








-
Creeping bentgrass (Agrostis stolonifera) is one of the perennial cool-season turfgrass species with an optimal temperature growth temperature range of 18 to 24 °C[1]. It is not only extensively used on the greens of golf courses located in temperate and frigid areas but also utilized in transition and subtropical zones due to the superior characteristics of fine texture and high tolerance to low mowing height[2]. However, the atmospheric temperature often rises beyond the temperature threshold for optimal growth of creeping bentgrass, especially during the summer season[3]. High temperature is able to restrict the normal physiological metabolic activities leading to a decrease in photosynthesis[4], respiration[5] and antioxidant metabolism[6] in creeping bentgrass. Long-term heat stress caused turf quality decline, plant growth inhibition and leaf senescence[7]. In creeping bentgrass, green leaf number and growth height decreased gradually due to high temperature stress[8]. In addition, plant density, tiller density, number of roots and root fresh weight also reduced which was attributed to heat stress[9].
Many approaches could be utilized to enhance heat tolerance in cool-season turfgrasses including breeding heat-tolerant cultivars[10], heat acclimation[11], cultivation and management measures[12] and chemical regulation[13]. Chemical regulation was reported to be one of the simplest and convenient methods to control plant growth and development by foliar application of exogenous substances to effectively mitigate heat stress injury in various plants, including perennial grass species. Among the majority of growth regulators, chitosan was previously documented to improve stress tolerance in many plants. Chitosan, a deacetylation product of chitin widely existing in plants, was natural, non-toxic and a biodegradable biopolymer extensively being used for food, medicine, agriculture and other fields[14, 15]. Chitosan emerged as being multifunctional as an elicitor in plants, which took part in seed germination, plant growth and development, biotic and abiotic stresses[16−18]. On the one hand, chitosan significantly promoted shoot growth and root development in cordyline (Cordyline terminalis) seedlings and increased total biomass in milk thistle (Silybum marianum)[19, 20]. On the other hand, chitosan could also alleviate detrimental effects of various abiotic stresses, such as cold, drought and salinity stresses, through regulating the physiological responses of plants. Chitosan has been widely demonstrated to foster positive impacts on enhancing cold tolerance in tea plants (Camellia sinensis) as well as mitigating osmotic stress in rice (Oryza sativa) through increasing shoot fresh and dry weights[21, 22]. In white clover (Trifolium repens), chitosan-improved drought tolerance associated with changes in endogenous hormones and antioxidant systems[23]. In addition, exogenous chitosan significantly promoted root length and plant height of maize (Zea mays) seedlings under salt stress and reduced content of reactive oxygen species and malondialdehyde[24].
Although previous studies have confirmed that chitosan is beneficial to stress tolerance in several plants, the function of chitosan in association with heat tolerance in perennial creeping bentgrass is not well understood. The objective of this study was to determine the optimal concentration of chitosan that could positively alleviate the adverse effects of heat stress and investigate the effects of exogenous chitosan on photosynthesis and cell membrane stability under heat stress. The results in this study would be useful to better understand the function of chitosan and provide effective approaches for mitigating high temperature stress in creeping bentgrass, especially in the summer season.
-
Creeping bentgrass seeds (A. stolonifera cv. 'Penn-A4') were planted in sand-filled polyvinyl chloride (PVC) pipes (10 cm in diameter and 25 cm in height). The plant material was grown in a greenhouse at 25/20 °C (day/night) for about 2 months. Plants were irrigated daily, maintained at a canopy height of approximately 4−5 cm by trimming every 2 d and fertilized once a week with water-soluble fertilizer (Scotts Miracle-Gro Company, USA). After establishment, plants were transferred to growth chambers (XBQH-1, Jinan Xubang, Jinan, Shandong Province, China) for acclimating 7 d before treatment initiation. The temperature of the growth chamber was set at 25/20 °C (day/night) with 14 h photoperiod and 60% relative humidity.
Experimental design and treatments
Experiment one - Selection of optimal chitosan concentration for improving heat tolerance
-
Plants were exposed to heat stress for 49 d in the growth chamber with treatment temperature of 38/28 °C (day/night) after one week of acclimation. This experiment was set as a completely randomized design with three biological replicates for each treatment. Different chitosan concentrations of 0, 50, 100 and 500 mg·L−1 as reported by Geng et al.[25] were respectively sprayed on the leaves at 0 d of treatment, and then applied every 7 d during the 49 d of heat stress. The treatment of deionized water without exogenous chitosan (0 mg·L−1) was set as the control. Chitosan (85% deacetylation degree) was dissolved in 1% (v/v) acetic acid and deionized water was added to achieve the desired concentrations. Creeping bentgrass was no longer trimmed but watered daily to ensure available water for plant growth during the experimental period. Plants were relocated randomly every day within the growth chamber to minimize the effects of environmental difference on treatments.
Growth indexes measurement
Shoot growth parameters
-
Turf quality was rated weekly on a scale from 1 to 9 based on texture, color, uniformity and density[26]. As a common index, turf quality represents overall turf performance with 1 being a completely dead turf and 9 being healthy turf with green and dense turf canopy. A rating of 6 indicates the minimal acceptable level of turf quality. The ratio of yellow leaves was calculated as the ratio of the number of yellow leaves with more than 50% of area being dry and showing chlorosis compared to the number of total leaves[27]. The number of yellow leaves and total leaves were counted on five labeled stolons once a week, respectively. Growth rate was calculated by measuring stolon length every week. Five stolons were randomly labeled in each PVC tube at 0 d of treatment and stolon length were measured using a ruler.
