









-
Terpenoids constitute a large group of metabolites and have highly diverse structures and functions. Terpenoid primary metabolites, such as plastoquinone, phytosterol, chlorophyll, carotenoids, and phytohormones, participate in respiration, membrane fluidity, photosynthesis, and regulation of growth and development[1−3]. Some secondary metabolites, such as phytoalexin, participate in allelopathic and plant–pathogen interactions[4]. All terpenoids are derived from two common precursors, isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), which are biosynthesized in plants through the cytosolic mevalonate (MVA) and plastidic methylerythritol phosphate (MEP) pathways[5]. The MVA pathway is involved in synthesis of sesquiterpene, triterpene, and sterols, while the MEP pathway produces monoterpene, diterpene, and other secondary metabolites[5]. Evidence suggests that there is metabolic flux between the two pathways via a metabolic network[6,7], but normal levels of the end products are maintained in disrupted pathways by the respective regulation.
The first step of the MEP pathway is catalyzed by 1-deoxy-D-xylulose-5-phosphate (DXP) synthase (DXS) and produces DXP from glyceraldehyde-3-phosphate and pyruvate. As known, a series of enzymatic reactions was further presented in the MEP pathway (Fig. 1). The gene encoding DXS enzyme was first identified in Escherichia coli and DXS homologs were subsequently found in model plants and crops, including Arabidopsis[8], barrel clover[9], tomato[10], rice[11], and maize[12], suggesting that DXS genes are highly conserved in plants. Studies to date show that DXS enzyme is encoded by a small gene family. Multiple DXS genes have been found in Medicago truncatula[9], Oryza sativa[11], Picea abies[13], Zea mays[12], Salvia miltiorrhiza[14], Aquilaria sinensis[15], and Artemisia annua[16], and two or three DXS genes in these plants cluster into three independent clades. It has been shown that DXS genes can also be divided into two types based on DXS function. DXS1 type is thought to be involved in primary metabolism (e.g. chlorophyll synthesis, photosynthetic processes), and mainly expressed in photosynthetic tissues[9,17−19]. Unlike DXS1 genes in other plants, studies in Aquilaria sinensis have shown that AsDXS1 is mainly expressed in the stems and were significantly induced by wound signals, indicating the involvement of AsDXS1 in sesquiterpene formation[15]. In addition, DXS1 gene also plays a role in carotenoid accumulation during fruit ripening in tomato and pepper[10,20]. On the other hand, DXS2 type enzyme is thought to be involved in biotic or abiotic resistance defenses and metabolism of special secondary metabolites. For instance, the expression of DXS2 in barrel clover (Medicago truncatula) and maize (Zea mays) make different influences in the apocarotenoid accumulation during mycorrhization[9,12]. The clade 3 of DXS gene, which is rarely identified in plants, often plays an interesting role in various plants. For example, DXS3 in rice has been suggested to also participate in defense responses[21]. In the accumulation of terpenes and linalool in 'Jumeigui' grape, only the higher transcript abundance of VvDXS3 showed significant correlation[22].
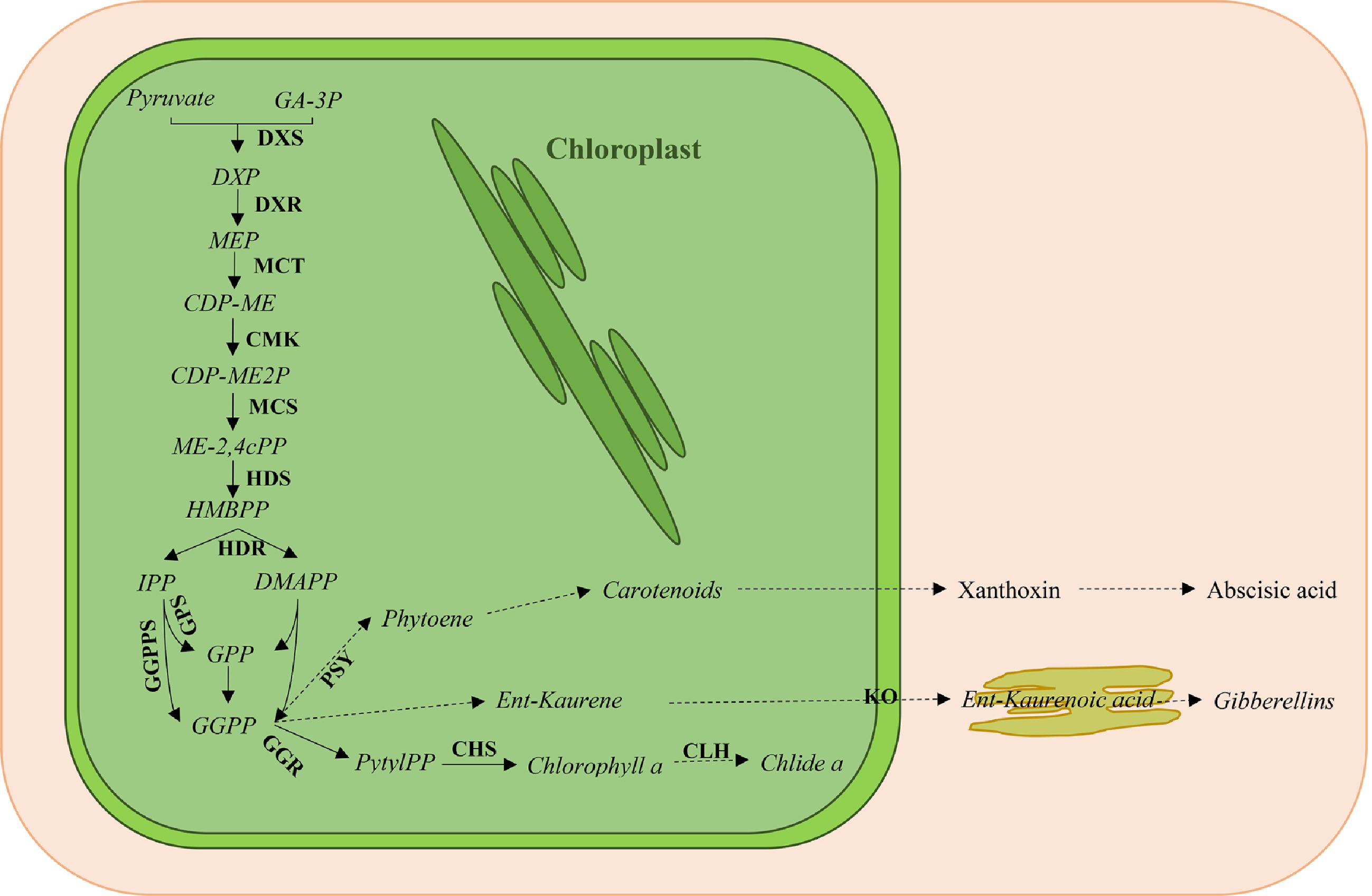
Figure 1. Overview of the plastidic methyl erythritol phosphate (MEP) pathway for isoprenoid biosynthesis. GA-3P, glyceraldehyde-3-phosphate; DXS, 1-deoxy-D-xylulose-5-phosphate synthase; DXP, 1-deoxy-D-xylulose-5-phosphate; DXR, 1-deoxy-D-xylulose reductase; MEP, methylerythritol phosphate; MCT, MEP cytidyltransferase; CDP-ME, 4-(cytidine 5’-diphosphate)-2-C-methylerythritol; CMK, CDP-ME kinase; CDP-ME2P, 2-Phospho-4-(cytidine 5’-diphospho)-2-C-methylerythritol; MCS, 2-C-methylerythritol 2,4-cyclodiphosphate (ME-2,4cPP) synthase; HDS, 1-hydroxy-2-methyl-2-butenyl 4-diphosphate (HMBPP) synthase; HDR, HMBPP reductase; IPP, isopentenyl diphosphate; DMAPP, dimethylallyl diphosphate; GPS, geranyl diphosphate (GPP) synthase; GGPPS, geranylgeranyl diphosphate (GGPP) synthase; PSY, phytoene synthase; GGR, geranylgeranyl reductase; PhytlPP, phytyl diphosphate; CHS, chlorophyll synthase; CLH, chlorophyllase; KO, ent-kaurene oxidase. Solid lines indicate a single enzymatic step, and dashed lines indicate several enzymatic steps.
Additionally, post-transcriptional and post-translational regulation are also important objectives of DXS functional mechanism research, as well as DXS gene expression and activity which are also regulated by external factors[8]. For example, Mansouri et al. found that DXS enzymatic activity decreased after application of gibberellic acid[23]. While DXS appear to have a universal function and evolutionary conservation, their functional action is species-specific. So, it is worth further exploring the metabolic regulation and movement for the isoprenoid product in plant growth and development.
While substantial progress has been made in understanding the function of the DXS in various plants (including model plants, crop and medicinal plants), little is known in the Poaceae family. There has been no research to date in the turfgrass field. Kentucky bluegrass, which belongs to the Poaceae family, is an important cool-season turfgrass in temperate and subarctic regions. Kentucky bluegrass has an aesthetic appearance and an excellent tolerance to low temperatures[24], but requires frequent mowing to maintain turf quality[25]. A preferred strategy is to use cultivars with shorter stature and good stress-resistance to reduce mowing frequency and improve quality. As previously reported, the MEP pathway and DXS genes play important roles in the biosynthesis of various metabolites, such as gibberellin (GAs), abscisic acid (ABA), and in regulating plant growth and development[7,26]. However, no DXS genes was isolated from Kentucky bluegrass, and little is known regarding the effects of DXS on growth and development in turfgrasses.
