









-
Carnation (Dianthus spp.) is popular ornamental flower; D. caryouphyllus and D. chinensis are widely planted and enjoyed by people worldwide. Flowers, as important ornamental plants, are widely studied. Varied flower shapes or florescence affect the economic value of ornamental plants. Thus, understanding the molecular mechanism of floral organ development and functional differentiation of these regulatory genes in carnation will aid in accelerating breeding improvement.
As early as 1991, the 'ABC model' of flower development was proposed in Arabidopsis thaliana and Antirrhinum majus[1]. In this model, class A, class B and class C genes were thought to regulate floral organ formation. Subsequently, the class E genes involved in the formation of floral organs were later discovered[2]. In Arabidopsis, four SEP genes were identified[3−5]: AtSEP1, AtSEP2, AtSEP3 and AtSEP4, all of which were expressed in the primordia of four whorl floral organs[6]. None of these four genes had any phenotype if they were individually mutated. However, in sep1/sep2/sep3 mutants, the petals, stamens and carpels of plants all changed into calyx sheet structures[2]. In the sep1/sep2/sep3/sep4 mutants, the normal floral primordium was absent, and all four floral organs became leaf like structures, suggesting that SEP genes were indispensable in controlling floral primordium formation[7−9].
Subsequently, the classical 'ABC model' of flower development was further improved with the discovery of the function of class E genes. The 'Floral quartets model'[10] was proposed, which showed that class A-, B-, C- and E- proteins interacted to regulate plant flowering and were continuously developing: AP1–AP1–SEP–SEP protein interactions were involved in sepal development, AP1–AP3–PI–SEP interactions determined petal development, AG–AP3–PI–SEP interactions determined stamen development, AG–AG–SEP–SEP interactions determined carpel development and AG–SHP–STK–SEP interactions determined ovule development[11]. In this model, every whorl of floral organ formation was regulated by at least one SEP protein, so class E genes were referred to as the 'glue' in the 'quartet model'[11,12].
In recent years, the function of class E genes has been reported in an increasing number of species. Previous studies have shown that the number of class E genes identified varies among different species; for example, two class E genes were identified in watermelon[13], four in Prunus mume[14], five in Oryza sativa[15] and ten in Brassica rapa[16]. These genes belonged to different subclades of class E genes, such as the SEP1/2, FBP9/23 and SEP4 subclades, which were derived from LOFSEP, which produced two consecutive gene copies in dicotyledons[6].
Previous studies have also found that the functions of SEP genes in many plants[17−23] are diverse, which may be involved in determining floral organ identity and plant architecture, fruit maturation and the transition from vegetative growth to reproductive growth processes. For example, in Phalaenopsis orchid, silenced PeSEP3 made the tepal a leaf-like organ. Downregulation of TM29 caused tomato parthenocarpic fruit development and floral reversion. However, in strawberry, FveSEP3 inhibited fruit growth in the absence of pollination and promoted fruit ripening[24]. These reports suggested that class E genes experienced functional redundancy and new functionalization in the process of evolution. However, to date, the roles of class E genes in flower development or whether they experience sub-functionalization and neo-functionalization in carnation remain unclear.
In this study, through transcriptome comparative analysis, we found that the expression of class E genes increased gradually in the first three stages (Sepal (S2), petal (S3), and stamen (S4) primordium development) after flowering initiation (S1). To explore the roles of class E genes in the development of flowers in carnation, six SEP-like genes were identified in D. chinensis. Then the expression patterns of these class E genes were analyzed by quantitative real-time PCR (qRT‒PCR). The interactions of class E proteins of D. chinensis were also investigated by yeast two-hybrid (Y2H) and bimolecular fluorescence complementation (BiFC) assays. In addition, the functions of SEP3 genes with different expression and interaction patterns were analyzed. This study demonstrated the role of class E genes in flower development, which lays a theoretical foundation for understanding the mechanism by which ABCE proteins in carnations regulate flower development and is of guiding significance for the directional improvement of carnation flower patterns.
-
D. chinensis 'L', a high-generation inbred line, were grown in an experimental field under natural conditions at Huazhong Agricultural University, in Wuhan, Hubei Province, China (30°28'36.5" N, 114°21'59.4" E). Six samples of different organs (stems and leaves during vegetative growth; sepals, petals, stamens, and pistils of flowers) were collected from D. chinensis 'L'. For each biological replicate, the samples were extracted and then immediately frozen in liquid nitrogen and stored at −80 °C until RNA extraction. Arabidopsis plants were grown under long-day conditions (16-h light/8-h dark cycle) at 22/21 °C day/night in an illumination incubator.
Microscopy and expression profiling analysis
-
Samples from different floral primordium development stages were identified under the microscope. Shoots were fixed and sectioned following previously described methods[25]. Transcriptome samples from six different flower developmental stages were sequenced and obtained (PRJNA574036). The RNA-seq reads were mapped to the new carnation genome[26] using HISAT2[27]. Principal component analysis (PCA) of the samples was performed using the prcomp function in R software. The expression levels of each gene in each RNA-seq library were calculated as the fragments per kilobase of exon model per million mapped fragments (FPKM). The average FPKM value across three biological replicates was calculated and represented in a heatmap.
Cloning of D. chinensis class E genes
-
Based on transcriptome data (PRJNA533533 and PRJNA574036)[28] and the two published genomes of the carnation[26,29], primers of the D. chinensis class E genes were designed specifically by Primer Premier 5.0 (Supplemental Table S1). Total RNA was isolated from flower buds of D. chinensis 'L' using EASYspin Pant RNA kit reagent according to the manufacturer's instructions. To remove potentially contaminating genomic DNA, RNA was treated with RNase−free DNase (Promega, USA). First strand complementary DNA (cDNA) was synthesized from 1 μg total RNA with the Prime Script TM RT Reagent Kit with gDNA Eraser (Takara, Otsu, Japan) following the manufacturer's instructions. All target fragments were cloned into the pMDTM18−T vector (TaKaRa, China) to transform DH5α Escherichia coli (Shanghai Weidi Biotechnology, China) and sequenced. The plasmids were extracted by Plasmid Miniprep Kit I (Biomiga, USA) and stored at −80 °C. The full-length coding sequences (CDS) of six DcSEP genes are shown in Supplemental Table S2.
