









-
Flowering transition is a process in which the apical meristem gradually develops from the vegetative meristem to the reproductive meristem under the control of complicated genetic networks in response to the internal regulatory factors and external environmental factors[1]. To date, some progress has been made in the study on the molecular mechanism of flowering transition in some plants such as Rosa[2]. Foucher et al. identified 13 potential flowering regulatory genes( RoRGA , RoSLY , RoSOC1 , MASAKO B3 , etc.) in R. hybrida 'Black Baccara' related to flower development, gibberellin, and photoperiodic pathways through EST data[3]. It was found that overexpression of FT genes into R. rugosa 'Bao White' can significantly reduce the juvenile phase of the plant and enable it to flower earlier[4]. By transcriptome sequencing, Dubois et al. found that transcripts of the flowering integrator RcSOC1 accumulate only during the growth stage and flower initiation stage, and are not expressed at the later stages of flowering[5]. Randoux et al. suggested that the expression of RoKSN was inhibited and resulted in the suppression of flowering when a high concentration of Gibberellic acid(GA) was applied to a season of flowering R. chinensis[6]. R. rugosa is a famous Chinese traditional flowering species with aromatic compounds and strong resistance to stress including cold, drought, pest, salt and alkali. However, the regulatory mechanism on the flowering transition of R. rugosa is still unclear. Accordingly, exploring the regulatory mechanism of flowering transition in R. rugosa can lay an important foundation for breeding new varieties with desired floral traits.
The process of flowering transition was regulated by the change of internal growth regulation substances, including carbohydrates and phytohormones[7]. Sugar molecules, such as sucrose, glucose, fructose, etc, are essential components of carbohydrates in plants, among which sucrose is the main form of long-distance carbohydrate transport in higher plants. As a flowering signal, sucrose is also involved in regulating transport efficiency, distribution of assimilated products, and the activation and repression of specific genes in plants[8]. Starch, which also belongs to carbohydrates, mainly generates sugar molecules through hydrolysis to provide the energy for the flowering transition in the plant. Relevant studies have shown that the decrease of amylose can exhibit a late-flowering phenotype[9]. Abscisic acid(ABA), Indole-3-acetic acid(IAA), and GA are all important phytohormones affecting plant growth and development, such as seed germination, stem elongation, flowering and fruiting, senescence and abscission, and dormancy, etc. Previous findings showed that the three phytohormones all play dual roles in regulating the flowering transition process of plants, namely promoting or inhibiting the flowering transition[10−12]. Light and temperature signals are important environmental factors affecting the flowering transition process. Light regulates plant flower bud differentiation and flowering via the photoperiod pathway, an essential pathway for flowering in the plant[13]. The light signal of photoperiod is perceived by leaf, converted into biological signals and then transmitted to the flowering-time genes downstream to regulate the flowering transition process[14]. Nowadays, many studies on the impact of temperature on plant flowering have focused on vernalization, a phase of low temperature that some flowering species must undergo before flowering[15]. The molecular mechanisms underlying the vernalization response in Arabidopsis thaliana are well understood[16].
In this study, R. rugosa 'Duoji Huangmei', a novel continuous flowering cultivar, was selected as plant material. We explored the changes in morphology and content of sugar, starch, and phytohormones of buds during the flowering transition process. Furthermore, we conducted RNA sequencing (RNA-seq) to analyze the expression patterns of DEGs involved in the pathways related to sucrose, starch, IAA, ABA, GA, photoperiod, and vernalization. Finally, the molecular regulatory networks of flowering transition in R. rugosa was predicted.
-
R. rugosa 'Duoji Huangmei' was a diploid cultivar obtained by interspecific hybridization of R. rugosa and R. xanthina, which blooms from April to September. The tested R. rugosa 'Duoji Huangmei' plants were planted in the Rose Germplasm Nursery, Forestry Experimental Station of Shandong Agricultural University, Tai'an, China (36°10′15″ N, 117°09′25″ E), where they grew under natural conditions. Branches with buds of different sizes from nine different plants were selected randomly and then taken back to the laboratory on February 20th, 2021. After measuring the length using a vernier caliper, the buds were cut longitudinally. For the two halves of each cut bud, one half was used for observation to determine the developmental stage under Olympus SZX2-ILLT stereoscopic microscope (Olympus, Japan) and the other half at different developmental stages were quickly put into different tubes with liquid nitrogen and then stored at −80 °C for physiological index determination and transcriptome analysis. There were three biological replicates for each sample with 60~150 buds.
Determination of sugar and starch content
-
To measure the content of sugar and starch, a total of 200 mg samples were ground and homogenized in 8 mL of deionized water and placed in a boiling water bath for 30 min. After cooling to room temperature, the residue was filtered for subsequent starch content determination. The extracting liquid was transferred to the volumetric flask (25 mL) and the volume was fixed by distilled water, of which 0.5 mL extract liquid was transferred into the test tube. After 10 min of adding 1.5 mL distilled water, 0.5 mL anthrone-ethyl acetate solution (2%, v/v) reagent and 5 mL concentrated sulfuric acid (98%, v/v), the absorbance at 630 nm was read in a Beckman DU 800 UV–visible spectrophotometer (Beckman Coulter, USA) and compared to glucose standards.
The residue after centrifugation was transferred into a test tube with 10 mL distilled water and placed in a boiling water bath for 15 min. After adding 2 mL of 9.2 mol·L−1 perchloric acid for 2 min and then cooling to room temperature, the residue was filtered and the extracting liquid was transferred to the volumetric flask (25 mL) and the volume was fixed by distilled water. A total of 2 mL extracted liquid was collected to measure starch content at 620 nm using glucose as standards.
Determination of endogenous phytohormones content
-
The content of endogenous phytohormones in the buds were measured by high-performance liquid chromatography-mass spectrometry (HPLC-MS). A total of 500 mg samples were extracted overnight at 4 °C with 10 times the volume of acetonitrile solution and 2 μL internal standard master mix. The supernatant was collected by centrifugate at 12,000 rpm for 5 min at 4 °C. The obtained precipitate was added to 5 times the volume of acetonitrile solution and then extracted twice to combine the obtained supernatant. By adding 35 mg of C18 filler and vigorously shaking for 30 s, the supernatant was taken by centrifugate at 10,000 rpm for 5 min at 4 °C. Finally, it was dried with nitrogen, reconstituted with 200 μL methanol, passed through a 0.22 μm organic filter membrane, and placed in a −20 °C refrigerator for on-machine detection.
The HPLC-MS/MS analysis was conducted on a Poroshell 120 SB-C18 reversed-phase column (2.1 mm × 150 mm, 2.7 μm) at a flow rate of 0.3 mL·min−1 and a column temperature of 30 °C. Methanol / 0.1% formic acid (A) and water / 0.1% formic acid (B) were used as the mobile phase in gradient elution mode. The separation was performed using the following gradient: 20% for 1 min, from 20% to 50% in 2 min, from 50% to 80% in 6 min, at 80% for 1.5 min, then declining from 80% to 20% in 6s and 20% for 2.9 min. The injection volume is 2 μL. The ESI source operation parameters were as follows: ion source, ESI+ and ESI−; source temperature, 400 °C; ion spray voltage, +4,500 V and −4,000 V; curtain gas, 15 psi; atomization gas pressure, 65 psi; auxiliary gas pressure, 70 psi. Endogenous phytohormones were analyzed using scheduled multiple reaction monitoring (MRM).
Transcriptome analysis
-
Total RNA was extracted using the Total RNA Isolation Kit (Vazyme, China) according to the manufacturer's instructions. The quantity and quality of total RNA were assessed using 1% agarose gels and a NanoPhotometer® spectrophotometer (IMPLEN, USA). The integrity and concentration of total RNA were assessed using an Agilent 2100 RNA Nano 6000 Assay Kit (Agilent Technologies, USA) and Qubit®3.0 Fluorometer (Life Technologies, USA).
The poly(A) mRNA was isolated from total RNA through a NEBNext® Ultra™ RNA Library Prep Kit (New England Biolabs, China). After construction and normalization, three cDNA libraries were sequenced on the Illumina Hiseq 6000 Sequencing platform (Anoroad, China) and 150 bp paired-end reads were generated. The raw data were processed by removing the low-quality sequences (reads with more than 50% Q < 19 bases), the adaptor-pollute sequences, and sequences with ambiguous base reads accounting for more than 5%.
To understand their functions, the obtained clean reads were aligned to the R. rugosa genome database (
http://eplantftp.njau.edu.cn/Rosa_rugosa/ ) by HISAT2 v2.1.0. The expression levels of genes were calculated as Fragments Per Kilobase of exon model per Million mapped fragments (FPKM), which eliminates the effect of sequencing depth and gene length on gene expression levels and permits direct data comparisons by the DESeq method[17]. The DEGs were identified if their log2|Fold Change| was over 1 with a q-value ≤ 0.05. And then the GO (Gene Ontology,http://geneontology.org/ ) and KEGG (Kyoto Encyclopedia of Genes and Genomes,www.kegg.jp ) enrichment according to the identified DEGs were analyzed. The key DEGs involved in flowering transition were identified and their expression patterns were analyzed.qRT-PCR verification
-
Quantitative real-time PCR (qRT-PCR) was carried out to verify the transcriptome data and analyze the expression patterns of 12 DEGs related to the flowering transition in R. rugosa 'Duoji Huangmei'. All actions were done on a CFX96 Real-time PCR System according to the instructions of the SYBR Green Pro Taq HS premixed qPCR kit (Accurate Biology, China). The amplification program was as follows: 30 s at 95 °C, 40 cycles of 5 s at 95 °C and 30 s at 60 °C. The relative expression level of differential genes was calculated by the 2−ΔΔCᴛ method with RrGADPH as an internal control. All primers used in this study are listed (Supplemental Table S1). All samples were repeated three times.
