










-
The reduction in seed vigor with aging significantly impacts crop production[1−3]. Seed longevity is determined by a complex interaction between genetic, environmental, and stochastic factors[4,5]. Environmental factors, such as moisture content, relative humidity, oxygen pressure, and storage temperature, can affect seed longevity. Mature orthodox seeds are desiccation tolerant and can endure extreme conditions such as high[6] and low temperatures[7]. Even in a low hydration state, seeds age at a readily manipulated rate, making them an appropriate model for studying longevity and aging[8,9]. Moreover, seed longevity varies widely even among isogenic individuals[5,10,11].
RNA integrity number (RIN) is a reliable marker for studying seed aging, as RNA is highly susceptible to oxidative damage[12−15]. In dry seeds, various stored mRNAs are essential for protein synthesis during the early stages of germination[13,16−18]. About 12,000-18,000 mRNAs with different functions are present in dry seed, and they have been connected to seed viability at different aging stages and seed resurrection after rehydration[19,20]. Rice seeds have been shown to contain 529 stored LLRs associated with germination capacity after inhibiting new mRNA synthesis during rehydration[21,22]. Notably, seed-stored mRNAs associated with monosomes undergo translational regulation during germination[23]. These LLRs and de novo transcribed mRNAs during rehydration are crucial to the germination process, with the former providing energy during the initial stages of germination and the latter accelerating energy production thereafter[21]. A shared locus with a positive allele was found, but the parental origin of the allele differed. Correlation analysis did not reveal any relationship between induced aging treatments and long-term storage[24−26]. However, most mRNAs showed a similar pattern of deterioration during both NAT and CDT[27]. The change in stored mRNA levels during seed aging showed that CDT seeds aged similarly to NAT seeds, but the degradation of stored mRNAs in CDT seeds occurred in a shorter time frame (years)[28].
Bran from bread wheat (Triticum aestivum 'Babbler') contains multiple outer layers of dead maternal tissues that cover living aleurone cells[29,30]. These outer layers with dead cells act as a protective barrier against degradation, while the aleurone layer mobilizes organic substrates from the endosperm during post-germinative growth. Thus, damage to stored RNA in the aleurone cells is expected to impact post-germination and early seedling development[31−33].
Previous studies analyzing transcriptome changes during seed storage have focused on the maturity stage after the drying phase[34]. Illumina sequencing has been used to detect stored mRNAs in dry, mature seeds, but this technology requires interruption of mRNAs before sequencing to ensure accuracy, while the length of mRNA in aging seeds (low RIN samples) can still exceed 8,000 bp[35]. As a result, short-read sequencing makes it impossible to determine whether the mRNAs were full-length in the aging seeds. However, the Oxford Nanopore platform, a third-generation technology, can directly sequence DNA/RNA with over 10 Kbps, enabling the detection of mRNA in aged seeds[36].
Our study focuses on the presence and integrity of mRNAs in aged wheat seeds. We evaluated the longevity of a diverse collection of wheat germplasms. We evaluated the germination percentage (GP) of the wheat cultivar Chinese Spring. Using MinION Nanopore sequencing, we identified full-length LLRs in wheat seeds, and next-generation sequencing (NGS) technology was further applied to investigate their expression patterns across different tissues. We identified the most stable LLRs in the embryo and the aleurone layer, including a seed-specific LLR which belongs to the LEA protein family. We further surveyed the stability of seven LLRs in Chinese Spring seeds and 18 other wheat varieties using RT-PCR and PCR amplification. We found three of the most stable LLRs identified in Chinese Spring were more stable in high longevity varieties than in short longevity varieties after aging, indicating their potential roles in seed longevity and germination. Overall, our study provides valuable insights into the mechanisms of seed longevity and may contribute to developing more effective seed storage and preservation strategies.
-
Twenty wheat (Triticum aestivum L.) varieties were subjected to CDT for 12 d for the purpose of germination assays and 25 d for PCR amplification (Supplemental Table S1). Chinese Spring seeds under NAT were harvested in 2013 (Guanghan, Sichuan, China; Natural Aging Treatment, NAT for 8 years), 2016 (Chongzhou, Sichuan, China; NAT for 5 years), 2018 (Chongzhou, Sichuan, China; NAT for 3 years), and 2020 (Chongzhou, Sichuan, China; NAT for 1 year). Chinese Spring seeds for CDT initiation were harvested in 2018 (Chongzhou, Sichuan, China) and then subjected to CDT for 5, 15, and 25 d in 2021.