Phylogenetic and alignment analysis of class E genes
-
A total of 52 class E genes from different species, including the AtAP1 gene as an outgroup, were used for phylogenetic analysis (Supplemental Table S3). Protein sequences were obtained from NCBI (
www.ncbi.nlm.nih.gov ) and previous reports[30]. The amino acid sequences were aligned with the DNAMAN v6.0.x program. A phylogenetic tree was constructed using MEGA v6.0[31] by the neighbor joining (NJ) method with 1,000 iterations for the bootstrap values.Gene expression analysis by qRT−PCR
-
Total RNA of each sample was extracted using an EASYspin Plant RNA kit reagent (Aidlab Biotechnologies, Beijing, China) according to the manufacturer's instructions. The specific primers of six DcSEPs for qRT−PCR were designed within the nonconservative C-terminal region using Primer Premier 5.0 software and are listed in Supplemental Table S4. The qRT−PCR was conducted using SYBR Premix Ex Taq (Takara, Beijing, China) and the ABI Prism 7500 Sequence Detection System (Applied Biosystems, Beijing, China). Each PCR was performed with three biological and three technical replicates. The housekeeping gene DcGAPDH was selected as an internal quantitative control (Supplemental Table S4). The relative expression values were calculated using the comparative CT(2−ΔΔCᴛ) method[32].
Paraffin section
-
Floral buds were divided into six developmental stages: Stage 1 (S1): the stage of floral initiation, S2: sepal primordium development stage, S3: petal primordium development stage, S4: stamen primordium development stage, S5: carpel primordium development stage, S6: late stage of differentiation of floral organ primordium. Samples of different flower bud differentiation stages were fixed overnight in fresh FAA (3.7% formaldehyde, 5% acetic acid, and 50% ethanol). Samples were finally embedded in paraffin for subsequent use of tolonium chloride as a dye. After the sections were sliced, they were observed and photographed under a Jiangnan NLCD500 microscope (Jiangnan, Nanjing, China).
Yeast two-hybrid assays
-
The GAL4-based Matchmaker Two-Hybrid System (Clontech) was used. Every full-length ORF of class E genes from D. chinensis was fused into pGADT7 or pGBKT7 to form the prey or bait constructs, respectively. The bait and prey plasmids were cotransformed into yeast strain AH109 and spotted on medium lacking leucine and tryptophane (SD/–Leu–Trp, Coolaber, Beijing, China). Protein interactions were tested on SD/–Leu–Trp–His–Ade plates (Coolaber, Beijing, China). X-α-Gal (5-Bromo-4-chloro-3-indolyl-α-Dgalactopyranoside) was used as a substrate to quantify the interaction affinity. Each combination was gradient diluted separately. To confirm the reliability of the results, at least three individual clones were used for each combination. The primers used were listed in Supplemental Table S5.
BiFC
-
To verify the reliability of the yeast two-hybrid assay results, the CDSs of DcSEP1 and DcSEP3-1, DcSEP3-2, DcSEP4-1 and DcSEP4-2 (without stop codon) were amplified with the primers (Supplemental Table S6) and cloned into the pGBKT7-gene and pGADT7-gene separately to create DcSEP4-2-YFPN, DcSEP1-YFPC, DcSEP3-1-YFPC, DcSEP3-2-YFPC and DcSEP4-1-YFPC constructs. The constructs carried by Agrobacterium tumefaciens GV3101 were used for the transfection of 5-week-old Nicotiana benthamiana leaves, according to the protocol described by Walter et al.[33]. After 2-day culture, the samples were observed with a fluorescence microscope (LEICA, DM2500).
Plant transformation
-
The pMD18-T vectors containing CDS of DcSEP3s genes were digested by restriction enzymes, and the target fragments were ligated into the corresponding sites of vector, modified from the binary vector pCAMBIA2300 containing the CaMV35S promoter, resulting in 35S:DcSEP3-1 and 35S:DcSEP3-2 constructions, respectively. All the constructed plasmids were confirmed by PCR and sequenced. The resulting plasmids were then transformed into the A. tumefaciens strain GV3101. The floral dip method in Arabidopsis was carried out as previously described[34]. The transformed seeds were screened on Murashige and Skoog (MS) agar with 50 μg·ml−1 kanamycin and 50 μg·ml−1 cefotaxime. T2 plants were used in this study.
Statistical analysis
-
Statistical significance was checked using GraphPad Prism version 9.0 for one-way ANOVA. And significant difference was shown at p < 0.01 (**).
-
The floral organ primordium of developing D. chinensis 'L' was divided into six typical stages: Stage 1 (S1): the stage of floral initiation, S2: sepal primordium development stage, S3: petal primordium development stage, S4: stamen primordium development stage, S5: carpel primordium development stage and S6: late stage of differentiation of floral organ primordium (Fig. 1a). The total RNA was isolated from the flowers of the six stages and sequenced using the Illumina platform, generating more than 21 million high-quality reads representing more than 6 Gbp in every sample. (Supplemental Table S7). Q30 values (sequencing error rate < 1%) ranged from 89.95% to 95.47%. The PCA plot showing clustering of three biological replicates of different stages of flower development was high, indicating that the dataset was reliable (Fig. 1b). Based on the recently published carnation genome[26], we aligned the sequencing reads to the reference genome and calculated the expression levels (FPKM) (Fig. 1c). The results showed that there were more than 2,000 genes with FPKM values > 60, more than 6,000 genes with FPKM values between 15 and 60, and more than 20,000 genes with FPKM values < 1 in each sample (Supplemental Table S8).
