









-
Coal is one of the most crucial energy sources worldwide. Chinese coal production increased from 3.41 billion tons in 2016 to 4.13 billion tons in 2021 (National Bureau of Statistics, 2022). In China, about 95% of the coal is from underground mining (Wang et al., 2020). This activity can lead to a decline in soil fertility, arable land reduction and environmental pollution in mining areas. Therefore, it is urgent to conduct ecological reconstruction of mining areas. Reclamation is an efficient solution to reconstruct the ecological environment in coal-mining subsidence area, and soil fertility quality restoration is critical to land reclamation. Soil organic carbon (SOC) is an important soil fertility quality indicator. Higher SOC usually indicates better soil quality (Bandyopadhyay & Maiti, 2019). Microbes in soil can assimilate CO2 and convert it into SOC (Antonelli et al., 2018). Carbon-fixing microbes play a crucial role in increasing SOC in barren soils where plant growth is limited (Su et al., 2013). Most of the carbon-fixing microbes are autotrophic. At present, six carbon-fixing pathways of autotrophic microbes have been elucidated (Fuchs, 2011), among which Calvin Benson-Bassham (CBB) cycle is the dominant pathway (Yu King Hing et al., 2019). Ribulose-1,5-bisphosphate carboxylase/oxygenase (RubisCO) is the key enzyme to control Calvin cycle rate, and has four (I, II, III, IV) forms (Tabita, 2004). Form I RubisCO was dominant in different soil environments and encoded by cbbL gene. The cbbL gene was highly conserved and widely present in the environment (Kusian & Bowien, 1997). Therefore, this gene is an effective biomarker to investigate autotrophic carbon-fixing microbial community in mining reclaimed soil.
Many studies carried out in coal-mining areas have investigated the effects of reclamation on the diversity and activity of bacteria, archaea and fungi (Hou et al., 2018; Li et al., 2018; Wang et al., 2020). However, few of them have focused on carbon-fixing microbes in coal-mining areas. Notably, Čížková et al. (2018) found that the carbon fixation potential and microbial biomass in reclaimed soil were significantly higher than in unreclaimed lignite mining soil. Moreover, reclamation time of coal-mining areas significantly affect the abundance of carbon-fixing Acidobacteria, Bacteroidetes, Cyanobacteria, Firmicutes and Proteobacteria, and these phyla positively correlated with SOC (Ma et al., 2022). In addition to SOC, other environmental factors, such as pH and total nitrogen (Liu et al., 2022), can significantly affect community structure of carbon-fixing microbes. Moreover, fertilization treatments also significantly altered cbbL-carrying bacterial community composition and diversity (Liu et al., 2022). However, the carbon-fixing microbial biomass, diversity and activity under different reclamation and fertilization treatments in reclaiming soil of coal-mining subsidence areas still remain unclear.
In this study, real-time quantitative PCR, ELISA and high-throughput sequencing were used to measure the effects on carbon-fixing microbial community under different reclamation and fertilization treatments were measured in an underground coal-mining subsidence area of Shanxi Province, northern China. We aim to: (1) determine the effects of reclamation and fertilization on the soil carbon-fixing microbial biomass, RubisCO activity and community structure in coal-mining subsidence areas, (2) reveal the main soil biophysicochemical factors that influenced soil carbon-fixing microbial community. We hypothesize that carbon-fixing microbial community composition will markedly change after reclamation and fertilization due to altered soil properties, and the fertilization treatments with manure is more conducive to improving the soil carbon-fixing potential in a coal-mining subsidence area. We hope our study would provide implications for the effective soil fertility improvement from the perspective of carbon-fixing microorganisms in coal-mining area, which may also be conducive to achievement of carbon neutrality in agricultural ecosystems.
-
The experiment was conducted in a reclamation field in a coal-mining subsidence area of Shanxi Yuci Guanyao Yong'an Coal Industry Co., Ltd. (37°50′19.97″ N, 112°48′21.58″ E) (Supplemental Fig. S1), Shanxi Province, northern China. This region has a temperate continental monsoon climate, with mean annual precipitation of 462 mm, 175 frost-free days and 9−10 °C mean annual temperature. A large area of this land subsided due to underground coal mining. To reuse the subsided land, after the discontinuation of gangue discharge in 2019, the gangue landfill area was covered with 1 m-thick soil and mechanically leveled. The covering soil is raw and classified as calcareous cinnamon soil (Calciustepts). Its physicochemical properties were as follows: soil organic matter (SOM) 3.20 g·kg−1, total nitrogen (TN) 0.21 g·kg−1, available phosphorus (AP) 1.48 mg·kg−1, available potassium (AK) 79.00 mg·kg−1, pH 8.34.
After the above engineering reclamation, the land was further biologically reclaimed since 2020 and five treatments were performed, including UL (uncultivated land), CK (maize cultivation without fertilization), NPK (maize cultivation with chemical fertilizer), M (maize cultivation with manure), MNPK (maize cultivation with co-fertilization of manure and chemical fertilizer). The manure was chicken manure compost containing 27.8% organic matter, 1.68% nitrogen, 1.54% P2O5 and 0.82% K2O. The chemical fertilizer contained urea (N, 46%), calcium superphosphate (P2O5, 12%) and potassium sulfate (K2O, 60%). The fertilizing quantity of each treatment is listed in Supplemental Table S1. Each treatment had three replicates, and each replicate plot was 10 m × 5 m = 50 m2 (n = 15). Maize (Zea mays Linn.) was sown in late April with a planting density of 60,000 ha−1 and harvested in late September in each plot.
A total of 15 topsoil samples (0−20 cm, five treatments × three replicates) were collected using the five-point mixed sampling method in each plot at maize harvest in September, 2020 (1-year reclamation). The samples were sieved through a 2 mm mesh sieve after removal of plant residues and detritus. Each sample was then subdivided and respectively stored at 4 °C (for enzyme analysis), −80 °C (for microbial molecular biological analysis), or room temperature (for soil chemical analyses).
Analyses of soil properties
-
All the chemical properties were measured using routine methods (Tedesco et al., 1995). Soil moisture (SM) was determined by oven drying at 105 °C until a constant weight. pH was measured using a 1:2.5 (w/v) soil-water slurry. TN and alkaline nitrogen (AN) were determined using semimicro-Kjeldahl method and alkali N-proliferation method, respectively. Total phosphorus (TP) was measured via the alkali fusion-Mo-Sb anti-spectrophotometric method. AP was extracted using sodium bicarbonate and measured via the colorimetric method. Total potassium (TK) and AK were respectively extracted using sodium hydroxide and ammonium acetate, and measured by flame photometry. SOC was determined using potassium dichromate volumetric method. Soil microbial biomass carbon (MBC) was determined using the chloroform-fumigation extraction method by Vance et al. (1987). Soil dissolved organic carbon (DOC) was extracted following the procedure by Zhu et al. (2015). Soil particulate organic carbon (POC) concentration was measured according to Cambardella & Elliott (1992). Soil easily oxidizable organic carbon (EOC) was measured using the potassium permanganate oxidation method (Blair et al., 1995).
Soil enzymatic activity analyses
-
The activity of soil catalase (CAT) was determined from titration of KMnO4 consumption (Lin, 2010). RubisCO enzyme activity of the soil samples was measured by immunoassay (ELISA) kit of RubisCO enzyme (Sangon Biotech, China) according to the manufacturer’s instructions.
DNA extraction, qPCR and sequencing of microbial cbbL gene
-
DNA of each sample was extracted from 0.25 g −80 °C stored soil using PowerSoil DNA Isolation Kit (QIAGEN, Germany). Then 1% agarose gel electrophoresis and a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA) were used to evaluate the quality and quantity of extracted DNA.
