20].
The primer pair used in this study (AMV4.5NF-AMDGR) was only 32% AM fungal specific. Contrastingly, Van Geel et al.[21] and Lumini et al.[22] found 72% and 76% AM fungal specificity with this primer pair, respectively. This difference may stem from the fact that they used different plant hosts and PCR parameters. Cui et al.[23] found many other organisms represented in their AM fungal Illumina study, similar to our study, though they do not mention any particulars on the other taxa found. Cao et al.[24] found only 24% AM fungal specificity with this primer pair, very similar to the specificity of our study. They also found large abundance of members of the Basidiomycota and Chytridiomycota[24]. A longer and more Glomeromycota-specific DNA barcode may improve the specificity and taxonomic resolution problems[25]. Additionally, this study highlighted the need for consistent, replicable workflows for sampling and bioinformatic analysis[26], as next-generation sequencing (NGS) and Illumina, in particular, are relatively new techniques for uncovering AM fungal diversity. Collaboration with experienced bioinformaticists, in particular, is necessary to ensure ‘real’ answers to important biological questions.
Interestingly, Ambispora spp. were found exclusively in the pine barrens, and Acaulospora spp. were found almost exclusively in the pine barrens sites, with only three OTUs observed in non-pine barrens ecosystems (Iowa). This distribution pattern corroborates the finding that Acaulospora and Ambispora may be more adapted to acidic soils compared to other AM fungi[27,28]. In the study by Oehl et al.[27], spores of Ambispora sp. were only observed from soil pH of 5.0, while Nicolson and Schenck[29] found only Acaulospora laevis spores in soil pH of 4.0−4.5, and Young et al.[28] found A. laevis to predominate in soils of pH 4.3−4.8. Castillo et al.[30] found Acaulospora sp. to also be the most common AM fungal species found in the acidic soils of Southern Chile (pH 5.5). Oehl et al.[31] reported Acaulospora sp. to be rare in conventional farmland and French et al.[32] found Acaulospora sp. in only natural sites, just as our study found. This study found switchgrass associated Ambispora spp. in samples strictly from soils of pH 4.8 and 5.0 (RP and CM, respectively) and most Acaulospora spp. from pH 4.8−5.2. Clark[33] summarizes the findings of other authors which found Glomus, Acaulospora, and Gigaspora to be the most predominant genera inhabiting acidic soils. This gives credence to our findings, since Glomus and Acaulospora were similarly found to be the dominant genera in this study and Gigaspora was much more dominant in the pine barrens (17,396 reads) compared to managed fields (9,335 reads) and the Iowa prairie (0 reads) (Table 2). It is likely that these species of AM fungi are more adapted to acidic soils. Additional experiments are needed to test this hypothesis.
Our results corroborate previous reports that anthropogenic land use impacts AM fungal communities[13,34]. While some studies found that intensive agricultural practices tend to yield lower AM fungal diversity compared to natural sites[11,35], others find surprisingly diverse communities in agricultural sites, particularly when fertilization and tillage are kept to a minimum[36]. Differences in these results could stem from differences in methodology (primer bias, NGS bioinformatic decisions, cloning limitations) or the extent and type of field management. The ‘managed’ sites surveyed in this study had low levels of fertilizers added several years prior to sampling, were mowed only once annually, and were polycultures, which may explain the relatively high AM fungal diversity. Additionally, a recent study by Garcia de Leon et al.[37] showed that anthropogenic impacts on AM fungal diversity are not always consistent in nature. They, instead, argue that anthropogenic land use causes AM fungal diversity to equalize over different sites, not simply increase or decrease in a consistent manner. This may help explain why some managed sites in our study (i.e. AF14) had a very diverse AM fungal community, while other managed sites (i.e. EC) had much less. Future studies must consider that anthropogenic change to a site may not drive AM fungal diversity in a consistent fashion, but rather, it may equalize the community diversity over a larger scale.
Certain AM fungi were more common in the managed field sites in this study. Diversispora spp. were more than four times more prolific in managed sites (358 reads) than in pine barrens sites (87 reads) (Table 2). Moora et al.[13] found Archaeosporaceae and Diversisporaceae (as well as Claroideoglomeraceae) to be indicator taxa for disturbed, agricultural sites. Therefore, it seems that these two clades could be associated less with acidic soils and more with disturbed sites. These could therefore be pioneer AM fungal clades.