Fresh Chinese Spring seeds were harvested in 2021 (45 d after flowering; NAT for 0 years) from plants grown in 15 cm pots in a thermostatic growth chamber with a controlled temperature of 20/12 °C (day/night) and a 16/8 h photoperiod. Then, the seeds were naturally air-dried as a control. All seeds were stored in a dry glass jar at −80 °C.
Aging treatment and seed germination assays
-
The initial moisture content can influence the longevity of seeds[5]. Thus, seed moisture content was determined using near-infrared transmittance (NIT; Foss-Tecator 1241, Foss, Högänas, Sweden). For CDT, 100 g of wheat seeds were wrapped in nylon bags (three replicates per accession) and subjected to a 43 °C temperature and 76% relative humidity (RH) in a climate chamber[11,37]. For NAT, the seeds were dried to a consistent moisture content and stored at room temperature for 1, 3, 5, and 8 years.
Seeds were germinated on germination paper and incubated in the dark at 20 °C. Each germination assay starts with 50 seeds with three replicates for each accession. After 7 d of imbibition, seeds were scored as germinated when the radicle emerged from the seed coat[38]. Ni is the number of germinated seeds on Day i, and the estimated germination indices were as follows[39]: (Germination percentage) GP: N7/50. After subjecting seed from each of the 20 different varieties to identical aging treatments, the ranking of seed longevity was determined by comparing the changes in germination percentage (ΔGP%) between non-aged seeds (NAT_0Y) and aged seeds (CDT_12D). ΔGP% was calculated as:
[(NAT_0Y_GP − CDT_12D_GP)/NAT_0Y_GP] × 100%.
The half inhibitory time is defined as the number of days required for the aging of seeds to reach a ΔGP% of 50%[40].
RNA extraction and RIN value assay
-
Twenty whole wheat seeds were mixed directly without tissue segmentation for RNA isolation, which was then used for Nanopore sequencing. As for Illumina RNA sequencing, each embryo and aleurone layer sample was separated from 10 and 20 dry seeds by hand-cutting, respectively. After the tissue was cut, it was placed directly under the microscope and slices frozen for observation (Supplemental Fig. S1a, S1b). For samples that were used for RNA extraction, we rapidly separated the tissues on dry ice. We immediately transferred them into liquid nitrogen for storage at −80 °C to prevent RNA degradation and ensure accurate downstream analysis. Real-Time Quantitative Polymerase Chain Reaction (qPCR) was employed to validate the expression of tissue-specific genes and provide the isolated tissue's purity (Supplemental Fig. S1c). RNA extraction followed the Nanopore and Next-generation sequencing (NGS) protocol. Total RNA was extracted with the Befitt kit (Invitrogen, California, USA). The quantity and quality of the extracted RNA were determined using a Nanodrop 2000 spectrophotometer (Thermo Scientific, USA) and verified using an Agilent 2100 bioanalyzer (Agilent Technologies, USA). Then RNA was stored at −80 °C for later use. The RNA Integrity Number (RIN) was calculated for RNA extracted from Chinese Spring, Aikang 58, and Zhengmai 366 seeds before and after aging.
Nanopore sequencing and identification of full-length LLRs
-
The poly(A) mRNAs Magnetic Isolation Module of VAHTS mRNA Capture Beads (Vazyme, Nanjing, China) was used to enrich mRNAs according to the manufacturer's protocol. Approximately 37.5 µg of total RNA was used for each sample. The final poly-A+ RNA concentration was measured using a Quantus Fluorometer (Promega Corporation, Madison, WI, USA) and checked by an Agilent 2100 bioanalyzer (Agilent Technologies, USA).
Synthesis of cDNA for sequencing was performed by following the strand-switching protocol from Oxford Nanopore Technologies. With the protocol, an incomplete cDNA sequence should arise from an incomplete or fragmented template[41]. According to Oxford Nanopore protocols, libraries were barcoded, pooled, and prepared for sequencing. Briefly, each library pool consists of two samples, CK and CDT_25D, and was sequenced on a MinION SpotON Flow Cell MK I (R9.4) (Oxford Nanopore Technologies, Oxford, UK). Sequencing data were obtained using Albacore 0.8.4 (Oxford Nanopore Technologies). Reads were de-multiplexed based on the barcode using porkchop 0.2.0 (
https://github.com/rrwick/Porechop , released 3/27/2017) with default settings. Blast (https://ftp.ncbi.nlm.nih.gov/blast/ ) was used to compare aligned reads and reference transcript lengths (iwgsc_refseqv1.1_ annotation_200916_HCLC_cds.fa) to identify transcripts with at least one sequence alignment in all samples. The integrity of the read was normalized between 0 and 100%, and an identity ≥ 90% was considered[36]. The sequence with the highest completeness was selected as the representative of the transcript, and this value represents the best transcript performance after decay.NGS and identification of stored mRNAs
-
The NGS was performed on equal molar amounts of the RNA libraries using the Illumina HiSeq-2000 and Hi-Seq Ten platforms by Berry Genomics Co., Ltd. Three sets of RNA-seq data were replicated and combined for analysis. The fluorescence image files were converted into short reads through base calling and stored in FASTQ format. The data processing followed the instructions provided by Berry Genomics in Beijing (China).