The abundance of carbon-fixing microbes and bacteria were quantified using absolute quantitative PCR (qPCR) with the cbbL gene primer (K2F: 5′-ACCAYCAAGCCSAAGCTSGG-3′, V2R: 5′-GCCTTCSAGCTTGCCSACCRC-3′) and bacterial 16S rRNA gene primer (338F: 5′-CCTACGGGAGGCAGCAG-3′, 518R: 5′-ATTACCGCGGCTGCTGG-3′), respectively (Rasche et al., 2011; Tolli & King, 2005). Consequently, the cbbL/16S rRNA gene ratio was calculated to reflect the proportion of carbon-fixing bacteria in soil bacterial community. The qPCR mixture (final volume, 20 μL) included 10 μL of 2× SYBR® Premix (Biomed, China), 0.8 μL of each primer, 1.4 μL of DNA template and 7 μL of ddH2O. The thermocycling conditions were: initial denaturation at 95 °C (2 min in cbbL, 10 min in 16S rRNA), followed by 40 cycles of denaturation at 95 °C (15 s in cbbL, 30 s in 16S rRNA) and annealing at 60 °C for 1 min. Then the melting curve was used to verify the amplification specificity. The standard curves for both genes were constructed using tenfold dilution series (ranging from 102 to 108) of the recombinant plasmids containing target gene fragments from the soil. The qPCR efficiency of the cbbL and 16S rRNA gene were 102% (R2 = 0.999) and 98% (R2 = 0.998).
Adequate amount of −80 °C stored soil samples were delivered to Shanghai Majorbio Bio-pharm Technology Co., Ltd (Shanghai, China) for sequencing of microbial cbbL gene with the primer set K2F/V2R on an Illumina MiSeq PE300 platform. The raw reads are available in SRA (Sequence Read Archive) database of NCBI with accession number PRJNA852533.
Data processing and statistical analyses
-
The raw cbbL gene sequencing reads were demultiplexed, quality-filtered by fastp version 0.20.0 and merged based on overlaps by FLASH version 1.2.7. Sequencing reads were assigned to each sample according to the unique individual barcodes. For further improvement of sequencing data quality, the original sequences were controlled and filtered by QIIME (Quantitative Insights into Microbial Ecology) software package. UPARSE standard pipeline (v7.0.1090,
http://drive5.com/uparse/ ) was utilized to cluster high-quality sequences into operational taxonomic unit (OTU) with a 97% similarity, and chimera was identified and removed (Edgar, 2013). The representative sequences of each OTU were compared with related sequences retrieved from NCBI (National Center for Biotechnology Information) database to assign a taxonomic classification using BLAST (Huang et al., 2021a).Alpha diversity analysis, including Sobs index (S), Shannon-Wiener diversity (H) (Wei et al., 2011), and Pielou index (J) (Yuan et al., 2016), were calculated using Microsoft Excel 2019 software. Sobs index was the observed OTU number (Qin et al., 2019). Linear discriminant analysis (LDA) effect size (LEfSe,
http://huttenhower.sph.harvard.edu/galaxy ) (Segata et al., 2011) was performed to screen enriched bacterial taxa in soils under different treatments.Principal component analysis (PCA) and biclustering heatmap analysis were used to identify the differences of the carbon-fixing microbial community among different treatments using ade4 package and ComplexHeatmap package of R (v.4.0.3), respectively.
The Vegan package (v2.5.2) in the R (v4.0.3) was used to conduct calculation of Variance inflation factors (VIF), Redundancy analysis (RDA) between the carbon-fixing microbial community structure and environmental variables, and variance partitioning analysis (VPA). VIF were used as the criterion for distinguish collinearity among explanatory variables, environmental variables with VIF > 5 were eliminated before RDA and VPA (Zhang et al., 2022). According to the RDA results, structural equation modeling (SEM) was constructed using IBM SPSS AMOS 24.0. Based on the influence and relationship among known factors, the model was fitted with the maximum likelihood estimation method. The fitness of the model was evaluated via low χ2/df (χ2/df < 3, the closer χ2/ df is to 1, the better is the model fit, P > 0.05), high goodness-of-fit index (GFI > 0.89), low root mean square error of approximation (RMSEA < 0.01, if RMSEA = 0, it means complete fitness of the model) and low akaike information criterion (AIC) (Shipley, 2000).
Microsoft Excel 2016 software was utilized for initial data analysis, SPSS 26.0 software was utilized for one-way ANOVA and multiple comparisons (Duncan post hoc test, P < 0.05).
-
Soil properties among different treatments are listed in Table 1. For basic physicochemical and enzymatic properties, compared with UL, TN was significantly increased in NPK, M and MNPK (P < 0.05), AP was significantly increased in NPK and MNPK (P < 0.05), TK was significantly decreased in the other four treatments (P < 0.05). The highest pH was in UL. Compared with CK, NPK, M and MNPK increased contents of TN, AP, AK and CAT, but decreased contents of SM, AN and TP. Compared with CK, TN and CAT were significantly higher in M and MNPK (P < 0.05).
Table 1. Soil biophysicochemical properties in different treatments
Index Treatments UL CK NPK M MNPK SM (g·kg−1) 0.17 ± 0.02ab 0.19 ± 0.02a 0.15 ± 0.01b 0.18 ± 0.01ab 0.15 ± 0.01b pH 8.27 ± 0.09a 7.86 ± 0.49a 7.85 ± 0.42a 8.15 ± 0.06a 8.14 ± 0.13a TN (g·kg−1) 0.27 ± 0.04c 0.32 ± 0.06bc 0.40 ± 0.07b 0.56 ± 0.01a 0.59 ± 0.06a AN (mg·kg−1) 304.05 ± 0.00ab 344.59 ± 11.50a 302.85 ± 30.84ab 315.97 ± 145.67ab 174.08 ± 99.02b TP (g·kg−1) 0.38 ± 0.00a 0.69 ± 0.46a 0.39 ± 0.20a 0.35 ± 0.28a 0.39 ± 0.20a AP (mg·kg−1) 5.09 ± 0.00b 10.93 ± 0.26ab 17.68 ± 5.80a 12.14 ± 0.80ab 18.65 ± 7.16a TK (g·kg−1) 5.61 ± 0.00a 3.72 ± 0.74b 2.88 ± 1.40b 3.20 ± 0.03b 4.01 ± 0.70b AK (mg·kg−1) 80.06 ± 0.00a 74.71 ± 8.34a 77.39 ± 8.34a 86.73 ± 10.08a 86.74 ± 15.17a CAT (ml·g−1) 2.15 ± 0.06a 1.52 ± 0.22b 1.65 ± 0.03b 2.22 ± 0.05a 1.97 ± 0.26a SOC (g·kg−1) 1.74 ± 0.00c 1.55 ± 0.08c 2.07 ± 0.17b 3.35 ± 0.24a 3.12 ± 0.04a MBC (mg·kg−1) 9.85 ± 0.45d 17.06 ± 1.50c 19.72 ± 0.58b 37.78 ± 2.29a 37.45 ± 0.82a DOC (g·kg−1) 0.09 ± 0.01a 0.09 ± 0.02a 0.08 ± 0.01a 0.08 ± 0.01a 0.08 ± 0.01a POC (g·kg−1) 0.24 ± 0.23a 0.28 ± 0.15a 0.52 ± 0.12a 0.60 ± 0.18a 0.71 ± 0.57a EOC (g·kg−1) 1.58 ± 0.22a 1.48 ± 0.58a 1.34 ± 0.35a 1.93 ± 0.46a 1.65 ± 0.69a Values indicate mean ± standard deviations (n = 3). Different letters in each row represent a significant difference among treatments (one-way ANOVA, P < 0.05). For soil carbon relevant properties, NPK, M and MNPK increased POC, and significantly increased SOC, MBC compared with UL and CK (P < 0.05). MBC was significantly higher in four other treatments than UL (P < 0.05).
RubisCO activity and cbbL gene abundance in different treatments
-
Soil RubisCO activity are depicted in Fig.1a and significant differences were observed among treatments. Compared with other treatments, M treatment significantly increased RubisCO activity (P < 0.05). RubisCO activity was the lowest in NPK and had no significant difference among UL, CK and MNPK (P > 0.05).

Figure 1.
(a) Soil RubisCO activity and (b) cbbL gene abundances in different treatments. Values are means (n = 3), and error bars represent standard deviation. Different lowercase letters above columns indicate significant differences (one-way ANOVA, P < 0.05) among treatments.