Overall, Glomus was, by far, the most common genus uncovered from all habitats. This is similar to findings in other Illumina AM fungal soil studies[23,38] and AM fungal root studies[39,40], as well as 454[41] and spore morphology studies[31]. However, Glomus is not monophyletic; this group needs further work to confirm the correct taxonomic nomenclature for the clade(s)[42]. The second-most abundant taxon in this study differed among site type, representing differences in complementary or competitive AM fungal species. Despite the fact that Glomus spp. are almost equally prolific at each site, the complementary AM fungi may be more adapted to local soil types or differentially adapted to land use intensity, as local adaptation of AM fungi has been shown to be an important factor determining AM fungal communities[43,44].
Iowa’s natural prairie’s switchgrass roots harbored the most diverse AM fungal community. The rarefaction curves evidencing this trend matched, overwhelmingly, with the diversity index findings, providing more than one source of evidence that this site has extensive AM fungal diversity. Other studies have similarly shown that grasslands have diverse AM fungal communities compared to forest habitats[13,45] and croplands[46]. However, some of the managed switchgrass sites in this study also contained high AM fungal diversity, such as AF17 and SO, corroborating the findings of Jansa et al.[47] and Hijri et al.[36] that managed lands can have surprisingly vast AM fungal diversity.
Despite uncovering many potentially undescribed, new AM fungal lineages, we must tread lightly on the path to asserting new taxa because the phylogenetic positions of many AM fungi are still in flux[48,49]. When carrying out ecological studies using solely or mostly molecular evidence (i.e. DNA sequences), our data are only as good as our reference databases. And like other recent studies on switchgrass AM fungi[50], many sequences captured in this study could not be identified using the current reference database. More work should be done to barcode herbarium specimens so that our databases have an adequate supply of sequences for previously described species[51]. To confirm these new pine barrens species, spore extraction from forest soils will be necessary. Ecological work on these species would determine whether these species are indeed important for plant tolerance to acidic, oligotrophic soils. Additionally, our AM fungal diversity and community composition findings rely on our limited and uneven sampling efforts, which can be problematic for downstream conclusions. Additional sampling would provide a more robust picture of community diversity in these regions.
-
This study is the first to detail the AM fungal communities associated with switchgrass roots in the pine barrens ecosystem using NGS methods. Glomus was the most prolific AM fungal genus inhabiting in all the sampled sites; however, different land use types were inhabited by different AM fungal communities. A significant finding of this study was that Acaulospora and Ambispora were almost exclusively associated with the pine barrens ecosystems. Through Illumina sequencing, this study further enhances the breadth of knowledge on the DNA sequence diversity of AM fungi and begins to uncover the AM fungal communities associated with pine barrens ecosystems. The NGS data also indicated that soil pH correlated with root AM fungal community composition and diversity. More research on switchgrass associated AM fungi across a larger geographic region would help gain insight into what makes switchgrass thrive in different regions and under different land uses.
-
Switchgrass roots and soil were collected in the summer months (June, July, and August) of 2014, 2016, and 2017 from several ‘natural’ and ‘managed’ switchgrass populations, in New Jersey (NJ), New York (NY) and Iowa (IO) in the United States. The natural sampling sites include: the Doolittle Prairie State Preserve (IO), IO; and five pine barrens sites: Wharton State Forest (WSF), NJ; Colliers Mills (CM), NJ; Federal Aviation Administration (FAA), NJ; Rocky Point (RP), NY; and Long Island Pine Barrens Preserve (LIPB), NY. The managed sites were: Somerset Research Field (SO), NJ; Adelphia Research Field (AF), NJ; and E.A.R.T.H. Center (EC), NJ. (see Supplementary Methods for more sampling and site information). Similar to the managed sites, the Iowa prairie consisted mostly of grasses and sedges. Also, the prairie’s soil had relatively high nutrient levels, much like some of the managed sites. However, the prairie has not been mowed in the recent past and is not fertilized like the managed sites. It has prescribed burns on a regular basis, which provides interesting comparison to the pine barrens.