Root growth indexes
-
Root length was measured with a scale on the last day of treatment. Shoot and root were separated, put into the oven at 105 °C for 30 min, and then dried at 80 °C to a constant weight. Shoot biomass and root biomass were weighed respectively to calculate root-shoot ratio.
Experiment two - Physiological responses with foliar application of chitosan
-
Following one week of acclimation to growth chamber conditions, plants were then exposed to four treatments in creeping bentgrass as follows: 1) control (control + H2O), plants sprayed with 10 mL deionized water under 25/20 °C (day/night); 2) only chitosan treatment (control + chitosan), plants with foliar application of 10 mL 100 mg·L-1 chitosan under non-stressed condition (25/20 °C, day/night); 3) high temperature treatment (H + H2O), plants were sprayed with 10 mL deionized water without the chitosan application under heat stress (38/28 °C, day/night) and 4) chitosan treatment and high temperature treatment (H + chitosan), plants were sprayed with 10 mL 100 mg·L−1 exogenous chitosan under heat stress (38/28 °C, day/night). Plants were treated with deionized water or chitosan every 7 d from the beginning at 0 d of treatment. The experiment was a completely randomized design with four biological replicates (four pots of plants) for each treatment.
Physiological parameter measurement
-
Physiological indicators, including photosynthetic rate (Pn), photochemical efficiency (Fv/Fm), electrolyte leakage (EL) and malondialdehyde content (MDA) were measured at 0, 21 and 42 d of treatments. Leaf Fv/Fm, the ratio of variable fluorescence (Fv) to maximum fluorescence (Fm), was measured with a fluorescence induction monitor (OPTI-Sciences, Hudson, USA) after acclimating in dark for 30 min with leaf clips. Leaf Pn was detected using a LI-6400 portable photosynthesis system (LI-COR, Lincoln, NE, USA). The analyzer was set at the PAR of 650 μmol∙m−2∙s−1, 400 µmol∙m−1 CO2 and block temperature of 25 °C. The fully-expanded leaves from each biological replicate were placed in the leaf chamber.
Leaf EL was used to indicate the cellular membrane stability following treatment and was calculated as (Cinitial/Cmax) × 100%[28]. Fresh leaves (0.2 g) were collected in each test tube containing 35 mL of deionized water. Leaf samples were placed on a shaker for 24 h to measure Cinitial by using a conductivity meter (Orion Star A212, Thermo Scientific Inc., Waltham, MA, USA). Samples were killed by being autoclaved at 121 °C for 20 min and shaken for another 24 h. The final conductance reading (Cmax) of killed leaves were then recorded.
MDA was measured according to the method documented by Xu et al.[29] with some modifications. Leaves (0.35 g) were ground and 3 mL of 50 mM cold PBS (pH = 7.8) and 1 mL 1 mM EDTA-Na2 were added to a test tube. Samples were centrifuged at 15000 g for 30 min at 4 °C, and then the supernatant was collected for MDA determination. Two mL of supernatant was mixed with 1 mL of reaction solution (20% trichloroacetic acid with 0.5% thiobarbituric acid) and then incubated at 95 °C for 30 min. The mixture was quickly cooled on ice and centrifuged at 10,000 g for 10 min at 4 °C. The supernatant was taken to determine absorbance at 450 nm, 532 nm and 600 nm by using a spectrophotometer (Ultrospec 2100 pro, Biochrom Ltd., Cambridge, UK).
Statistical analysis
-
Statistical analysis was performed using IBM SPSS Statistics (SPSS 21.0; SPSS Inc., Chicago, IL, USA). ANOVA analysis was used to determine whether exogenous chitosan had a significant impact on creeping bentgrass. Duncan multiple comparison was used to analyze the significant difference of turf quality, ratio of yellow leaves, growth rate, root length, root-shoot ratio, biomass and physiological indexes (Pn, Fv/Fm, EL and MDA) among different treatments at the confidence level of 0.05. The values of all column charts were expressed as mean ± standard error (SE) at the level of 0.05.
-
Turf quality was gradually decreased after high temperature treatment during the experimental period (Fig. 1a). Plants maintained the higher turf quality with foliar application of chitosan from 14 to 49 d compared with the untreated control under heat stress, and difference between different concentrations of chitosan (50, 100 and 500 mg·L−1) was not significant (Fig. 1a). At 49 d of heat stress, turf quality treated with exogenous chitosan was higher than the control by 17.5%, 15.8% and 11.9% under concentrations of 50, 100 and 500 mg·L−1, respectively. The shoot phenotypes with exogenous chitosan under heat stress were consistent with measurement that plants with chitosan application showed obviously superior performance (Fig. 1b).