Previously, our work reported that DXS1 homologous gene expression level is significantly lower in the dwarf mutant than in the wild-type[27]. The dwarf mutant was obtained from F3 plants derived from 'Baron' seeds exposed to space environment on the satellite which usually results in abundant and non-directional mutations. Additionally, there are differences in leaf color, plant height and other traits between mutant and wild-type plants. Therefore, we studied the relationship between DXS1 gene and plant growth, subsequently understanding the physiological differences between mutant and wild-type plants. In this study, we (a) isolated and characterized a DXS1 gene from Kentucky bluegrass, (b) identified its regulatory effects on the biosynthesis and metabolism of various isoprenoids, and (c) used an underexpressing transgenic line to examine the functional role of PpDXS1 in plant growth and development.
-
Three DXS homologous unigenes were selected from the P. pratensis transcriptome database (NCBI accession number: SRA315988) and then assembled for a fragment sequence for putative P. pratensis DXS gene. RACE primers were designed against the candidate sequence and used to clone the putative DXS ORF frame from Kentucky bluegrass 'Baron' cultivar. An ORF of 2139 bp was produced and named PpDXS1. Sequence alignments revealed that PpDXS1 amino acid sequence was very similar to DXS1 proteins from other plants (Supplemental Fig. S1a), including Hordeum vulgare (97% identity), Aegilops tauschii (97% identity), Brachypodium distachyon (95% identity), Setaria italica (90% identity), and Oryza brachyantha (90% identity).
PpDXS1 amino acid sequence and structure were examined further to elucidate functional features. The PpDXS1 protein contained 712 amino acid residues and had a predicted molecular mass of 76.7 kDa and deduced isoelectric point of 6.5. Protein subcellular localization of PpDXS1 was predicted in the chloroplast by the WoLF PSORT server, which was consistent with previous studies in various plants[19,28]. Additional primary structure features are shown in Supplemental Fig. S1. The crystal structure of PpDXS1 was not available in the SWISS-MODEL database. Available protein sequences were therefore used with the Expasy server to deduce a 3D model for PpDXS1, with a model for AtDXS also constructed for comparison. Highly conserved functional residues in PpDXS1 and AtDXS were predicted using the InterPro server. Residues 62–259 were highly conserved and formed a thiamine diphosphate binding pocket. Additional common domains important for biological function were also identified (Fig. 2). Differences between AtDXS and PpDXS1 were observed from 648/654 residues onwards (Fig. 2; shown in red and yellow). Therefore, it is speculated whether the difference in DXS1 protein structure among different species will perform different functions in plants.
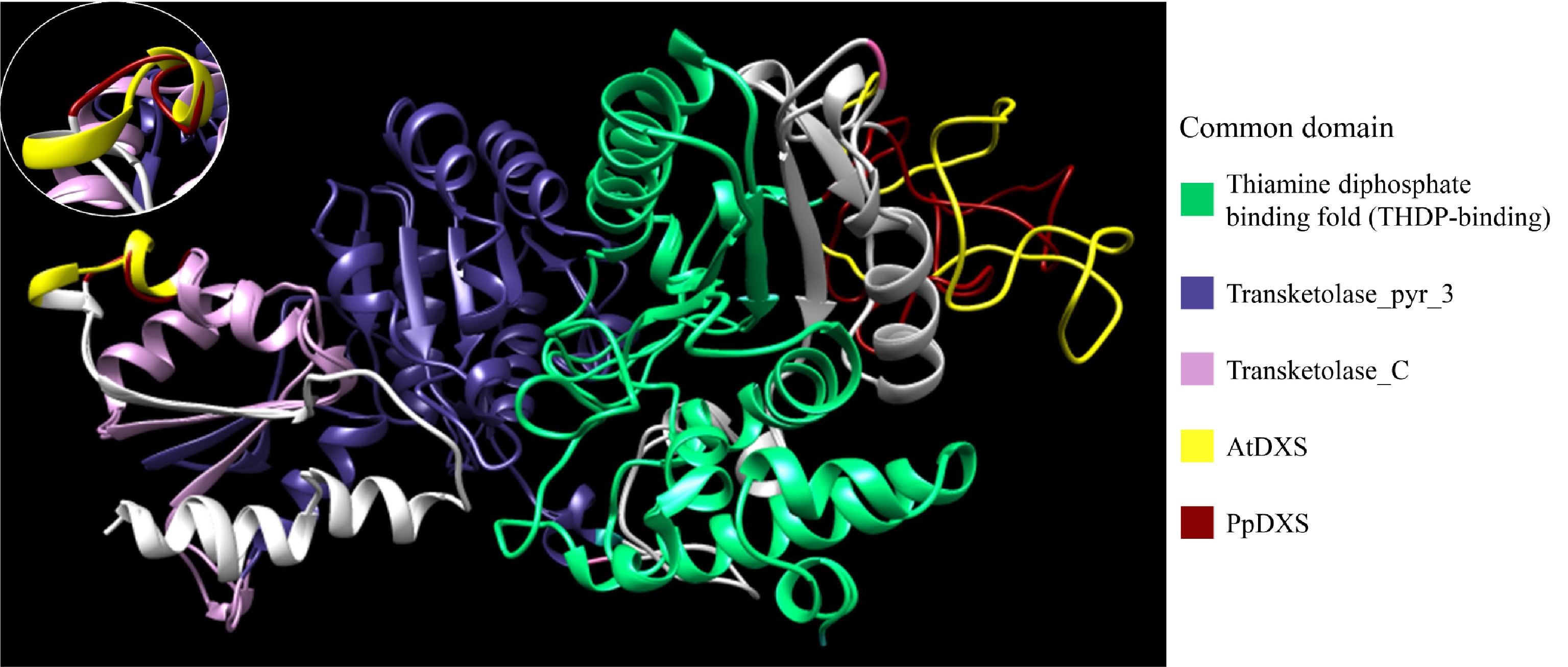
Figure 2. Expasy structure prediction models of PpDXS and AtDXS. Model images were generated using Chimera and conserved domains were analyzed using the InterPro server. Residues 62–259 (PpDXS) and 74–273 (AtDXS) represent the THDP-binding domain (green). Residues 387–552 (PpDXS) and 399–564 (AtDXS) represent the thansketolase_pyr_3 domain (blue). Residues 566–698 (PpDXS) and 578–701 (AtDXS) represent the thansketolase_C domain (pink). Differences between the PpDXS and AtDXS model are shown in red (PpDXS) and yellow (AtDXS).
Phylogenetic analysis of DXS amino acid sequences among plants
-
PpDXS1 was shown to have a close relationship with Aegilops tauschili, Brachypodium distachyon and other Poaceae plants (Fig. 3). The phylogenetic tree grouped into three independent clades of DXS proteins conserved among plants. The first group referred to DXS1 proteins from a variety of dicots and monocots. However, a DXS-like protein from Arabidopsis (At3g21500) also belongs to this group and its function is still unknown[19, 29]. The clade 2 proteins are mainly involved in secondary metabolism and includes Oryza sativa DXS2 together with representatives from woody plants (e.g. Picea abies and Ginkgo biloba)[11,13,30]. Finally, the clade 3 proteins include Arabidopsis DXS3, rice DXS3, maize DXS3 and agarwood DXS3. As shown, the third branch is the distant phylogenetic DXS group. In this study, PpDXS1 belonged to the DXS1 clade, but PpDXS2 probably exists in Kentucky bluegrass because of its polyploidy. In rice and maize, two DXS1 proteins are also clustered into clade 1 and have been reported to participate in primary metabolism[11,12], while BdDXS1 in Brachypodium distachyon and AetDXS1 protein in Aegilops tauschili had not been reported for functions. DXS1 protein not only participates in photosynthesis in most plants, but has also been shown to be involved in sesquiterpene metabolism and plant protection in agarwood (Aquilaria sinensis) and potato (Solanum tuberosum L.)[15,18,31]. It is implied that DXS1 proteins among species perform conserved yet distinct functions. So, our findings in DXS1 would be beneficial to closely related Poaceae members associated with regulation of plant growth and development.
