









-
Endosperm is the main part of the plant seed. It is not only the source of nutrition but also the integration of seed growth and development[1]. The endosperm is localized between the embryo and the seed coat. In most mature seeds, the mutual signaling process between the testa and the endosperm seems to coordinate the normal growth process of the seed and is very important for the normal development of the seed[2]. Early proliferation of the endosperm is associated with final seed size, and alterations in the rate and duration of cell division in the endosperm are considered a biotechnological strategy to alter seed size[3]. Molecular genetic studies of early endosperm development, primarily in Arabidopsis, have yielded an intriguing array of findings, including genomic imprinting and epigenetic mechanisms, auxin signaling, microtubule formation and development. A range of transcription factors affecting endosperm development have also been identified in different kinds of crops[4,5].
Different from dicotyledonous plants, the seeds of monocotyledonous plants consist of the embryo, endosperm and pericarp, with the endosperm as the main part; the endosperm is also the main part of the accumulation of reserve substances during seed development[6,7]. Molecular studies on the early development of monocot endosperm have mainly focused on maize, rice and wheat. Among them, in maize, McClintock's classic study[8] provided strong data on endosperm cytogenetics and development, and more recently, a large amount of relevant data has been obtained using molecular biology[9]. In wheat[10] and barley[11], functional analyses have defined key genes related to starch biosynthesis in endosperm and their association with different components and structures of starch over the past decade. Much progress has been made in the correlation of physicochemical properties.
Among many plant seeds, coconut has not only the largest seed volume but also the most unique endosperm structure and development process. The development of coconut fruit and the formation of coconut copra is a very special process: after double fertilization, the embryo sac of the coconut fruit gradually expands, and the initial embryo sac is filled with clear liquid, in which suspensions of different sizes of free nuclei exist[12]. As the fruit develops, a large number of free spherical cells form in the center of the cellular organization, with free nuclei present in the initial liquid. Subsequently, these cells and free nuclei migrate toward the periphery of the embryo sac and adhere to the surface of the endothelium. Cells first aggregate at the bottom of the embryo sac and gradually grow upward to the top of the embryo sac to the germination hole of the embryo. Bacterial nuclear divisions in the endosperm are frequent in the early-formed coconut endosperm cell layer, but mitosis becomes more common as cell differentiation continues. The basal cell layer in the endosperm adjacent to the endothelium remains meristematic, eventually forming the mature endosperm composed of radially elongated cells, which we see as copra[13,14]. However, in the more than half a century since the completion of the cell morphology study of coconut meat formation, there has been no new substantial progress in the molecular biological mechanism of coconut meat development[15].
ATAC-seq (Assay for Transposase Accessible Chromatin using sequencing) is the use of sequencing to study chromatin accessible to transposases[16]. The open region of the sample genomic DNA is cut by using the Tn5 transposase carrying the next-generation sequencing adapter, and the digested DNA fragments are enriched for high-throughput sequencing and bioinformatics analysis. Based on this technology, open regions of the genome can be quickly, simply and efficiently achieved. Compared with MNase-seq, DNase-seq, and FAIRE-seq, ATAC-seq technology also has the advantages of low sample requirements for library construction, high detection sensitivity, and good reproducibility[17]. Moreover, ATAC-seq can reflect the chromatin state at the genome-wide level, discover key transcription factors that regulate biological processes, predict the downstream functional genes of transcription factors and analyze the chromatin opening of single cells[18].
In this paper, using the solid endosperm of coconut as material, we obtained chromatin opening data of the genomes at four different developmental stages (7, 8, 9, and 10 months after pollination) by ATAC-seq and RNA-seq technologies. Through the combined analysis of the results of ATAC-seq and RNA-seq, a considerable number of combinations containing transcription factors and downstream regulatory genes were identified. Among them, one transcription factor (CnGATA20) and its downstream regulated gene (CnOLE18) were selected for further research. Finally, the predicted regulatory relationship was verified through yeast one-hybrid, protoplast transient expression and dual-luciferase reporter assays. The results provide a new strategy and research foundation for identifying important functional genes and elucidating the regulatory network of metabolism in the development of coconut endosperm.
-
RNA libraries of coconut endosperm from four different developmental periods were constructed, and high-throughput ATAC-seq and RNA-seq were performed by Romics. Ultimately, 121.0, 118.8, 115.7, and 115.0 million reads were generated from CO1, CO2, CO3, and CO4 tissue by ATAC-seq, respectively. For RNA-seq, 57.7, 54.5, 50.1, and 48.5 million reads were obtained from CO1, CO2, CO3, and CO4 tissue, respectively. After aligning the ATAC-seq reads to the coconut genome, we found that 71.9%, 72.4%, 70.1%, and 75.5% of all reads from CO1, CO2, CO3, and CO4 were uniquely mapped to the genome, which was more than sufficient to successfully identify accessible chromatin regions in coconut (Table 1). The rates of uniquely mapped reads in the RNA-seq were 92.0%, 92.4%, 93.2%, and 93.2%, which shows that the quality of sequencing conforms to the quality control standard (Table 2).
Table 1. Statistics of ATAC-seq sequencing results.
Statistics CO1 CO2 CO3 CO4 All 121020906 118840434 115711432 115008357 UnMapped 507153 497831 2057646 4584387 Mapped 120513753 118342603 113653786 110423970 MappedRate 0.996 0.996 0.982 0.96 UniqueMapped 86960478 86007908 81107124 86868484 UniqueMappedRate 0.719 0.724 0.701 0.755 MultiMapped 33553275 32334695 32546662 23555486 The CO1, CO2, CO3 and CO4 represent four different stages of coconut endosperm development: 7, 8, 9, and 10 months, respectively; All: The number of all reads involved in the comparison, that is, the number of filtered reads (clean data); UnMapped: Number of reads not matched to the genome; Mapped: Number of reads matched to the genome; MappedRate: Mapped Reads/All; UniqueMapped: Number of reads uniquely matched to the genome; UniqueMappedRate: Unique Mapping/Mapped Reads; MultiMapped: Number of reads multiple matched to the genome. Table 2. Statistics of RNA-seq sequencing results.