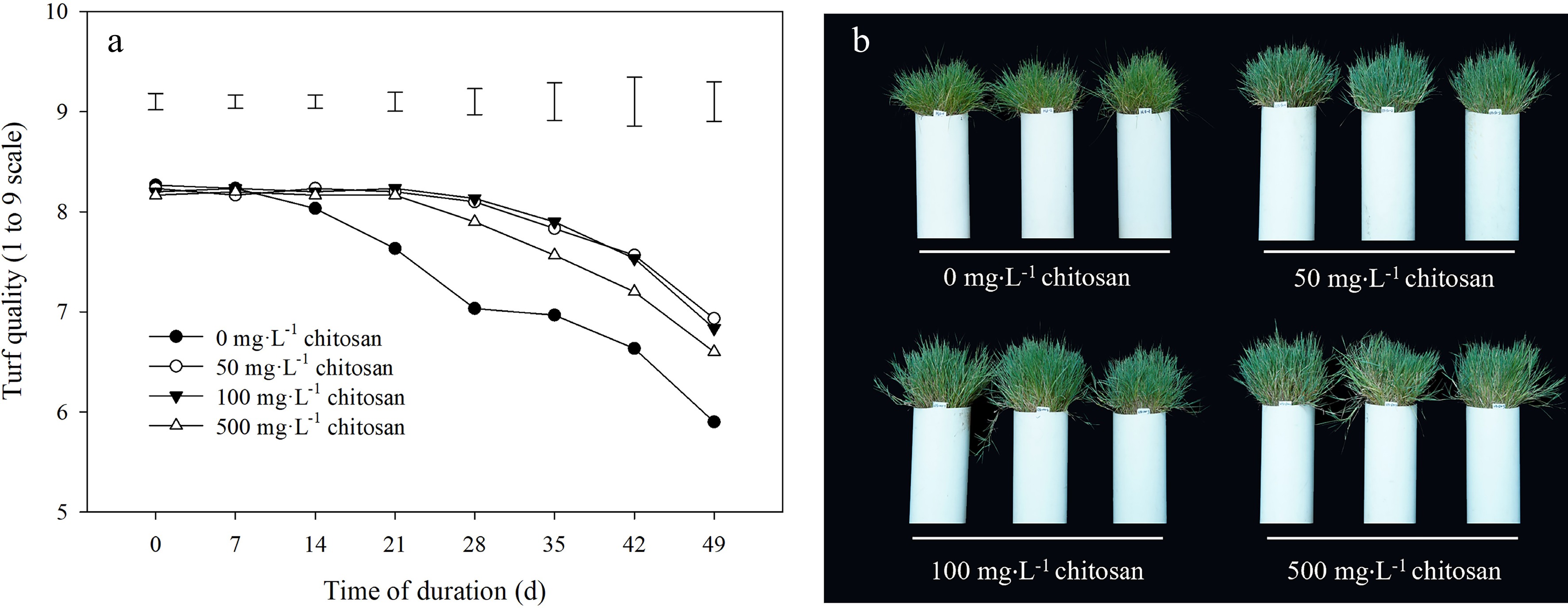
Figure 1.
Effects of different chitosan concentrations on (a) turf quality and (b) shoot phenotypes in creeping bentgrass under heat stress. Vertical bars indicate significant difference based on LSD values at 0.05 level for the comparison among treatments.
High temperature led to a rapid increase in the ratio of yellow leaves during the experimental period (Fig. 2a). The lower degree of ratio of yellow leaves in creeping bentgrass applied with chitosan than untreated control was manifested at 7 and 21 d. There was no significant effect on ratio of yellow leaves observed by exogenous application of different chitosan concentrations at 28 d through 49 d of heat stress (Fig. 2a). Foliar application of chitosan did not result in a significant effect on growth rate (Fig. 2b).
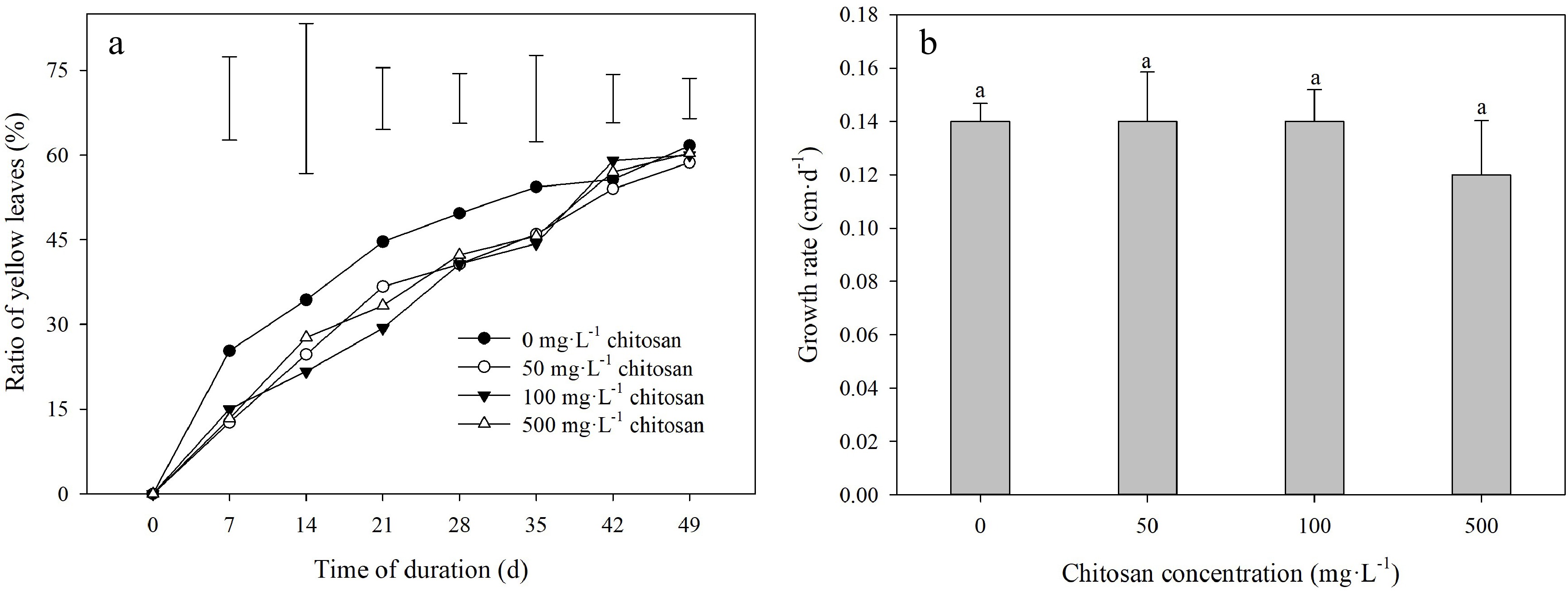
Figure 2.