-
The bud length increased continuously during the flowering transition process of R. rugosa 'Duoji Huangmei' (Fig. 1a). The flowering transition process was divided into three stages, namely vegetative growth stage (S1), flowering transition stage (S2), and sepal primordial differentiation stage (S3). The bud length was less than 0.3 cm and the growth cone was pointed conical at S1 (Fig. 1b). Subsequently, the bud length increased to 0.3 cm ~ 0.5 cm, and the top of the growth cone gradually changed to oval at S2 (Fig. 1c). When the bud length was greater than 0.5 cm, the sepal primordium began to bulge, and the flowering transition process entered S3 (Fig. 1d).
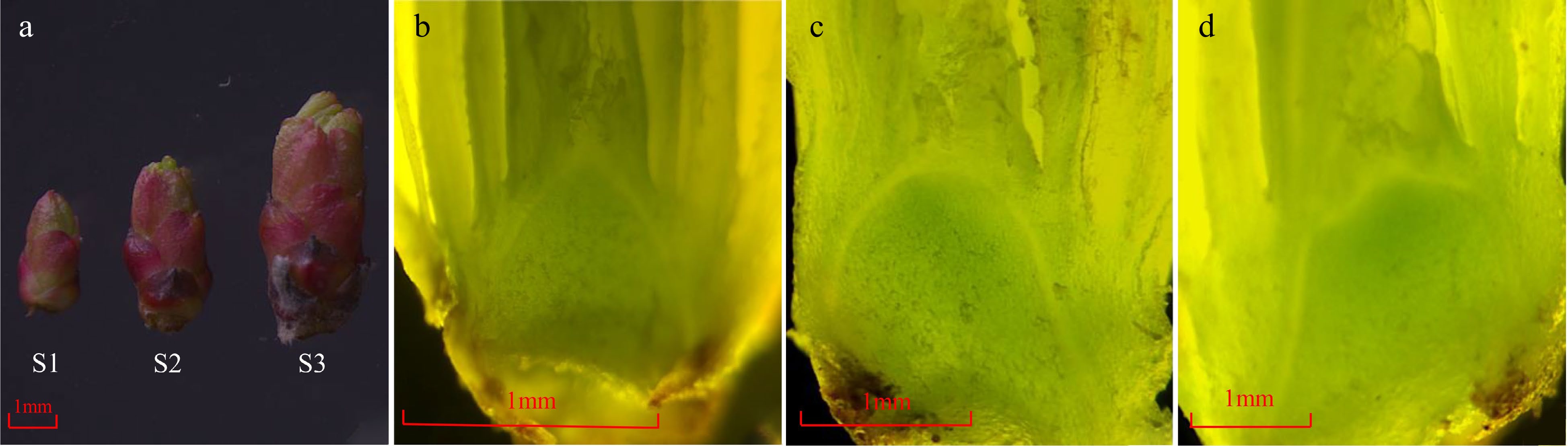
Figure 1.
The morphology changes of buds during the flowering transition process in R. rugosa ‘Duoji Huangmei’. (a) The external morphology of buds at different development stages. (b) The growth cone was pointed conical at S1. (c) The growth cone was oval at S2. (d) The sepal primordium began to bulge at S3.
Content changes of sugar and starch
-
Both sugar and starch content showed up-down patterns, peaking at S2 (Fig. 2a, b). The content of sugar had the highest level at S2 (122.85 mg·g−1), and similar levels at S1 (94.37 mg·g−1) and S3 (91.19 mg·g−1). The content of starch increased from 5.55 mg·g−1 (S1) to 9.65 mg·g−1 (S2), and then decreased to 7.79 mg·g−1 at S3 stage (Fig. 2b). In addition, the content of sugar was about 13 times higher than that of starch at the same stage.
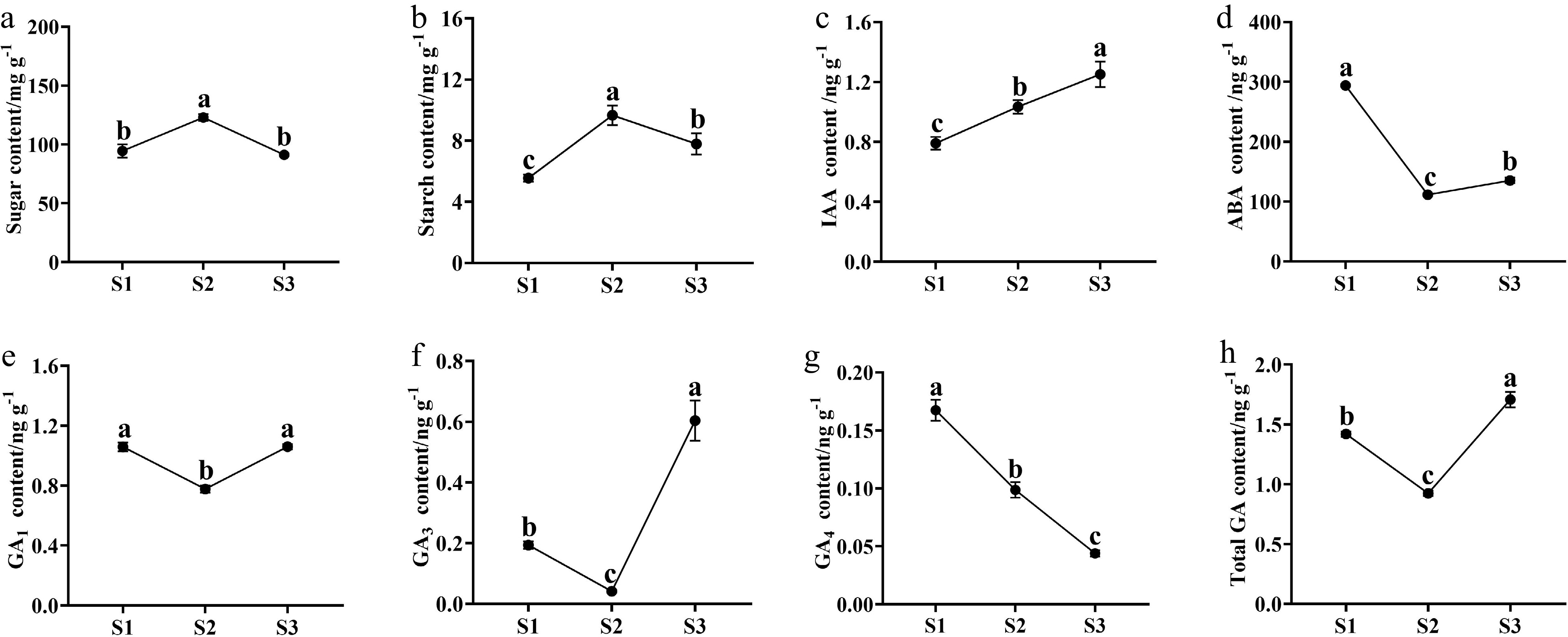
Figure 2.
The content changes of sugars, starch, and endogenous phytohormones in buds during the flowering transition process of R. rugosa 'Duoji Huangmei'. (a) Sugar content. (b) Starch content. (c) IAA content. (d) ABA content. (e) GA1 content. (f) GA3 content. (g) GA4 content. (h) Total GA content. The different small letter for each content indicated significant difference between germplasms at ɑ = 0.05.
Content changes of endogenous phytohormones
-
The content of endogenous phytohormones presented diverse trends during the flowering transition of R. rugosa 'Duoji Huangmei'. The IAA concentration increased continuously with the contents from 0.79 ng·g−1 at S1 to 1.03 ng·g−1 at S2 and 1.25 ng·g−1 at S3 (Fig. 2c). The ABA content decreased 62.08% from S1 (293.92 ng·g−1) to S2 (111.47 ng·g−1) and then kept a gentle trend to S3 (135.22 ng·g−1; Fig. 2d). The content of GA1 and GA3 had similar trends with valley values at S2. The GA1 content maintained higher levels at S1 (1.05 ng·g−1) and S3 (1.06 ng·g−1) than that at S2 (0.77 ng·g−1; Fig. 2e). However, the GA3 content was much higher at S3 than the content at S1 and S2 (Fig. 2f). GA4 content decreased continuously from 0.17 ng·g−1 at S1 to 0.10 ng·g−1 at S2, and 0.04 ng·g−1 at S3 (Fig. 2g). Taken together, total GA content showed higher levels at S1 (1.42 ng·g−1) and S3 (0.92 ng·g−1), and the lowest level at S2 (1.71 ng·g−1; Fig. 2h).
Global analysis of RNA-seq data
-
Nine cDNA libraries were constructed to explore the key genes associated with the flowering transition in R. rugosa 'Duoji Huangmei'. And the raw data were deposited at the NCBI Sequence Read Archive (SRA) under accession numbers SRR21783417−SRR21783425. A total of 412,576,223 clean reads were obtained after removing the adaptor and low-quality reads (Supplemental Table S2). The Q20 and Q30 base rates were more than 96.45% and 91.17%, respectively. The average GC content was about 47.19%. The mapping rate of each library ranged from 76.28% to 77.51%. These results indicated that the sequencing quality was sufficient for further analyses.
GO and KEGG analyses of DEGs
-
There were 3676 DEGs identified from the compare combine of S2 vs S1, which included 2174 up-regulated genes and 1502 down-regulated genes (Fig. 3a). Taking S2 as control, a total of 1227 genes expressed differentially in S3, with more down-regulated genes (972) than up-regulated genes (255). Three thousand six hundred and six DEGs had different expression patterns between S3 and S1, including 2031 up-regulated genes and 1575 down-regulated genes. Five thousand three hundred and thirty two DEGs were found in three compare combinations, of which 417 DEGs were present in all combinations (Fig. 3b).