Chinese Spring seeds were divided into aleurone layers and embryos after undergoing NAT (0, 1, 3, 5, and 8 years) and CDT (5, 15, and 25 d) and then sent for sequencing, respectively. Quality control was based on the Q30 (> 80%), GC content (50%~60%), and sequence duplication levels of the clean data. Principal component analysis (PCA) and a correlation heatmap were performed on all samples to demonstrate the reproducibility and usability of the data. Transcripts per million (TPM) calculations were performed by Kallisto[42] and compared to the reference transcriptome. The overlapping genes from CDT and NAT samples with a detectable transcripts level (TPM ≥ 1) were identified as candidates for LLRs. The average TPM values of the three replicates were calculated as the transcript level of genes in each sample (except A25_Embryo_1).
After TPM normalization, genes with a detectable transcripts level (TPM ≥ 1) or not detectable (TPM < 1) were identified. The genes that were significant differences in transcript levels (log2 fold change (FC) ≥ 1 or log2FC ≤ −1, FDR < 0.05) were analyzed by the EdgR package (version 3.18.1) of the Trimmed Mean of M-values (TMM) algorithm[43]. After the data are normalized to equalize expression level distributions between samples, the stable transcripts attain elevated read counts in degraded samples[44]. For each most stable LLR, read depth was plotted against the base pair position to establish the distribution of sequence lengths, which was used to analyze the degradation of the transcripts. The data for other tissues (anther, leaf, pistils, root, shell, and stem) and for the developing embryo (2−38 d) and developing endosperm (8−32 d) were obtained from WheatGmap (
www.wheatgmap.org ) and Wheatomics[45].Sequence analysis of the most stable LLRs
-
The wheat gene promoter, 5' untranslated regions, 3' untranslated regions, coding sequence, and protein sequences were extracted from the IWGSC Genome (GFF, GTF) in NCBI (
www.ncbi.nlm.nih.gov ) and TBtools (GXF sequences extract function)[46]. The sequence 2000 bp upstream of the start codon was used for cis-acting element analysis by Plantcare (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/ ). MEME was used to identify the enriched motifs in the coding sequence and protein sequences (https://meme-suite.org/meme/ ). Briefly, the classical mode was selected for motif discovery. Sequences were uploaded into the primary sequence box. The motif length was set to 6–9 bp. The average GC% content and coding sequence length of the most stable LLRs were analyzed by Notepad++, and the frequencies of the background genes (high-confidence genes) were also calculated.Functional analysis of LLRs
-
For Gene Ontology (GO) enrichment analysis, TGT (
https://wheat.cau.edu.cn/TGT/ ) was used to assess the LLRs with log2FC < 0 and LLRs with log2FC ≥ 1 and FDR < 0.05 set, respectively. The function of proteins was retrieved using UniProt (www.UniProt.org ) and WheatOmics databases[45].qPCR and RT-PCR analyses
-
The primers of genes used for assessing tissue separation purity and screening for stored mRNAs through qPCR and RT-PCR can be found in Supplemental Table S2. The total RNA of the wheat root, stem, leaf, embryo, and aleurone layer was reverse-transcribed into cDNA using HiScript® III RT SuperMix (Vazyme). One microgram of RNA was incubated with a 4× gDNA wiper mix at 42 °C for 2 min to remove genomic DNA. Then, 5× HiScript III qRT SuperMix was added to the reaction and incubated at 37 °C for 15 min, followed by 85 °C for 5 s to synthesize cDNA. The cDNA mixture was diluted 1:4 with sterile H2O. The qPCR experiment was carried out using the ChamQ Universal SYBR qPCR Master mix (Vazyme) as the reaction reagent. The reaction mixture, comprising 12.5 μl of the master mix, 0.4 μM of each primer, and 1 μl of cDNA, was prepared to a final reaction volume of 20 μl. The real-time qPCR was performed on a Bio-Rad CFX96 Real-Time system, and the TBtools (Simple q-PCR summary) software was used to calculate the relative expression levels. The total RNA of 19 of 20 wheat varieties (except ZM121, for which seeds were not enough for this assay) was reverse-transcribed into cDNA for PCR amplification. PCR products were separated by electrophoresis in 3% agarose gels and stained with ethidium bromide. The gel images were obtained with a BioDoc-It imaging document system and used without any modifications (except for cropping to show the DNA band). The gene amplification data were converted into a heat map, where successful amplification in at least two out of three replicate experiments is shown in dark blue, and unsuccessful amplification is in light blue.