The sampled switchgrass variety in the managed sites was 'Kanlow', a lowland ecotype, because it is the same ecotype as the switchgrass found in the natural sites in this study. AF and LIPB were sampled two times, AF in 2014 and 2017 (AF14, AF17), and LIPB in 2014 and 2016 (LIPB14, LIPB16) in order to confirm unexpected findings from initial microscopic observations. Therefore, nine total sites were sampled, while eleven total site-years were sampled. For simplicity, the word 'site' is used in place of 'site-year'. Ten plants’ roots were collected from WSF, CM, FAA, RP, LIPB14, LIPB16, IO, and EC. Six whole plant roots were collected from SO, AF14, and AF17; this was because these research plots had limited supply of switchgrass available. Individual plant root samples were collected no less than 3 meters apart at each site, in order to avoid clonal ramets[52]. Roots were collected from 15−20 cm below the soil surface. A total of 98 plant root samples were collected.
One pooled soil sample was collected at each site from 15−20 cm below the soil surface, around the switchgrass roots. Samples were kept on ice before analysis. Pooled soil samples for each site were analyzed for chemical makeup, pH, and other soil features by Spectrum Analytic Inc. (Washington Court House, OH) (Supplementary Table S1). Exchangeable Al (Exch. Al), phosphorus (P), potassium (K), nitrate (NO3), and calcium (Ca) were extracted by Mehlich-3 (ICP) and are reported in ppm. Total N (N) and soil organic matter (SOM) are reported as percentages. Total aluminum (Al) is reported in mg/Kg. Cation exchange capacity (CEC) is reported as meq/100 g soil. Root samples were rinsed under running water to rid the roots of excess soil. Then they were surface sterilized by washing with 95% ethanol for 30 s, 0.825% NaOCl solution for 2 min, and 70% ethanol for 2 min. Lastly, they were rinsed with sterile water 3−5 times and stored at −80°C. In order to confirm the plant host identity, DNA from the leaf sheath of a host plant sample from each site was confirmed to be P. virgatum (see Supplementary Methods for additional information).
Observation of AM fungal colonization
-
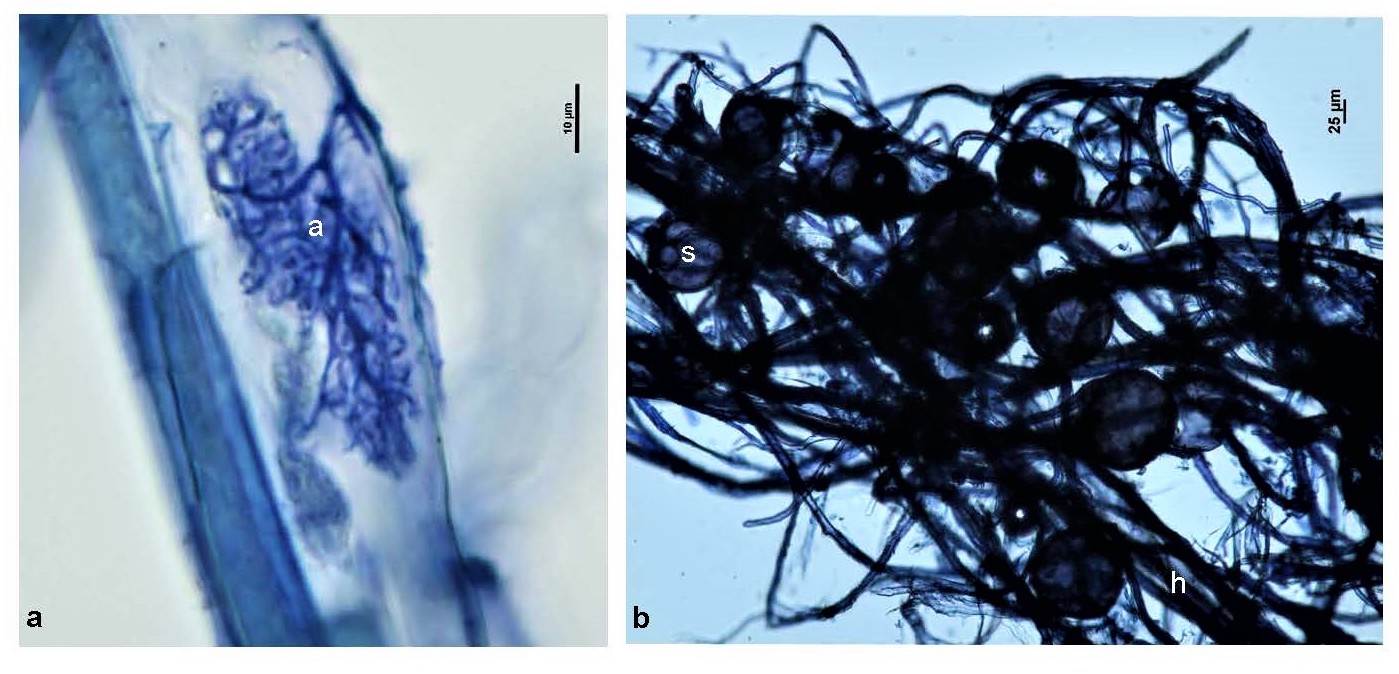
Switchgrass root samples were first visualized under a Nikon Eclipse 80i microscope to observe whether AM fungal colonization was present in the roots. To do this, roots were stained with 0.05% aniline blue following a modified version of the procedures of Grace and Stribley[53]. Arbuscules, hyphae, and spores were observed at 600X magnification and photographed using a Nikon DSFi-1 camera (Supplementary Fig. S1).