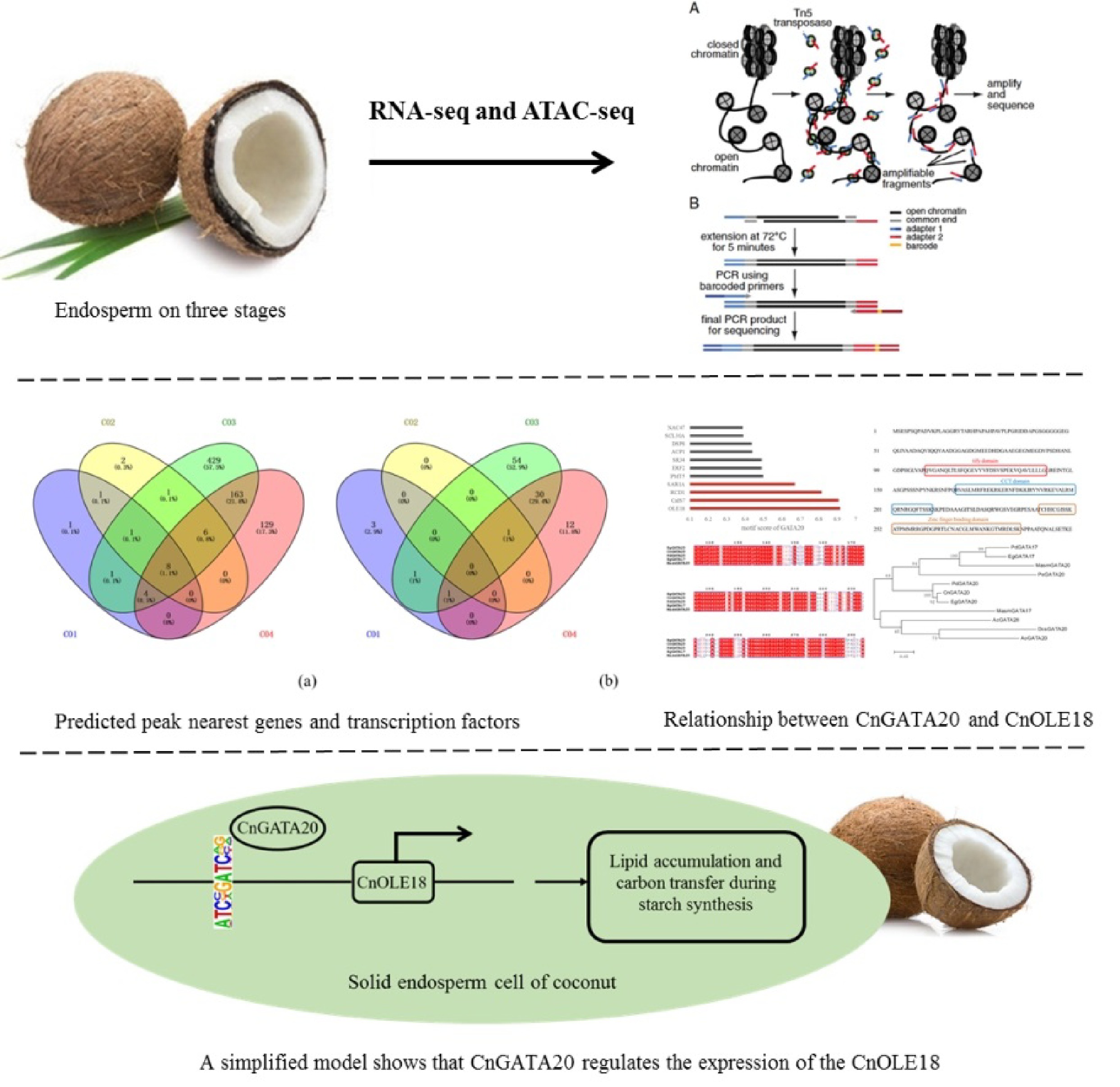
Statistics CO1 CO2 CO3 CO4 All 57678112 54506264 50073176 48519010 UnMapped 2087496 1596700 1389366 1368720 Mapped 55590616 52909564 48683810 47150290 MappedRate 0.964 0.971 0.972 0.972 UniqueMapped 53064910 50372992 46682864 45236198 UniqueMappedRate 0.92 0.924 0.932 0.932 MultiMapped 2525706 2536572 2000946 1914092 The CO1, CO2, CO3 and CO4 represent four different stages of coconut endosperm development: 7, 8, 9, and 10 months, respectively; All: The number of all reads involved in the comparison, that is, the number of filtered reads (clean data); UnMapped: Number of reads not matched to the genome; Mapped: Number of reads matched to the genome; MappedRate: Mapped Reads/All; UniqueMapped: Number of reads uniquely matched to the genome; UniqueMappedRate: Unique Mapping/Mapped Reads; MultiMapped: Number of reads multiple matched to the genome. To determine the transcription factors that play a regulatory role according to the chromatin open region and understand how transcription factors regulate downstream genes, the results of ATAC-seq and RNA-seq were analyzed together to determine the regulatory patterns of transcription factors. We overlapped the downregulated differential genes in RNA-seq with the genes related to the chromatin open region with a weakened signal in ATAC-seq. At the same time, the upregulated differential genes in the RNA-seq and the related genes in the chromatin open region with enhanced ATAC-seq signals were analyzed by overlap. Among the downregulated differentially expressed genes between the different groups, 26,437 and 368 genes were common to ATAC-seq and RNA-seq, respectively. The common ATAC-seq and RNA-seq genes were 32, 36 and 9, respectively, among the upregulated differentially expressed genes between the different groups (Fig. 1). By combining analysis of the regulation of the transcription process from DNA to RNA, genes and transcription factors (TFs) that possibly act in coconut endosperm development were identified on a large scale.
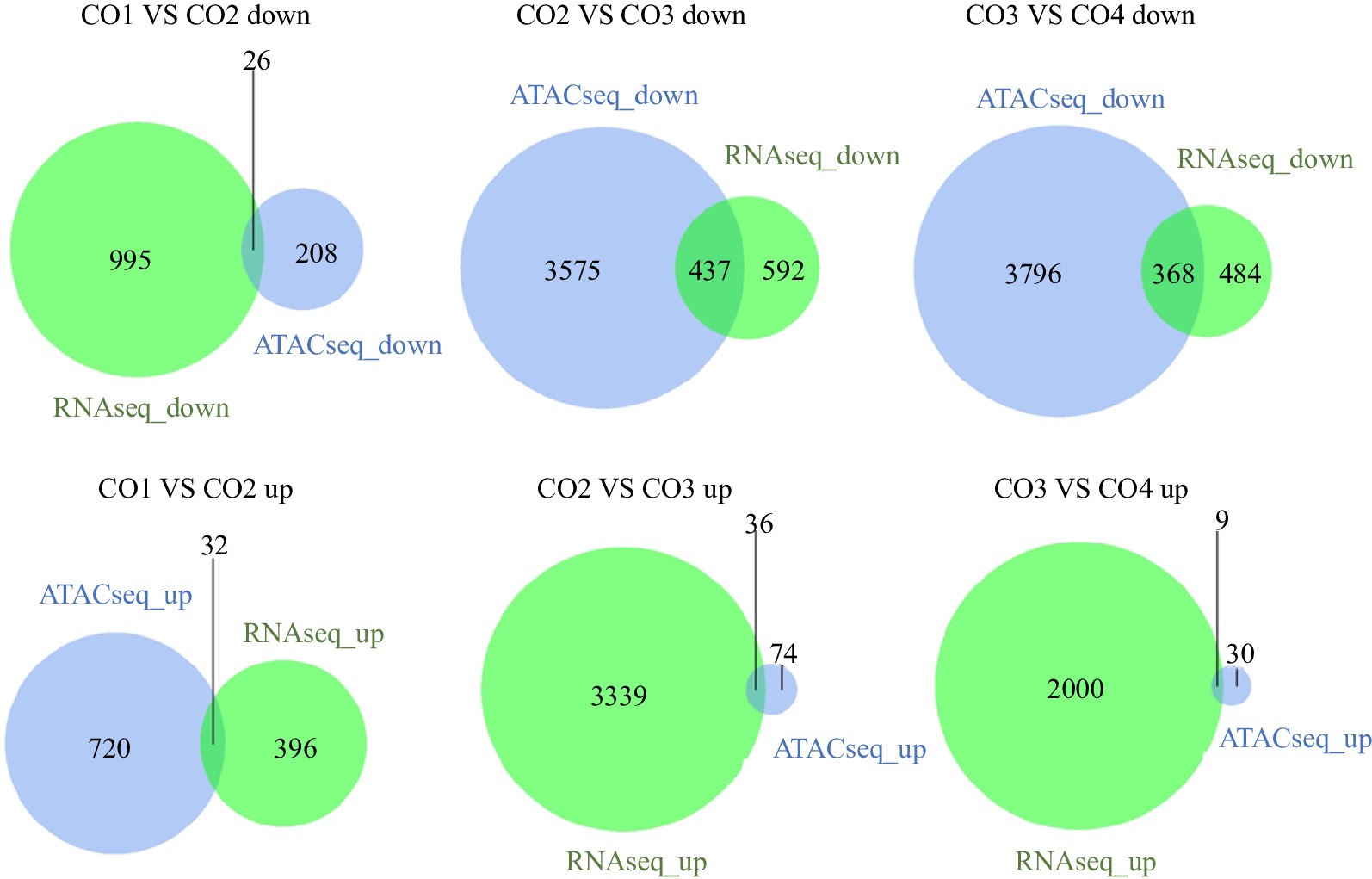
Figure 1.