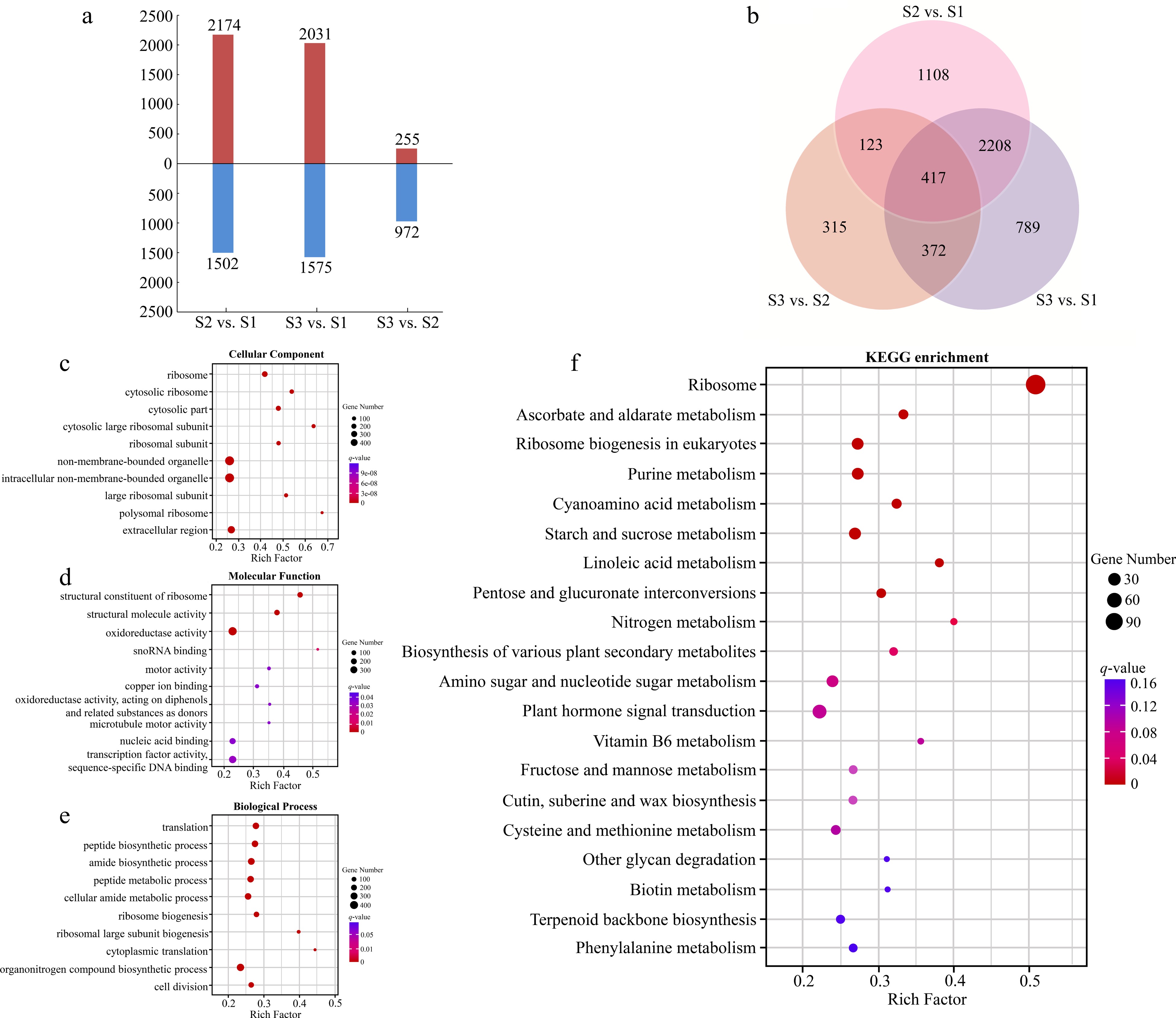
Figure 3.
Analyses of DEGs at three stages of flowering transition in R. rugosa. (a) Numbers of DEGs in each comparison. (b) Venn diagram analyses of differential and stage-specific expression genes in each comparison. (c) The GO terms of DEGs in the cellular components. (d) The GO terms of DEGs in the molecular functions. (e) The GO terms of DEGs in the biological process. (f) KEGG analysis of DEGs.
For the GO annotation, 3109, 3053, and 2930 DEGs were enriched in cellular component (CC), molecular functions (MF), and biological processes (BP), respectively. The majority of DEGs of CC category were assigned into 'Ribosome' (GO:0005840, 164 DEGs), 'Cytosolic ribosome' (GO:0022626, 92 DEGs), and 'Cytosolic part' (GO:0044445, 103 DEGs; Fig. 3c). For the MF category, the three most abundant structural constituents were 'Structural constituent of ribosome' (GO:0003735, 145 DEGs), 'Structural molecule activity' (GO:0005198, 168 DEGs), and 'Oxidoreductase activity' (GO:0016491, 394 DEGs; Fig. 3d). The three most abundant sub-categories divided from the BP category were 'Translation' (GO:0006412, 183 DEGs), 'Peptide biosynthetic process' (GO:0043043, 195 DEGs), and 'amide biosynthetic process' (GO:0043604, 192 DEGs; Fig. 3e).
By KEGG analysis, 794 DEGs were enriched in 132 metabolic pathways (Fig. 3f). Among them, the three most significant pathways for enrichment are 'Ribosome' (ko03010, 119 DEGs), 'Ascorbate and aldarate metabolism' (ko00053, 16 DEGs), and 'Ribosome biogenesis in eukaryote' (ko03008, 28 DEGs). In addition, we also found two pathways related to the flowering transition, namely 'Starch and sucrose metabolism' (ko00500, 25 DEGs) and 'Plant hormone signal transduction' (ko04075, 41 DEGs).
Expression analysis of sucrose- and starch-related DEGs
-
A total of 17 DEGs involved in starch and sucrose metabolism and transport were identified during the flowering transition process of R. rugosa 'Duoji Huangmei', which including five DEGs related to sugar transport and 12 DEGs related to sugar and starch metabolism, respectively (Fig. 4). There was one down-regulated DEG (RrTPS: evm.TU.chr6.439) and 11 up-regulated DEGs in the combination of S2 vs S1. In the combination of S3 vs S2, the expression of all nine DEGs exhibited downward trends. In addition, there were four DEGs identified from both S2 vs S1 and S3 vs S2, i.e., RrSUT14 (evm.TU.chr2.691), RrTPS (evm.TU.chr6.439), RrTPP (evm.TU.chr3.4724), and RrBAM3 (evm.TU.chr5.2267). Among them, except the expression level of RrTPS decreasing continuously, the other three DEGs showed up-down expression patterns.
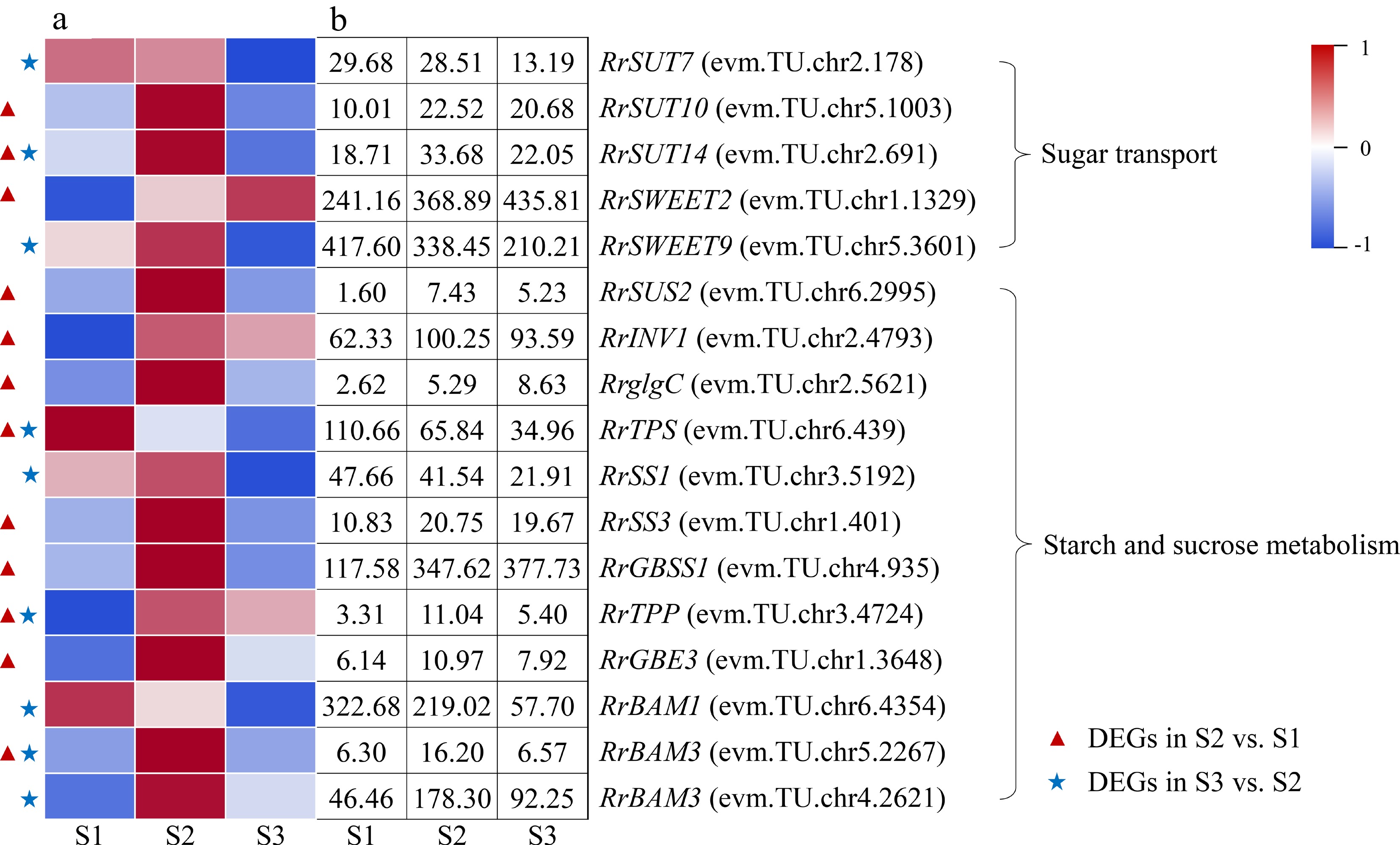
Figure 4.