DNA extraction, PCR, Illumina sequencing
-
After confirming AM fungal presence in the roots, DNA sequencing was necessary for understanding the AM fungal community diversity present, as microscopic root observations cannot inform on species identity. For DNA extraction, stratified subsampling across all root systems within a site was implemented in order to gauge average AM fungal diversity present in that site. Stratified subsampling was done three times to provide triplicate DNA samples for each of our 11 sites, totally 33 samples (11 × 3 = 33)[54]. A total of 0.125 g of subsampled roots were used for each extraction. Liquid nitrogen was used to freeze and grind each sample of roots into a fine powder. DNA was then extracted from all 33 samples using a DNeasy PowerSoil DNA Isolation Kit (Qiagen Germantown, MD), according to manufacturer’s instructions. DNA concentrations were checked with a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA).
The primer pair AMV4.5NF/AMDGR[55] was used to amplify the variable region of the small subunit rRNA gene for AM fungi. Primers were designed for Illumina by attaching Nextera XT adapters (Illumina, San Diego, CA), which attach on one end to Illumina MiSeq adapters and on the other end, to our AM fungal primers. PCR was conducted with a mixture of 0.5 µl each of forward and reverse primers, 12.5 µl Taq 2X Master Mix (New England BioLabs, Maine), 1 µl template DNA, with PCR grade water added to a total volume of 25 µl. The PCR parameters were: 95 °C for 2 min, then 35 cycles at 95 °C for 45 s, 52/55/58 °C for 45 s, and 72 °C for 1.5 min, with a final extension of 72 °C for 5 min. Three annealing temperatures were used to maximize the amplification of AM fungal species[56]. Each of the 33 samples were run at these three annealing temperatures, totaling 99 PCR reactions. PCR reactions from the three annealing temperatures were pooled together per site, as well as, for each site sampled a second year (AF and LIPB), leaving 33 samples in total. Gel electrophoresis confirmed bands in the majority of the samples and no bands in the negative controls.
PCR products were cleaned up with an Agencourt AMPure XP kit (Beckman Coulter, Brea, CA) following manufacturer’s instructions. A secondary PCR was run to attach Nextera indices and Illumina adapters to each sample, using a Nextera XT Index Kit (Illumina, San Diego, CA). Each reaction contained 25 µl NEBNext High Fidelity 2X PCR Master Mix, 5 µl clean, primary PCR amplicon, 5 µl Nextera XT index 1 primer, 5 µl Nextera XT index 2 primer, and 10 µl PCR grade water, yielding a 50 µl total reaction volume. The secondary PCR conditions were 95 °C for 3 min, then 8 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s. The final extension was 72 °C for 5 min. Secondary PCR products were cleaned again using the Agencourt AMPure XP kit. The DNA concentration of each reaction was checked using a Qubit dsDNA BR Assay Kit (Thermo Fisher Scientific, Inc., Waltham, MA). All samples were normalized via dilution to 4 nM (= 1.33 ng/µl), pooled in equal volumes, and sequenced on Illumina MiSeq with 2 X 300 bp paired-end reads using the MiSeq Reagent Kit v3 (600 cycle). These steps followed standard protocols according to manufacturer’s instructions.