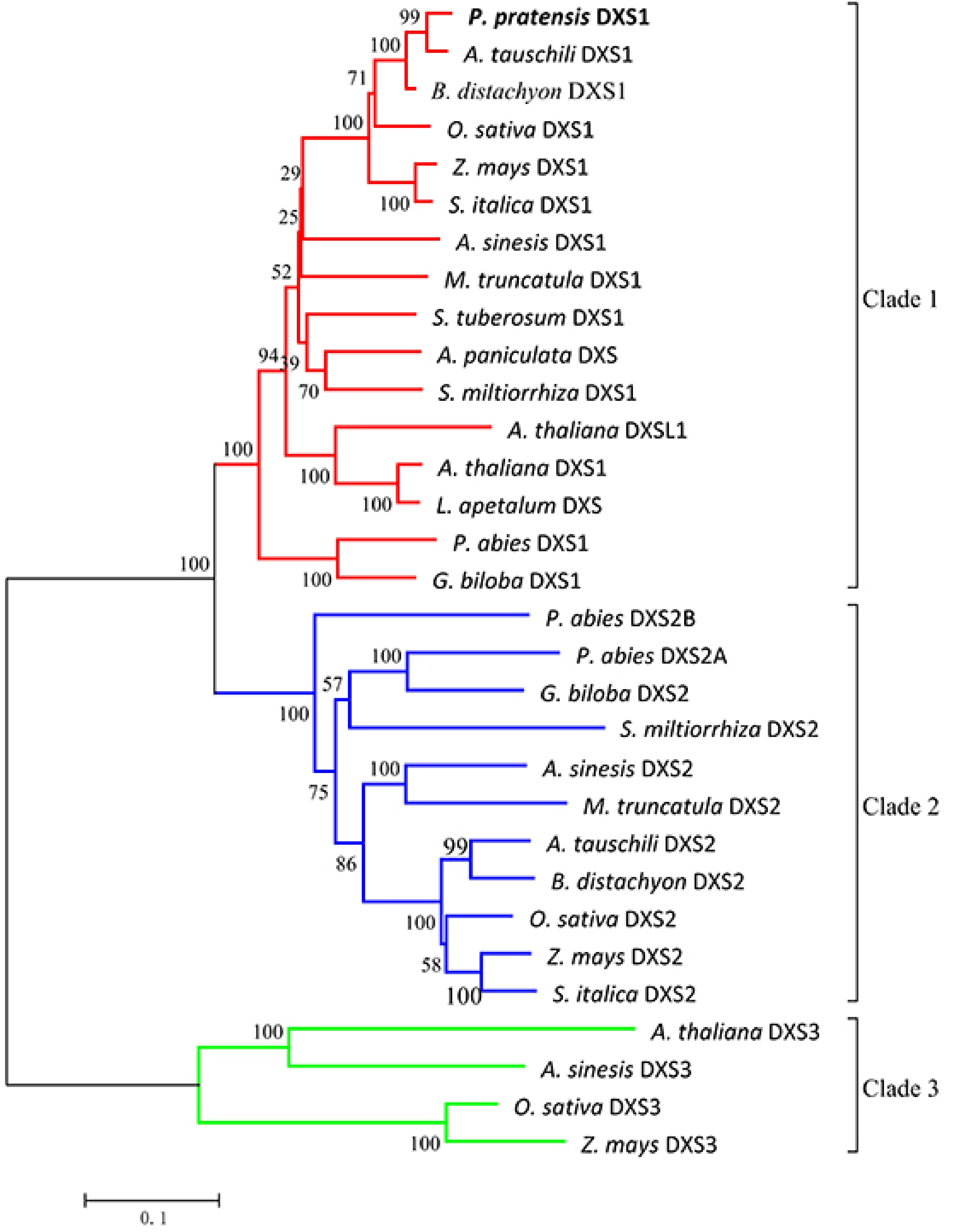
Figure 3. Phylogenetic tree of plant DXS proteins. The tree was constructed with the neighbor-joining method and the JTT model using MEGA 5.0 software with 1,000 bootstrap values. The TAIR or GenBank accession numbers of DXS amino acid sequences used for phylogenetic analysis are as follows: Arabidopsis thaliana (DXS1 clade At4g15560, DXSL1 clade At3g21500, DXS3 clade At5g11380); Oryza sativa (DXS1 clade NP_001055524.1, DXS2 clade NP_001059086.1, DXS3 clade BAA83576.1); Zea mays (DXS1 clade NP_001157805.1, DXS2 clade NP_001295426.1, DXS3 clade HQ113384.1); Aquilaria sinensis (DXS1 clade AFU75321.1, DXS2 clade AHI62962.1, DXS3 clade AFU75320); Andrographis paniculata (DXS, AAP14353.1); Lepidium apetalum (DXS, KU314760.1); Medicago truncatula (DXS1 clade CAD22530.1, DXS2 clade CAN89181.1); Picea abies (DXS1 clade ABS50518.1, DXS2A clade ABS50519.1, DXS2B clade ABS50520.1); Salvia miltiorrhiza (DXS1 clade ACF21004.1, DXS2 clade ACQ66107.1); Solanum tuberosum (DXS1 clade NP_001275130.1); Ginkgo biloba (DXS1 clade AAS89341.1, DXS2 clade AAR95699.1); Aegilops tauschili (DXS1 clade XP_020161542.1, DXS2 clade XP_020164020.1); Brachypodium distachyon (DXS1 clade XP_003568467.1, DXS2 clade XP_003557443.1); Setaria italica (DXS1 clade XP_004962111.1, DXS2 clade XP_004955719.1). Poa pratensis (DXS1 clade MG257788).
Isolation and analysis of the PpDXS upstream region
-
The 5’ upstream region of PpDXS1 gene (1,649 bp) were also isolated from Kentucky bluegrass (Supplemental Fig. S2). Cis-acting elements of the 5’ upstream region were predicted using the PlantCARE and MAT-INSPECTOR databases. The predicted elements corresponded to light response, hormone metabolism, and stress defense regulation (Fig. 4). Relevant response elements included the ABRE element (ABA response), CGTCA-motif (related to MeJA response), GARE-motif (gibberellin response), box-W1 (fungal responsive element), G-box and L-box (involved in light responsiveness), and protein binding sites (HD-ZIP3, and MBS). The impacts of the different motifs and elements on inducible PpDXS expression and function in Kentucky bluegrass were examined further.
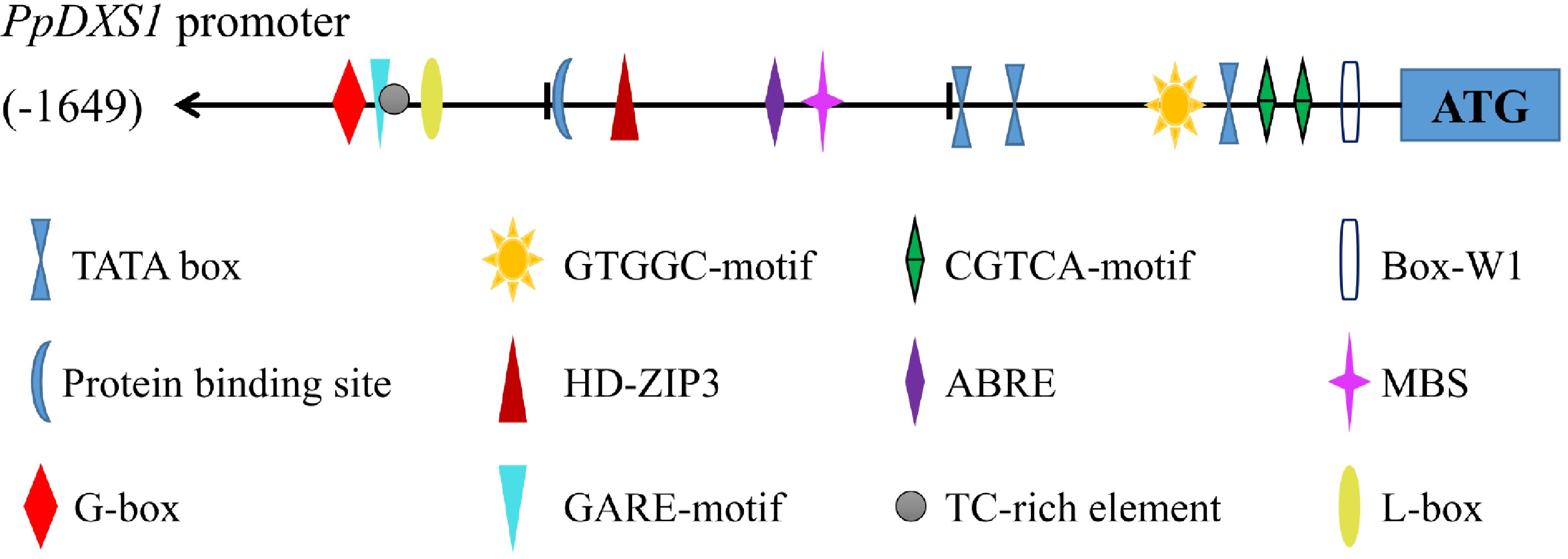
Figure 4. PpDXS1 promoter from P. pratensis. Putative cis elements are labeled as shown, Element positions are shown proportional to the full length of the promoter.
Expression analysis of the PpDXS1 in different tissues in P. pratensis
-
To gain insight into the expression pattern of PpDXS1, several tissues at a range of growth stages were examined in a quantitative real-time PCR experiment. As shown in Fig. 5, PpDXS1 transcripts were detectable in different tissues (roots, leaf sheaths, and leaves), but accumulated predominantly in green tissues including young and mature leaves. The transcript abundance of PpDXS1 was highest in mature plants, followed by young leaves. It is evidenced that the PpDXS1 gene belonged to DXS1 type (consistent with phylogenetic analysis) and may be involved in photosynthesis. Additionally, the transcript level of PpDXS1 was lower at the heading stage than in the developing leaves.