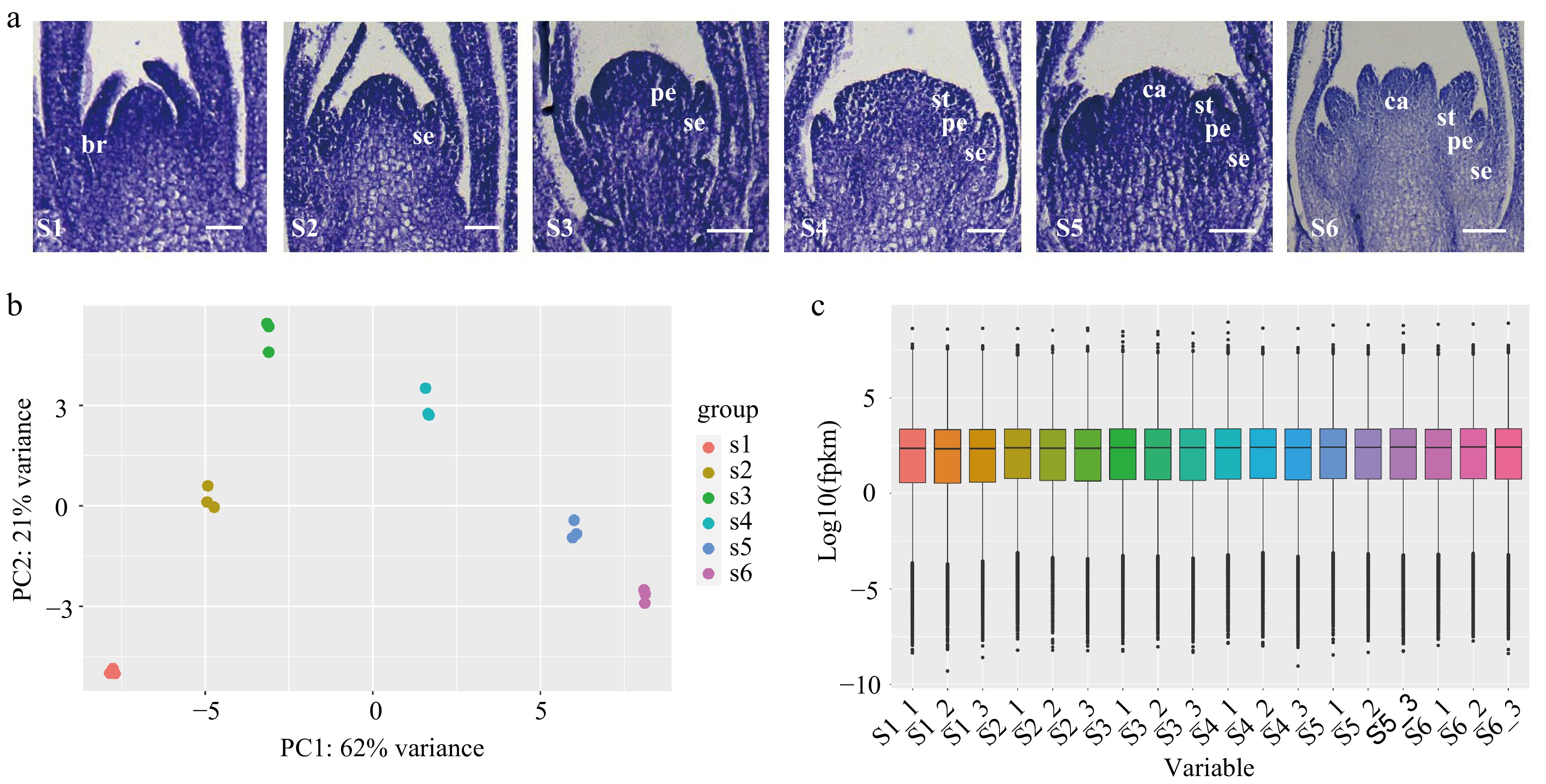
Figure 1.
Transcriptome sequencing of flowers at different floral organ primordium developmental stages in D. chinensis. (a) Floral organ primordium at different developmental stages. (b) Principal component analysis (PCA) analysis of different samples. (c) The expression in floral organ primordium at different stages. S1: the stage of floral initiation. S2: Sepal primordium development stage. S3: Petal primordium development stage. S4: Stamen primordium development stage. S5: Carpel primordium development stage. S6: Late stage of differentiation of floral organ primordium. br. bract; se. sepal; pe. petal; st. stamen; ca. carpel.
Dynamic comparative analysis of differentially expressed genes (DEGs) during floral organ primordium development
-
We conducted a comparative analysis of the DEGs using five combinations of stages that represented major changes in floral organ primordium development (S2_vs_S1, S3_vs_S2, S4_vs_S3, S5_vs_S4 and S6_vs_S5). There were 976 (S2_vs_S1), 1,398 (S3_vs_S2), 1,335 (S4_vs_S3), 1,521 (S5_vs_S4), and 410 (S6_vs_S5) upregulated DEGs identified, and 411, 1,539, 966, 1,792, and 845 down-regulated DEGs identified, respectively (Fig. 2a). Among them, MADS-box genes were only present in upregulated DEGs with floral organ primordium development. Especially during the first three developmental stages (S2–S4) after flower initiation (S1), there were a greater number of MADS-box DEGs (Fig. 2b). In contrast to other MADS-box genes, we found that class E genes were only differentially expressed in the first two comparison groups (S2_vs_S1, S3_vs_S2) (Fig. 2c), which suggested that class E genes may play important roles in the development of the sepal primordium and petal primordium.