Bioinformatic analysis
-
Removal of sequencing adapters, PCR primers and low-quality bases was performed through the CLC Genomics Workbench v8.5.1[57]. Then, using the same software, forward and reverse reads were merged and any non-merged reads (with no overlap) were discarded since full overlap was expected on the 250 bp expected sequence size. Quality control parameters were set to reject any sequences < 100 bp long. Sequences were de-replicated using the “fastx_uniques” command in USEARCH 9.0 [58]. Sequences were sorted by size and singleton sequences (those with abundance of < 2) were discarded from further analysis using the “sortbysize” and “minsize” commands, respectively, in USEARCH 9.0 [58]. Singletons were removed because they were likely artifacts of the amplification process[59]. Sequences were then pooled together (in order to allow for downstream statistical analyses) and clustered into operational taxonomic units (OTUs) at 97% similarity using the “cluster_otus” command in USEARCH 8.0[58]. AM fungal reads were clustered at 97% similarity, as this threshold has been deemed appropriate for separating AM fungal taxa to the morphospecies-level in prior studies[14,22]. Sequences were compared against the MaarjAM database[60] and chimeras detected via the “uchime2_ref” command in USEARCH 9.0[61]. All remaining OTUs were subjected to a Basic Local Alignment Search Tool (BLAST) against the National Center for Biotechnology Information (NCBI) nucleotide database using the “query” and “db” commands in BLAST+ 2.5.0. All OTUs were observed in MEGAN Community Edition 6.6.7[62] with the default lowest common ancestor (LCA) parameters (minimum score of 50.0, minimum support percent of 0.01, and with the minimum-complexity filter off). All OTUs with BLAST matches to Glomeromycotan fungi were kept for further analysis. All AM fungal OTUs with matches of “uncultured Glomeromycota”, “uncultured Glomeromycetes”, or “uncultured Glomerales” (i.e. all matches above family level) were subjected to manual queries against the MaarjAM database and NCBI database for further inspection, similar to the methods of Schlaeppi et al.[63]. Efforts were made to identify all AM fungal OTUs to the genus level. Comparison across the family or genus level is appropriate because AM fungal families are considered a phylogenetically significant level when it comes to ecosystem function[64]. Sequences representing each AM fungal clade detected in this study, as well as, Acaulosporaceae and Ambisporaceae sequences, which were endemic to the pine barrens sites, were uploaded to GenBank under accession numbers MH908518-MH908579.
Statistical analysis
-
Because the sequencing process results in unequal sequencing depth among samples[65], OTU abundance data were resampled before analysis using the median number of reads from all 33 samples (21,049) (Table 1, Supplementary Data S1)[66]. This was done with the ‘rrarefy’ function in vegan 2.4−4, using RStudio 1.1.419[67]. This method has been previously shown to reduce bias due to varying sequencing depths among samples while still maintaining important OTU information[13,41]. Raw OTU and read totals are shown in Table 1, but subsequent mention of OTU and read abundance use the rarefied data. In order to determine whether sufficient number of samples (reads) were obtained, rarefaction curves were drawn using the ‘ggiNEXT’ function in the iNEXT package[68] using RStudio 1.1.419[69]. To compare AM fungal diversity between samples, three diversity indices (Shannon, Simpson, and Fisher’s α) were calculated for each site with the ‘diversity’ and ‘fisher.alpha’ functions in vegan 2.4−4, using RStudio 1.1.419[67,69].
Soil pH values were scaled prior to downstream analyses using the root-mean-square (‘scale’ function, without centering in RStudio 1.1.419)[69]. Pearson’s correlation coefficient (r) was used to test for correlation between soil pH and AM fungal diversity (Shannon, Simpson, and Fisher’s α diversity indices). Correlations and corresponding significance values were calculated using the rcorr() function in the Hmisc package using RStudio 1.1.419[69].
Analysis of similarity (ANOSIM), permutational multivariate analysis of variance (PERMANOVA), and nonmetric multidimensional scaling (NMDS) were conducted to explore the effects of land use and soil pH on AM fungal communities among the sites sampled in this study. Functions ‘anosim’, ‘adonis’, and ‘metaMDS’ were used for these analyses, respectively, in vegan 2.4−4 using RStudio 1.1.419[67,69]. Bray-Curtis dissimilarity measurements were used to determine community similarity between groups. To explore the effects of land use and soil pH, on AM fungal communities, we broke the sites down into categories. For comparing different land uses on AM fungal communities, we categorized our samples into three categories: ‘pine barrens’, ‘managed’, and ‘Iowa’. Iowa was separated for this comparison because its soil type (pH and micro- and macro-nutrient levels) and land use (native prairie) is dramatically different. Next, to explore the effects of soil pH, we separated our samples into two pH categories: samples with soil pH ≥ 6.0 (AF14, AF17, EC, IO) versus samples with soil pH < 6.0 (CM, WSF, LIPB14, LIPB16, FAA, RP, SO). Soil pH of 6.0 was chosen as the cutoff value because aluminum toxicity becomes detrimental to plant growth when soil acidity falls below 5.5−6.0 ( http://www.spectrumanalytic.com).