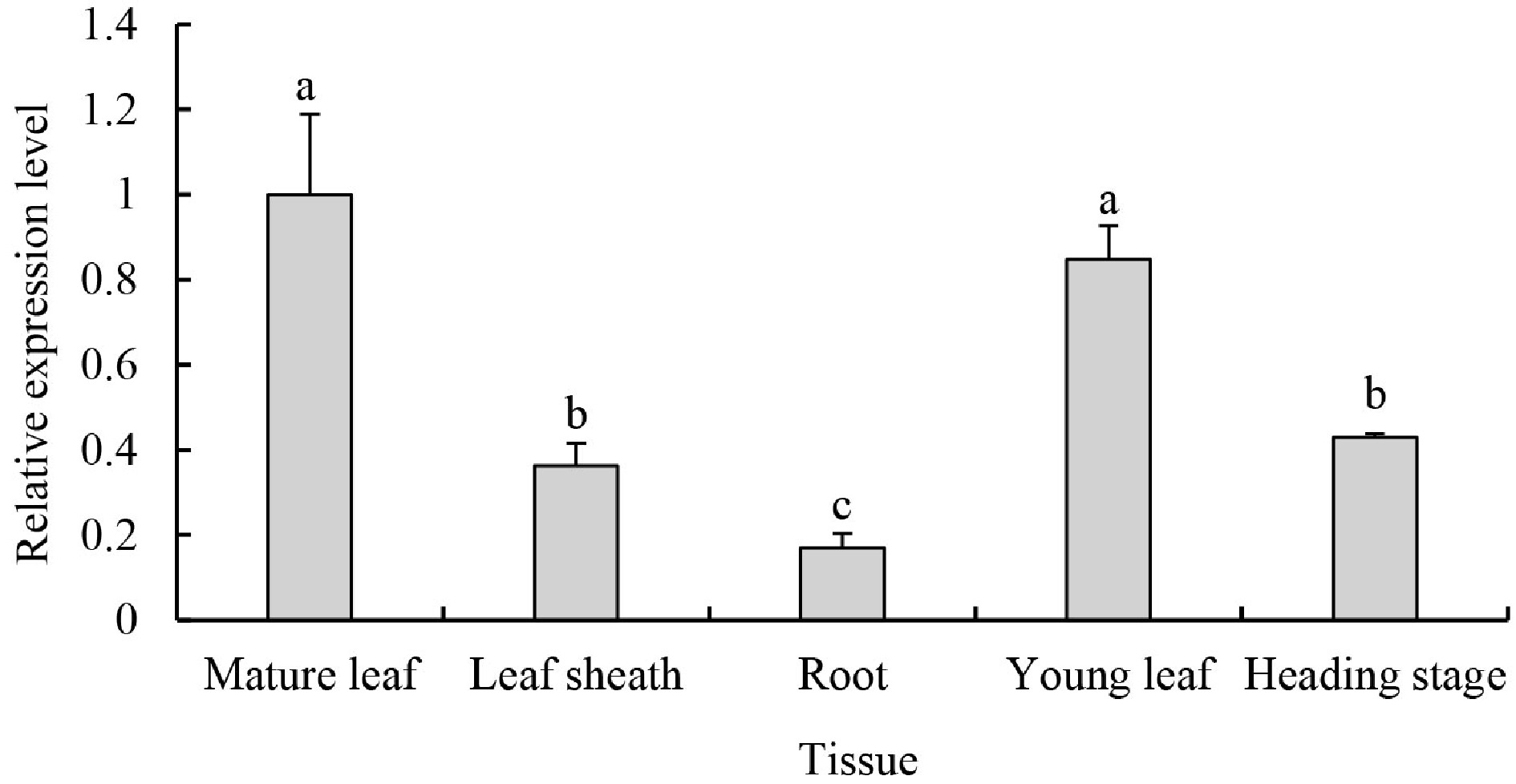
Figure 5. PpDXS1 expression analysis in P. pratensis. Quantitative RT-PCR analysis of PpDXS1 gene expression. Fully expanded leaves (named as mature leaf), leaf sheaths and roots at vigorous growth stage were used for gene organ/tissue-specific expression analysis. For the time-series gene expression analysis, young leaves at early growth stage (named as young leaf), mature leaves at vigorous growth stage (similarly named as mature leaf), and old leaves at heading stage (named heading stage) were used. All qRT-PCR reactions were performed from triplicate biological samples. The
$2^{-\Delta \Delta} $ Cᴛ method was used to calculate the fold expression relative to the control (mature leaf). Means of three replicates ± standard error is shown. Bars superscripted with different letters are significantly different at p < 0.05.Regulation of different inducible factors on PpDXS1 gene expression in P. pratensis
-
DXS is the rate-limiting enzyme in the MEP pathway, which is involved in the formation of diverse terpenoids, including important phytohormones for plant growth. Results shown in Fig. 4 show that the response elements and motif of ABA (ABRE), JA (CGTCA-motif), GA(GARE-motif), and fungal (box-W1) were identified in the PpDXS1 promoter sequence. Therefore, we examined the effects of exogenous factors on PpDXS1 expression in P. pratensis. Plants were exposed to foliar applications of GA3, ABA, JA, or pathogen inoculation, and the relative expression level of PpDXS1 were determined in the leaves collected 0, 3, 6, and 12 h after treatment (Fig. 6).
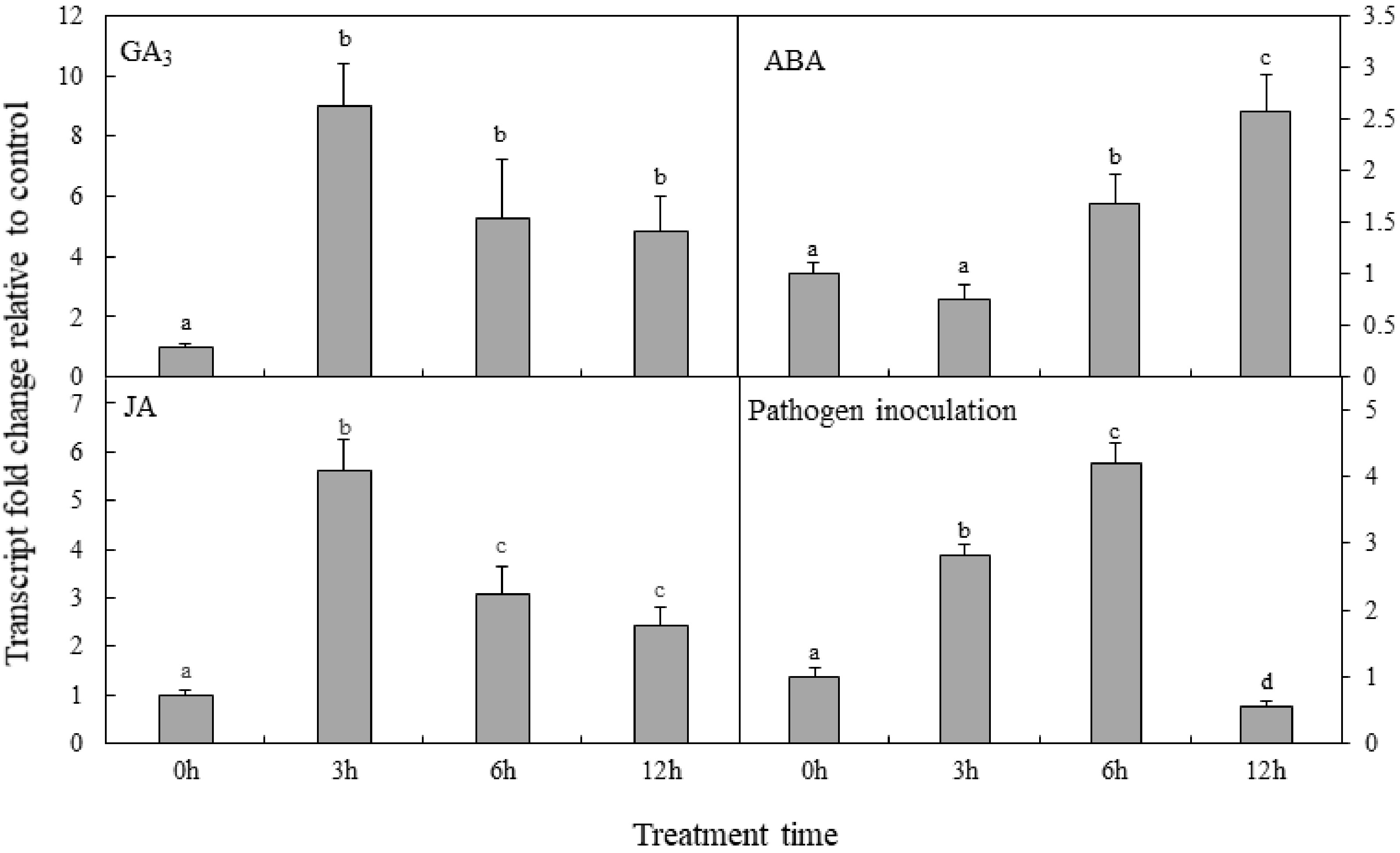
Figure 6. Analysis of PpDXS1 expression after exogenous phytohormone treatment or pathogen inoculation in P. pratensis. Quantitative real-time PCR analysis of relative PpDXS1 expression after foliar exposure to gibberellic acid (GA3), abscisic acid (ABA), jasmonate (JA), and pathogen inoculation. All qRT-PCR reactions were performed from triplicate biological samples. The
$2^{-\Delta \Delta} $ Cᴛ method was used to calculate the fold expression relative to the control (0h-point). Mean of three replicates ± standard error is shown. Bars superscripted with different letters are significantly different at p < 0.05.As shown in Fig. 6, GA3 treatment had a positive effect on PpDXS1 gene expression and the level peaked 3 h after application. Meanwhile, increased transcript level of PpDXS1 6 h and 12 h after ABA treatment was observed, while the fold change of ABA regulation has a lower value, compared to GA3 treatment. It is predicted that exogenous ABA have a slight impact on PpDXS1 gene expression, compared with GA3 treatment. After JA treatment, PpDXS1 expression was induced and peaked at the 3h-point (5.6-fold). A similar response was seen after pathogen inoculation. Gene expression was stimulated, and peaked at 6 h after inoculation (4.2-fold). On the other hand, it is indicated that the inductive effect of exogenous JA was faster than the pathogen infection for P. pratensis.