Effects of different chitosan concentrations on (a) ratio of yellow leaves and (b) growth rate in creeping bentgrass under heat stress. Vertical bars indicate significant difference based on LSD values at 0.05 level for the comparison among treatments. Different lowercase letters represent significant difference between different treatments during the experimental period (P < 0.05). Error bars represent standard error (SE).
Root growth
-
At the end of the experiment, root length was longer than the control by 31.5% and 47.7%, respectively, in foliar chitosan-treated creeping bentgrasses with both 50 and 100 mg·L−1, but the difference between 500 mg·L−1 chitosan and control was not significant under heat stress (Fig. 3a). The effects of treatment with application of chitosan on root growth could be well reflected by root phenotype (Fig. 3b).
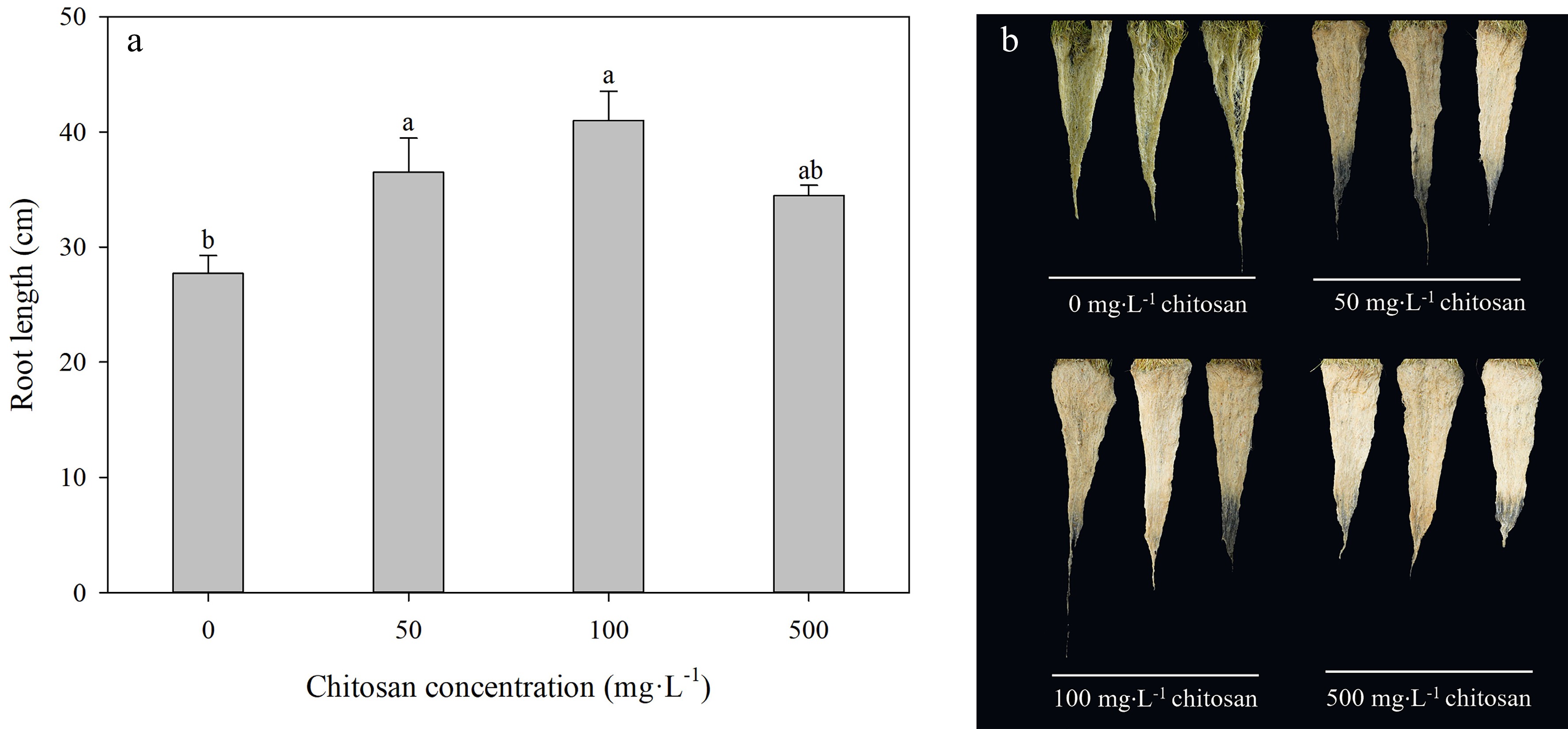
Figure 3.