Overlap of genes that have the same trend in RNA-seq and ATAC-seq. ATACseq Down represents the number set of related genes in the chromatin open region with weakened ATAC-seq signal. RNAseq Down indicates the number set of down regulated differential genes of RNA-seq. ATACseq Up represents the number set of related genes in the chromatin open region enhanced by ATAC-seq signal. RNAseq Up represents the number set of up-regulated differential genes of RNA-seq. CO1 vs CO2 down/up: Compared with the CO2, the expression of genes in the CO1 is significantly down/up regulated.
Mapping accessible chromatin in endosperm cells
-
According to the ATAC-seq results, the functional regions of the whole genome were divided into promoter-TSS (promoter to transcription start site, default range is 1 kb upstream to 100 bp downstream of the gene transcription start site), TTS (transcription termination site, default range is 100 bp upstream to 1 bp downstream of the gene transcription termination site), exon, intron, and intergenic area. It is well known that TFs bind to the transcription start sites (promoter-TSSs) of genes and regulate the transcription of upstream and downstream genes. Therefore, we mainly focused on the peaks in the promoter-TSS area. As a result, the genomic distributions of peaks located in the promoter-TSS region were 1.52%, 1.61%, 11.99% and 5.69% in different periods of coconut development (Supplemental Table S1). This finding indicates that the degree of chromatin accessibility at the genome level was the highest in CO3, and the degree of chromatin accessibility in CO4 was lower than that in CO3 but higher than those in CO1 and CO2.
For the four developmental periods, the peaks located in intergenic regions were the highest in all functional regions, with values of 92.83%, 92.57%, 77.89%, and 81.86%, respectively. Intergenic regions account for a large proportion of the genome, so more peaks are obtained than other regions, but most of these peaks do not contain real regulatory factor-binding sites (Fig. 2). Moreover, the genomic distribution of peaks was very similar between CO1 and CO2, with approximately 6% of peaks located in the TTS, exon and intron regions. The genomic distribution of peaks was very similar between CO3 and CO4, with approximately 10% of peaks located in the TTS, exon and intron regions. The TTS regions may be downstream regions for transcription, and regulatory factors binding to the exon or intron regions of genes may play a role in the variable splicing process of the genes.
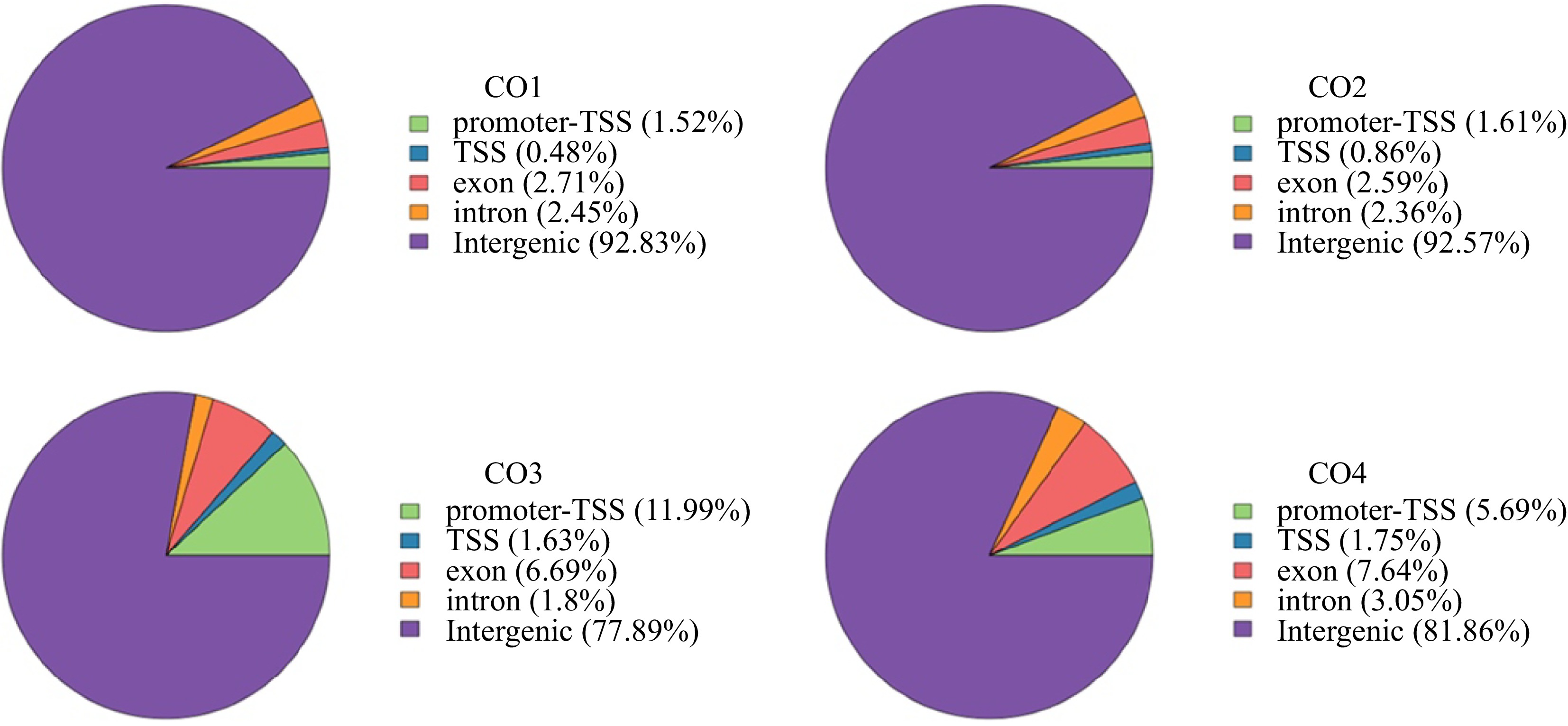
Figure 2.
Distribution of Peak in functional regions. Proportion of peaks regions matched to elements in the coconut genome at CO1, CO2, CO3, and CO4 stages, respectively. Promoter-TSS: The region from the promoter to the transcription start site ranges from 1 kp upstream of the gene start point to 100 bp downstream of the gene start point. TTS: Transcription termination sites, ranged from 100 bp upstream of the gene endpoint to 1 kp downstream of the gene endpoint.