Effect of PpDXS1 underexpression on plant height, chlorophyll and hormone content in P. pratensis
-
To test the role of PpDXS1 in regulating the terpenoid biosynthetic pathway in P. pratensis, three transgenic lines with antisense expression of PpDXS1 gene and empty vector (CK) of Kentucky bluegrass were obtained by screening and identification.. In order to further determine the positive plants, western blot analysis was carried out for the extracted protein from transgenic plants and the CK plant (Fig. 7a). The result showed that the PpDXS1 protein level (the band of 76 kDa) decreased in the transgenic antiDXS1-56/-102 lines, except the antiDXS1-101 strain which was not used for further research (Fig. 7a and supplemental Table S1). It is indicated that DXS1 protein accumulation could be decreased by antisense expression of the PpDXS1 gene. It should be noted here that there is another band below the target band (i.e. 74 kDa) in Fig. 7a, which indicated that the second band could be the DXS2 protein of Kentucky bluegrass as the specificity of the PpDXS1 polyclonal antibody in this study was not very high.
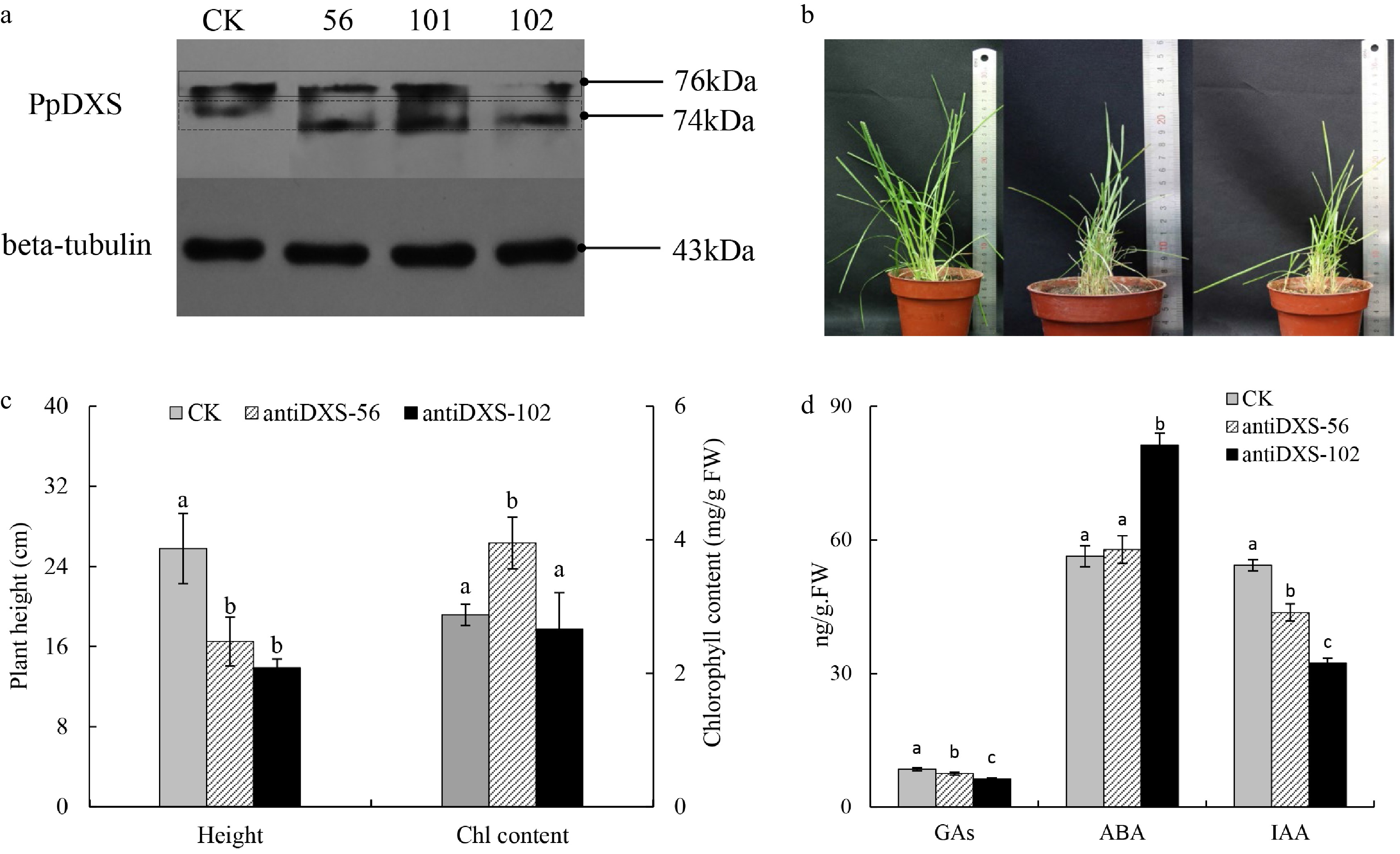
Figure 7. Comparison of CK and transgenic lines underexpressing PpDXS1 in Poa pratensis. (a) Western analysis of PpDXS1 protein levels in mature leaves of CK and antiDXS1-56, -101, and -102. (b) The photos of CK, antiDXS1-56 and antiDXS1-102 transgenic plants (left to right). (c) Plant height and total chlorophyll content of CK, antiDXS1-56 and antiDXS1-102 transgenic plants. Bars superscripted with different letters are significantly different at p < 0.05. (d) Phytohormone content of CK, antiDXS1-56 and antiDXS1-102 transgenic plants. Means of three replicates ± standard error is shown. The data were subjected to ANOVA test to determine the LSD between CK (control) and transgenic lines (antiDXS1-56 and antiDXS1-102) at p < 0.05.
Additionally, transgenic lines displayed the phenotype of reduced plant height. As shown in Fig. 7b and c, Plant height was reduced in transgenic lines by approximately 40% relative to CK. For chlorophyll, the total pigment content of transgenic antiDXS1-56 is significantly higher than that of CK, but the difference between antiDXS1-102 and CK is not significant (Fig. 7c).
Differences in phytohormones content between transgenic lines and the control plant were also analyzed. All DXS1-transformed lines showed decreased GAs and IAA content compared with that of the control (Fig. 7d). By contrast, the accumulation of ABA in the transgenic plant were increased. There are different accumulation among GAs, IAA and ABA content in transgenic P. pratensis, which may be related to the various regulation of DXS1 gene. In general, it is illustrated that PpDXS1 is an important gene in the regulation of phytohormone and chlorophyll accumulation.
Underexpression of PpDXS1 gene regulated the differential gene expression involved in hormone anabolism in transgenic P. pratensis
-
To study the effects of PpDXS1 underexpression on plant growth and development, RNA-seq analysis was used to compare transcriptional level in CK and an underexpressing antisense line (antiDXS1-102). Differential gene expression analysis revealed a total of 3,800 genes whose expression significantly differed (log2 fold change ≥ 1 and q value < 0.005) between antiDXS1-102 and CK, 1845 and 1955 of which were up- and down-regulated in antiDXS1-102, respectively (Supplemental Fig. S3a). The differentially expressed genes (DEGs) were involved in diverse biological processes such as terpene synthase activity, protein kinase activity, and metabolic processes (Supplemental Fig. S3b, c). On the basis of enrichment analysis and related keywords of KEGG pathway and GO terms, some DEGs between CK and antiDXS1-102 line were summarized in Fig. 8a. Firstly, one DXS unigene was down-regulated in the transgenic line, while other related isoprenoid biosynthetic genes (e.g. FPS and SPS) are up-regulated. Secondly, the expression of genes related to auxin biosynthesis and signal transduction were affected, for example the transcripts encoding auxin responsive factor 9 (ARF9) was down-regulated. Similarily, the GA20ox2 gene involved in gibberellin biosynthesis was down-regulated in the transgenic line. Conversely, ABA biosynthetic genes were enriched in the transformed line compared to CK, such as phytoene synthase (PSY) gene and 9-cis-epoxycarotenoid dioxygenase 1 (NCED1) gene. Finally, in the pathway of chlorophyll biosynthesis and metabolism, the expression level of geranylgeranyl reductase (GGR) genes and chlorophyllide a oxygenase (CAO) genes that promoted chlorophyll a (or side chain) synthesis were increased. The CLH gene which directs chlorophyll degradation were down-regulated in the antiDXS1-102 line, while the increase of chlorophyll content in the antiDXS1-102 line was not significant. These observations were consistent with the above result of phenotype and physiological characteristics in the transgenic line, which indicate that PpDXS1 directly influences the production of physiological metabolites, particularly those producing GA, IAA, and ABA at the end of the MEP pathway.