Soil cbbL gene abundance was depicted in Fig. 1b and was significantly the highest in M (2.07 × 107 copies/g dry soil, P < 0.05), which was consistent with RubisCO activity. Moreover, the ratio of cbbL/16S rRNA in M was also significantly the highest (3.37%) (Supplemental Fig. S2). The lowest cbbL gene abundance was in CK (7.92 × 105 copies/g dry soil). Compared with CK, cbbL gene abundance was increased 4.18, 25.18 and 8.74 times in NPK, M and MNPK, respectively. But there was no statistical difference among UL, CK, NPK and MNPK treatments.
Alpha diversity and composition of carbon-fixing microbial community in different treatments
-
A total of 6,140,983 raw sequences were obtained. Each sample contained 34931 high quality sequences after quality filtering and subsampling (normalizing the sequence number according to the minimum sample). All the sequences were further classified into 17 phyla, 43 classes, 84 orders, 160 families, 361 genera and 3719 OTUs. The rarefaction curve (Supplemental Fig. S3) showed that the current sampling depth included most carbon-fixing microbial taxa in the samples and was sufficient for further analyses.
Based on sequencing data, the α diversity indices (Sobs Richness, Shannon-Weaver Diversity, Pielou Evenness indices) of the carbon-fixing microbes in different treatments are depicted in Supplemental Fig. S4. The highest values of Sobs, Shannon-Weaver and Pielou indices were in MNPK, and Sobs index was significantly higher than the other four treatments (P < 0.05). The Shannon-Weaver and Pielou indices were higher in NPK, M and MNPK than CK. They suggested that fertilization increased soil microbial diversity and evenness, especially in the MNPK, and MNPK also significantly improved soil microbial richness (P < 0.05).
Furthermore, carbon-fixing microbial community composition at phylum level and class level are shown in Fig. 2a & b, respectively. The dominant phyla were Proteobacteria (73.26%−86.23%), Cyanobacteria (5.58%−17.69%) and Actinobacteria (2.16%−5.40%), comprising 91.15%−96.00% of the sequences. The dominant classes were Alphaproteobacteria (43.07%−70.18%), Betaproteobacteria (11.68%−21.60%), Gammaproteobacteria (1.77%−8.55%), Cyanophyceae (4.81%−15.14%), Cyanobacteria (0.64%−1.96%) and Actinobacteria (1.93%−5.04%), comprising 90.36%−95.78% of the sequences. It is worth noting that carbon-fixing fungi and archaea were also detected, which were not reflected in the figure due to relatively low abundance. In addition, the top 10 dominant genera were Bradyrhizobium, Rhodopseudomonas, Noviherbaspirillum, Cyanobium, Variovorax, Devosia, Marichromatium, Mesorhizobium, Nitrobacter and Thermoleptolyngbya, comprising 66.84%−88.26% of the sequences.
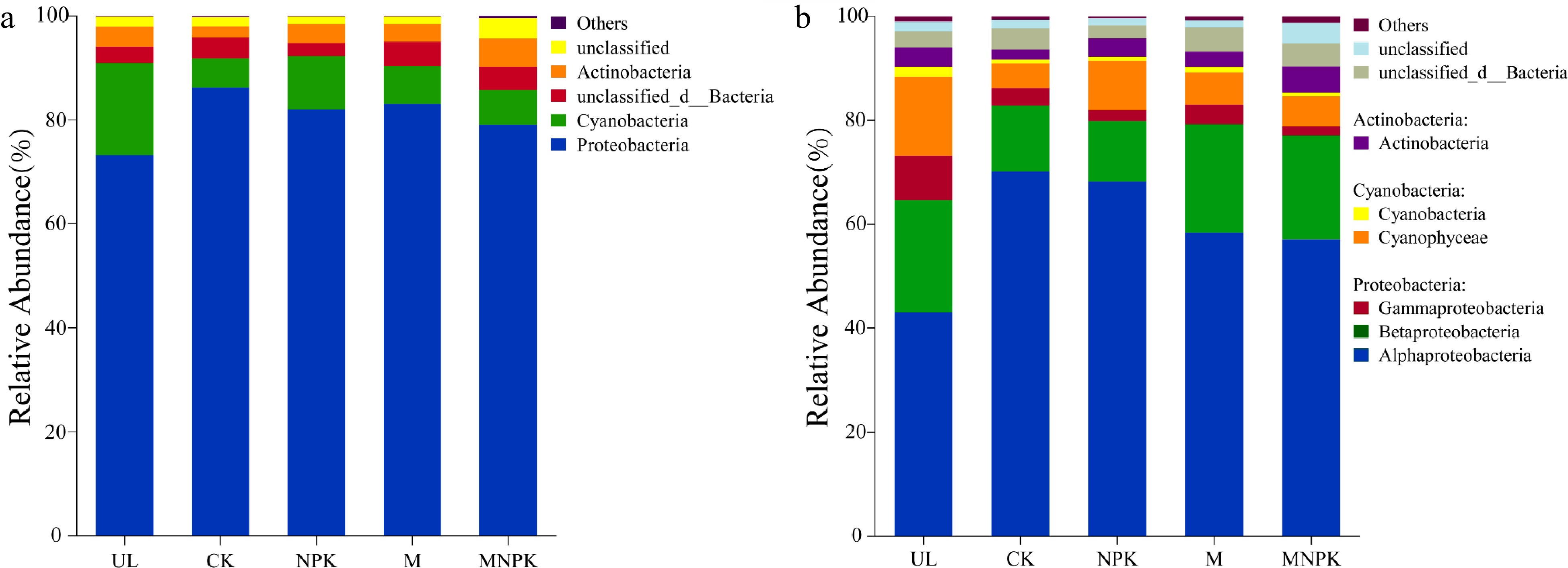
Figure 2.
Taxonomic composition of carbon-fixing microbial communities in soils at (a) phylum level and (b) class level.
Supplemental Table S1 showed that the relative abundances of the phyla Proteobacteria, Cyanobacteria and Actinobacteria were significantly different among treatments. Compared with UL, the other four treatments increased Proteobacteria, while significantly decreased Cyanobacteria (P < 0.05). NPK, M and MNPK increased Cyanobacteria and Actinobacteria, but decreased Proteobacteria compared with CK.
Supplemental Table S2 showed that the relative abundances of the classes Alphaproteobacteria, Gammaproteobacteria, Cyanophyceae, Cyanobacteria and Actinobacteria were significantly different among treatments. Compared with UL, the other four treatments increased Alphaproteobacteria, but significantly decreased Gammaproteobacteria, Cyanophyceae and Cyanobacteria (P < 0.05). NPK, M and MNPK increased Cyanobacteria and Actinobacteria, but decreased Alphaproteobacteria compared with CK.
LefSe analysis was applied to ascertain the biomarkers, which were significantly enriched carbon-fixing microbial taxa in certain treatments (Fig. 3a). There were 17 biomarkers with LDA score > 3.5, 16 of which belonged to Proteobacteria phylum. Therefore, reclamation mainly influenced the assembly of proteobacteria, which was consistent with the results of carbon-fixing microbial community composition (Fig. 2a). Compared with UL, class Alphaproteobacteria was significantly enriched in four other treatments; family Bradyrhizobiaceae was significantly enriched in NPK and MNPK; family Comamonadaceae was significantly enriched in M and MNPK; family Phyllobacteriaceae, genus Mesorhizobium and genus Nitrobacter were significantly enriched in MNPK; family Devosiaceae and genus Devosia were significantly enriched in CK. Compared with four other treatments, five biomarkers were significantly enriched in UL, namely class Gammaproteobacteria, order Chromatiales, family Chromatiaceae and genus Marichromatium, i.e. these taxa were significantly decreased in all bioreclamation treatments (CK, NPK, M and MNPK).