Expression of starch- and sucrose-related DEGs. (a)The expression heat map of DEGs. (b) The FPKM values of DEGs. BAM: Beta-amylase; GBE3: 1,4-alpha-glucan-branching enzyme 3; GBSS1: Granule-bound starch synthase 1; glgC: Glucose-1-phosphate adenylyltransferase; INV1: insoluble isoenzyme CWINV1-like; SS1: Starch synthase 1; SUS2: Sucrose-phosphate synthase 2; SUT: Sugar transport protein; SWEET: Sugars will eventually be exported transporter; TPP: Trehalose-phosphate phosphatase; TPS: Trehalose-phosphate synthase
Expression analysis of IAA-related DEGs
-
One DEG involved in IAA metabolism (RrYUCCA2: evm.TU.chr5.4871) and two DEGs related to IAA transport (RrAUX1: evm.TU.chr5.185; RrPIN1: evm.TU.chr4.173) were discovered and presented up-regulated expression during the flowering transition process of R. rugosa 'Duoji Huangmei' (Fig. 5). A total of 11 DEGs involved in IAA signal transduction were distinguished. However, the expression trends of DEGs related to IAA signal transduction were complex, which was different from the highly consistent expression trends of DEGs associated to IAA metabolism and transport. In the combine of S2 vs S1, five DEGs related to IAA signal transduction showed up-regulated trends, i.e., RrTIR1 (evm.TU.chr4.4951), RrIAA13 (evm.TU.chr6.4429), RrSAUR76 (evm.TU.chr2.1179), RrSAUR78 (evm.TU.chr2.5004), and RrGH3.6 (evm.TU.chr1.3329), and three DEGs related to IAA signal transduction showed down-regulated trends, namely RrIAA16 (evm.TU.chr7.3988), RrIAA17 (evm.TU.chr4.5430) and RrSAUR32 (evm.TU.chr3.3449). In the combination of S3 vs S2, only one DEGs related to IAA signal transduction (RrGH3.1: evm.TU.chr2.2246) presented up-regulated expression trend and two DEGs related to IAA signal transduction (RrIAA17: evm.TU.chr2.4338; RrARF: evm.TU.chr4.5433) showed down-regulated expression trends.
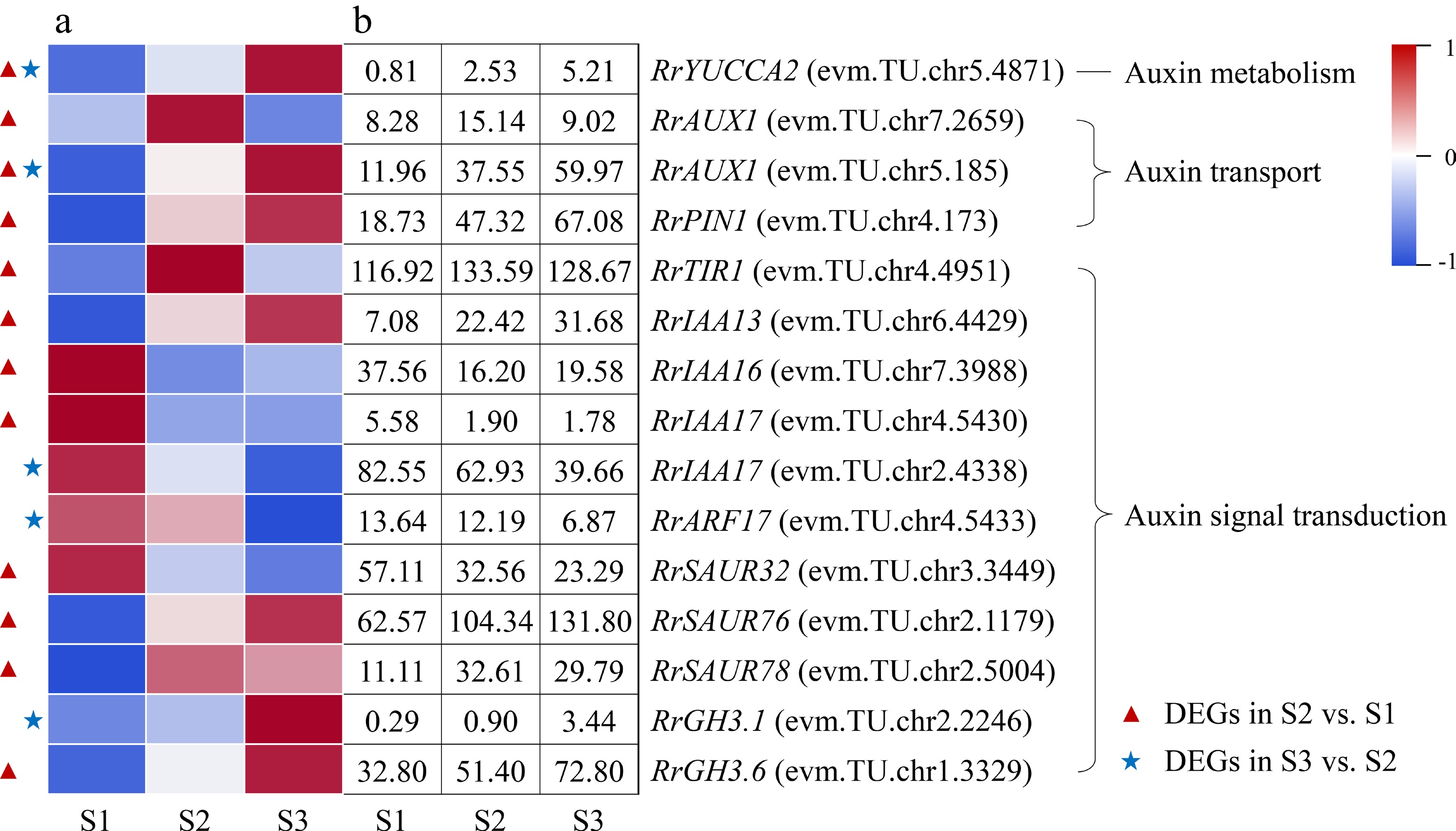
Figure 5.