Effects of different chitosan concentrations on (a) root length and (b) root phenotypes in creeping bentgrass under heat stress. Different lowercase letters represent significant difference between different treatments during the experimental period (P < 0.05). Error bars represent standard error (SE).
The positive effects in both shoot biomass and root biomass were observed due to the foliar application of chitosan under heat conditions (Fig. 4). Shoot biomass significantly increased with chitosan treatments of 50, 100 and 500 mg·L−1 by 59.9%, 95.0% and 93.3% higher than the control, respectively (Fig. 4a). The chitosan-induced increases with concentrations of 100 and 500 mg·L−1 in shoot biomass were greater than that of 50 mg·L−1 chitosan. The effect of exogenous chitosan on root biomass was similar to that of shoot biomass. Plants treated with the chitosan application of 50, 100 and 500 mg·L−1 had significantly larger root biomass by 4.2-fold, 9.0-fold and 10.6-fold than the control at the end of the experiment, respectively. Chitosan-treated plants with 50, 100 and 500 mg·L−1 resulted in a significant increase in root-shoot ratio by 3.0-fold, 4.0-fold and 5.1-fold compared with the control, respectively (Fig. 4b). Furthermore, root-shoot ratio applied by 500 mg·L−1 chitosan was significantly higher by 53.2% than 50 mg·L−1 chitosan.
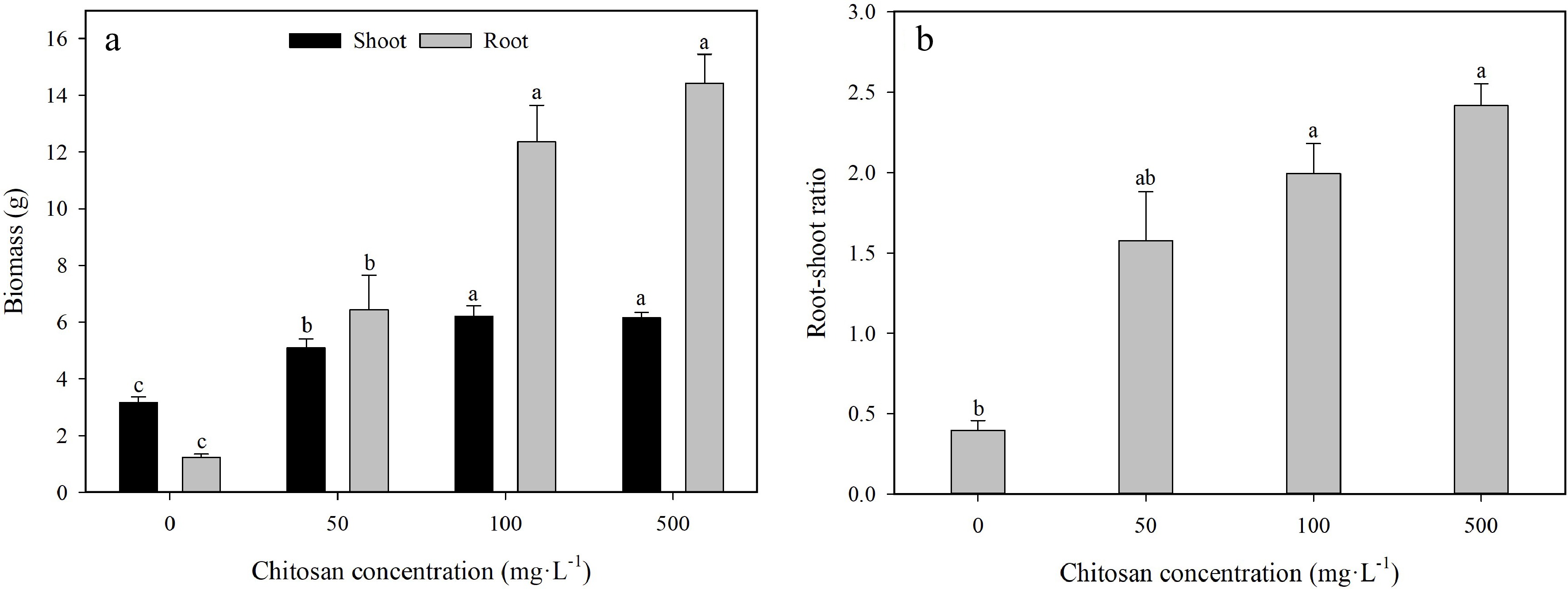
Figure 4.
Effects of different chitosan concentrations on (a) biomass and (b) root-shoot ratio in creeping bentgrass under heat stress. Different lowercase letters represent significant difference between different treatments during the experimental period (P < 0.05). Error bars represent standard error (SE).