Statistical analysis
-
All analyses were performed in WPS and GraphPad Prism with one-way ANOVA (Tukey's test) in homogeneous groups, assuming significant differences when p < 0.05 and p < 0.01.
-
The longevity of 20 wheat varieties was evaluated based on their seed germination percentage (GP) after 12 d of CDT (Fig. 1a). Near-infrared transmittance measurements showed increased seed moisture content after CDT (Supplemental Fig. S2a), but no significant correlation was found between moisture content and GP. Ten of the varieties showed high seed longevity (ΔGP% ≤ 50%), indicating that the aging treatment time was lower than half of the inhibitory time[40] (Fig. 1a, for the definition of 'inhibitory time' see the Materials and methods section, 'Aging treatment and seed germination assays'). These results suggest that genetic differences among the wheat varieties may significantly contribute to the variation in GP (Fig. 1b).
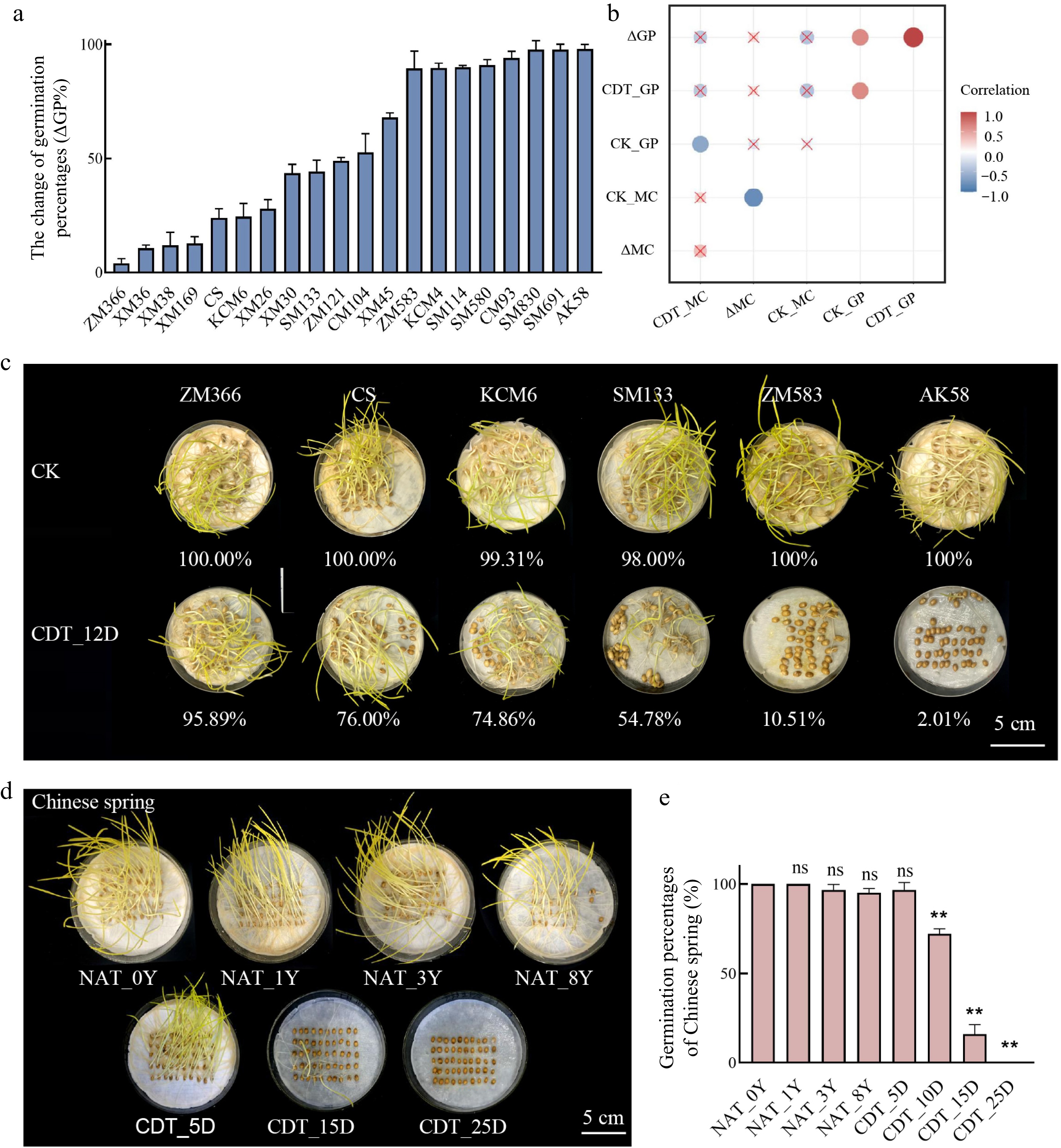
Figure 1.