Analyses of chromatin accessibility and gene expression
-
Transcription factors tend to bind to specific DNA sequences that usually have highly similar nucleotide sequence patterns, and each transcription factor has a motif with a target DNA sequence. Thus, transcription factors possibly bound in the open chromatin can be obtained by retrieving the motif predicted by each open region. On the basis of the motifs of the promoter-TSS region in four periods, we identified the type and proportion of the TF family that may play a regulatory role in different developmental stages. These TFs are mainly typical plant regulatory factors, such as NAC, homeobox, MYB, GATA and WRKY, with only the specific proportion being different in the four periods. To identify transcriptionally regulated genes critical for development, we also generated Venn diagrams using motifs of these transcription factors. Comparison of all motifs from different TF families showed a unique and common family of transcription factors between different developmental stages, indicating development-specific regulatory programs (Fig. 3).

Figure 3.
Categorization of motif in Promoter-TSS region. Proportion of transcription factors corresponding to motif in Promoter-TSS region of coconut genome at CO1, CO2, CO3 and CO4 stages, respectively.
Identification of the dynamic epigenetic regulation of CnGATA20
-
To further identify the regulatory factors related to coconut endosperm development, we also annotated the peak nearest genes of the promoter-TSS region (Fig. 4a) and identified the unique and common genes and transcription factors between different periods in a Venn diagram (Fig. 4b). After analyzing all intergroup-specific TFs in the four samples, we noticed that CnGATA20 was specifically upregulated during CO3, and CO3 was an important period of coconut endosperm development. Therefore, it is speculated that CnGATA20 may be a key transcription factor in the development of coconut endosperm. According to the position information of peaks corresponding to motifs, we can predict the possible binding site of CnGATA20 on the chromosome and find the gene regulated by the transcription start site located near the site. We found that CnGATA20 had the highest motif score at the promoter region of CnOLE18 (Fig. 5a). Therefore, we speculated that CnOLE18 may be the target gene regulated by CnGATA20. Meanwhile, IGV (Integrative Genomics Viewer) revealed that the chromatin accessibility of CnOLE18 increased during CO3, which is consistent with the fact that CnGATA20 may regulate coconut endosperm development during the same period (Fig. 5b).
Figure 4.
Venn diagrams of Peak nearest genes and transcription factors in Promoter-TSS region. (a) Distribution of Peaks nearest genes in Promoter-TSS region of coconut genome, Venn diagrams shows the overlap between CO1, CO2, CO3 and CO4 stages , respectively. (b) Distribution of Peaks nearest transcription factors in Promoter-TSS region of coconut genome, Venn diagrams show the overlap between CO1, CO2, CO3 and CO4 stages, respectively.

Figure 5.
(a) The motif score of CnGATA20 at transcription initiation sites of different genes. (b) The IGV view shows chromatin accessibility of CnOLE18 in Promoter-TSS region at CO1, CO2, CO3 and CO4 stages.
Gene cloning and sequence analysis of CnGATA20
-
The open reading frame (ORF) cDNA sequence of CnGATA20 was isolated from coconut endosperm by PCR amplification. Sequence analyses revealed that CnGATA20 contains a 981 bp ORF that encodes a protein containing 326 amino acids. CnGATA20 has three typical conserved domains: a 34-aa (amino acid) tify domain, a 44-aa CCT domain, and a 44-aa zinc finger-binding domain (Fig. 6a). We constructed two phylogenetic trees using MEGA 6.0 to determine the phylogenetic relationships between CnGATA20 and GATA proteins from other plant species (Fig. 6b). Phylogenetic analysis suggested that CnGATA20 is closely related to EgGATA20 and PdGATA20, indicating possible functional and evolutionary similarity among them. In addition, multiple sequence alignment was performed to show the high sequence conservation between CnGATA20 and other GATA from Elaeis guineensis, Phoenix dactylifera and Musa acuminata (Fig. 6c).
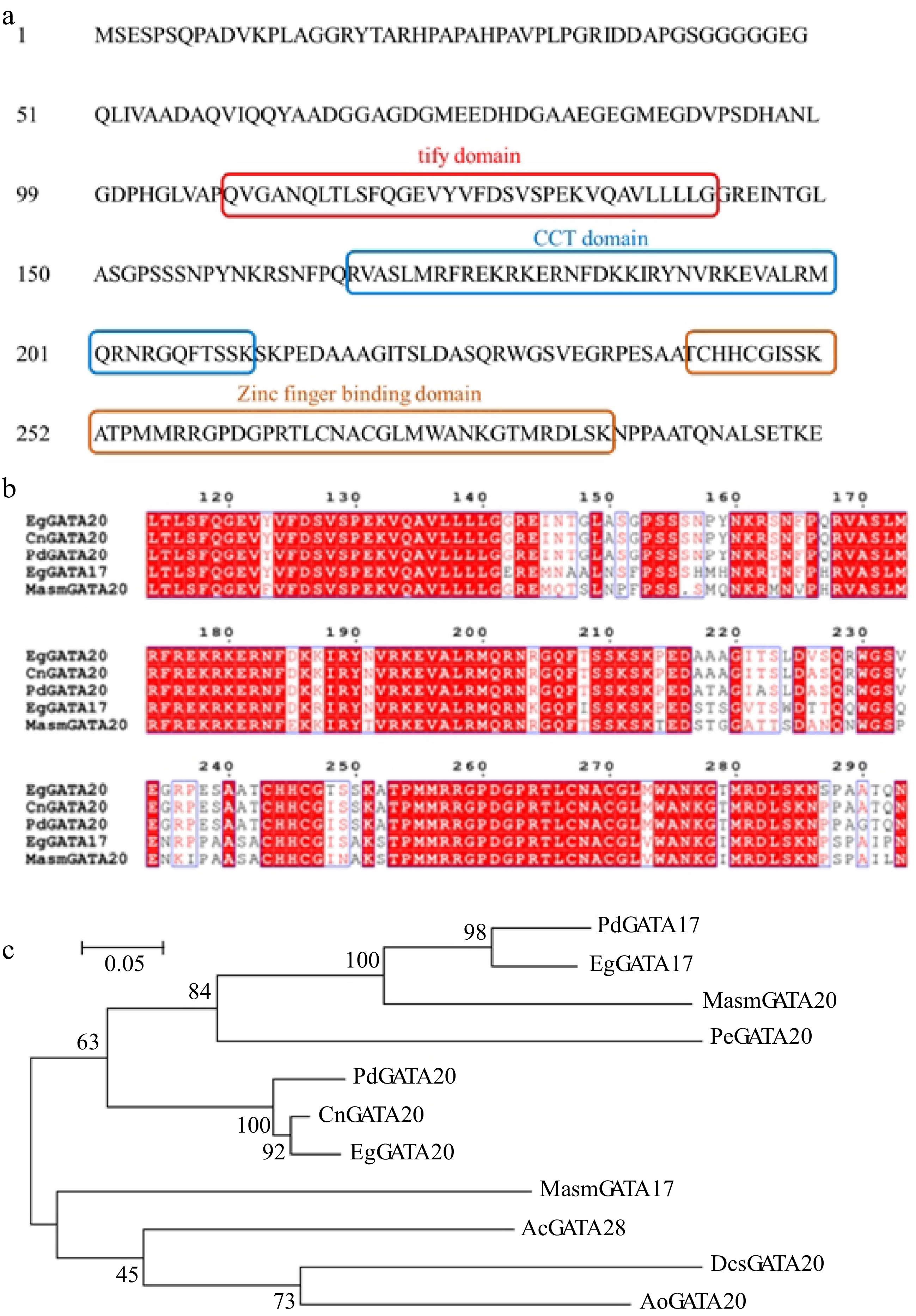
Figure 6.