Expression of IAA-related DEGs. (a) The expression heat map of DEGs. (b) The FPKM values of DEGs. ARF: Auxin response factor; AUX1: Auxin 1; GH3: Gretchen hagen 3; PIN1: PIN-formed acicular protein 1; SAUR: Small auxin-up RNA protein; TIR1: Transport inhibitor response protein 1;YUCCA2: YUCCA flavin-containing monooxygenase 2
Expression analysis of ABA-related DEGs
-
Two DEGs involved in ABA metabolism (RrNCED3: evm.TU.chr1.2533; RrAO2: evm.TU.chr4.1894) were identified during the flowering transition process of R. rugosa 'Duoji Huangmei', which both decreased sharply in the combination of S2 vs S1 (Fig. 6). There were nine DEGs related to ABA signal transduction in the combination of S2 vs S1, of which eight DEGs showed sharply down-regulated expression trends except RrPYL4 (evm.TU.chr7.2783). Furthermore, eight DEGs associated to ABA signal transduction were selected from the combination of S3 vs S2, which showed slight downward expression patterns from S2 to S3. In addition, four DEGs, namely RrPYR1 (evm.TU.chr4.2796), RrPP2C8 (evm.TU.chr3.5236), RrPP2C16 (evm.TU.chr2.987), and
RrABF (evm.TU.chr5.5115), were present in both S2 vs S1 and S3 vs S2. 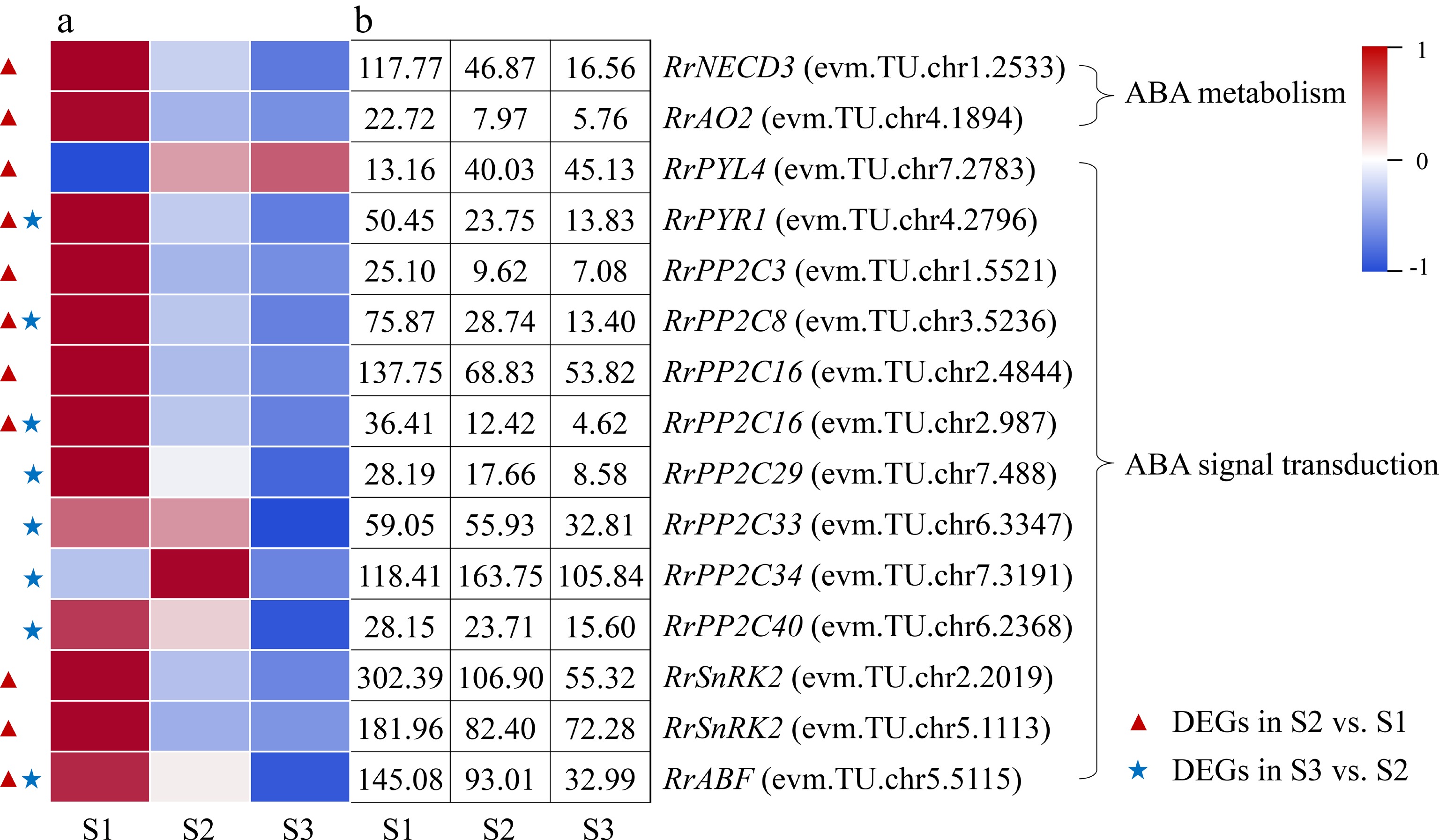
Figure 6.
Expression of ABA-related DEGs. (a) The expression heat map of DEGs. (b) The FPKM values of DEGs. ABF: ABA responsive element binding factor; AO2: Aldehyde oxidase 2; NECD3: 9-cis-epoxycarotenoid dioxygenase 3; PP2C: Protein phosphatase 2C; PYL4: Pyrabactin resistance-like 4 protein; PYR1: Pyrabactin resistance 1 protein; SnRK2: Sucrose nonfermenting 1-related protein kinase 2
Expression analysis of GA-related DEGs
-
Two DEGs associated with GA metabolism, RrGA2ox1 (evm.TU.chr1.2534) and RrGA3ox1 (evm.TU.chr2.4322), were distinguished and both exhibited down-regulated expression trends during the flowering transition process of R. rugosa 'Duoji Huangmei' (Fig. 7). And the former one was identified as DEG in combinations of both S2 vs S1 and S3 vs S2, which expression continued to decline. A total of 14 DEGs related to GA signal transduction were identified during the flowering transition process. Interestingly, 12 of 14 DEGs were present in the combination of S2 vs S1, of which nine DEGs showed up-regulated expression trends and three DEGs (RrGID1A: evm.TU.chr7.3415; RrSnakin-2: evm.TU.chr5.4611; RrSCL1: evm.TU.chr5.2181) showed downward patterns. Unlike the combination of S2 vs S1, there were only six DEGs identified in the combine of S3 vs S2, and all of them showed down-regulated expression trends.
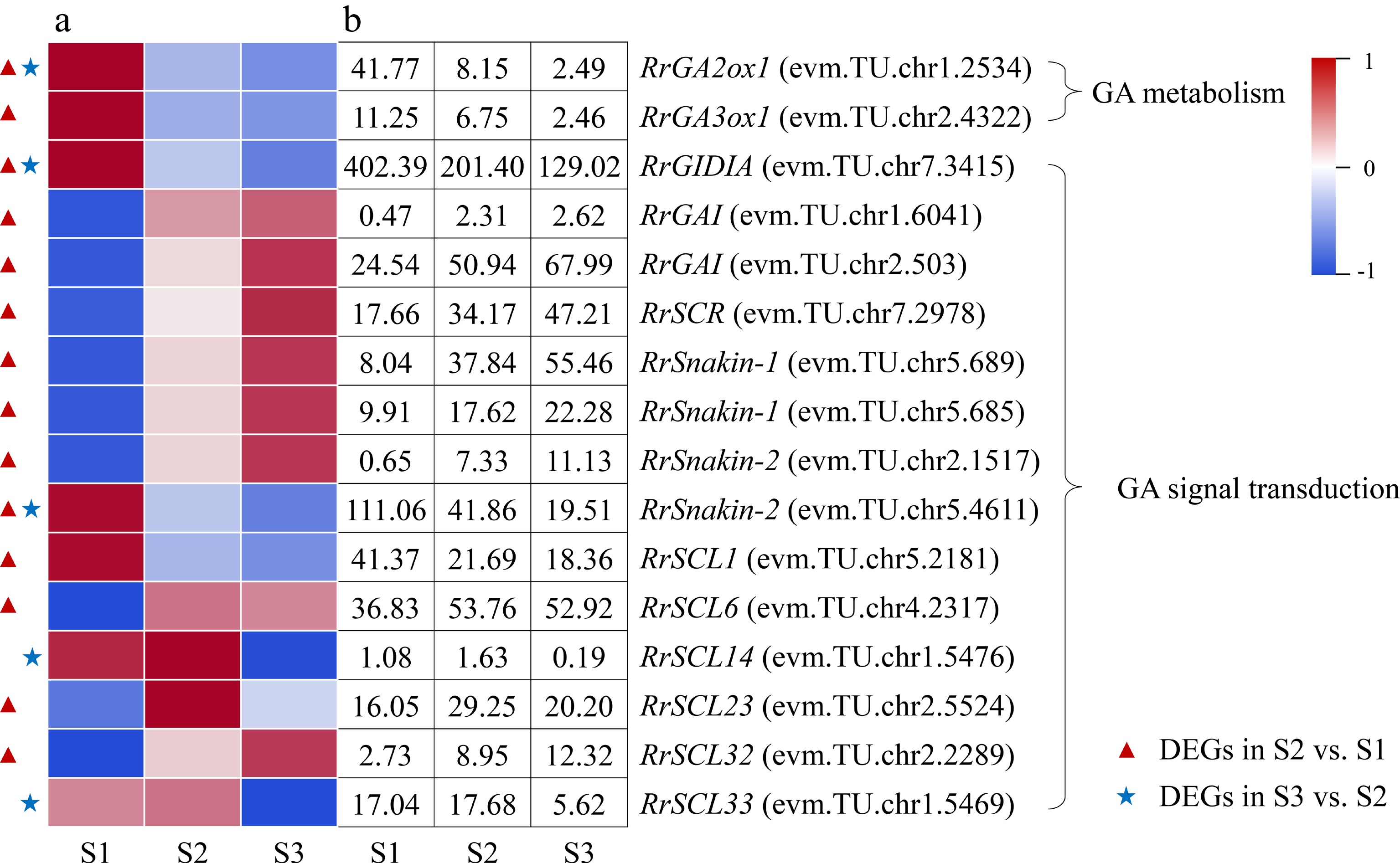
Figure 7.
Expression of GA-related DEGs. (a) The expression heat map of DEGs. (b) The FPKM values of DEGs. GA2ox1: Gibberellin 2-beta-dioxygenase 1; GA3ox1: Gibberellin 3-oxidase 1; GAI: GA insensitive protein; GID1A: GA insensitive dwarf 1 protein; SCL: Scarecrow-like protein; SCR: Scarecrow protein
Expression analysis of photoperiod- and vernalization-related DEGs
-
A total of seven DEGs associated with photoperiod were annotated during the flowering transition process of R. rugosa 'Duoji Huangmei', of which five and three DEGs were found in the combination of S2 vs S1 and S3 vs S2, respectively (Fig. 8). Four of the five DEGs in the combination of S2 vs S1, i.e., RrBBX21 (evm.TU.chr2.4992), RrCOL2 (evm.TU.chr3.5220), RrCOL4 (evm.TU.chr1.3253), and RrCOL16 (evm.TU.chr6.1098) showed down-regulated expression trends, and one DEG (RrCOP1: evm.TU.chr2.4992) had an up-regulated expression pattern. One of the three DEGs in the combination of S3 vs S2, RrCOP1 (evm.TU.chr1.6863), had an up-regulated expression trend, while the other three DEGs showed down-regulated expression sharply. In addition, two DEGs involved in vernalization (RrVRN1: evm.TU.chr3.5381; RrVIN3: evm.TU.chr4.4724) were identified, which only presented in the combination of S2 vs S1 and showed up-regulated expression. It must be mentioned that the florigen gene FT is a critical regulator in the flowering transition of plants. One RrFT gene was annotated in this study, i.e., evm.TU.chr6.761, whose expression level presented an up-down trend during the flowering transition process.