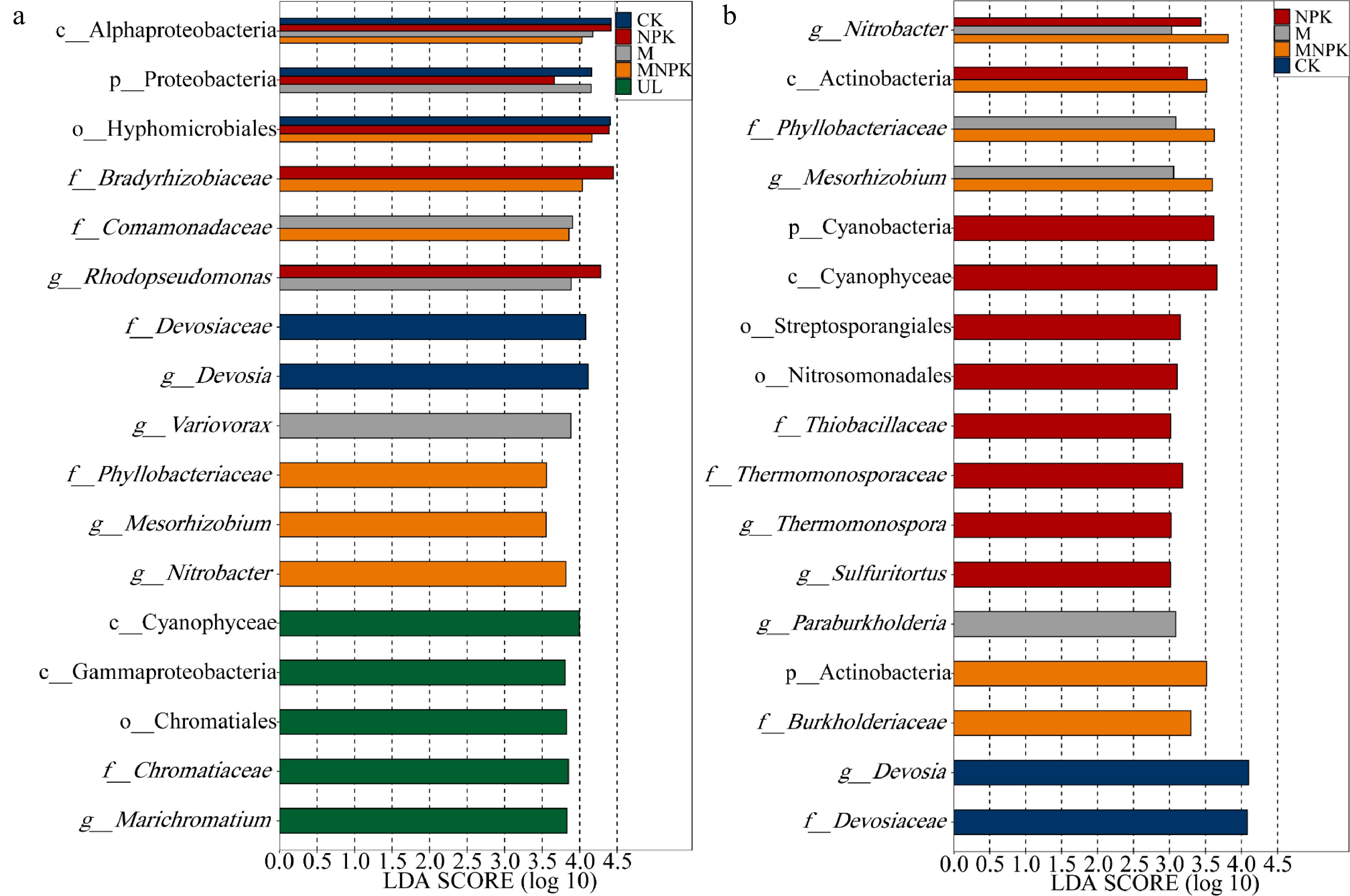
Figure 3.
Linear discriminant analysis (LDA) effect size analysis determined biomarkers (a) between UL and other treatments (CK, NPK, M and MNPK) and (b) between CK and fertilization treatments (NPK, M and MNPK). The LDA score indicates the effect size and ranking of each differentially abundant taxon (P < 0.05, LDA score > 3.5, a) (P < 0.05, LDA score > 3.0, b). The ordinate is the taxon with significant difference among groups, and the abscissa is a bar chart to visually show the LDA log score of each taxon. Blue, red, gray and orange bars represent the biomarkers in CK, NPK, M and MNPK having significantly greater abundances than in UL, respectively (a). Green bars represent the biomarkers in UL having significantly greater abundances than in all the other four treatments (a). Gray orange and red bars represent the biomarkers in M MNPK and NPK having significantly greater abundances than in CK, respectively (b). Blue bars represent the biomarkers in CK having significantly greater abundances than in all the other three treatments (b).
There were 17 biomarkers with LDA score > 3.0 (Fig. 3b), 10, five and two of them respectively belonged to Proteobacteria, Actinobacteria and Cyanobacteria phylum. Therefore, fertilization mainly influenced the assembly of these three phyla, which was consistent with the results of taxa composition mentioned in Fig. 2a. Compared with CK, genus Nitrobacter was significantly enriched in NPK M and MNPK; family Phyllobacteriaceae and genus Mesorhizobium were significantly enriched in M and MNPK; class Actinobacteria was significantly enriched in NPK and MNPK; Phylum Cyanobacteria, class Cyanophyceae, family Thiobacillaceae, genus Sulfuritortus, family Thermomonosporaceae and genus Thermomonospora were significantly enriched in NPK. Compared with NPK, M and MNPK, family Devosiaceae and genus Devosia were significantly enriched in CK, which means these taxa were significantly decreased in all fertilization treatments (NPK, M and MNPK).
Carbon-fixing microbial community structure in different treatments
-
Afterwards, PCA and biclustering heatmap analysis were applied to explore β-diversity of carbon-fixing microbes. PCA biplot showed that 35.93% of total variance in carbon-fixing microbial community structure was explained by the first two axes (Fig. 4). It also showed that five treatments were clearly differentiated into three clusters, UL and CK as cluster 1, M and MNPK as cluster 2, and NPK as cluster 3 along the PC2 axis, which indicated that the carbon-fixing microbial community structure among the three clusters was different. The score in PC2 axis significantly differed among each cluster (P < 0.05).
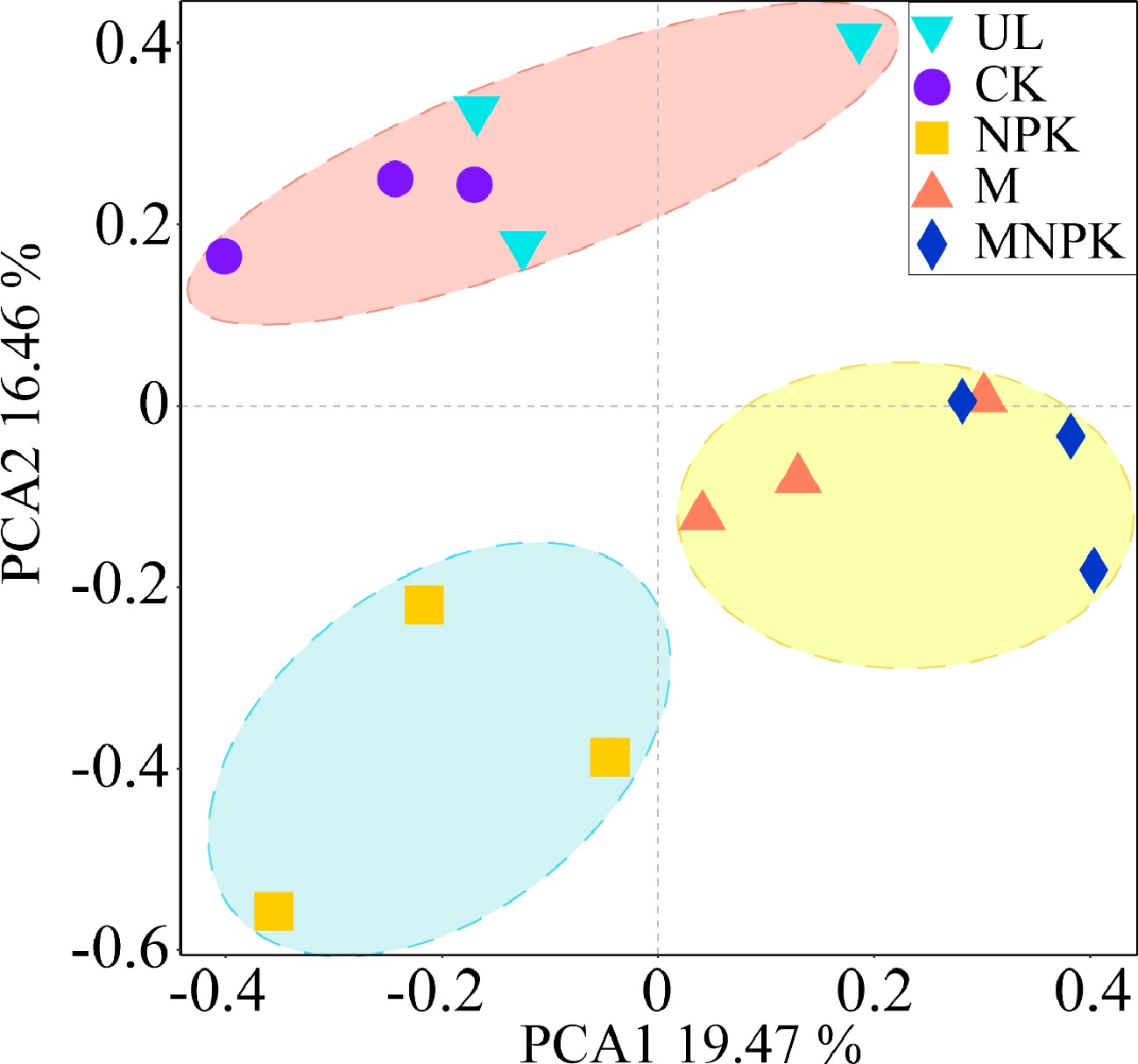
Figure 4.