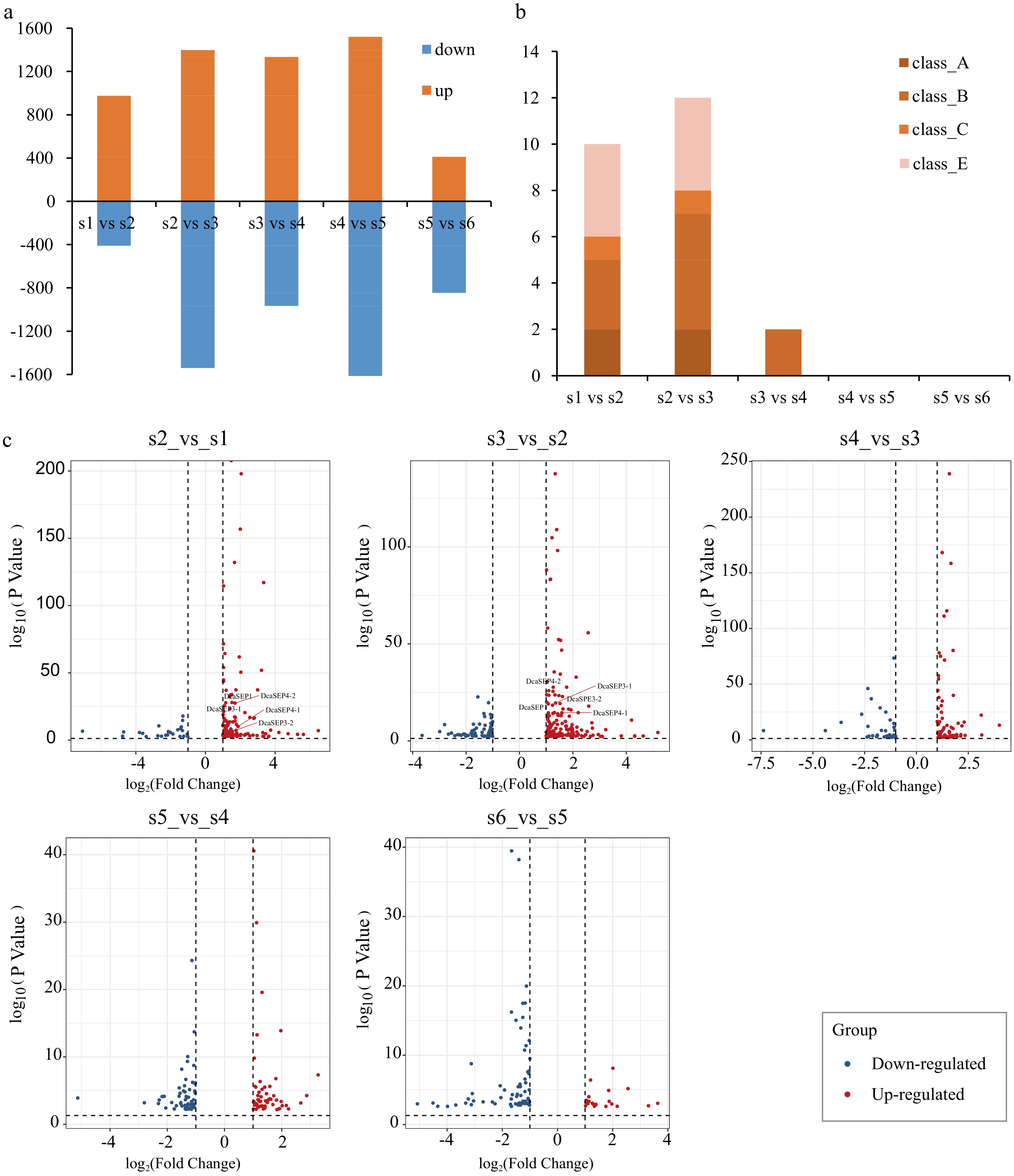
Figure 2.
Analysis of differential expression genes in different floral organs primordium development stages. (a) Bar graph showing differential expression genes (DEG) number of up-regulated and down-regulated in different pair comparisons (S2_vs_S1, S3_vs_S2, S4_vs_S3, S5_vs_S4 and S6_vs_S5). (b) The number of ABCE class MADS-box genes with differentially expressed in different comparison. (c) The DEGs of class E genes in each different comparison are displayed. Red represents up-regulated genes and blue represents down-regulated genes.
Identification and evolutionary analysis of DcSEP genes
-
Based on the MADS-box genes identified from two carnation genomes, genome_v0 and genome_v1[26,29], we identified the known class ABCE genes and found that the number of class ABC genes was the same in the two carnation genomes. The number of class E genes was six in genome_v0[29] published in 2014 and five in genome_v1 published in 2022[26]. To further confirm the members of class E genes in D. chinensis, the E genes were amplified using cDNA from D. chinensis as a template. Then, six DcSEP genes were identified and amplified in D. chinensis. Finally, a total of 15 full-length ABCE genes were obtained. Then, a phylogenetic tree was constructed by using the MADS-box proteins from D. chinensis and other species (Fig. 3). Referring to the naming of D. caryophyllus proteins[30], the corresponding D. chinensis genes were designated AP1 (DcAP1), FUL (DcFUL), AP3 (DcAP3-1 and DcAP3-2), PI (DcPI and DcPI2), TM6 (DcTM6), AG (DcAG1 and DcAG2), and SEP (DcSEP1, DcSEP3-1, DcSEP3-2, DcSEP4-1, DcSEP4-2, and DcSEP4-3).