Photosynthetic rate and photochemical efficiency
-
No significant difference was detected in Pn and Fv/Fm in both chitosan-treated and untreated plants under non-stressed condition (Fig. 5a, b). High temperature resulted in a rapid decline in Pn and Fv/Fm during the experimental period. However, Pn in chitosan-treated plants was significantly higher than that without chitosan application by 74.8% at 21 d of heat stress (Fig. 5a). At 42 d of treatment, plants without foliar application of chitosan under heat stress had a negative Pn value which was significantly lower than that of chitosan-treated plants. For Fv/Fm, plants with exogenous chitosan had significantly lower decline than chitosan-untreated plants at 21 and 42 d of heat stress (Fig. 5b).
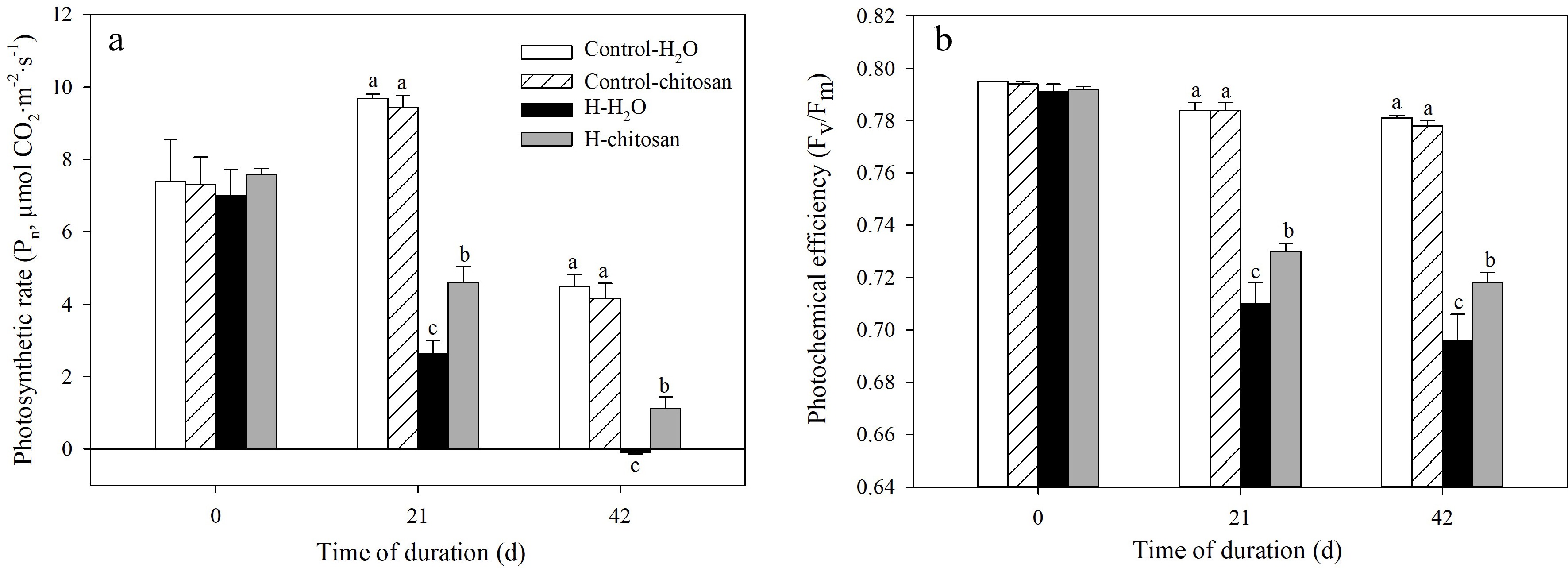
Figure 5.
Effects of chitosan on (a) photosynthetic rate and (b) photochemical efficiency in creeping bentgrass. Control + H2O, plants were sprayed with 10 mL deionized water under 25/20 °C (day/night); Control + CTS, plants were treated foliar application with 10 mL 100 mg·L−1 chitosan under non-stressed condition (25/20 °C, day/night); H + H2O, plants were sprayed with 10 mL deionized water without chitosan under heat stress (38/28 °C, day/night); H + CTS, plants were applied with exogenous chitosan (10 mL, 100 mg·L−1) under heat stress (38/28 °C, day/night). Different lowercase letters represent significant difference between different treatments during the experimental period (P < 0.05). Error bars represent standard error (SE).
Cell membrane stability
-
EL and MDA content were maintained at a normal level under non-stressed conditions and no significant difference was found among different treatments (Fig. 6a, b). High temperature stress significantly increased EL and MDA content compared to control with or without chitosan application during the treatment time. Under heat stress, plants treated with chitosan application had a significantly lower EL by 40% and 41% than chitosan-untreated plants at 21 and 42 d of treatment, respectively (Fig. 6a). In addition, there was no significant difference between chitosan-treated and untreated plants at 21 d of heat stress, but MDA content in plants treated with chitosan declined by 25% compared with plants without chitosan application at 42 d (Fig. 6b).