Principal component analysis (PCA) of the carbon-fixing microbial community in different treatments.
Biclustering heatmap were also applied to explore the community structure of carbon-fixing microbes under different treatments (Fig. 5). Vertical clustering of heatmap showed that five treatments were classified into two clusters: M and MNPK were clustered together, while UL, CK and NPK formed a second cluster, and the second cluster was divided into two subclusters, UL and CK as a subcluster, NPK as another subcluster, which was consistent with PCA. Horizontal clustering of heatmap showed that the carbon-fixing microbial community structure differed among treatments. Compared with UL and CK, Actinomadura was increased in NPK, M and MNPK. Compared with the other three treatments Variovorax, Hydrogenophaga, Novosphingobium and Polumorphum harbored a higher relative abundance in M and MNPK. Pseudonocardia relative abundance was higher in NPK and MNPK than in three other treatments. Compared with four other treatments, Mesorhizobium, Nitrobacter, Cupriavidus, Rhodoferax, Rhodoblastus, Mycobacterium, Sphingomonas and Sinorhizobium were more abundant in MNPK. The relative abundance of Noviherbaspirillum, Marichromatium and Thermoleptolyngbya were higher in UL than in four other treatments.
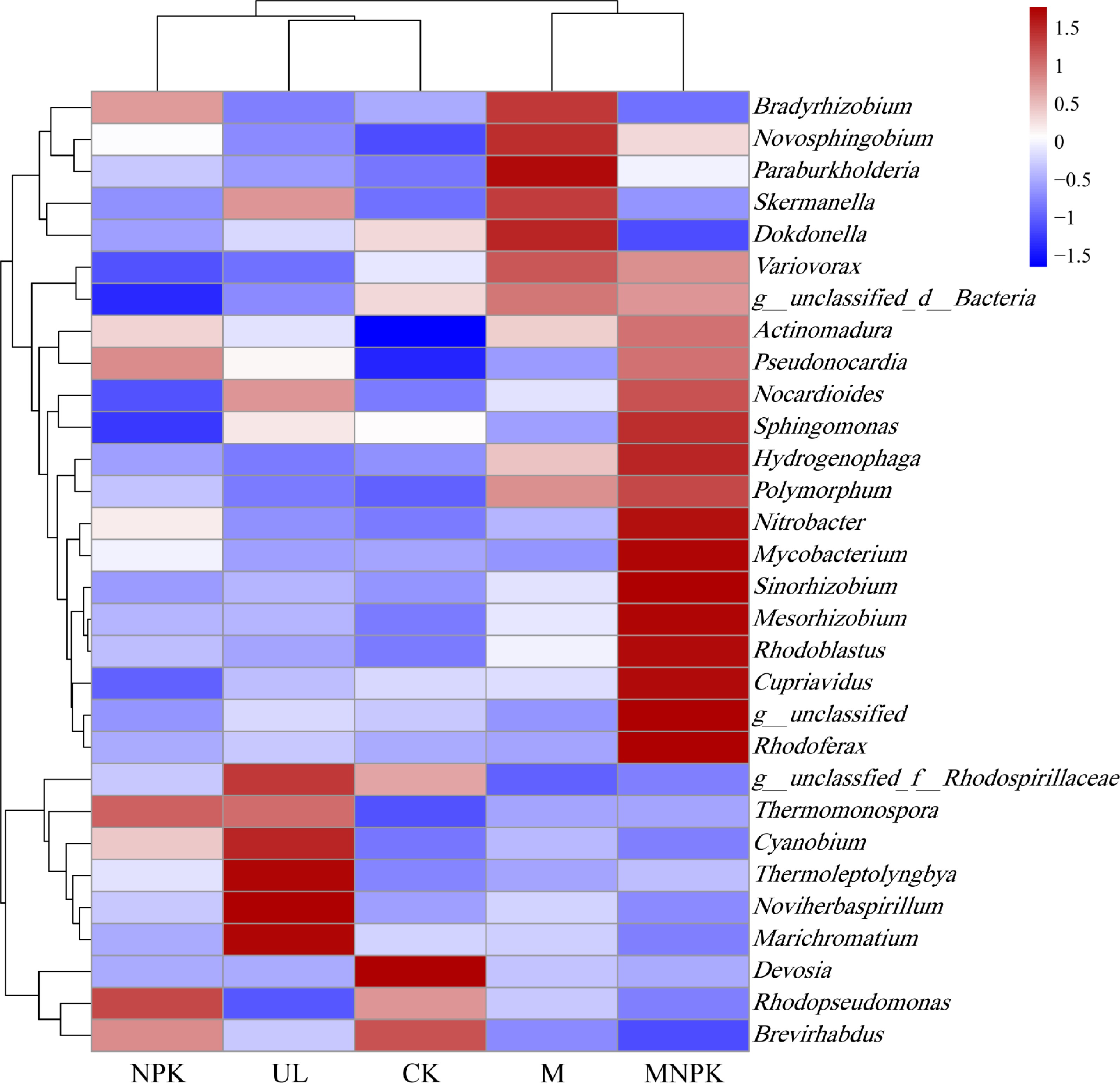
Figure 5.
Biclustering heatmap of the carbon-fixing microbial distributions of the top 30 abundant genera is present in different treatments. The color intensity of the color lumps represents the abundance of the carbon-fixing microbial genera in different treatments, with red representing higher abundance and blue representing lower abundance.
Correlations between the carbon-fixing microbial communities and soil properties
-
RDA were utilized to investigate the correlation of soil properties with soil carbon-fixing microbial community structure. Soil properties with VIF > 5 were removed before RDA to avoid the effect of collinearity. Then, eight variables, including CAT, EOC, AP, DOC, AK, SM, TP and AN were screened out for RDA.
The RDA (Fig. 6a) showed that the first two axes explained 41.93% and 11.45% of the total variance in soil carbon-fixing microbial community, respectively. The score in RDA2 axis significantly differed among treatments (P < 0.05). M and MNPK were clustered together and obviously separated from UL, CK and NPK, which was consistent with PCA and biclustering heatmap results. The soil properties with the highest explanatory proportion of soil carbon-fixing microbial community structure were SM (r = 0.6288, P = 0.001), CAT (r = 0.5508, P = 0.006), TP (r = 3972, P = 0.048), DOC (r = 0.3875, P = 0.041).