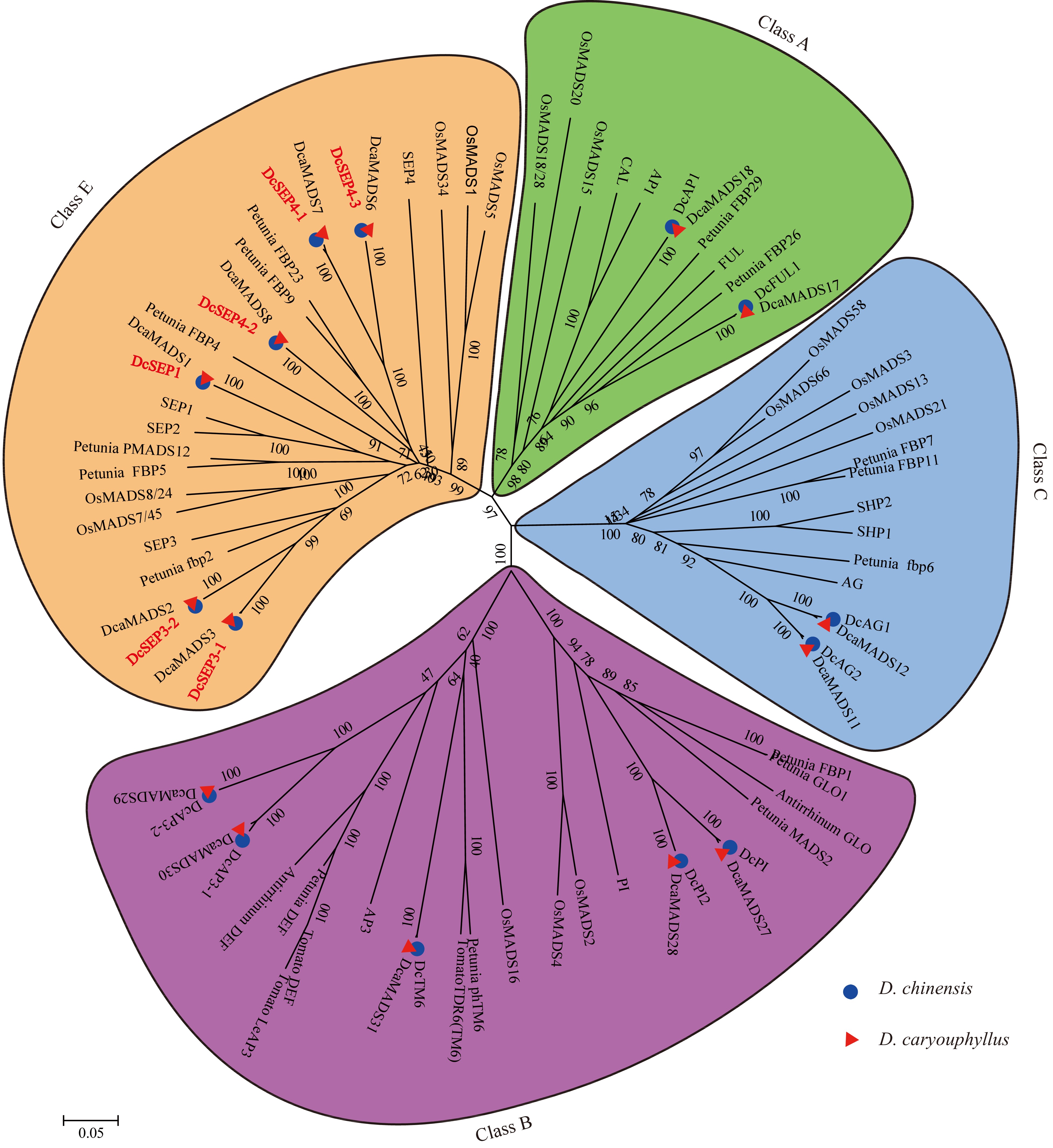
Figure 3.
The phylogenetic tree of the class A-, B-, C- and E- proteins. The subgroups are indicated by different colors.
To further identify the subclade of class E genes, these class E amino acid sequences were aligned which showed that they all had the conserved MADS domain and K domain as well as the typical SEPI and SEPII terminal motifs (Supplemental Fig. S1). The similarity of these class E amino acid sequences in D. chinensis and D. caryophyllus was between 95.86 and 100% (Supplemental Table S9). In addition, we found that the number of class E genes in different published species was different (Table 1, Supplemental Fig. S2). Among them, the number of both SEP1/2/4 and SEP3 subgroup members varied, such as in the SEP3 subgroup, three members in B. rapa, two members in Triticum aestivum and only one member in Citrullus lanatus and P. mume. This result indicated the difference in class E genes among different species.
Table 1. The number and references of class E homologous genes in different species.
Expression of the genes related to flower development in D. chinensis
-
To investigate the molecular mechanism underlying floral organ identity, the expression patterns of these genes were assayed using transcriptome data and qRT–PCR (Fig. 4). We applied the FPKM value obtained via transcriptome profiling to generate a heatmap for the DcMADS-box gene expression patterns during floral development (Fig. 4a). The results revealed that DcAP1 and DcFUL1 were expressed at the early stage of floral development (S1), in which the shoot apical meristem (SAM) transformed into flower meristem and the bract primordium differentiated, and their expression level increased gradually with the development of floral organs. Regarding class B, DcPI and DcPI2 (PI homologs), DcAP3-1 and DcAP3-2 (AP3 homologs), and DcTM6 were all expressed from the petal primordium at stages 3–6, but the expressions of DcAP3-1 and DcPI2 were low and their expression level increased from S4. DcAG1 began to be expressed after flowering initiation (S1), but DcAG2 began to be expressed after the emergence of the stamen primordium (S4). DcSEP3-1 was expressed in sepal and petal primordia at stages 3 and 4, while DcSEP3-2 had lower expression than DcSEP3-1, and its expression activation stage was later than that of DcSEP3-1. The expression pattern of DcSEP4-2 was similar to that of class A genes and DcSEP1 and they are grouped together. The expression pattern of DcSEP4-1 was similar to that of DcSEP3-2 and expressed from petal primordium development. Overall, ABCE genes exhibited dynamic expression patterns in different flower development stages. In addition, the qRT–PCR results also showed that the gene expression patterns of class E genes were different in the tissues and organs of D. chinensis (Fig. 4b–c).