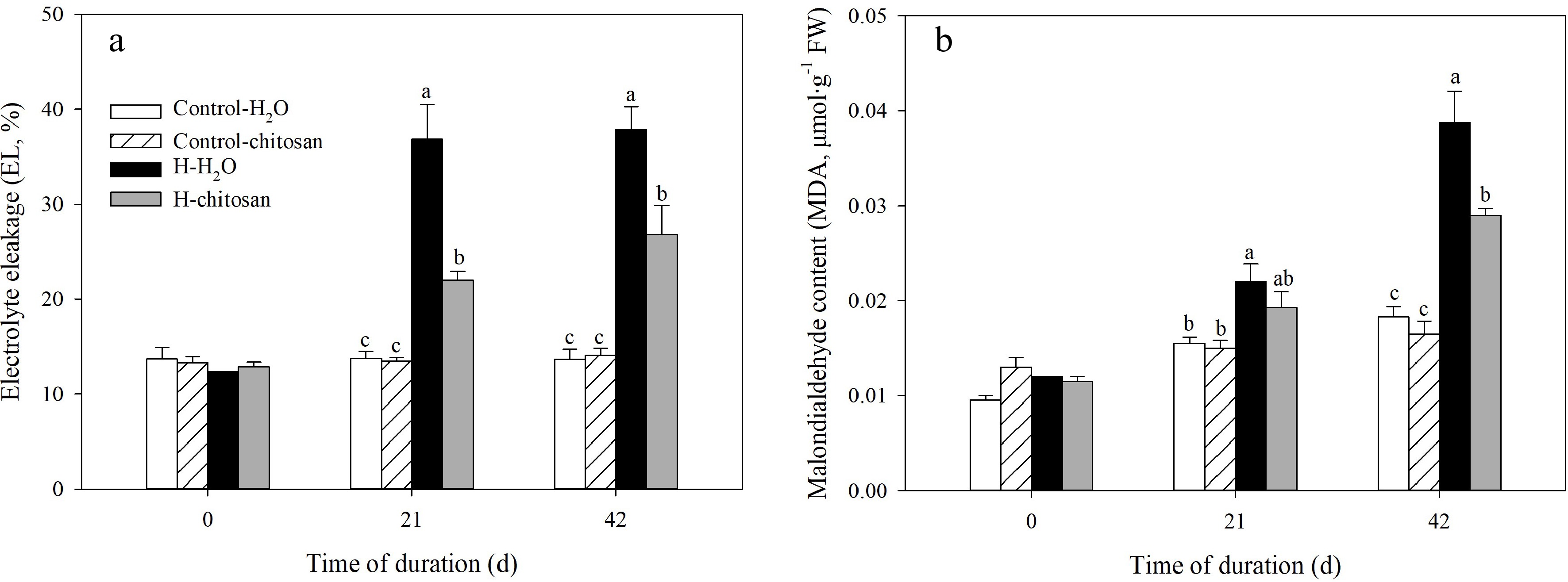
Figure 6.
Effects of chitosan on (a) electrolyte leakage and (b) malondialdehyde content in creeping bentgrass. Control + H2O, plants were sprayed with 10 mL deionized water under 25/20 °C (day/night); Control + CTS, plants were treated foliar application with 10 mL 100 mg·L−1 chitosan under non-stressed condition (25/20 °C, day/night); H + H2O, plants were sprayed with 10 mL deionized water without chitosan under heat stress (38/28 °C, day/night); H + CTS, plants were applied with exogenous chitosan (10 mL, 100 mg·L−1) under heat stress (38/28 °C, day/night). Different lowercase letters represent significant difference between different treatments during the experimental period (P < 0.05). Error bars represent standard error (SE).
-
High temperature, as one of the major abiotic stresses restricting the growth of cool-season turfgrass, is an important and difficult problem for turfgrass management, especially during summer[3]. Heat stress induced the limitation of physiological and metabolism in various turfgrass species[30−32]. The heat-induced damage also occurred in the morphology and growth of plants, especially when turfgrass species suffered continuous severe heat stress[33]. It was previously found that heat-tolerant species usually had better growth performance and physiological characteristics than heat-sensitive plants in creeping bentgrass[9, 34]. In the present study, exogenous chitosan with 100 mg·L−1 significantly enhanced thermotolerance in association with superior plant growth indexes (Figs 1, 3 & 4), photosynthesis (Fig. 5) and cell membrane stability (Fig. 6) compared with the untreated plants under heat stress in creeping bentgrass as discussed below.
Shoot growth and development were directly associated with turf quality in turfgrass. Studies have shown that high temperature stress would reduce turf quality and increase the ratio of yellow leaves in creeping bentgrass[35]. Turf quality and canopy height also significantly decreased due to heat stress in perennial ryegrass (Lolium perenne) [36]. Chitosan serves as not only a plant growth regulator to promote growth and development of various plants, but also an abiotic stress tolerance inducer[37−39]. In our study, foliar application of chitosan improved turf quality and shoot biomass in response to heat stress during the experimental period (Figs 1 & 4a). Similar result was also found by Younas et al.[40] that growth and yield characteristics of maize including shoot dry weight, cob weight and grain yield were improved by applying chitosan under water deficit conditions. Furthermore, chitosan treatment improved growth and quality in soybean sprouts (Glycine max)[41], increased shoot height and number of nodes in potato (Solanum tuberosum)[42]. Chitosan-induced increase in turf quality and biomass in this study indicated that chitosan could enhance heat tolerance and promote plant growth in creeping bentgrass under high temperature conditions.