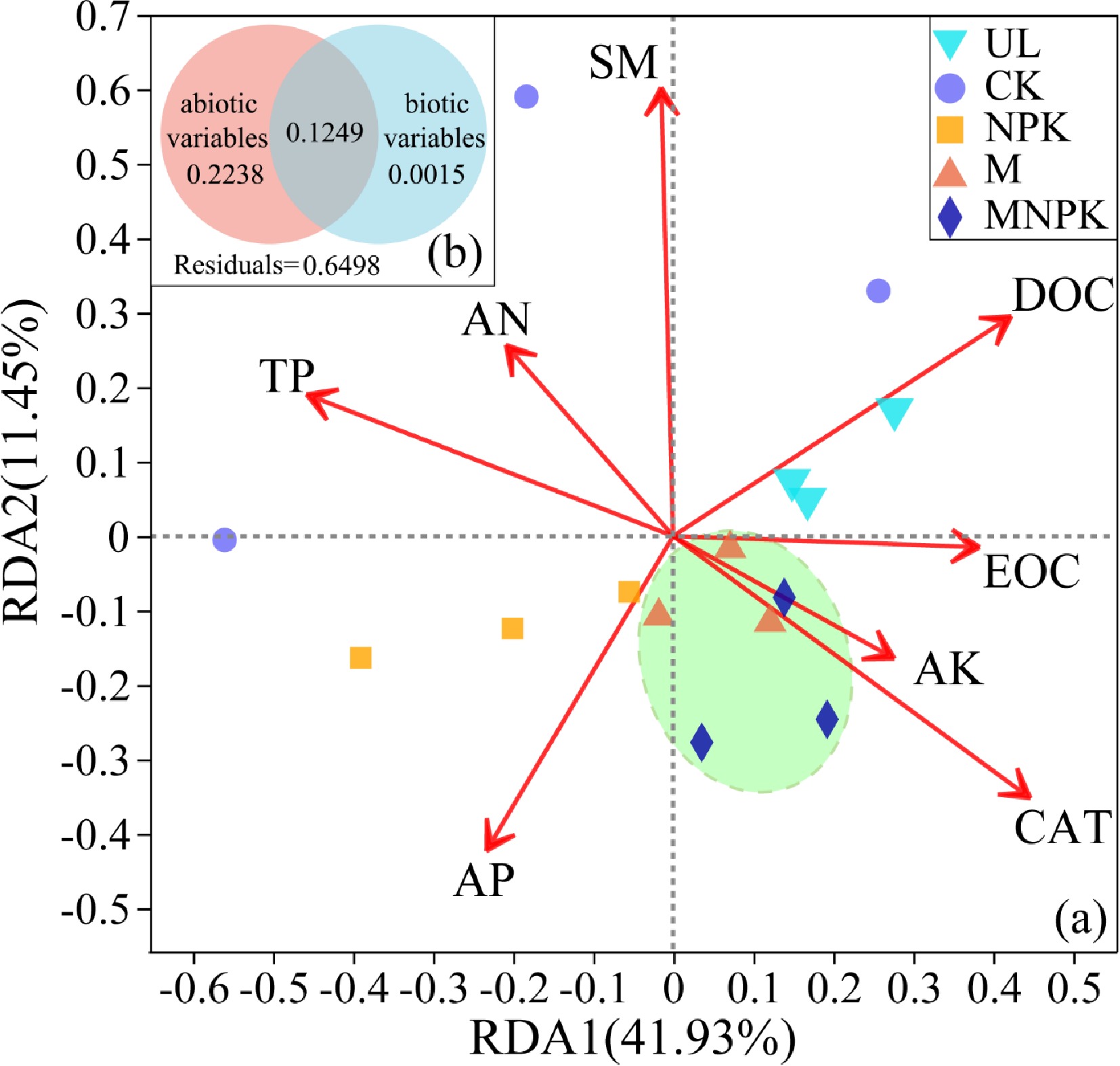
Figure 6.
(a) Redundancy analysis (RDA) linking carbon-fixing microbial communities with environmental variables in different treatments. Arrows represent the correlation between the soil properties and carbon-fixing microbial communities. Variables that are angled at more than 90° of each other have the least correlation. The length of the arrow represents the correlation. Variables that have arrows extended in opposite directions correlate negatively to each other. (b) Diagrams explaining variance partitioning (VPA) show the relative contribution of ecological drivers with VIF < 5 to soil carbon-fixing microbial community structure. The abiotic variables include EOC, AP, DOC, AK, SM, TP and AN; the biotic variables include CAT. The numbers are the percentages of the total variables explained by the factors.
To further unveil the relationship between carbon-fixing microbial community and environmental variables, the explanatory variables included in RDA were divided into two groups (abiotic variables and biotic variables) for VPA. The VPA (Fig. 6b) showed that abiotic variables were the more important factors affecting carbon-fixing microbial community assembly. The abiotic variables and biotic variables explained 22.38% and 0.15% of the variation in the carbon-fixing microbial community, respectively. Moreover, 64.98% of variation remained unexplained.
Based on RDA and PCA, the dominant soil properties (CAT, DOC, TP and EOC) and fertilization treatments with different manure application doses that affected the structure of carbon-fixing microbial community, were further included in SEM analysis. SEM was applied to reveal the causal relationship among soil properties, treatments (manure application dose), RubisCO activity, community composition (loading score on the first PCA axis) (Li et al., 2015), cbbL gene abundance, α diversity (Shannon-Weaver diversity index) (Zhang et al., 2021) and soil carbon accumulation (Fig. 7). It also used to further explain and quantify the contribution of key factors influencing both carbon-fixing microbial community and soil carbon fixation.
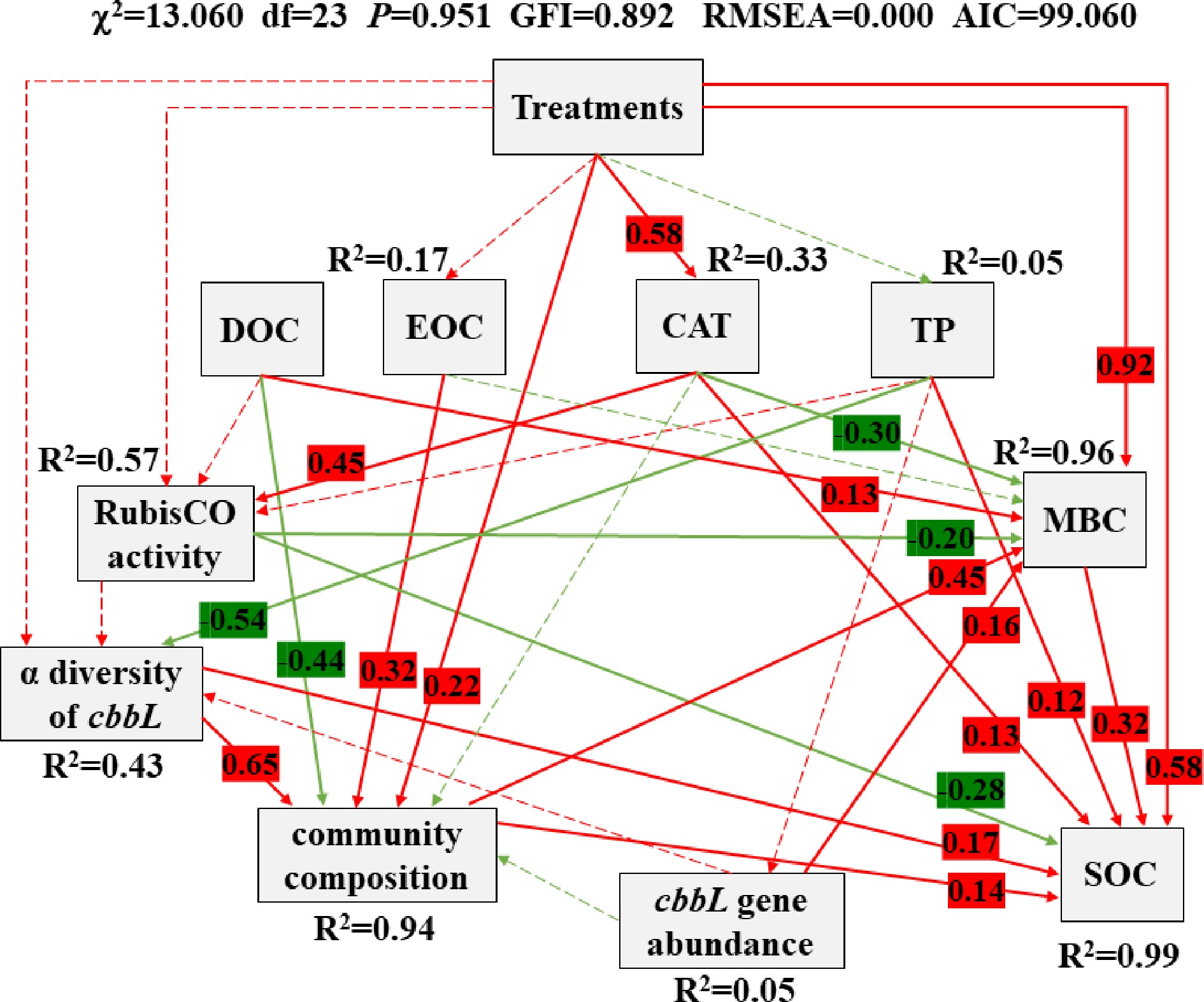
Figure 7.