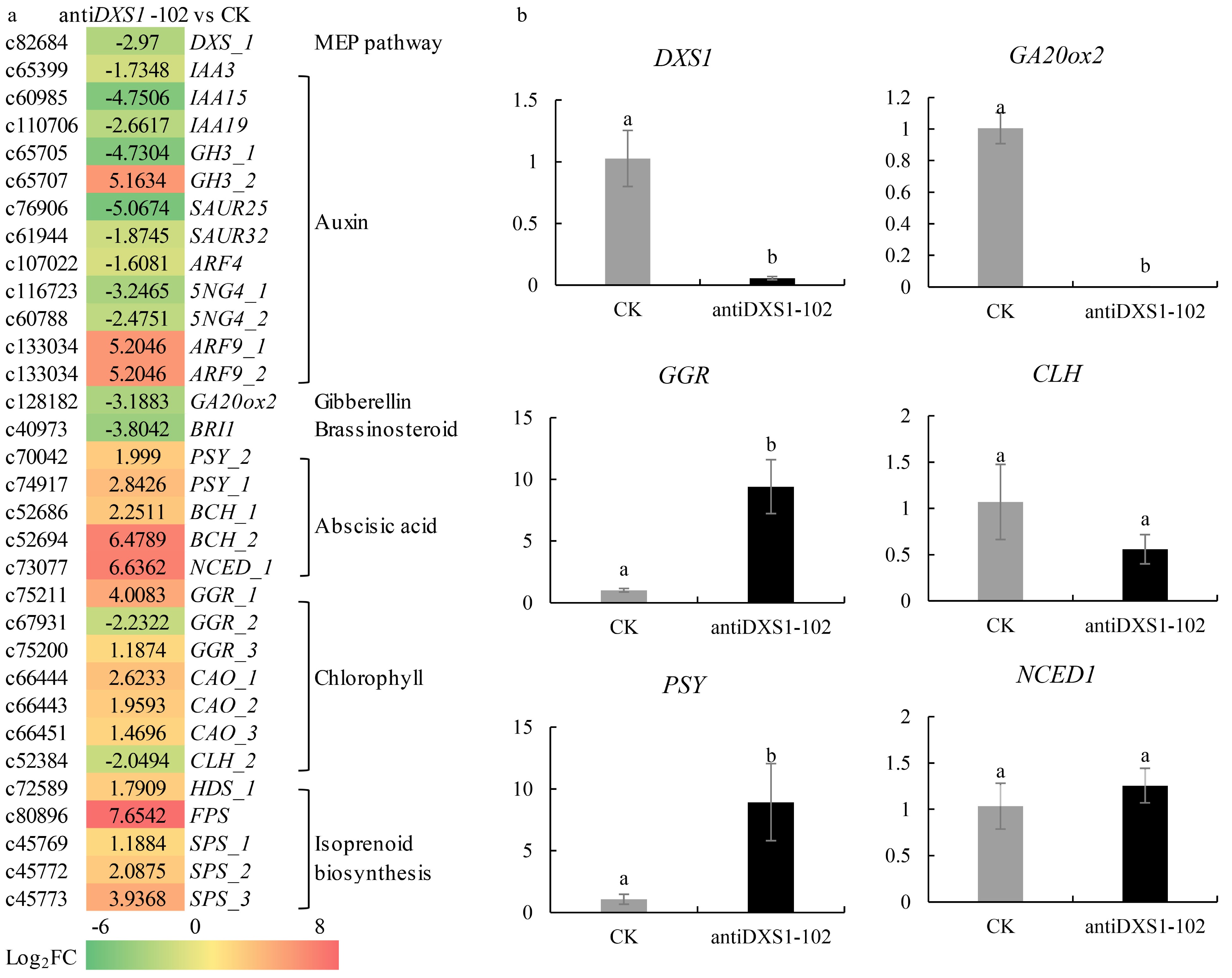
Figure 8. Differential expression analysis of related genes in CK and antiDXS1-102. (a) Heatmaps of differentially expressed genes (DEGs). DXS, 1-deoxy-D-xylulose-5-phosphate synthase; IAA3/15/19, auxin-responsive protein 3/15/19; GH3, indole-3-acetic acid-amido synthetase GH3 gene family; SAUR25/32, auxin-responsive SAUR protein family; ARF9, auxin responsive factor 9; GA20ox2, gibberellin 20 oxidase 1; BRI1, brassinosteroid-insensitive 1; PSY, phytoene synthase; BCH, beta-carotene hydroxylase; NCED1, 9-cis epoxycarotenoid dioxygenase 1; GGR, geranylgeranyl reductase; CLH, chlorophyllase; CAO, chlorophyllide a oxygenase; HDS, 1-hydroxy-2-methyl-2-butenyl 4-diphosphate (HMBPP) synthase; FPS, farnesyl diphosphate synthase; SPS, solanesyl-diphosphate synthase. Upregulation and downregulation in antiDXS1-102 compared to CK are shown using a color scale for log2 (antiDXS1-102/WT ratio) values. On the left side of the color block is the Unigene ID from the transcriptome database, and on the right side is the gene annotation in the database. (b) qRT-PCR results of selected six DEGs. The transformed line of empty vector (CK) is the control and the value is one. The
$2^{-\Delta \Delta} $ Cᴛ method was used to calculate the value of fold change compared to the control (CK, the transformed line of empty vector). All qRT-PCR reactions were performed from triplicate biological samples. Mean of three replicates ± 1 standard error is shown. Bars with superscript letters are significantly different at p < 0.05.To verify the RNA-seq data and further investigate the above DEGs, quantitative real-time PCR assays were performed for selected genes with significantly different transcription levels between CK and antiDXS1-102, namely DXS1, GA20ox1, GGR, CLH, PSY1, and NCED1. Expression of DXS1 and GA20ox1 was lower in antiDXS1-102 than in CK (Fig. 8b), indicating that antisense expression of PpDXS1 impacted the lower transcription of genes involved in GA synthesis. Meanwhile, GGR gene, involved in the biosynthesis of phytyl diphosphate and chlorophyll a, exhibited higher expression in the antiDXS1-102 line. Higher expression of PSY was seen in antiDXS1-102 than in CK, which corresponded with the RNA-seq results.
-
The MEP pathway, also known as the non-mevalonate route, was identified as an alternative terpenoid biosynthesis pathway in Eubacteria and higher plants[32]. There has been substantial progress in the identification of DXS genes, the first rate-limiting enzyme of the MEP pathway[18,33,34]. Although DXS1 function has been studied in many plants, little is known about the Poaceae family and in particular turfgrasses. In this study, a 2139 bp ORF and the upstream region of the PpDXS1 gene were isolated from P. pratensis. To our knowledge, this is the first report of DXS1 isolation from turfgrass, and the first study to examine metabolic regulation of isoprenoid biosynthesis in this plant.
DXS1 has also been annotated in Aegilops tauschii (AetDXS1) and Brachypodium distachyon (BdDXS1) with its nucleotide sequence released in NCBI, which show high similarity to PpDXS1 as found in this study, however, the physiological functions of AetDXS1 and BdDXS1 have not been previously reported. In this study, it was found that PpDXS1 possesses a conserved motif with a thiamine diphosphate binding site and transketolase motif. Bioinformatic subcellular localization prediction suggested that PpDXS1 is located in the chloroplast. In the present study, the PpDXS1 gene expression occurred in the leaves, leaf sheath, and root with the highest transcription level in leaves (Fig. 5). In Medicago truncatula and tobacco plants, DXS1 gene was also relatively higher in leaves and stems than in the roots[9,35]. As shown in the results of the phylogenetic tree and expression assay in different tissues (Fig. 3 & 5), it is indicated that PpDXS1 gene is probably in clade 1 of the DXS gene family, and is mainly expressed in leaves. Previous studies also suggest that DXS1 type are responsible for the primary metabolism, including chlorophyll biosynthesis and photosynthetic process, and mainly performed the function in leaves[12,36]. Additionally, Lois et al. reported that DXS1 is probably involved in fruit ripening and sesquiterpene formation[10]. However, in the recent report by García et al., it showed that DXS1 gene plays a key role in plant development and survival at the early growth stage besides being related to fruit carotenoid synthesis[37]. Therefore, it is indicated that DXS1 perform conserved but species-specific function, and it is important to explore the concrete roles of DXS isoforms in the regulation of plant growth and development.