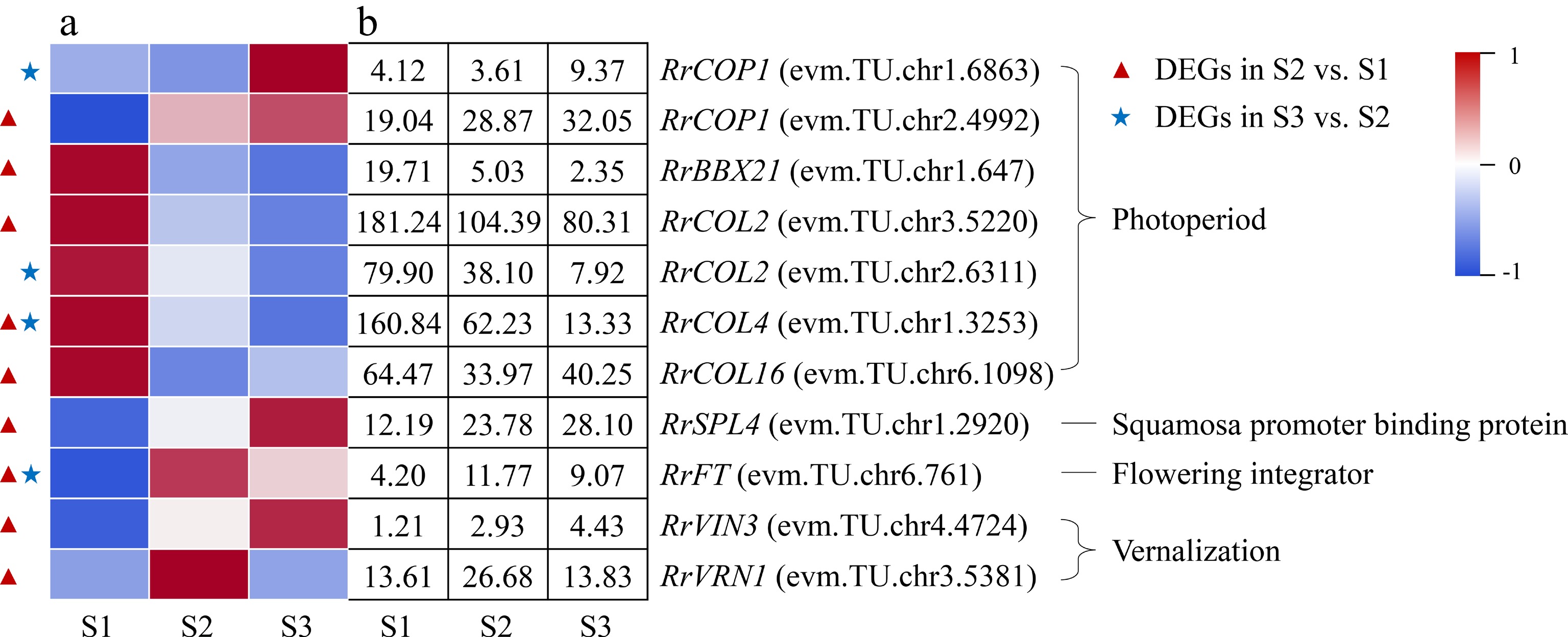
Figure 8.
Expression of photoperiod- and vernalization-related DEGs. (a) The expression heat map of DEGs. (b) The FPKM values of DEGs. BBX21: B-box 21; CO: CONSTANS; COL: CONSTANS-like; COP1: Constitutive Photomorphogenesis 1; FLC: Flowering Locus C; FT: Flowering locus T protein; SPL4: Squamosa promoter-binding-like protein 4;VIN3: Vernalization insensitive protein 3; VRN1: Vernalization protein 1
qRT-PCR validation
-
In this experiment, a total of 12 genes related to the starch and sucrose metabolism (RrSUT14, RrTPP, RrTPS), IAA metabolism and signal transduction (RrYUCCA2, RrAUX1), GA metabolism and signal transduction (RrGA2ox1, RrGID1A), ABA metabolism and signal transduction (RrPYR1, RrPP2C16, RrABF), photoperiod (RrCOL4), and the florigen gene RrFT were selected for qRT-PCR verification. As shown in Fig. 9, the expression trends of these genes were similar to those of transcriptome sequencing, and the correlation coefficients were all greater than 0.87. The results showed that the RNA-seq data were accurate and reliable.
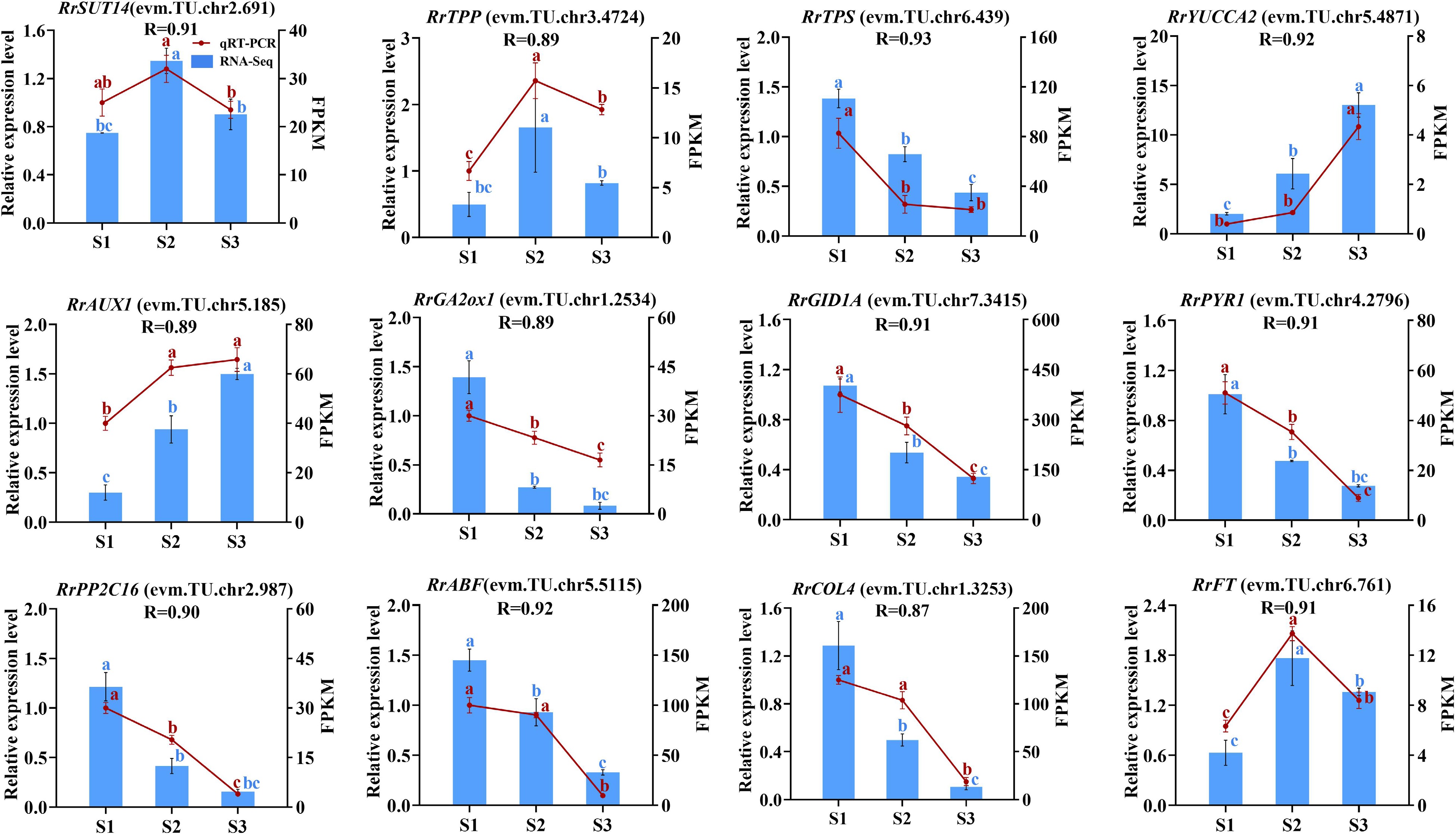
Figure 9.
The correlation of 12 genes between RNA-seq and qRT-PCR data. Letters on the error line indicate significant differences. The different lower case letters for each expression level indicated significant difference between germplasms at ɑ = 0.05.