Structural equation model (SEM) shows the causal influences of treatments, DOC, EOC, CAT, TP, RubisCO activity, α diversity of cbbL, community composition (carbon-fixing microbial community), cbbL gene abundance, SOC and MBC. Positive and negative effects are respectively showed in red and green, and significant and non-significant effects are showed with solid and dashed arrow lines, respectively. The standardized coefficients are marked above each path (only marked significant effect paths) and indicate the expected impact of a unit standard-deviation change at one node on units of standard-deviation change in connected nodes. R2 values represent the proportion of the variance explained for each endogenous variable.
This model is completely consistent with our causal hypothesis (χ2 = 13.060, df = 23, P = 0.951, GFI = 0.892, RMSEA = 0.000, AIC = 99.060) and it could explain 99%, 96%, 94%, 57% and 43% variance of the SOC, MBC, community composition, RubisCO activity and α diversity of cbbL, respectively. SOC was directly affected by treatments, MBC, TP, CAT, RubisCO activity, α diversity of cbbL and community composition and indirectly affected by DOC (standardized indirect effects = −0.15) and EOC (standardized indirect effects = 0.06). MBC was directly affected by treatments, CAT, DOC, RubisCO activity, community composition and cbbL gene abundance. Community composition was directly affected by treatments, EOC, DOC and α diversity of cbbL.
-
This study was defined to assess the restoration and responses of carbon-fixing microbial communities in coal-mining areas after different treatments. Among the different treatments, the comparison between UL and CK reveals the variations of carbon-fixing microbial communities before and after reclamation without fertilization. This study revealed significant increase of MBC after reclamation. This could be attributed to the increase of exogenous carbon and other nutrients needed for microbial growth, which promotes the proliferation of microbes, and leads to the increase of MBC after reclamation. Moreover, microbial residues, crop litter, roots and exudates were decomposed and transformed into humus, which promoted soil aggregates formation and changed soil structure, increased microbial proliferation and MBC.
Additionally, Proteobacteria, Cyanobacteria and Actinobacteria were dominant phyla of carbon-fixing microbial communities under different treatments, which suggests that these were the major contributors to soil microbial carbon fixation, which was similar to the results of Badger & Bek (2008). LefSe analysis (Fig. 3) further suggested the importance of Proteobacteria and Cyanobacteria phyla in the soil reclamation process, Cao et al. (2020) and Ma et al. (2022) have shown similar results in their studies on Chinese coal-mining soils. In addition, compared to unreclaimed soil (UL), our study revealed that reclamation (CK) significantly enriched Proteobacteria and Devosia (P < 0.05), while significantly reduced Cyanobacteria and Marichromatium (P < 0.05). This may be because reclamation enriched soil nutrient such as TN and AP (Table 1), and Proteobacteria is classified as copiotrophs, which prefer nutrient-rich conditions (Lienhard et al., 2013). Devosia, a potential plant-associated nitrogen-fixing bacterium, enriched within the root endosphere (Sun et al., 2021), thereby there may be a link between the enrichment of Devosia and the increase of TN and AN with the growth of plants after reclamation. Cyanobacteria evolved and thrived in low-nutrient systems and oligotrophic systems, respectively (Reinl et al., 2021), the significant increase of other microbes reduced the proportion of Cyanobacteria after reclamation. Marichromatium, a photosynthetic bacterium, could utilize variety of carbon, nitrogen and sulfur sources (Parag et al., 2013), thereby Marichromatium has higher relative abundance in uncultivated land because of its adaptability to barren environments. Moreover, Zhang et al. (2017) reported that organic carbon source type would also affect Marichromatium abundance, so we speculated that reclamation may also alter the organic carbon composition, which provides a new direction for further research.
The cbbL gene abundance, RubisCO activity and α diversity did not significantly differ before (UL) and after reclamation (CK). Moreover, PCA and biclustering heatmap showed that carbon-fixing microbial community structure was similar in CK and UL, which was inconsistent with other studies (Li et al., 2014). Cao et al. (2020) reported that soil microbial diversity and construction were strongly influenced by reclamation time. Soil improvement effect was positively correlated with soil reclamation time. Therefore, the above results of this study maybe because reclamation time in our study is too short to show obvious improvement effect of reclamation. Above all, reclamation even without fertilization in the initial stage could contribute to significant shifts in dominant taxa and MBC, and a trend of ecological recovery.
Effects of fertilization on carbon-fixing microbial communities
-
Previous studies have shown that one-year of reclamation with fertilization does improve soil fertility (Cheng, 2022; Gao et al., 2021). Different fertilization treatments greatly influence various fractions of carbon as well as carbon fixation (Wang et al., 2020). In our study, the comparison of M, NPK and MNPK with CK could reveal the effect of fertilization on carbon-fixing microbial communities. Our study showed that the reclamation with addition of fertilizers (NPK, M and MNPK) increased POC, SOC and MBC (P < 0.05) (Table 1), which was consistent with Anandakumar’s study (Anandakumar et al., 2022) in semi-arid areas of India. Carbon-fixing microbial community composition was significantly affected by fertilization (Figs 5 & 6). This may be because fertilization increased the nutrient elements required by carbon-fixing microbes, and thereby altered carbon-fixing microbial community structure. Moreover, Devosia was significantly decreased, while Nitrobacter was significantly enriched after fertilization (P < 0.05) (Fig. 3b). Xu et al. (2017) reported that Devosia was strongly positively correlated with AN. Therefore, it might because AN was the highest in CK (Table 1), Devosia was more suitable for growth and reproduction under CK treatment. Nitrobacter fixed carbon through Calvin cycle and may play a crucial role in coupling soil carbon (C) and nitrogen (N) cycles (Wang et al., 2019). These bacterial taxa are related to soil N cycle, which imply that influence of N cycle cannot be neglected in the study of soil C cycle, and it is essential to further simultaneously investigate microbes involved in soil C and N cycle.
In addition, it should be noted that there were significant separations of the carbon-fixing microbial community structure between the chemical fertilizer treatment (NPK) and manure and manure with chemical fertilizer treatments (M and MNPK) based on Figs 5 & 6. Moreover, MBC, SOC and TN were significantly higher in manure treatments (M and MNPK), compared with chemical fertilizer, which was consistent with Huang et al. (2021b). The application of manure (M) significantly increased carbon-fixing microbial biomass, the cbbL/16S rRNA ratio and RubisCO diversity. It may be because that mining area soil is relatively barren, and manure can increase organic matter more directly and effectively (Dennis et al., 2010), higher organic carbon content promoted facultative autotrophic bacterial growth and resulted in a high carbon-fixing microbial biomass (Yuan et al., 2012). Compared with CK, the application of manure with chemical fertilizer (MNPK) significantly increased richness of carbon-fixing microbial community, which was similar with Ding et al. (2016). This is because the combination of manure and chemical fertilizer not only supplemented the input of organic carbon, improved the availability of nutrients and water retention capacity, but also improved soil physical properties, which greatly stimulated carbon-fixing microbial community and activity (Guo et al., 2010), increased carbon-fixing microbial richness. However, RubisCO activity was significantly reduced by chemical fertilizer (NPK) compared with non-fertilized treatments (CK), but the carbon-fixing microbial biomass and α diversity was not significantly affected, which was consistent with the report by Jing et al. in China (Jing et al., 2021). This may be due to the application of chemical fertilizer leads to the great increase in soil phosphorus (P) (Table 1), which limit or co-limit the growth of soil autotrophic microorganisms (Yuan et al., 2015). In summary, compared with CK, the application of manure (M) improves the carbon-fixing microbial activity and abundance significantly, thereby promotes soil carbon fixation and ecological restoration in the mining area, while the application of manure with chemical fertilizer (MNPK) is more conducive to the improvement of carbon-fixing microbial diversity.