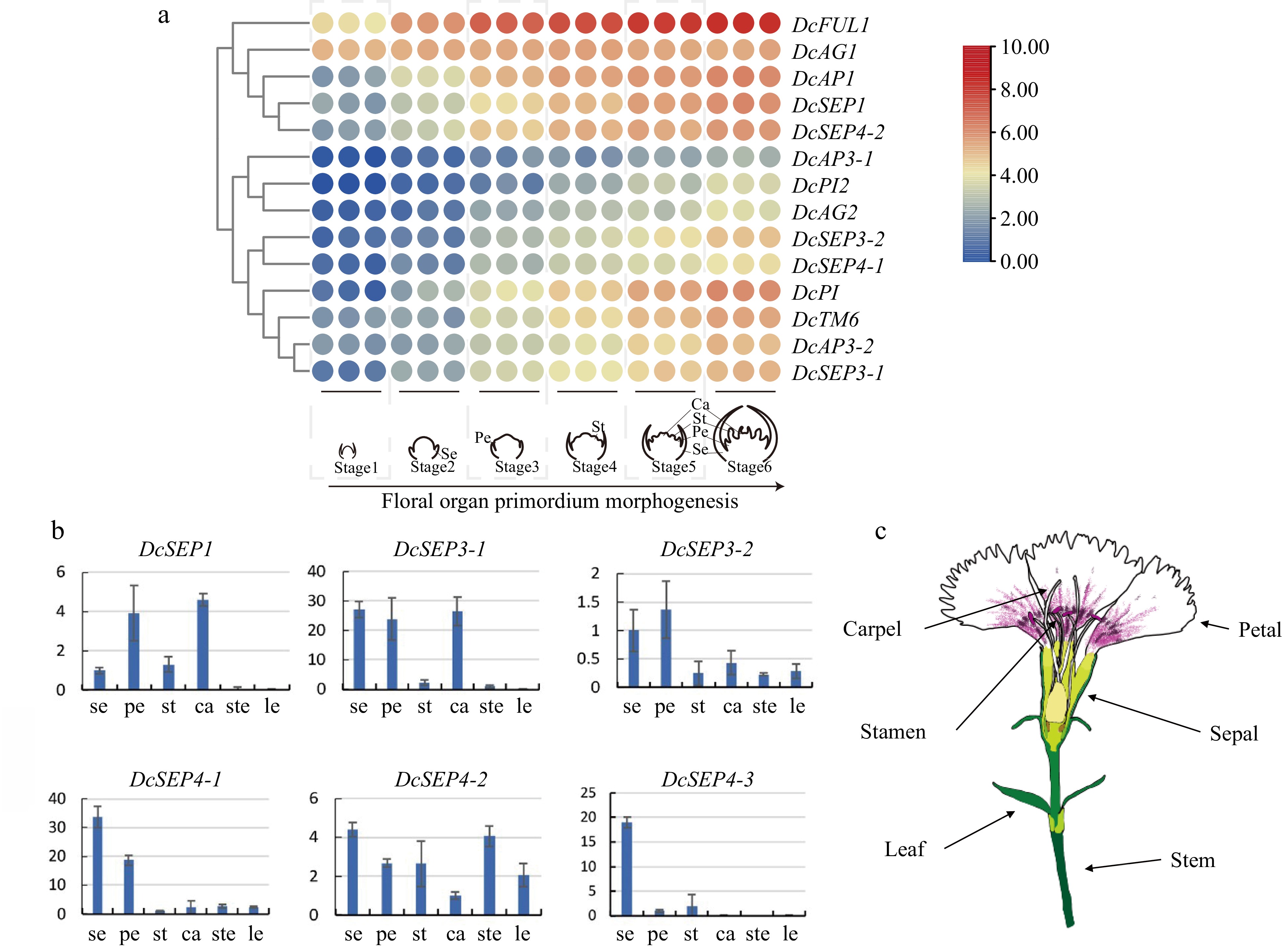
Figure 4.
Analysis of expression in D. chinensis E class genes at different tissues and stages. (a) The spatial expression patterns of class E genes in developing flowers of D. chinensis as revealed by RNA-seq. (b) The expression of six class E genes of D. chinensis in different organs were detected by qRT–PCR. Error bars indicate the collective standard deviations of three biological replicates and three technical replicates. se, sepal. pe, petal. st, stamen. ca, carpel. ste, stem. le, leaf. (c) The different organs of D. chinensis.
Protein interaction of class E genes in D. chinensis
-
To clarify how homologous or heterologous dimers can be formed among six class E proteins and other MADS-box proteins in D. chinensis, the yeast two-hybrid method was used in this study to analyze the interaction patterns of these proteins (Fig. 5 and Supplemental Fig. S3). None of the selected proteins were self-activated (Supplemental Fig. S4).

Figure 5.
The interaction statistics of class E proteins with other class A−, B−, C genes in D. chinensis. '+' in light orange represents weak interaction; '++' in orange represents moderate interaction; '+++' in deep orange represents relatively strong interaction; '++++' in brown represents strong interaction; '−' in bule represents that there is no detectable interaction of proteins, '\' in grey represents did not cover in this study.
The results showed that the class E proteins DcSEP3-1, DcSEP3-2, DcSEP4-2 and DcSEP4-3 interacted with more proteins than DcSEP1 and DcSEP4-1 (Fig. 5 and Supplemental Fig. S3). The DcSEP3-1 protein interacted with one class A protein (DcAP1), while DcSEP3-2 and DcSEP4-3 interacted with the other class A protein (DcFUL1). DcSEP4-2 interacted with two class A proteins. DcSEP4-2 lightly interacted with DcAP1 and had a relatively strong interaction with DcFUL1. Four class E proteins (DcSEP3-1, DcSEP3-2, DcSEP4-2 and DcSEP4-3) all interacted with DcPI and DcPI2 of class B. Among them, DcSEP3-2 still interacted with DcTM6 of class B, and DcSEP4-2 interacted with DcAP3-1. For the interaction with class C genes, DcSEP3-1, DcSEP3-2, DcSEP4-2 and DcSEP4-3 all interacted with DcAG1 and DcAG2. Moreover, the proteins of class E not only interacted with class A-, B- and C- proteins but also, they interacted with their own proteins to form homologous dimers, such as DcSEP3-1, DcSEP3-2, DcSEP4-2 and DcSEP4-3 (Fig. 5 and Supplemental Fig. S3). To verify the reliability of the results, four combinations of DcSEP4-2 interactions with other proteins (DcSEP1, DcSEP3-1, DcSEP3-2 and DcSEP4-1) were selected for BiFC experiments (Supplemental Fig. S5) and showed similar results to those of Y2H, suggesting that the two methods being mutually supportive. Overall, compared other subclade, the interactions of the SEP3 subclade were the richest in all the protein interactions of class E genes. It is speculated that SEP3 subclade functions are more important. Moreover, the two genes (DcSEP3-1 and DcSEP3-2) belong to the same subclade have different interaction patterns, which suggesting that they play different roles in flower development.