(a) Three typical conserved domains in CnGATA20. (b) Comparison of deduced amino acid sequences of CnGATA20 with GATA genes from Elaeis guineensis, Phoenix dactylifera and Musa acuminata subsp. Malaccensis by Clustal X 2.0. (c) Phylogenetic analysis of CnGATA20 and related proteins from other plant species. Phylogenetic analysis of CnGATA20 and 10 other plants GATA protein sequences obtained from the NCBI database. PdGATA17: Phoenix dactylifera, XP_008789602.2; PdGATA20: Phoenix dactylifera, XP_008811434.1; EgGATA17: Elaeis guineensis, XP_010913727.1; EgGATA20: Elaeis guineensis, XP_010929337.1; MasmGATA17: Musa acuminata subsp. Malaccensis, XP_009417492.1; MasmGATA20: Musa acuminata subsp. Malaccensis, XP_009384454.1; PeGATA20: Phalaenopsis equestris, XP_020572684.1; AcGATA28: Ananas comosus, OAY66115.1; DcsGATA20: Dioscorea cayenensis subsp. rotundata, XP_039144785.1; AoGATA20: Asparagus officinalis, XP_020275468.1.
Subcellular localization and yeast one-hybrid assay
-
The p35MK1-CnGATA20-GFP fusion construct was generated and transferred into the leaves of N. benthamiana to identify the subcellular localization of CnGATA20 in plants. As shown in Fig. 7a, we found that GFP fluorescence signals overlapped with the nuclei stained by 4',6-diamidino-2-phenylindole (DAPI). These results suggested that CnGATA20 localized to the nucleus. Moreover, a yeast one-hybrid assay was performed to verify the interaction between CnGATA20 and the CnOLE18 promoter. pHIS2.1-pro53 and pGADT7-53 (pHIS2.1-pro53+pGADT7-53) were cotransferred into yeast Y187 as a positive control, and pHIS2.1-proCnOLE18+pGADT7-53 was used as a negative control. The yeast strains cotransformed with proCnOLE18+pGADT7-CnGATA20 were cultured on selective medium containing 2 mM 3-AT. The positive control survived well, while the negative control was unable to grow (Fig. 7b). These results indicated that the CnGATA20 protein could interact with the CnOLE18 promoter in the yeast system.
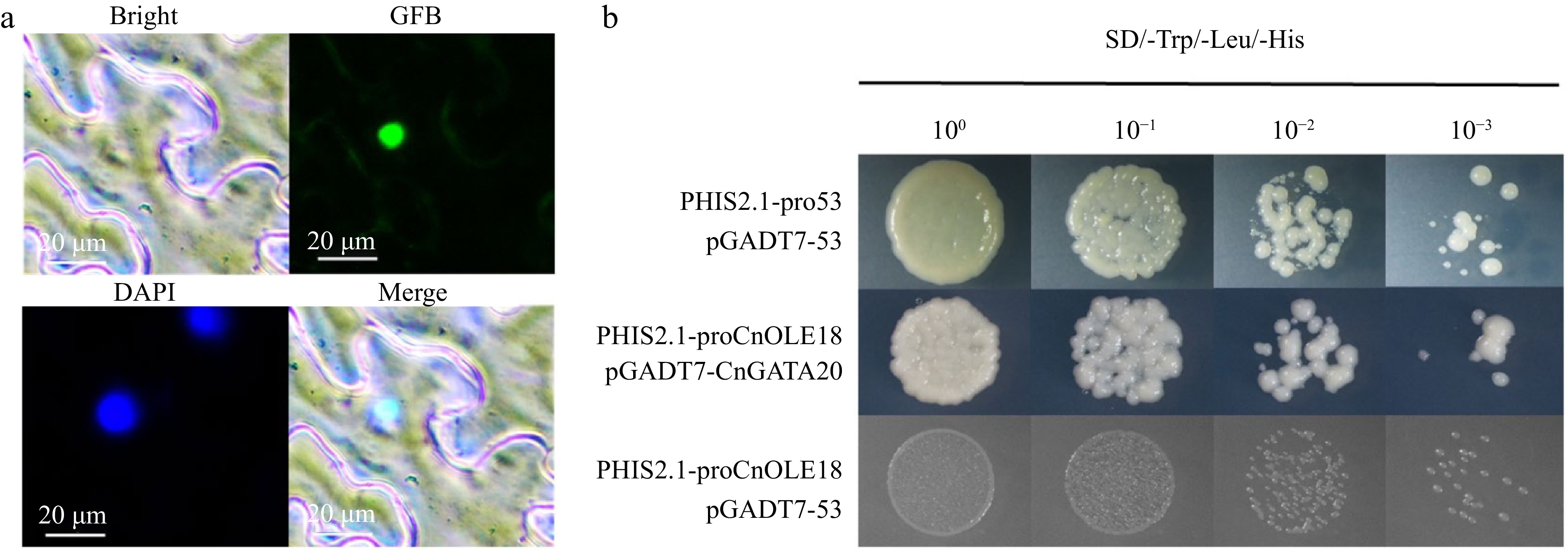
Figure 7.
Subcellular localization and yeast one-hybrid of CnGATA20. (a) Bright-feld, GFP, DAPI and merged images of the CnGATA20 protein fused with GFP in N. benthamiana leaves, Scale bar, 20 μm. (b) PHIS2.1-pro53+pGADT7-53 was used as positive control, the PHIS2.1-proCnOLE18+pGADT7-53 was served as the negative control. SD-Trp/Leu/His, SD medium without Trp, Leu, His, or Leu supplemented with 3-AT at a concentration of 2 mM.
Transient expression and dual-luciferase analysis
-
Protoplasts isolated from the tender leaves of coconut seedlings were used to confirm the regulatory relationship between CnGATA20 and CnOLE18. According to the PEG-mediated transfection method, the pGreenII62-SK-CnGATA20 and pGreenII62-SK plant expression vectors were introduced into fresh coconut leaf protoplasts. The protoplasts were collected for qRT–PCR analysis after incubation in weak light for 16 h. As shown in Fig. 8, analysis of the fluorescence quantification data showed that the expression level of CnOLE18 was significantly increased when CnGATA20 was overexpressed in protoplasts. Therefore, these data indicated that CnGATA20 positively regulates the expression of CnOLE18 (Fig. 8a).