Heat stress also leads to injury of belowground root as well as the aboveground shoot. Roots are the main organs for acquiring water and mineral nutrients from the soil in plants[43]. However, root system are more sensitive to heat stress in comparison to shoots[31]. In general, root growth was positively correlated with heat tolerance as reported in various cool-season turfgrass species, such as in tall fescue (L. arundinaceum), perennial ryegrass and creeping bentgrass[10,44,45]. Hence, root growth played a crucial role in enhancing thermotolerance in turfgrass species in response to heat stress. In our study, foliar application of chitosan led to the significant promotion of root length, root-shoot ratio and root biomass compared with the untreated control (Figs 3 & 4). The positive effects of chitosan in thermotolerance through facilitating plant roots have been widely documented in several previous studies. For example, chitosan alleviated adverse effects of salt stress through inhibiting the decline in root length in maize seedlings[24]. Chitosan also mitigated the suppression of root growth in maize under cadmium stress as reflected by higher root length, root surface and root volume[46]. Those results suggested that chitosan played a positive role during root growth under high temperature stress.
Various physiological activities in plants were limited by high temperature of which photosynthesis as one of physiological processes is sensitive to external temperature. Heat-induced negative effects were observed when creeping bentgrass was subjected to heat stress, as reflected by reduction in chlorophyll content, Fv/Fm and Pn as well as inhibition in rubisco activity and rubisco activation state involving in photosynthesis[1, 47]. In this study, a significant decrease in both Fv/Fm and Pn was detected in plants due to imposition of heat stress, but exogenous chitosan effectively alleviated the decline in Fv/Fm and Pn (Fig. 5). It has also been found that chitosan pretreatment significantly mitigated the adverse effect on Fv/Fm in creeping bentgrass under salt stress[25]. Application of irradiated chitosan significantly increased chlorophyll content and Pn as well as enhanced drought tolerance of sugarcane (Saccharum spp.)[48]. Similarly, the result in this present study demonstrated that chitosan alleviated severe damage in photosynthetic system caused by high temperature. Furthermore, high temperature also led to electrolyte leakage and membrane lipid peroxidation resulting from cell membrane damage[49]. In our study, both EL and MDA content in plants without chitosan treatment significantly increased, but chitosan application significantly inhibited the increase in EL and MDA content under high temperature stress (Fig. 6). This result was also reported by other studies in several plant species, such as in potato[42], thyme (Thymus daenensis)[17] and edible rape (Brassica rapa)[50] indicating that exogenous chitosan contributed to the promotive effects on cell membrane stability and integrity and improved heat tolerance in creeping bentgrass.
In summary, foliar application of chitosan significantly enhanced heat tolerance as reflected through promoting both shoot and root growth in creeping bentgrass as shown by higher turf quality, root length, root-shoot ratio and biomass. Furthermore, chitosan application also could inhibit the decline in photosynthesis and maintain cell membrane stability. The mechanisms involved in chitosan-induced thermotolerance at the molecular level under heat conditions need to be further investigated in our next studies. This study will provide new insight for turf managers to enhance heat tolerance of perennial turfgrass species, especially during the summer season in cool-season grasses.
This research was supported by the Chongqing Key Laboratory of Germplasm Innovation and Utilization of Native Plants (XTZW2021-KF04) and Innovation and Promotion of Forestry Science and Technology of Jiangsu Province (LYKJ[2021]29).
-
The authors declare that they have no conflict of interest.
- Copyright: © 2022 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Li Q, Li R, He F, Yang Z, Yu J. 2022. Growth and physiological effects of chitosan on heat tolerance in creeping bentgrass (Agrostis stolonifera). Grass Research 2:6 doi: 10.48130/GR-2022-0006 

Growth and physiological effects of chitosan on heat tolerance in creeping bentgrass (Agrostis stolonifera)
- Received: 14 August 2022
- Accepted: 17 October 2022
- Published online: 27 October 2022
Abstract: High temperature is one of the major abiotic stresses limiting growth and development of cool-season grass species, but chitosan could effectively enhance heat tolerance and improve plant growth. The objective of this study was to determine the optimal concentration of chitosan that could alleviate heat stress in creeping bentgrass (Agrostis stolonifera) and investigate the effects of exogenous chitosan on photosynthesis and cell membrane stability under heat stress. Under heat stress (38/28 °C, day/night), different chitosan concentrations of 0, 50, 100 and 500 mg·L−1 were applied on the leaves of creeping bentgrass (cv. 'Penn-A4'). Foliar application of chitosan exhibited the positive effects on plant growth and the optimal concentration was 100 mg·L−1 which significantly improved turf quality, root length, root-shoot ratio as well as shoot and root biomass. Chitosan-treated plants subjected to high temperature stress had a lower decline in photosynthetic rate and photochemical efficiency as well as less increase in electrolyte leakage and malondialdehyde content. The results demonstrate that chitosan-improved heat tolerance as reflected by the superior growth performance of both shoot and root, photosynthesis and cell membrane stability in creeping bentgrass under heat stress.
-
Key words:
- Creeping bentgrass /
- Exogenous substances /
- Heat stress /
- Growth