-
In higher plants, sugar and starch play roles as both energy substances and flowering signals to regulate the floral transition process[18]. The content of sugar and starch both presented up-down or continuous increased trends during flowering transition process[19, 20], which predicted that the accumulation of sugar and starch probably can promote early phase of flowering transition in plants. In this study, we found that the content of sugar and starch both showed up-down patterns (Fig. 2a, b), consisted with the expression patterns of most related DEGs showng up-regulated in the combination of S2 vs S1 (Fig. 4). In addition, three DEGs (RrSWEET2: evm.TU.chr1.1329; RrSUT10: evm.TU.chr5.1003; RrSUT14: evm.TU.chr2.691) involved in sugar transport presented up-regulated expression in the combination of S2 vs S1. Previous studies showed that over expression of MdSUT2 and SWEET10 in A. thaliana resulted in early flowering, and the flowering transition of pSuT mutants was delayed significantly[21−23]. Accordingly, we speculated that the flowering transition process from S1 to S2 in R. rugosa was promoted by the co-expression of three DEGs involved in sugar transport and ten DEGs related to sugar and starch metabolism, such as RrSUS2 (evm.TU.chr6.2995), RrglgC (evm.TU.chr2.5621), RrTPS (evm.TU.chr6.439), etc. In addition, the expression of all nine DEGs were downward trends in the combination of S3 vs S2, including three sugar transport-related genes (RrSWEET9: evm.TU.chr5.3601; RrSUT7: evm.TU.chr2.178; RrSUT14: evm.TU.chr2.691) and six sugar and starch metabolism-related genes (RrTPS: evm.TU.chr6.439; RrTPP: evm.TU.chr3.4724; RrSS1: evm.TU.chr3.5192; RrBAM1: evm.TU.chr6.4354; RrBAM3: evm.TU.chr4.2621; RrBAM3: evm.TU.chr5.2267). This indicated that the flowering transition process from S2 to S3 may not require large amounts of sugar and starch. Previous studies showed that the expression of genes related to sugar and starch metabolism also gradually increases during the flowering transition process in Malus domestica and Fragaria × ananassa. This indicated that Rosaceae plants may regulate the flowering transition process by accumulating sufficient sugars and starch[24, 25].
IAA metabolism, transport, and signal transduction during the flowering transition process of R. rugosa
-
For most plants, the content of IAA presented continuous increased or up-down trends during flowering transition process and exogenous IAA treatment can promote or restrain plant flowering transition[12, 26]. In this study, the content of IAA increased continually during the floral transition process (Fig. 2c). Relevant studies also indicated that IAA content also continued to increase during the flowering transition in R. odorata var. gigantea and Crocus sativus[27, 28]. And only one DEG involved in IAA biosynthesis, RrYUCCA2 (evm.TU.chr5.4871), was identified and its expression level presented continually increased trend during the whole flowering transition process, which was consistent with the change in the content of IAA (Fig. 5). IAA was synthesized in the cytoplasm, but its content in cells was determined by multiple IAA transporters, such as PINs, AUX1/LAXs, and ABCBs, etc[29]. In this study, the expression levels of one output transport DEG (RrPIN1: evm.TU.chr4.173) and two input transport DEGs (RrAUX1: evm.TU.chr5.185; RrAUX1: evm.TU.chr7.2659) increased in the combination of S2 vs S1. Accordingly, we speculated that the increased expression levels of RrPIN1 and RrAUX1 may induce the spatiotemporal variation of IAA content in cells and then affect the expression of IAA responsive genes in IAA signal transduction pathway. IAA signal transduction pathway was consisted of IAA receptor (TIR1/AFB), transcriptional repressor (AUX/IAA), IAA response factor (ARF) and downstream target genes[30]. In this study, the expression of IAA receptor gene RrTIR1 (evm.TU.chr4.4951) was up-regulated in the combination of S2 vs S1, which was probably promoted by the change of IAA content. Subsequently, three transcriptional repressor DEGs with differential expressions (RrIAA13: evm.TU.chr6.4429; RrIAA16: evm.TU.chr7.3988; RrIAA17: evm.TU.chr4.5430) transduced IAA signals to RrSAUR32 (evm.TU.chr3.3449), RrSAUR76 (evm.TU.chr2.1179), RrSAUR78 (evm.TU.chr2.5004), and RrGH3.6 (evm.TU.chr1.3329) via non-differential expressed IAA response gene RrARF during the early stage of flowering transition.
Taken together, the changes of IAA content during flowering transition of many plants have a strong regularity, which indicates that IAA may be closely related to flowering transition. Many genes involved in IAA metabolism, transport and signal transduction pathways have also been identified, but have not been proved to directly affect the flowering transition process. Therefore, the effect of IAA-related genes on flowering transition is worthy of further study.
ABA metabolism and signal transduction during flowering transition process of R. rugosa
-
Relevant studies have shown that the content of ABA presented down-up trends during the floral transition process and exogenous ABA treatment can restrain plant flowering transition in most plants[31]. In this study, we found that the ABA content decreased 62.08% from S1 to S2 and then kept a gentle up trend to S3 (Fig. 2d), which was consistent with many Rosaceae plants, such as R. odorata var. gigantea and Prunus avium[27, 32]. Furthermore, the expression levels of two ABA synthesis-related genes (RrNECD3: evm.TU.chr1.2533; RrAO2: evm.TU.chr4.1894) decreased significantly and exhibited no significant difference in the combination of S2 vs S1 and S3 vs S2, respectively, which was consistent with the change of ABA content (Fig. 6). Subsequently, ABA signals were transducted via PYR/PYL/RCAR-PP2C-SnRK2 pathway to activate downstream targets such as transcription factors and ion channels, triggering an ABA response[33]. In this study, we found that the expression trends of most DEGs involved in the PYR/PYL/RCAR-PP2C-SnRK2 signal transduction pathway were consistent with ABA content. Up to now, no other genes involved to PYR/PYL/RCAR-PP2C-SnRK2 signal transduction pathway have been proved to directly affect the flowering transition process, except of BrABF3 in Brassica rapa var. chinensis which promotes flowering through the direct activation of CO transcription[34]. Related studies indicated a potential regulatory role for ABA signalling in the flowering time of Eriobotya japonica[35]. Therefore, direct evidence is needed to prove that ABA content and genes related to ABA metabolism and signal transduction have direct impact on flowering transition of R. rugosa.
GA metabolism and signal transduction during flowering transition process of R. rugosa
-
The total GA content presented down-up or up-down trends during flowering transition process in different plants and exogenous GA treatment can regulate plant flowering transition[28, 36]. In this study, total GA content showed the lowest level at S2 (Fig. 2h), which was consistent with the results in Phalaenopsis aphrodite and Nelumbo nucifera[37,38]. However, GA content showed a decreasing trend during the flowering transition in some Rosaceae plants[39−40]. It was noteworthy that GA3 content presented a similar trend to total GA content (Fig. 2f), and we found that exogenous GA3 treatment of different concentrations demonstrated significant effect on flowering transition of R. rugosa (in process), which indicated that GA3 played an important role in GA effect on the flowering transition of R. rugosa.
Moreover, GA3ox catalyzes the production of active GA, while GA2ox catalyzes the conversion of active GA into inactive GA[41]. In this study, the down-regulated expression of RrGA3ox1 (evm.TU.chr2.4322) may induce the decrease of GA content from S1 to S2, and the down-regulated expression of RrGA2ox1 (evm.TU.chr1.2534) perhaps increased the GA content in the combine of S3 vs S2 (Fig. 7). Previous studies showed that GA content can affect the content of DELLA protein, which in turn led to changes in the expression of related genes inhibited or promoted by DELLA, showing GA signal response and regulating plant flowering transition finally[42]. In this study, there were 11 and four DEGs related to GA signal transduction in the combination of S2 vs S1 and S3 vs S2, respectively, which indicated that GA signal transduction were more complicated in the early stage of flowering transformation of R. rugosa. We speculated that the reduction in total GA content and the down-regulated expression of one GA receptor protein gene (RrGID1A: evm.TU.chr7.3415) increased the content of DELLA protein by promoting the expression of three DELLA family genes (RrGAI: evm.TU.chr1.6041; RrGAI: evm.TU.chr2.503; RrSCR: evm.TU.chr7.2978). After that, DELLA protein promoted the expression of four SCL family genes (RrSCL1: evm.TU.chr5.2181; RrSCL6: evm.TU.chr4.2317; RrSCL23: evm.TU.chr2.5524; RrSCL32: evm.TU.chr2.2289) and inhibited the expression of one GASA family gene (RrSnakin-2: evm.TU.chr5.4611) from S1 to S2.
Photoperiod- and vernalization-related genes expression during the flowering transition process of R. rugosa
-
Photoperiod induces floral transition in plants by the changes in circadian rhythms. CO is an important element of the plant photoperiod regulatory pathway, which integrates light and circadian clock signals and delivers them to the flowering integron FT to regulate plant flowering transition[43]. However, CO family genes play diversified roles, namely promoting, inhibiting, or no effect on flowering transition[44, 45]. COP1 promotes CO degradation at the post-transcriptional level[46]. In this study, the up-regulated expression of two COP1 genes (evm.TU.chr2.4992; evm.TU.chr1.6863) and the down-regulated expression of four COL genes (evm.TU.chr3.5220; evm.TU.chr1.3253; evm.TU.chr6.1098; evm.TU.chr2.6311) may cause a continuous decrease in CO protein content during flowering transition process, which suggested that CO protein acted as an inhibitor to the flowering transition in R. rugosa.
Temperature is an important factor affecting plant flowering, and most plants require a certain period of low-temperature induction before flowering, namely vernalization. The VRN gene play an important role during the vernalization process, which can inhibit the expression of FLC, an essential gene in the vernalization pathway, thereby promote flowering[47]. In this study, there was no DEG related to vernalization in the combination of S3 vs S2, which suggested that vernalization only acts at the early stage of flowering transition of R. rugosa. In the combination of S2 vs S1, the expression of both RrVRN1 (evm.TU.chr3.5381) and RrVIN3 (evm.TU.chr4.4724) showed up-regulated trends, but the expression of RrFLC genes were not detected. It was indicated that RrVRN1 (evm.TU.chr3.5381) perhaps affected the flowering transition of R. rugosa by promoting directly the expression of RrFT (evm.TU.chr6.761), which was consistent with Triticum aestivum[48].