On the basis of prediction analysis on the 5’ upstream region which often contains binding sites of regulatory factors, PpDXS1 gene expression increased after treatment with GA3 and ABA. Similarly, Yang et al. reported that ABA treatment upregulated SmDXS transcription in Salvia miltiorrhiza hairy roots[38]. Interestingly, the fold change of GA treatment were generally higher than those of other induced treatments, which indicated that GA3 had more effect on PpDXS1 expression than ABA in P. pratensis. Also, plants produce secondary metabolites to adapt to unfavorable conditions, such as pathogen attack. JA and fungal infection act as signals to induce secondary metabolite biosynthesis by elevating expression of related genes. For example, StDXS1 transcript levels were lower in Solanum tuberosum with symptoms of late blight (caused by the oomycete Phytophthora infestans), but expression was induced up to 12 h post-infection[33]. Similarly, exogenous JA treatment stimulated the GrDXS expression in rose-scented geranium[20]. In our study, PpDXS1 transcript expression in leaves after JA treatment was highest at the 3h-point, increasing up to 6 h after pathogen inoculation. It is indicated that the pathogen infection has longer inductive effect of the expression of PpDXS1 compared with the JA treatment. Although these changes are only observed in the leaves, it can not be ignored that the low level of PpDXS1 gene expression is correlated with the pathogen inoculation and the accumulation of phytoalexin[21].
DXS, as the rate-limiting enzyme of the MEP pathway, regulates the flux of the plant MEP pathway but how does it affect the biosynthesis of downstream terpenoids? The genetic transformation experiment described in the study was designed to evaluate the effects on plant growth and isoprenoid abundance. In this work, GAs and IAA levels were decreased, but higher accumulation of ABA occurred in the underexpression mutant compared to the CK plants of Kentucky bluegrass. In Arabidopsis, plants with DXS either overexpressed or suppressed different levels of the final isoprenoid products[39]. In brief, the total chlorophyll and ABA contents of DXS-suppressed plants were significantly lower than those of the control plants, but the expression of GA4 gene was up-regulated, and the overexpressed plants were the opposite. However, the GA content showed a higher level in Arabidopsis overexpressing the DXS1 gene from Morus notabilis[40]. Also, it is found that overexpression of DXS1 from potato (Solanum tuberosum) resulted in a higher accumulation of chlorophyll and decreased ABA and GA4 content in the transgenic Arabidopsis lines compared to control plants[37]. Although various research of homologous or heterologous expression found that overexpression or underexpression of DXS1 was linked to the same changes in isoprenoid levels[20], it was not clear whether lower expression of DXS necessarily led to decreased production of all isoprenoid end products, which merited further investigation.
Aside from the measured changes in phytohormones content in this work, the significant decrease was observed in plant height of the antiDXS1-102 mutant compared to CK. These differences are likely due, in part, to observed changes in IAA and GA levels as changes in these hormones result in dwarf phenotype (Fig. 7d). Although DXS is the first rate-limiting enzyme in the MEP pathway, additional limiting and regulatory enzymes or genes also play indispensable roles in regulation of downstream terpenoids. For example, geranylgeranyl diphosphate synthase can also control the flow of intermediates[41]. So, to further identify the regulatory and control points of DXS1 in the MEP pathway, RNA-seq was used to examine the expression of key MEP and related isoprenoid biosynthetic pathway genes in CK and antiDXS1-102 plants, to determine the effects of lowered PpDXS1 levels on isoprenoid synthesis. DEG analysis revealed that genes involved in GA and IAA biosynthesis and signal transduction pathways were downregulated in the antiDXS1-102 mutant compared to CK, which is consistent with the GA and IAA content. The antiDXS1-102 plant exhibited enhanced expression of ABA biosynthetic genes, again consistent with the ABA content. The state of the MEP pathway or intermediates were assumed to be in equilibrium with these decreased or increased downstream terpenoids. On the other hand, the overexpression or suppression of PpDXS1 gene may influence the content of downstream isoprenoids, but each of these post-IPP biosynthetic pathways may have its own set of rate-limiting and regulatory steps[5].
-
In the present study, functional analysis of DXS1 in P. pratensis revealed that underexpression of PpDXS1 on the MEP pathway have different impacts on terpenoid biosynthesis and physiology, including decreased plant height, endogenous GAs and IAA production, but promoted ABA accumulation, as well as the expression levels of related genes. This work provides not only important guidance to further study the role of DXS1 in response to environmental stresses in turfgrass, but also an addition to the knowledge of DXS1 enzyme in plants in general and Poa pratensis in particular.
-
Kentucky bluegrass cultivar ‘Baron’ was used as wild type (WT). WT plants were grown in soil-sand-perlite (1:1:1, v/v) in SC-10 single-cell containers (12 cm diameter × 10 cm depth) at 25 °C with 14/10 h day/night photoperiod. In the WT growth process, young leaves at early growth stage, mature leaves at vigorous growth stage, and old leaves at heading stage were used for gene expression analysis of the time-series sample. In addition, fully expanded leaves, leaf sheaths and roots at vigorous growth stage were also used for gene organ/tissue-specific expression analysis. Plants were also foliar sprayed with 30 mg/L gibberellic acid (GA3), 10 mg/L abscisic acid (ABA), 535 mg/L jasmonate (JA), and a spore suspension of Puccinia graminis[42]. The leaf samples were collected 3, 6, and 12 h after treatment and used for gene expression analysis.
Cloning the PpDXS1 sequence of ORF and promoter region
-
Candidate nucleotide sequences of DXS unigenes from relatives of P. pratensis were used in Blastn searches of P. pratensis transcriptome sequences (NCBI accession number: SRA315988). Three candidate DXS unigenes were assembled and specific primers were designed to amplify the middle fragment. Then, specific primers (Table 1) for amplification of 5′ and 3′ fragments using a SMART RACE cDNA Amplification Kit (Clontech, USA). Fragments were amplified and 10 clones of full ORF sequence were confirmed by sequencing and blast. Finally, PpDXS1 gene ORF sequence of one consistent clone are deposited in GenBank (accession numbers: MG257788).
Table 1. Nucleotide sequences of gene-specific primers
Primer name Sequence (5'-3') Description 3'-GSP CGACGACCTCATCACCATCCTCCG 3'-RACE primer 5'-GSP GTCTTGGTGCCCTTGACCTCCCG 5'-RACE-out-primer 5'-NEST TCCGCCTATTTGCTTCGTCACTCC 5'-RACE-in-primer DXS-F ATGGCGCTCTCGACGACGTTCT ORF-F DXS-R CTAAACATTCTGCACCGTCATG ORF-R antiDXS-F(BstEII) GGGTNACCATGGCGCTCTCGACGACGTTCT Primers for vector construction antiDXS-R(BglII) GAAGATCTAACATTCTGCACCGTCATG SP1 CCGTCGGTCTGCCGCATCGTC Primer for promoter amplification SP2 TCGCTTGTCCTGAGGGGTGTTG SP3 TGAGGGACAGGTTCTTCATGTGGA Genomic DNA was extracted from WT leaves using a modified CTAB method. To clone the promoter region of PpDXS, high-efficiency thermal asymmetric interlaced PCR were performed using degenerate primers and specific nested-primers (Table 1, Supplemental Table S2) designed against the PpDXS ORF sequence. Amplified fragments produced by the nested PCR were cloned and then sequenced. Sequences (shown in Supplemental Fig. S2) that extended upstream of PpDXS were isolated and used for the further analysis. The 5′ upstream sequence was analyzed for putative cis-acting regulatory elements using the Plant CARE database.
Bioinformatics analysis of PpDXS1
-
DXS1 protein sequences were collected using the BLASTp program and were aligned using the ClustalW program (http://www.ddbj.nig.ac.jp) with standard parameters. The physical and chemical characteristics of the amino acid sequence were conducted in ProtParam program (http://web.expasy.org/protparam) and the subcellular localization were analyzed by TargetP 1.1 Server. The phylogenetic tree was generated by the neighbor-joining method on the MEGA5 software and bootstrap values were obtained from 1,000 replicates[43]. Signal peptide and transmembrane topological structures were predicted by SignalP (http://www.cbs.dtu.dk/services/SignalP) and TMHMM 2.0 server (http://www.cbs.dtu.dk/services/TMHMM). Crystal structure of PpDXS and AtDXS were modeled using the Expasy server and conserved protein domains were analyzed by searching the deduced amino acid sequences against the NCBI Conserved Domain Database (CDD, http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). The subcellular localization was predicted using the WoLF PSORT server.
Relative expression analysis
-
Total RNA from root, leaf sheath and leaf were obtained with TRIzol reagent kit (Invitrogen, USA) using 100~500 mg tissue homogenized in liquid nitrogen. Its quantity and purity were assessed using the NanoDrop 2000 (Thermo, USA) and reverse transcriptional reaction was carried out with 0.5 μg total RNA using PrimeScriptTM RT reagent Kit (Perfect Real Time) (Takara, Japan) according to the supplier’s instruction.