DcSEP3s of D. chinensis play different roles in the regulation of flower development
-
To further investigate the roles of the two DcSEP3s in flower development, the constructed vectors containing DcSEP3-1 and DcSEP3-2 were transformed into Arabidopsis. Transgenic plants were obtained by screening. The relative gene expression levels of these transgenic plants and wild-type Arabidopsis were analyzed (Fig. 6). Ectopic expression of DcSEP3-1 and DcSEP3-2 strongly influenced flowering time and plant architecture. Phenotypic analysis of transgenic lines showed that overexpression of DcSEP3-2 genes in Arabidopsis resulted in early flowering, smaller rosettes, dwarfism, abnormal floral organs and the number of rosette leaves decreased significantly compared with the wild type (Fig. 6 and Supplemental Figs S6 & S7). The transgenic line overexpressing DcSEP3-1 only showed an early-flowering phenotype (Fig. 6 and Supplemental Fig. S7). All these results further suggested that two DcSEP3 genes of D. chinensis were sub-functionalized.
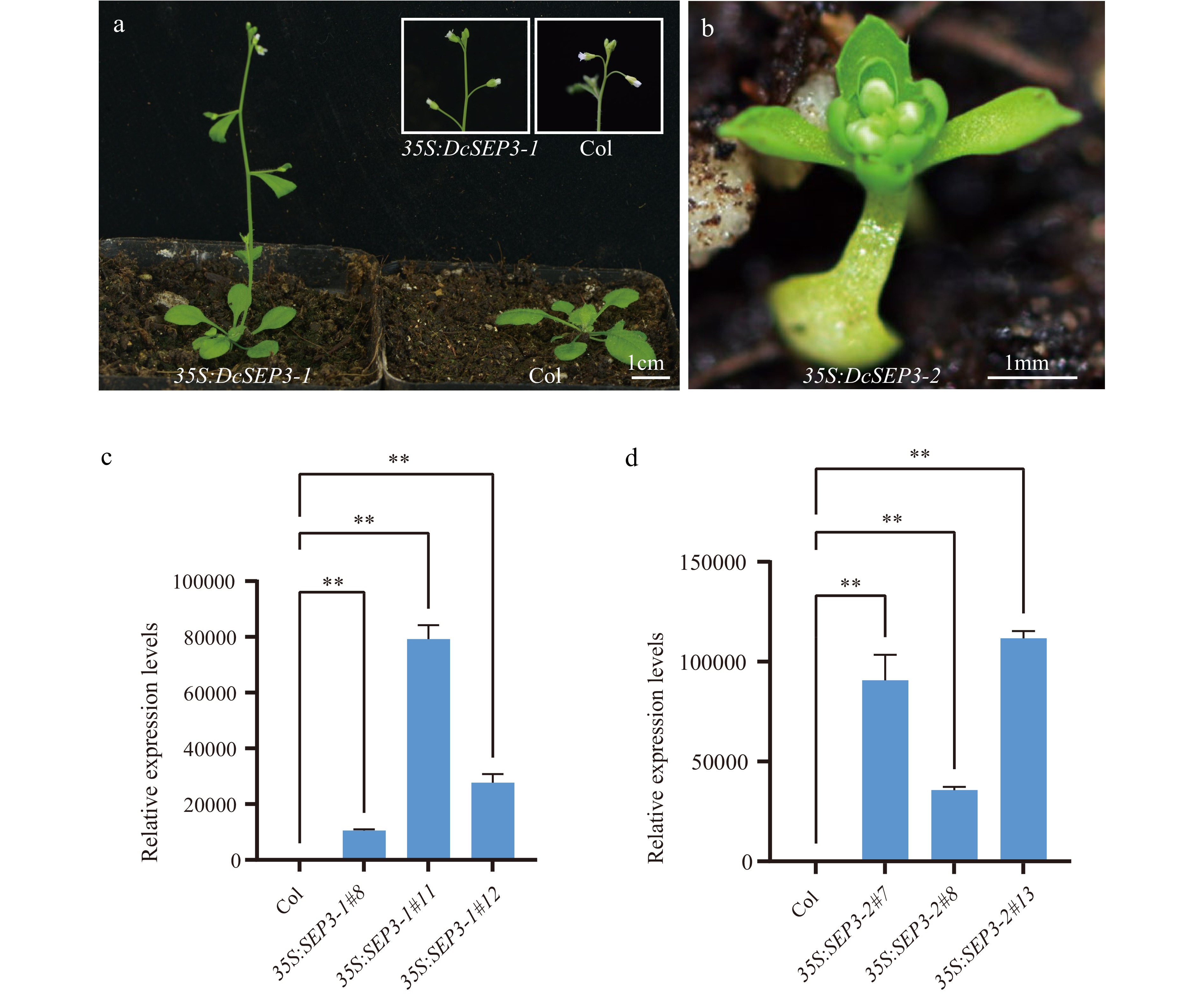
Figure 6.
Phenotype and expression analysis of D. chinensis class E genes overexpression in A. thaliana. (a) Transgenic plant of 35S:DcSEP3-1 (left), Columbia (Col) Arabidopsis (right). (b) Transgenic plant of 35S:DcSEP3-2. (c) Expression of DcSEP3-1 in transgenic lines. (d) Expression of DcSEP3-2 in transgenic lines. Data represent the mean ± SE from three biological replicates, and AtEF1a was used as internal control. Significant difference was shown at p < 0.01 (**).