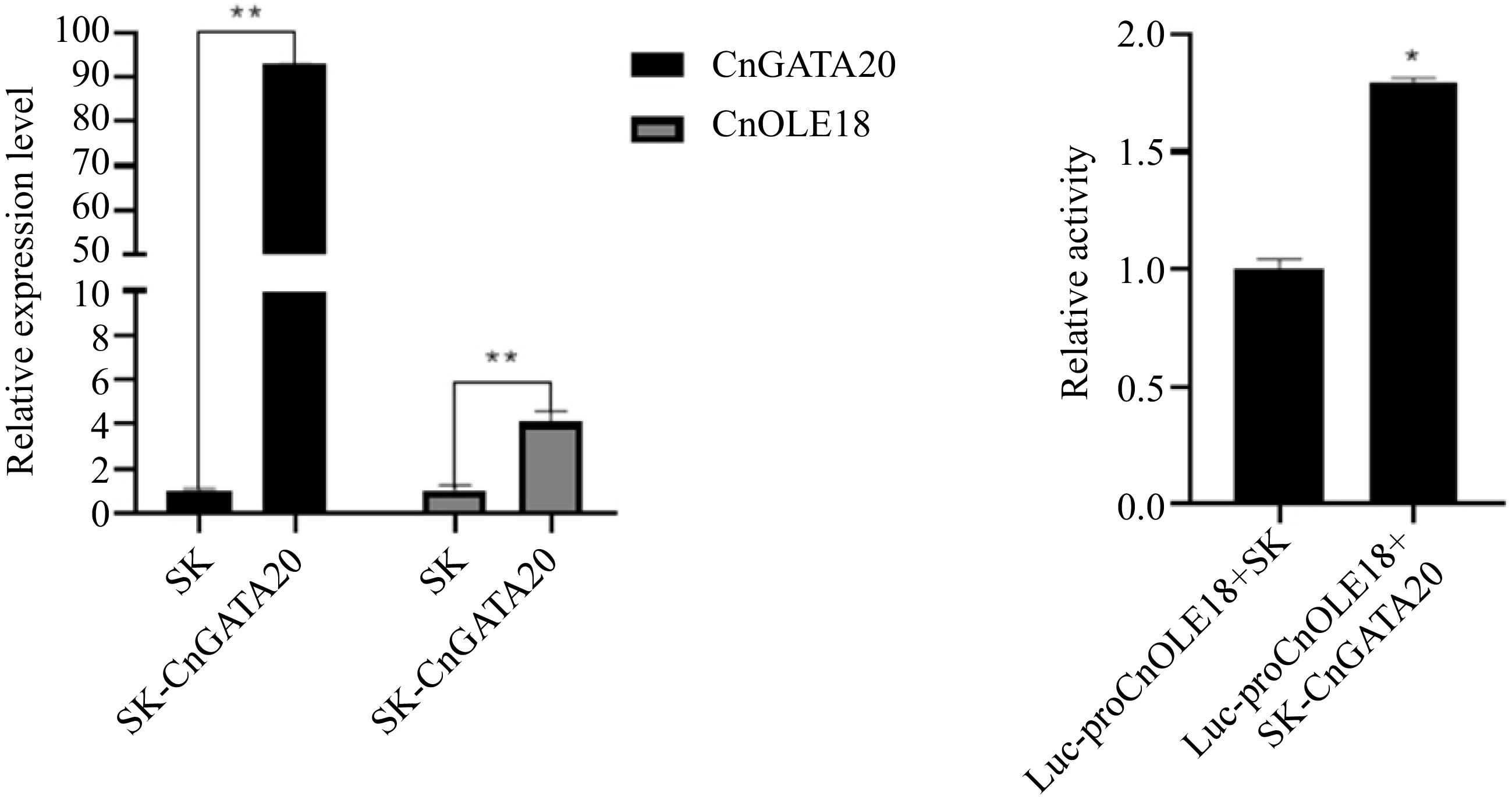
Figure 8.
Transient and dual-luciferase reporter assay. (a) Transient overexpression of GnGATA20 in the protoplasts of coconut, SK: Empty vector (control). (b) Dual-luciferase reporter assay of CnGATA20 in Cocos nucifera L. protoplast. Quantification of the fluorescence intensity of Luc-proCnOLE18+SK and Luc-proCnOLE18+SK-CnGATA20 . Three biological triplicates were used for every sample. The values are means ± SD. ** represents a highly significant difference (P < 0.01) using Student's t-test.
To further identify the interactions between CnGATA20 and the promoters of CnOLE18 in vivo, we conducted transient assays in coconut protoplasts using a dual-luciferase reporter system. pGreenII62-SK+pGreenII0800-Luc-proCnOLE18 were cotransferred into protoplasts as a negative control. The results showed that the cotransformation of pGreenII62-SK-CnGATA20+pGreenII0800-Luc-proCnOLE18 increased the relative LUC/REN ratio by nearly 1.79-fold (Fig. 8b). Thus, we verified that CnGATA20 enhances the transcription of CnOLE18 by directly interacting with their promoters.
-
In this paper, we identified the open chromatin regions (OCRs) and group differences in OCRs during the development of coconut solid endosperm (copra). Meanwhile, by integrating ATAC-seq data with RNA-seq results of copra at four different developmental stages (7, 8, 9, and 10 months after pollination), the regulatory relationship pathways between multiple transcription factors and functional genes were identified. Among them, in the process of screening CnOLE18 as the target gene, the CnGATA20 binding site was identified. Moreover, the regulatory pathway by which CnGATA20 positively regulates the expression of CnOLE18 was confirmed by the detection experiments of yeast one-hybrid, protoplast transient expression and a dual-luciferase reporter system (Fig. 9). In addition to discovering and verifying the regulatory pathways of GATA and OLE genes, based on ATAC-seq and RNA-seq in this project, a large number of potential regulatory relationships between transcription factors and functional genes in coconut were detected via bioinformatics analysis at the genomic level, and for the first time, ATAC-seq technology was applied to the identification and analysis of important transcription factors during coconut endosperm development. Compared with previous studies, the strategy of ATAC-seq combined with RNA-seq will provide a new research method for exploring the regulation at both the transcriptional and posttranscriptional levels during coconut endosperm growth and development. The follow-up research will be more efficient and targeted strategically than previous studies on individual transcription factors. Moreover, the obtained ATAC-seq data will also be useful for integration with further genomic analyses as well as other epigenetic information in coconut.
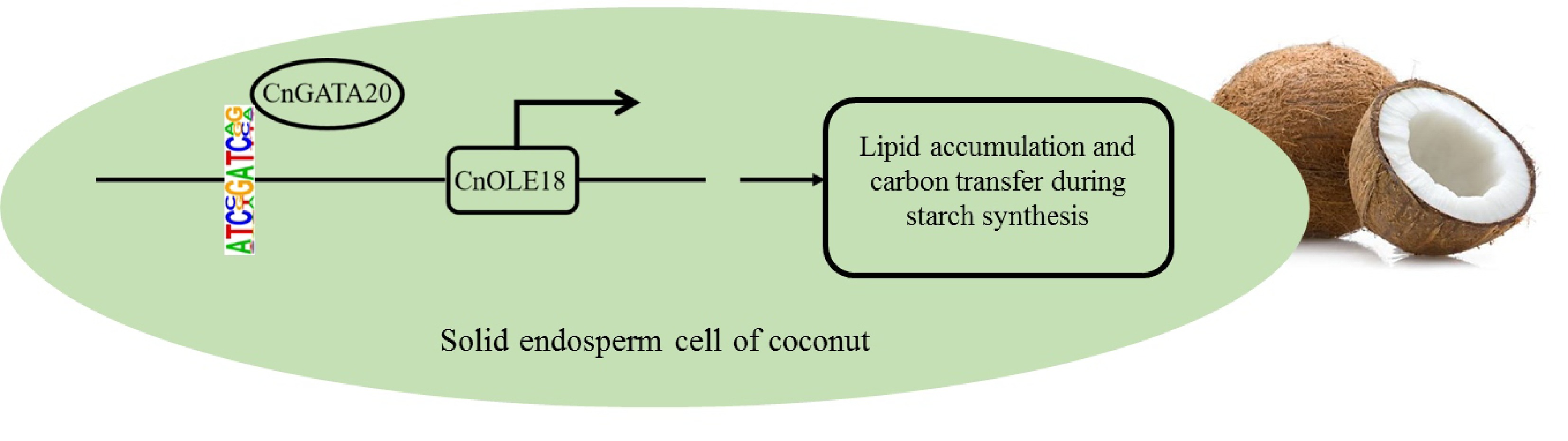
Figure 9.