Effects of soil properties and manure application dose on carbon-fixing microbial communities
-
Environmental factors affecting soil carbon storage remain largely unknown, particularly in mining reclamation ecosystems, where biophysicochemical properties were key factors indirectly affecting variation in soil carbon storage by affecting the biomass, diversity and community structure of carbon-fixing microbes (Li et al., 2018). This study indicated that SM CAT, TP and DOC were the key factors significantly influencing soil carbon-fixing microbial community structure based on RDA. The effect of SM and CAT on carbon-fixing microbial community structure were significant and greater than that of TP and DOC.
This study showed that even the differences of DOC and EOC did not reach statistically significant levels after reclamation and fertilization, they still exert marked effect on the variations of soil carbon-fixing microbial community. Carbon-fixing microbes are sensitive to the DOC and EOC content (Li et al., 2020), thereby DOC and EOC directly influence microbial composition. Our study revealed that the changes in DOC and EOC caused by reclamation and fertilization can affect carbon-fixing potential through affecting carbon-fixing microbial community composition. Relative to carbon-fixing microbial abundance and diversity, DOC and EOC made a greater contribution to the alteration of microbial carbon-fixing potential, which was consistent with previous research in Chinese Loess Plateau (Xiao et al., 2018).
RubisCO activity was positively correlated with CAT, which was because CAT accelerates the decomposition of H2O2 and other harmful substances, thereby promoting the conversion of substances and energy in the soil and providing favorable environment for the survival of carbon-fixing microbes (Ma et al., 2022). A great deal of research revealed that SOC was positively correlated with RubisCO activity (Tang et al., 2015; Techtmann et al., 2012; Yuan et al., 2011), which was opposite with our study result. This may be because at the beginning of the reclamation, new equilibrium relationship between SOC fixation and mineralization has not been established, and SOC has not reached the stage of gradual accumulation.
SEM results indicated that in addition to soil properties, fertilization treatments with different manure application doses also affected carbon-fixing microbial community composition. This may due to application of manure could loosen soil, improve soil aeration, enhance carbon-fixing microbial activity, and change carbon-fixing microbial community composition (Shao et al., 2019). SOC and MBC were positively correlated with fertilization treatments with different manure application doses, which was because manure was rich in organic matter, SOC increased with the increase of manure application dose. Moreover, the application of manure promoted the decomposition of original SOC by priming effect, increased the carbon and other nutrients needed for microbial growth, thereby promotes microbial proliferation and increases MBC (Martín-Lammerding et al., 2015). Our study showed that microbial evolution trends in the complex environment of mining areas was not completely consistent with other studies. This is because there are other factors not included that altered carbon-fixing microbial community structure (Fig. 7), and soil types and soil characteristics varied in different regions, it is difficult to obtain an entirely consistent effect pattern of soil biophysicochemical properties on carbon-fixing microbial communities in different regions.
Overall, the results confirmed the hypothesis that carbon-fixing microbes play a crucial part in soil carbon sequestration in barren coal mining areas even after only a short-term reclamation and fertilization. The results also highlighted reclamation and fertilization significantly altered carbon-fixing microbial diversity and community, and improved potential ecosystem function. Our future research will further explore the isolation of carbon-fixing microbial strain and its application as microbial fertilizer in reclaimed soil of coal-mining subsidence areas, which would further facilitate soil fertility improvement in coal mining subsidence area.
-
Our study revealed that reclamation even without fertilization in the initial stage could bring significant shifts in MBC and dominant taxa (i.e., Proteobacteria, Cyanobacteria, Devosia and Marichromatium) and showed a trend of ecological recovery. Moreover, reclamation with fertilization significantly increased SOC and MBC (P < 0.05) and significantly altered carbon-fixing microbial community composition. Among these fertilization treatments, the application of manure (M) is more conducive to increasing carbon-fixing microbial abundance, the cbbL/16S rRNA ratio and RubisCO activity in the current short-term reclamation. SM, CAT, TP and DOC were the key factors significant influencing soil carbon-fixing microbial community structure, which provided a theoretical basis for improving the carbon-fixing potential. Reclamation and fertilization could significantly influence carbon-fixing microbial community structure (P < 0.05) and increase soil carbon storage due to altered soil properties and manure application dose. The fertilization treatments with manure were more conducive to improving the soil carbon-fixing potential. These findings contributed to improving soil fertility and accelerating ecological restoration and reconstruction in the mining area.
This work was financially supported by the National Natural Science Foundation of China (41907215), the Incentive Research Foundation of Shanxi Province for Recruited Doctoral Talents (SXYBKY2018009), the Science and Technology Innovation Foundation of Shanxi Agricultural University (2018YJ24) and the Natural Science Research Project of Shanxi Province (202103021224171).
-
The authors declare that they have no conflict of interest.
- Supplemental Fig. S1 Schematic diagram of the geographic location of the study site.
- Supplemental Fig. S2 16S rRNA abundance (a) and cbbL/16S rRNA gene ratio (b) in different treatments. Values are means (n=3), and error bars represent standard deviation. Different lowercase letters above columns indicate significant differences (one-way ANOVA, P<0.05) among treatments.
- Supplemental Fig. S3 Rarefaction curves of operational taxonomic unit (OTU) numbers.
- Supplemental Fig. S4 Sobs Richness Index (a), Shannon-Weaver Diversity Index (b), Pielou Evenness Index (c) charts of the carbon-fixing microbial community. The error bars represent standard deviation. Different lowercase letters above columns indicate difference (one-way ANOVA, P<0.05) among treatments.
- Supplemental Table S1 The relative abundance of dominant phylum in different treatments.
- Supplemental Table S2 The relative abundance of dominant classes in different treatments.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Shang Y, Wu M, Zhang J, Meng H, Hong J, et al. 2023. Nutrient enhanced reclamation promoted growth, diversity and activities of carbon fixing microbes in a coal-mining subsistence land. Soil Science and Environment 2:2 doi: 10.48130/SSE-2023-0002 

Nutrient enhanced reclamation promoted growth, diversity and activities of carbon fixing microbes in a coal-mining subsistence land
- Received: 02 December 2022
- Accepted: 03 March 2023
- Published online: 03 April 2023
Abstract: Carbon-fixing microbes can potentially improve soil fertility. However, the potential and function of carbon-fixing microbes remains largely uninvestigated in reclaimed soil of coal-mining subsidence areas. In this study, treatments included UL (uncultivated land), CK (maize cultivation without fertilization), NPK (maize cultivation with chemical fertilizer), M (maize cultivation with manure), MNPK (maize cultivation with manure and chemical fertilizer) after 1-year reclamation in a typical coal mining subsidence area. Quantitative PCR, enzyme-linked immunosorbent assay (ELISA) and high-throughput sequencing were employed to investigate the topsoil carbon-fixing microbial biomass, RubisCO activity and community composition. The results showed that the dominant taxa (i.e., Proteobacteria, Cyanobacteria, Devosia and Marichromatium) were significantly changed after reclamation (P < 0.05). Carbon-fixing microbial community structure in fertilization treatments (NPK, M and MNPK) obviously differed from non-fertilizer treatments (UL and CK). Soil organic carbon and microbial biomass carbon were significantly higher in fertilization treatments than non-fertilizer treatments (P < 0.05). M significantly increased RubisCO activity and cbbL gene abundance (P < 0.05), MNPK significantly increased carbon-fixing microbial richness (P < 0.05). Carbon-fixing microbial community structure was strongly influenced by soil moisture, catalase, total phosphorus and dissolved organic carbon. Some environmental factors indirectly influenced SOC by affecting carbon-fixing microbial biomass, diversity and community structure. Our study implies that even short-term (1-year) reclamation and fertilization could significantly influence carbon-fixing microbial community structure and promote soil carbon accumulation, and the fertilization treatments with manure (M and MNPK) were more conducive, which indicated that carbon-fixing microbes were greatly conducive to improve soil fertility in reclaimed mining areas and achieve carbon neutrality.
-
Key words:
- coal-mining /
- reclaimed soil /
- fertilization /
- cbbL gene /
- carbon-fixing microbes /
- RubisCO activity