Quantitative real-time PCR was carried out using a SYBR Green assay (Takara, Dalian, China) on a Bio-Rad CFX96 System (Roche, USA). Each 25 μL assay contained 12.5 μL SYBR Premix Ex Taq, 2 μL cDNA and 100 nM of each primer (Supplemental Table S2). For exogenous application, the relative mRNA abundance was calculated by using the comparative CT method (
$2^{-\Delta\Delta} $ Cᴛ) and normalization to 18S gene. The analysis included three biological replicates and three technical replicates for each sample. The data were statistically analyzed via the SPSS21.0 software, and subjected to one-way analysis of variance (ANOVA) to determine the least significant difference (LSD) among the treatments at p < 0.05.Genetic transformation and regeneration of Kentucky bluegrass
-
Antisense oligonucleotides have been used for more than a decade to downregulate gene expression[44]. So, in the study, an RNA antisense-expressing cassette targeting the PpDXS1 gene was placed under the control of the CaMV 35S promoter and NOS terminator in the pCAMBIA1301 vector. The PpDXS1 gene was amplified from a pGEMT-easy plasmid clone using gene-specific full-length primers that incorporated BglII and BstEII restriction sites in the forward and reverse primers, respectively (Table 1). After amplification and restriction digestion, the PpDXS1 gene was inserted into the entry plasmid pCAMBIA1301 to form recombinant plasmid pCAMBIA1301-antiDXS (Supplemental Fig. S4).
Embryogenic calli (approximately 100 small pieces of callus) were placed as a 2.5 cm diameter monolayer in a 5.0 cm Petri dish containing subculture medium (MS basal medium plus 30 g/L sucrose, 1 mg/L 2,4-D, 0.5 mg/L 6-BA, and 0.2 M mannitol) for 4–8 h of osmotic treatment prior to bombardment. Gold particles (0.6 μm, Bio-Rad, CA) were DNA-coated essentially as described by Ha et al.[45]. The bombardment mixture contained 0.6 mg gold particles and 1 μg plasmid DNA per shot. Bombardment was carried out using a Biolistic PDS-1000/He Particle Delivery System (Bio-Rad, Supplemental Fig. S5) with a target distance of 6 cm. Bombarded calli were incubated on subculture medium overnight.
Callus bombarded with pCAMBIA1301-antiDXS1 and pCAMBIA1301 empty vector were incubated in subculture medium for 7 d and then transferred to selected regeneration medium containing 100 mg/L hygromycin and a certain amount of 6-BA and KT. After two selection rounds with 100 mg/L hygromycin under dark conditions, hygromycin resistance callus was transferred to regeneration medium supplemented with 50 mg/L hygromycin and incubated under a 16/8-h (light/dark) photoperiod. Fully recovered plantlets were transferred to containers for further root development and, finally, green plants were transferred to soil in a greenhouse. Transformed plants of pCAMBIA1301-antiDXS1 were identified in T0 plants by PCR with three pairs of specific primers for the hygromycin gene (Hyg-F/R, 517 bp), 35S promoter regions (35S-F/R, 195 bp) and compound primers (35S-F and antiDXS1-F), see details in Supplemental Table S2. And the transformed plants of empty vector (used as controls, namely CK) were identified by two pairs of primers of hygromycin gene and 35S promoter regions.
Protein extraction and western blot analysis
-
Total plant protein extracts were obtained from 50–100 mg of fresh tissue. Samples were ground in liquid nitrogen, suspended in 500–1,000 μL ice-cold RIPA buffer supplemented with 1 mM PMSF, and ultrasonicated. Resuspended samples were then centrifuged at 12,000 rpm for 10 min at 4 °C, and then the supernatant was recovered and re-centrifuged. Supernatant protein concentration was determined using the bicinchoninic acid (BCA) method. Proteins were separated using SDS-PAGE and then electrotransferred to polyvinylidene fluoride membranes (PALL, America). Membranes were incubated overnight at 4°C with primary antibody (diluted 1:500) then with horseradish peroxidase-conjugated secondary antibody (diluted 1:5,000) for 2 h at room temperature. The objective protein is a polyclonal antibody on the basis of designed immunogen fragments according to the predicted DXS protein sequence and synthesized by Abmart (Shanghai, China). Imaging and quantification were performed using Quantity One (Bio-Rad).
Analysis of plant height, endogenous hormone and chlorophyll content in transformed plants
-
Firstly, plant height of one CK and two antiDXS1-transformed lines was recorded and five replications were set in each pot. Then, fresh samples comprising the terminal bud and young leaves (0.3–1 g) from plants at the same stage of development were collected and used to determine endogenous hormone content. Levels of total GAs, ABA, IAA, and JA were determined using the ELISA method according to the manufacturer’s protocol (supplied by the Crop Chemical Control Laboratory at China Agricultural University). Chlorophyll was extracted by soaking 50 mg of fresh leaves in 8 mL acetone (95%, v/v) for 72 h in the dark, followed by spectrophometric quantification at 470, 645, and 663 nm (Beckman, CA, USA). Total chlorophyll content was calculated using the Arnon method[46]. These data were statistically analyzed via the SPSS21.0 software, and subjected to one-way analysis of variance (ANOVA) to determine the least significant difference (LSD) between CK and antiDXS1 transgenic plants at p < 0.05.
Transcriptional expression analysis by RNA-sequencing
-
Kentucky bluegrass leaf blades were collected from the control (CK) and transformed plant of target gene (antiDXS1-102) and used for sequencing. Total mRNA was extracted using TRIzol Reagent (Invitrogen, USA) according to the manufacturer’s instructions, and cDNA library construction and normalization were performed as described previously[47]. Total RNA samples were sequenced using the Illumina HiSeq platform. Cleaned and qualified reads were assembled de novo using Trinity software, as described previously[48], and mapped to reference transcriptome libraries using RSEM (v 0.7) alignment. Read counts of transcripts with a reciprocal match to the reference transcriptome were extracted and calculated using FPKM (expected number of Fragments per Kilobase of transcript sequence per Million base pairs sequenced) values for gene expression analysis. The matrix of read counts was used with the DESeq R statistical package to identify transcripts with significant expression differences between antiDXS-102 and CK (FDR < 0.05). Log2 (FPKM-antiDXS-102/FPKM-CK ratio) values were shown in heatmaps representing expression profiles of DEGs.
- All authors gratefully acknowledge the Forestry Central Laboratory of Beijing Forestry University for providing the research facilities. This study was supported by the National Natural Science Foundation of China (No. 31302016).
- The authors declare that they have no conflict of interest.
- Supplemental Fig. S1 Bioinformative analysis for PpDXS1 protein. (a) Amino acid sequence alignment of DXS1. (b) Prediction of function domain. (c) Prediction of signal peptide. (d) Prediction of subcellular localization. (e) Prediction of transmembrane topological structures.
- Supplemental Fig. S2 The promoter sequence of PpDXS1 from Kentucky bluegrass.
- Supplemental Fig. S3 Differential expression genes of antiDXS1-102 vs CK. (a) Volcanic map of differential expression genes of antiDXS1-102 vs CK. (b) GO enrichment of down-regulated differentially expressed genes on antiDXS1-102 vs CK. (c) GO enrichment of up-regulated differentially expressed genes on antiDXS1-102 vs CK.
- Supplemental Fig. S4 The map of recombinant plasmid pCAMBIA1301-antiDXS1.
- Supplemental Fig. S5 Biolistic PDS-1000He Particle Delivery System.
- Supplemental Table S1 Analysis of gray value in the western blot.
- Supplemental Table S2 The other primers for the study.
- Copyright: © 2021 by the author(s). Exclusive Licensee Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Gan L, Chao Y, Han L, Yin S. 2021. Underexpression of PpDXS1 gene decreased plant height and resulted in altered accumulation of phytohormones in Kentucky bluegrass. Grass Research 1: 9 doi: 10.48130/GR-2021-0009

Underexpression of PpDXS1 gene decreased plant height and resulted in altered accumulation of phytohormones in Kentucky bluegrass
- Received: 26 March 2021
- Accepted: 22 September 2021
- Published online: 19 October 2021
Abstract: 1-deoxy-D-xylulose-5-phosphate synthase (DXS) catalyzes the first and rate-limiting step of the plastidic 2-C-methyl-D-derythritol-4-phosphate (MEP) pathway which regulates the synthesis of terpenoids, such as gibberellins, abscisic acid, and chlorophyll. The objective of this study was to determine the functional role of PpDXS1 in plant growth and development in Kentucky bluegrass (Poa pratensis L.). The PpDXS1 gene has a 2139 bp open reading frame that encodes a polypeptide of 712 amino acids with a calculated molecular weight of 76.7 kDa. PpDXS1 gene expression was the highest in leaves. Moreover, the PpDXS1 promoter contained several hormone response elements and gene expression was induced by exogenous treatment with gibberellin, abscisic acid, jasmonate, and pathogen infection. Functional analysis indicated that underexpression of PpDXS1 gene in Poa pratensis decreased plant height and endogenous gibberellin and indole acetic acid production, but promoted abscisic acid accumulation. Furthermore, transcriptome analysis and qRT-PCR results showed that the expression levels of related genes involved in the phytohormone biosynthesis and signal transduction were differentially regulated by PpDXS1 in transgenic Poa pratensis. Overall, these results indicated that PpDXS1 has strong effects on plant height and accumulation of phytohormones, and provided a preliminary understanding of molecular characterization, expression and function of PpDXS1 in Poa pratensis.
-
Key words:
- DXS1 /
- MEP pathway /
- Terpenoids /
- Plant height /
- Hormones /
- Underexpression /
- Poa pratensis