To further understand the phylogenetic diversity represented in this AM fungal survey, a phylogenetic tree was built to represent the AM fungal clades represented in this study. To generate the tree, 1−2 representative sequences from each clade obtained in this study were aligned with reference sequences from the MaarjAM and NCBI databases. Sequences were aligned with MUSCLE[70] in MEGA 6.06[71]. The best model to fit our data was found to be the Tamura 3-parameter model with gamma distribution[72]. A maximum likelihood tree was then built in MEGA 6.06[71] with 1000 bootstrap replications, using the tree with the highest log likelihood (−2482.7931). Bootstrap values > 70% are shown. Branch lengths represent the number of substitutions per site ( Fig. 2).
-
This research was financially supported by the National Science Foundation (grant number DEB 1452971), Rutgers Alberts Research Awards in Biodiversity, Rutgers Center for Turfgrass Science and the New Jersey Agricultural Experiment Station to N. Zhang, the National Military Family Association, and the Garden Club of America. We thank Howard S. Friedman for advice on statistical methodologies and Raphael Kurlansik for his help developing select R scripts. We also thank Ehud Zelzion for assisting with quality filtering of DNA sequence data.
- The authors declare that they have no conflict of interest.
- Supplementary Table S1 Sampling site information. Site information includes: site name, year(s) sampled, type of landscape (managed, pine barrens, or prairie), and associated soil properties. Nutrients measured in parts per million (ppm) represent extracted nutrients.
- Supplementary Data S1 AM fungal OTUs (rarefied) and their abundance (number of reads) at each of 11 sites. Sites are abbreviated: AF14 - Adelphia Research Field 2014, AF17 - Adelphia Research Field 2017, CM - Colliers Mills, EC - EARTH Center, IO - Iowa, FAA - Federal Aviation Administration, LIPB14 - Long Island Pine Barrens 2014, LIPB16 - Long Island Pine Barrens 2016, RP - Rocky Point, SO - Somerset Research Field, WSF - Wharton State Forest. Numbers after site names represent the site triplicate number (1, 2, or 3) used in DNA extractions. A taxon name is provided for each AM fungal OTU.
- Supplementary Methods Arbuscular mycorrhizal fungal communities associated with switchgrass in the acidic, oligotrophic pine barrens ecosystem.
- Supplementary Fig. S1 Morphological structures of AM fungi observed with P. virgatum L. 708 roots. (a) A = arbuscule. (b) H = extraradical hyphae, S = extraradical spores.
- Copyright: © 2021 by the author(s). Exclusive Licensee Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Bindell M, Luo J, Walsh E, Wagner NE, Miller SJ, et al. 2021. Arbuscular mycorrhizal fungal communities associated with switchgrass ( Panicum virgatum L.) in the acidic, oligotrophic pine barrens ecosystem. Grass Research 1: 2 doi: 10.48130/GR-2021-0002

Arbuscular mycorrhizal fungal communities associated with switchgrass (Panicum virgatum L.) in the acidic, oligotrophic pine barrens ecosystem
- Received: 14 January 2021
- Accepted: 09 February 2021
- Published online: 04 March 2021
Abstract: Arbuscular mycorrhizal (AM) fungi contribute globally to ecosystem services and play an important role in sustainable crop production. However, it is unclear which factors contribute most to their colonization and community structure at different sites, particularly in understudied ecosystems. This study investigated the AM fungal communities associated with switchgrass (Panicum virgatum L.) in the understudied acidic and oligotrophic pine barrens ecosystem using next-generation sequencing. Switchgrass was also sampled from agroecosystems, as well as, from a native prairie for comparison. The pine barrens switchgrass harbored a distinct AM fungal community − Acaulospora and Ambispora were almost exclusively found in the pine barrens sites, and some of these species may represent undescribed taxa. Glomus was the most ubiquitous AM fungal genus recovered from all sites. This study suggests differences in the AM fungal community structure under different soil properties and land uses. This is the first sequence-based report of the AM fungal communities in the pine barrens ecosystem.
-
Key words:
- AM fungi /
- microbiome /
- switchgrass
{kind=link}