-
Class E genes play significant roles in floral organ development, and every whorl of floral organ formation is regulated by at least one SEP protein[7,36]. In our results, the expression patterns of the six class E genes were different not only at different floral organ primordium developmental stages, but also in different tissues based on the transcriptome data and qRT–PCR analysis. For example, DcSEP1 and DcSEP4-2 were expressed from the S2, and DcSEP3-1 was expressed from the S3. DcSEP1 was highly expressed in petals and carpels which was similar to that of GRCD3 in Gerbera hybrida[37] and SlaSEP1 in Silene latifolia[38], while, DcSEP3-1 was highly expressed in sepals, petals, and carpels. In addition, through evolutionary analysis, the expression patterns of genes in the same subclade may be the same or different, which indicates that the evolution of class E genes in D. chinensis is complex. Especially, the expression pattern of the DcSEP3-2 gene was different from that of DcSEP3-1, and it was mainly detected in sepals and petals, although they were all SEP3 homologs. This was different from what has been reported in Arabidopsis; AtSEP3 was mainly expressed in the inner three whorls, and the expression level was highest in petals[9]. Besides, we found that the transgenic lines of same subclade genes also showed different phenotype, such as the transgenic lines overexpressing DcSEP3-1 and DcSEP3-2. The results suggested that the two DcSEP3 genes of D. chinensis were subfunctionalized. A similar phenomenon has been found in other species. For example, in the woody plant Platanus acerifolia, overexpression of PlacSEP3-1 in Arabidopsis showed slightly early flowering or slightly more cauline leaves, unlike PlacSEP3-2, which showed severe phenotypic changes[39]. In marigold, overexpression of TeSEP3-2 and TeSEP3-3 led to early flowering in Arabidopsis, which was different from that of TeSEP3-1[40]. These reports have shown that genes in the same subclade may undergo subfunctionalization. In addition, different subclades may have undergone multiple evolutionary events. For example, in G. hybrida, GhGRCD5 plays a unique role in regulating petal development, while, GhGRCD1 regulates stamen development[41]. In addition, in orchids, overexpression of the PeSEP3 gene leads to transgenic Arabidopsis plants showing severe phenotypes, such as early flowering and much smaller plant size, while overexpression of PeSEP1 shows no obvious change in phenotype[20].
In addition to the comparison of gene expression patterns and transgene experiments to predict whether the gene form the same subclade has been subfunctionalized, the pattern of protein interaction belonging to the same subclade is also good evidence. Previous studies have found that functionally redundant proteins have the same interaction pattern and may have shared interaction partners when performing their function[42,43]. For example, in Arabidopsis, there is functional redundancy between AtSEP1 and AtSEP3 proteins, and the interaction patterns of these two proteins are extremely similar[44]. In our study, the interaction patterns of different subclades showed a variety of results: DcSEP3-1 and DcSEP3-2 had different interaction patterns. The protein interaction patterns of DcSEP4-1 were different from that of DcSEP4-2 and DcSEP4-3, while DcSEP4-2 had a similar interaction pattern with DcSEP4-3. In our results, we found that DcSEP3-1 and DcSEP3-2 were different not only in their interaction patterns but also in their expression patterns and gene functions. Therefore, we speculate that the two genes belonging to the SEP3 subclade in Dianthus may experience sub-functionalization. However, the genes belonging to other subclades showed more complex patterns of interaction, such as the DcSEP4 subclade, and there is much work to be done to elucidate which evolutionary events these genes have undergone.
This work was supported by the National Natural Science Foundation of China (No.32072607).
-
Xiaopeng Fu is the Editorial Board member of journal Ornamental Plant Research. She was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and her research group.
-
# These authors contributed equally: Xiaoni Zhang, Quanshu Wu
- Supplemental Table S1 Primers for CDS amplification of class E genes.
- Supplemental Table S2 The CDS of class E genes.
- Supplemental Table S3 Protein sequences for phylogenetic tree.
- Supplemental Table S4 Primers for qRT-PCR.
- Supplemental Table S5 Primers for Yeast two‐hybrid of class E genes.
- Supplemental Table S6 Primers for BiFC of class E genes.
- Supplemental Table S7 Transcriptome sequencing data statistics.
- Supplemental Table S8 Distribution of gene expression in transcriptome samples at differentdevelopmental stages.
- Supplemental Table S9 Summery of class E genes in D. chinensis and D. caryophyllus.
- Supplemental Fig. S1 Alignment of D. chinensis (Dc) class E amino acid sequences with A. thaliana (At) and B. vulgaris (Bv).
- Supplemental Fig. S2 The phylogenetic tree of the class E genes.
- Supplemental Fig. S3 Yeast interaction analysis of E proteins of D. chinensis.
- Supplemental Fig. S4 Protein self-activation assay of class E genes.
- Supplemental Fig. S5 BIFC analysis of E proteins of D. chinensis.
- Supplemental Fig. S6 Rosettes number analysis of transgenic lines overexpressing DcSEP3-2.
- Supplemental Fig. S7 Phenotype analysis of transgenic lines of 35S:DcSEP3-1 and 35S:DcSEP3-2.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang X, Wu Q, Lin S, Li D, Bao M, et al. 2023. Identification and characterization of class E genes involved in floral organ development in Dianthus chinensis. Ornamental Plant Research 3:5 doi: 10.48130/OPR-2023-0005 

Identification and characterization of class E genes involved in floral organ development in Dianthus chinensis
- Received: 11 January 2023
- Accepted: 09 February 2023
- Published online: 15 March 2023
Abstract: The SEPALLATA (SEP) gene, as a 'glue' for the 'floral quartets model', plays an important role in floral organ development by forming tetramers with class A-, B-, and C- genes. The functional differentiation of class E genes has been reported in different species. Carnation (Dianthus spp.) is a world-famous economic flower that has been extensively used in landscaping, but the roles of SEP genes in carnation are unclear. Here, we found that the class E genes of D. chinensis cultivar 'L' showed different expression patterns during floral organ primordium development by transcriptome analysis. Combined with quantitative real-time PCR, its tissue and specific stage expression patterns were also different in different subclades. In addition, a yeast two-hybrid experiment was carried out to explore the interaction patterns of class E genes with other class A-, B-, and C- genes. Only DcSEP3s and DcSEP4s proteins interacted with all three classes of A-, B-, and C- proteins, and interestingly, is that DcSEP3-1 only interacted with the DcAP1 protein of class A, while the DcSEP3-2 protein only interacted with DcFUL1. Transgenic experiments showed that overexpression of DcSEP3-2 genes in Arabidopsis resulted in early flowering, smaller rosettes, dwarfism and abnormal floral organs. The transgenic line overexpressing of DcSEP3-1 only showed an early flowering phenotype. All these results indicated that the two DcSEP3s of class E genes in D. chinensis may undergo sub-functionalization. These findings advance our understanding of the molecular mechanisms of flower development in carnation.
-
Key words:
- SEP /
- Dianthus chinensis /
- Floral organ development /
- Protein interaction /
- Specific expression