A simplified model shows that CnGATA20 regulates the expression of the CnOLE18 gene, which controls the accumulation of lipid in endosperm of coconut.
GATA transcription factors belong to the family of zinc finger proteins, and their DNA-binding regions generally contain a highly conserved type IV zinc finger structure C-X2-C-X17-20-C-X2-C[19]. This structure is widely found in different kinds of plants, and it can affect seed germination and seedling growth, plant leaf development, and plant flowering time and play an important role in abiotic stress responses[20]. Currently, approximately 30 members of the GATA transcription factor family have been identified in plants[21]. In Arabidopsis, two GATA transcription factors (GNC and GNL) are important transcription targets downstream from ARF2 in the control of greening, flowering time, and senescence[22]. These two GATA factors also act as negative regulators of flowering by directly repressing SOC1 expression. In rice, a GATA transcription factor named OsGATA16 positively regulates and improves cold resistance at the seedling stage in rice by inhibiting cold-related genes such as OsWRKY45-1, OsSRFP1 and OsMYB30[23]. Among them, the unique GATA23 of Brassica and the unique HANGATA domain of monocots proved that the function of the B-GATA family was further expanded during plant evolution and played a dominant role in embryonic and flower development[24]. Although a series of GATA-like transcription factors have been found in different kinds of plant tissue[25], their main functions and regulatory mechanisms are rarely reported in the process of endosperm development in plant seeds, especially in palmae plants. Considering the diversity of GATA-like TF functions, in-depth research will affect the genetic improvement and variety breeding of palm plants in different aspects (including growth, quality, and stress resistance).
In mature seeds of oil crops, lipids are stored in specialized structures called oil droplets[26]. During the final stage of seed maturation, the intracellular water is gradually depleted, and the cells face a gradual shrinkage of the cell membrane; however, these oil droplets are still able to function as separate individuals and do not fuse with each other[27]. The stable existence of oil droplets in seeds is due to a layer of oil body proteins embedded in the surfaces of the oil droplets. Oil body proteins can regulate the size of liposomes and ensure that the plant is successfully dehydrated during the maturation process[28]. Oil body proteins are a class of proteins attached to the surfaces of oil bodies, including oleosin, caleosin (Sop1), steroleosin (Sop2) and a protein that has not been fully characterized (Sop3), of which oleosin is the most abundant type of oil body protein[29]. The current study suggests that OLE1 protects against the accumulation of lipids in tissues by reducing fatty acid (or TAG) degradation and carbon transfer in starch synthesis[30]. In Arabidopsis, insertion of an OLEOSIN T-DNA into the GBSS1 gene resulted in a significant reduction in amylose and a substantial increase in oil content in leaves of transgenic plants[31]. However, there is currently no report on the regulation of OLE gene expression in plant seeds, especially in the endosperm. The analysis of the OLE gene regulatory pathway via GATA-like transcription factors will also provide an important research strategy for regulating lipid accumulation in the endosperm of coconut.
-
Four different developmental periods of coconut (Cocos nucifera L.) fruits (after pollination: 7 months (CO1), 8 months (CO2), 9 months (CO3) and 10 months (CO4), respectively) were used as experimental material in this study. All analyzed coconut endosperms were collected from the Germplasm Resource Garden of Hainan University (Hainan, China). Each set of endosperm tissues at a particular stage was isolated from the coconut fruits. The solid endosperm was immediately frozen in liquid nitrogen and then stored at −80 °C for further analysis. Tobacco (Nicotiana benthamiana) used for subcellular localization was grown at 25 ºC with a photoperiod of 16 h light and 8 h darkness.
ATAC-seq, RNA-seq and data analysis
-
For the ATAC-seq and RNA-seq assays, samples were prepared from three biological replicates of coconut endosperm in four different developmental periods. Four samples were submitted to Romics (Shanghai, China) for library preparation and high-throughput sequencing services. All ATAC-seq clean data were mapped to the reference genome of coconut using BWA, which is fast alignment software for short sequences indexed by the BWT (Burrows–Wheeler Transformation) algorithm. Then, the chromatin open region was obtained by calculating the reads significantly enriched regions (Peak) on the reference genome using the statistical method. To visualize the ATAC-seq signal, IGV (Integrative Genomics Viewer) was applied to visualize the ATAC-seq enrichment of four samples in different periods. STAR (Spliced Transcripts Alignment to a Reference) was used for RNA-seq sequence alignment analysis, and FPKM was used to normalize the original expression of genes.
Identification of transcription factors and genes related to coconut endosperm development
-
Coconut endosperm development-related transcription factors were identified as follows: difference peaks among the predicted transcription factor-binding sites on the background sequence (genome DNA sequence) and the proportion of all transcription factor-binding sites were employed to speculate the key transcription factors in endosperm development. The transcription factors with the highest proportions and the most frequent occurrences in each overlap group were selected for further analysis. Additionally, to identify the genes that play an important role in the development of coconut endosperm, we screened and identified the differentially expressed genes in GO (Gene Ontology) and KEGG pathway enrichment analyses based on the results of ATAC-seq and RNA-seq overlap analysis. Genes with functions involved in endosperm development, metabolic processes, responses to stress and fatty acid biosynthesis were the focus. Furthermore, to find the sequence motif enriched in the promoter of each gene, the average odds score was used to calculate the score value of each sequence. A statistical test (Fisher's exact test) was then used to identify the enriched TF motif with a significant p value.
Gene cloning, sequence alignment and phylogenetic analysis
-
Total RNA from the coconut endosperm tissues was isolated with the CTAB-LiCl (hexadecyl trimethyl ammonium bromide-lithium chloride) technique[32]. A HiScriptIII® 1st Strand cDNA Synthesis Kit (+gDNA wiper) (Vazyme, Nanjing, China) was used to obtain first-strand cDNA. The coding sequences of the genes were downloaded from the GIGA DB Dataset (
http://gigadb.org/dataset/100347 ), and then Clustal Omega3 and MEGA 6.0 were used for amino acid alignment and construction of the phylogenetic tree.Subcellular localization of transcription factors in Nicotiana benthamiana
-
For subcellular localization analysis in N. benthamiana, the full-length cDNAs of CnGATA20 without the termination codon were cloned into the p35MK1-GFP vector driven by the CaMV 35S promoter. The p35MK1-CnGATA20 fusion expression vector was transformed into Agrobacterium tumefaciens GV3101. The GV3101 strains containing the above constructs were used to infect the leaves of 1-month-old N. benthamiana for transient expression. The infected tobacco plants were cultured for 32−36 h in the dark at room temperature. The GFP signal was detected using a confocal laser scanning microscope (TCS SP8, Leica, Heidelberg, Germany) within the excitation and emission wavelengths of BA534/55 nm.