Hypothetical gene regulatory network construction
-
The regulatory network of flowering transition in higher plants is complex and comprehensive with the effect of many factors. The sugar and starch can both provide energy and act as a flowering signal to regulate the flowering transition pathway. IAA, ABA and GA can regulate the flowering transition both independently and in interaction with each other. Relevant studies indicated that the expression of TPS is induced by ABA[49]. In combination with the dynamic changes of ABA, the continuous decrease of RrTPS expression level may be influenced by ABA content. Trehalose-6-phosphate(T6P) can act as a flowering signal to indirectly activate the expression of SPL, and then SPL indirectly activates the expression of FT[50]. The expression of RrSPL and RrFT were up-regulated from S1 to S2, which indicated that there may be a positive regulatory relationship among T6P, RrSPL, and RrFT. The IAA-inducible protein SAUR inhibits the activity of PP2C-type protein phosphatase to regulate the plant growth and development process, which makes IAA to interact with ABA to regulate flowering transition in plants[51]. Up-regulated expression of RrSAUR and down-regulated expression of RrPP2C from S1 to S2 indicated that RrPP2C perhaps was negatively regulated by RrSAUR. The interaction between IAA and ABA is also reflected in the regulatory role of ARF on ABF. ARF can regulate downstream IAA-responsive genes and can regulate the expression of ABF in the ABA signaling transduction pathway, which in turn affects ABA signaling[52]. The expression trends of RrARF and RrABF were consistent during the flowering transition in R. rugosa, suggesting that down-regulation of RrARF expression drives down-regulation of RrABF expression. DELLA can bind directly to CO through its CCT structural domain and prevent CO from binding to the FT promoter[42]. BBX21 can interact with COP1 to regulate plant photomorphogenesis[53]. From S1 to S2, RrDELLA and RrCOP1 were up-regulated in expression, and RrCO was down-regulated in expression, which indicated that RrCO may be repressed by RrCOP1 and RrDELLA. In A. thaliana, VRN1 and VIN3 are involved in suppressing the negative regulation of FT by FLC. However, RrFLC with any expression levels in this study was not detected, which may be induced by suppression of RrVRN1 and RrVRN3 to RrFLC. Related studies indicated that VRN1 can act directly with FT[48]. The expression trends of RrVRN1 and RrFT were consistent from S1 to S2, indicating that the up-regulated expression of RrFT is due to the direct effect of RrVRN1. In addition, RrFT can integrate signals from multiple pathways, such as starch and sugar metabolism, vernalization and photoperiodic, to regulate the flowering transition of R. rugosa as a central gene.
Based on the above results and previous research results, we constructed a hypothetical gene regulatory network map of flowering transition in R. rugosa 'Duoji Huangmei' (Fig. 10). In the network map, we proposed that 74 DEGs related to starch and sucrose (RrSUT7/10/14, RrSWEET2/9, RrSUS2, RrglgC, RrSS1/3, RrGBSS1, RrGBE3, RrBAM1,RrBAM3, RrINV1, RrTPS, RrTPP), IAA (RrYUCCA2, RrPIN1, RrAUX1, RrTIR1, RrIAA13/16/17, RrSAUR32/76/78, GH3.1/3.6), ABA (RrNECD3, RrAO2, RrPYR1, RrPYL4, RrPP2C3/8/16/29/33/34/40, RrSnRK2, RrABF), GA (RrGA2ox1, RrGA3ox1, RrGID1A, RrGAI, RrSCR, Rrsnakin-1/2, RrSCL1/6/23/32/33), photoperiod (RrBBX21, RrCOL2/4/16, RrCOP1), and vernalization (RrVRV1, RrVIN3) did not act independently but interact with each other to regulate flowering transition of R. rugosa. Moreover, we found that more DEGs presented in the combination of S2 vs S1 compared with the combination of S3 vs S2. Accordingly, we speculated that the gene regulatory network from S1 to S2 was more complicated during flowering transition of R. rugosa.
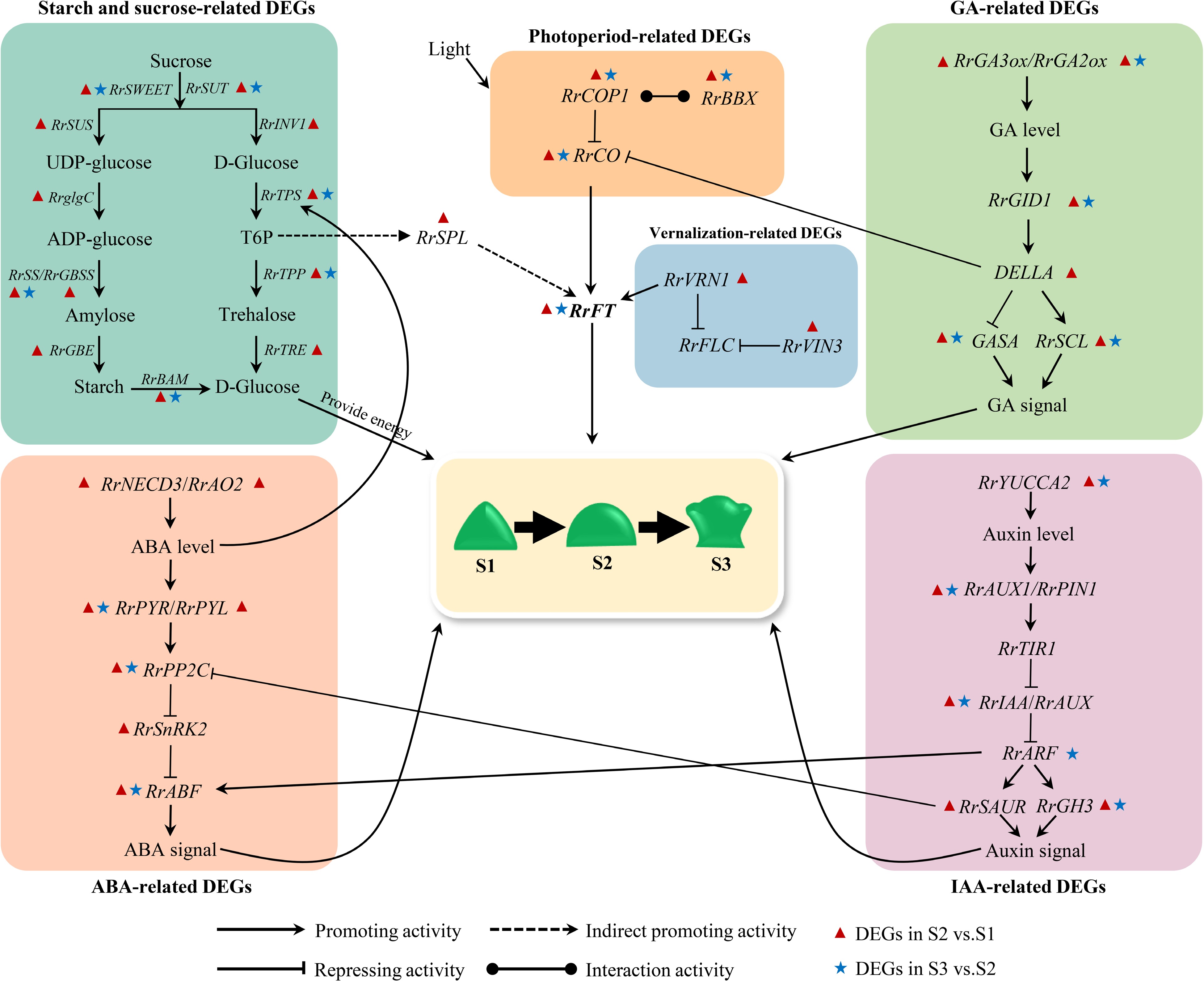
Figure 10.
Hypothetical model for the gene regulatory network of flowering transition in R. rugosa.
This project was funded by the Shandong Agricultural Seeds Engineering Project (2020LZGC011) and the National Science Foundation of China (NSFC) (31870688).
-
The authors declare that they have no conflict of interest.
-
# These authors contributed equally: Xiaobin Wang, Fei Zhao
- Supplemental Table S1 The list of qRT-PCR primer sequences.
- Supplemental Table S2 The quality information of transcriptome data.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang X, Zhao F, Wu Q, Xing S, Yu Y, et al. 2023. Physiological and transcriptome analyses to infer regulatory networks in flowering transition of Rosa rugosa. Ornamental Plant Research 3:4 doi: 10.48130/OPR-2023-0004 

Physiological and transcriptome analyses to infer regulatory networks in flowering transition of Rosa rugosa
- Received: 26 October 2022
- Accepted: 09 February 2023
- Published online: 27 February 2023
Abstract: Rosa rugosa is a famous Chinese traditional flowering species with high economic value. Flowering transition is an important process in plant growth and development. Although characterization of the flowering transition process has made great progress in some plants such as model plants, the process in R. rugosa has not been rigorously characterized to establish a mechanism. In this study, the changes of buds during flowering transition in R. rugosa ‘Duoji Huangmei’ were analyzed through transcriptomic sequencing combined with morphological and physiological index determinations. Results showed that with the morphology changes of buds, both sugar and starch content showed a similar up-down pattern while phytohormones content displayed various trends, which implied that sugar, starch and phytohormones might play diverse roles during flowering transition in R. rugosa. Moreover, a total of 4363 differentially expressed genes (DEGs) were identified at three developmental stages. Among them, 74 DEGs were involved in metabolism, transport, and signal transduction of sugar, starch, and phytohormones, as well as photoperiod and vernalization response. We proposed that these DEGs were not regulated independently but interacted with each other to construct a gene-gene network to regulate flowering transition of R. rugosa, and the regulatory network from vegetative growth stage (S1) to flowering transition stage (S2) was more complicated. These results further enriched the study of flowering transition in Rosa and lay an important foundation for breeding new varieties with desired floral traits.
-
Key words:
- Rosa rugosa /
- Flowering transition /
- Transcriptome sequencing /
- DEGs