Protoplast isolation and transformation
-
The tender basal sections of the youngest leaves collected from coconut seedlings were used for protoplast isolation. After enzymatic treatment, the purified protoplasts were harvested for further transient transformation as previously described[33]. The CnGATA20 overexpression plant vector for protoplast transformation was obtained by cloning the ORF of CnGATA20 into pGreenII62-SK. The pGreenII62-SK and pGreenII62-SK-CnGATA20 vectors were transformed into protoplasts with PEG solution (40% (w/v) PEG 4000, 0.4 M mannitol and 100 mM CaCl2). Protoplast transformation was carried out as previously described.
Protoplast RNA extraction and qRT-PCR
-
Protoplast total RNA was extracted from protoplasts using TRIzol® Reagent (Ambion®, Life Technologies, USA). First-strand cDNA was synthesized using the HiScript® III 1st Strand cDNA Synthesis Kit (+gDNA wiper) (Vazyme, Nanjing, China). RT-PCR was carried out using 2 × Q3 SYBR qPCR Master Mix (Universal) (TOLOBIO, Shanghai, China) with a CFX Connect Real-Time System (Bio-Rad, USA). All the gene-specific primer pairs were designed using Oligo 7.0 and are shown in Supplemental Table S2. The relative expression levels of all the genes were calculated from 2−ΔΔCᴛ values using β-actin as an endogenous control. Meanwhile, each sample had a negative control (ddH2O as template) and three replicates.
Promoter cloning and dual-luciferase transient expression assay
-
The promoter of CnOLE18 was obtained from the genomic DNA of coconut endosperm and cloned into the pGreenII0800-Luc vector as a reporter vector (pGreenII0800-Luc-proCnOLE18). The effector vector was pGreenII62-SK-CnGATA20, and the vector pGreenII62-SK (empty vector) was used as the negative control[34]. For transient expression, reporter vector and effector vector plasmids (10 µg each) were cotransformed into 200 μL of freshly isolated protoplasts according to the method described by Sun et al.[33]. Then, the LUC and REN luciferase activities were detected by the Dual-Luciferase Assay Kit (Promega, USA) and measured using a Synergy HT (BioTek, USA) following the manufacturer's instructions. The activity levels of reporter genes were compared by calculating the ratio of LUC to REN activity and normalized to the negative control. For each pair of vectors, at least six transient assay measurements as biological repeats were performed.
Yeast one-hybrid assay
-
For the yeast one-hybrid assay, the promoter of CnOLE18 was cloned into the pHIS2.1 vector to create the bait construct pHIS2.1-proCnOLE18. The CDS of CnGATA20 was inserted into the pGADT7 vector, and the pGADT7-CnGATA20 construct was used as the prey vector[35]. Then, the bait vector was introduced into Y187 yeast cells following the manufacturer's instructions (Weidi, Shanghai, China) and screened by growth on selective synthetic medium without tryptophan (SD/Trp). The prey vector was then transformed into yeast cells containing bait vector using the LiAc-PEG method[36]. pHIS2.1-pro53+pGADT7-53 was used as a positive control, and pHIS2.1-proCnOLE18+pGADT7-53 served as a negative control. The cotransformed yeast strains were grown on selective synthetic medium without tryptophan and histidine (SD/Trp-His).
After detecting the background histidine expression of the Y187 pHIS2.1-proCnOLE18 strain, it was restrained with 3-amino-1,2,4-triazole (3-AT) at the appropriate concentration. Then, the yeast strains were cotransformed with proCnOLE18+pGADT7-CnGATA20, cultured to an optical density of 0.5 at 600 nm and diluted to 10−3, 10−2, 10−1 and 100, and an aliquot (5 µL) of each dilution was inoculated onto selective medium lacking Trp, His and Leu with 3-AT (SD/Trp-His-Leu+3-AT). If the yeast grew on the medium, it indicates that TF interacts with the gene promoter.
Statistical analyses
-
Each sample is presented as the mean ± SD with three independent biological replicates. GraphPad Prism 8.0 software was used for all statistical tests. Significant differences between groups were determined by Student's t test. Differences were considered statistically significant when P ≤ 0.05 and highly significant when P < 0.01.
This research was supported by the Hainan Province Science and Technology Special Fund (No. ZDYF2022XDNY148) and National Key R&D Program of China (No. 2018YFD1000500).
-
The authors declare that they have no conflict of interest.
-
accompanies this paper at (https://www.maxapress.com/article/doi/10.48130/TP-2022-0008)
-
Received 20 July 2022; Accepted 26 September 2022; Published online 12 October 2022
-
Chromatin opening data of the genomes at four different developmental stages of endosperm of coconut were obtained by ATAC-seq and RNA-seq technologies
A considerable number of combinations containing transcription factors and downstream regulatory genes were identified
Regulation between transcription factor (CnGATA20) and its downstream regulated gene (CnOLE18) were verified
The results provide a new strategy for elucidating the regulatory network of metabolism in the development of coconut endosperm
-
# These authors contributed equally: Li Gao, Yaning Wang
- Supplemental Table S1 Promoter-TSS peaks with annotation of genes.
- Supplemental Table S2 List of gene cloning and qRT-PCR primers.
- Copyright: © 2022 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Gao L, Wang Y, Guo Q, Li D. 2022. Identification and functional analysis of transcription factors related to coconut (Cocos nucifera L.) endosperm development based on ATAC-seq. Tropical Plants 1:8 doi: 10.48130/TP-2022-0008 

Identification and functional analysis of transcription factors related to coconut (Cocos nucifera L.) endosperm development based on ATAC-seq
- Received: 20 July 2022
- Accepted: 26 September 2022
- Published online: 12 October 2022
Abstract: Coconut (Cocos nucifera L.) is a member of the palm tree family (Arecaceae) and the only living species of the genus Cocos. In this paper, the regulatory relationship pathways between multiple transcription factors and functional genes were identified by combining ATAC-seq and RNA-seq in coconut endosperm at four different developmental stages (fruit after pollination: 7 months, 8 months, 9 months and 10 months, respectively). The results indicated that the peaks detected in the promoter-TSS area accounted for the largest proportion (11.99%) in the third stage. These results suggest that the chromatin open region of cells in this period is more functional and that there are more functional genes with active transcription. In addition, a large number of potential regulatory relationships between transcription factors and functional genes were detected via bioinformatics analysis at the genomic level. Among them, CnGATA20 was predicted to be an important transcription factor with a binding site on the promoter region of the CnOLE18 gene. The regulatory pathway by which CnGATA20 positively regulates the expression of CnOLE18 was further confirmed by yeast one-hybrid, protoplast transient expression and dual-luciferase reporter system experiments. The results provide a new research strategy for exploring the regulation at both the transcriptional and posttranscriptional levels during coconut endosperm growth and development.
-
Key words:
- Coconut (Cocos nucifera L.) /
- ATAC-Seq /
- Endosperm /
- GATA transcriptional factor /
- Oleosin