The germination percentage (GP) of different wheat accessions. (a) The change of germination percentages of 20 wheat varieties after 12 d of controlled deterioration treatment (CDT) (ΔGP%). Data are the means ± standard deviations (SDs) based on three biological replicates. (b) A correlation heatmap between moisture content and GP. CK_GP indicates the GP of non-aged seeds; CDT_GP indicates the GP of seeds that CDT for 12 d; CK_MC indicates the moisture content of seeds before aging; CDT_MC indicates the moisture content of seeds after CDT for 12 d; ΔGP suggests the change of germination during CDT; ΔMC indicates the change in moisture content of seeds during CDT. (c) The GP of six wheat varieties after CDT_12D; bar = 5 cm. (d), (e) The GP of Chinese Spring seeds under NAT and CDT; bar = 5 cm; compared to NAT_0Y seeds; ns, not significant; **, p < 0.01, one-way ANOVA with Tukey's test.
The varieties with different seed longevity were selected to investigate changes in GP after CDT. Chinese Spring was chosen for subsequent studies due to its wide use in cytogenetic analysis and moderate longevity in our study. In Chinese Spring seeds, the GP of NAT seeds decreased slowly during aging, while the viability of CDT seeds decreased significantly. After 25 d of CDT, the seeds nearly lost their germination capacity (Fig. 1d, e). Therefore, the CDT method lets us obtain seeds of different ages quickly.
Correlation between seed longevity and RNA integrity in embryos and aleurone
-
The relationship between seed longevity and RNA integrity number (RIN) was analyzed using total RNA from the embryo and aleurone layer of Chinese Spring seeds under NAT (0, 1, 3, 5, and 8 years) and CDT (5, 15, and 25 d). The mean RIN was significantly reduced, particularly in the embryo under CDT conditions (Fig. 2a). GP was positively correlated with RIN in both the aleurone layer (R2 = 0.70) and embryo (R2 = 0.45) in Chinese Spring seeds (Fig. 2b). The RIN values were compared between the high longevity cultivar Zhengmai 366 and the low longevity cultivar Aikang 58 after 12 d of CDT, and a reduction in GP was associated with a decrease in RIN value (Supplemental Fig. S2b). These findings suggest that stored mRNAs in embryos and the aleurone layer contribute to seed longevity and germination.

Figure 2.
The RNA integrity (RIN) assays of Chinese Spring seeds. (a) RIN of Chinese Spring seeds (embryo and aleurone) under natural aging treatment (NAT) for 1, 3, and 8 years (compared to NAT_0Y) and controlled deterioration treatment (CDT) for 0, 5, 15, and 25 d (compared to CDT_0D). CDT for 0-d seeds was NAT for 3 years seeds. ns, not significant; *, p < 0.05; **, p < 0.01 one-way ANOVA with Tukey's test. (b) Correlation analysis between RIN and germination percentage of Chinese Spring seeds under NAT (0, 1, 3, 8 years) and CDT (0, 5, 15, 25 d) (Pearson's R2).
Identification of the full-length mRNAs by Nanopore sequencing
-
We used a long-read Nanopore sequencing platform to detect full-length mRNAs in aged seeds. The mRNAs were disrupted into small fragments before Illumina sequencing, so we chose Nanopore sequencing to obtain a more complete picture of the mRNA landscape in aged seeds. We analyzed samples from Chinese Spring seeds that were either NAT_0Y or subjected to CDT_25D. We obtained 205,991 and 115,606 sequences mapped to the reference transcriptome (iwgsc_ refseqv1.1_ annotation_200916 _HCLC_ cds. fa), respectively (Supplemental Table S3). We then kept 40,818 and 20,848 full-length stored mRNAs with identity ≥ 90% (Supplemental Table S3). A significantly higher amount of full-length mRNAs was detected in NAT_0Y (16,787 transcripts) than in the CDT_25D seeds (4,611 transcripts), suggesting that the mRNAs were largely degraded under CDT for 25 d (Supplemental Table S3). We identified 3,083 common full-length transcripts in NAT_0Y and CDT_25D seeds as LLR candidates (Supplemental Table S4, Fig. 3a).
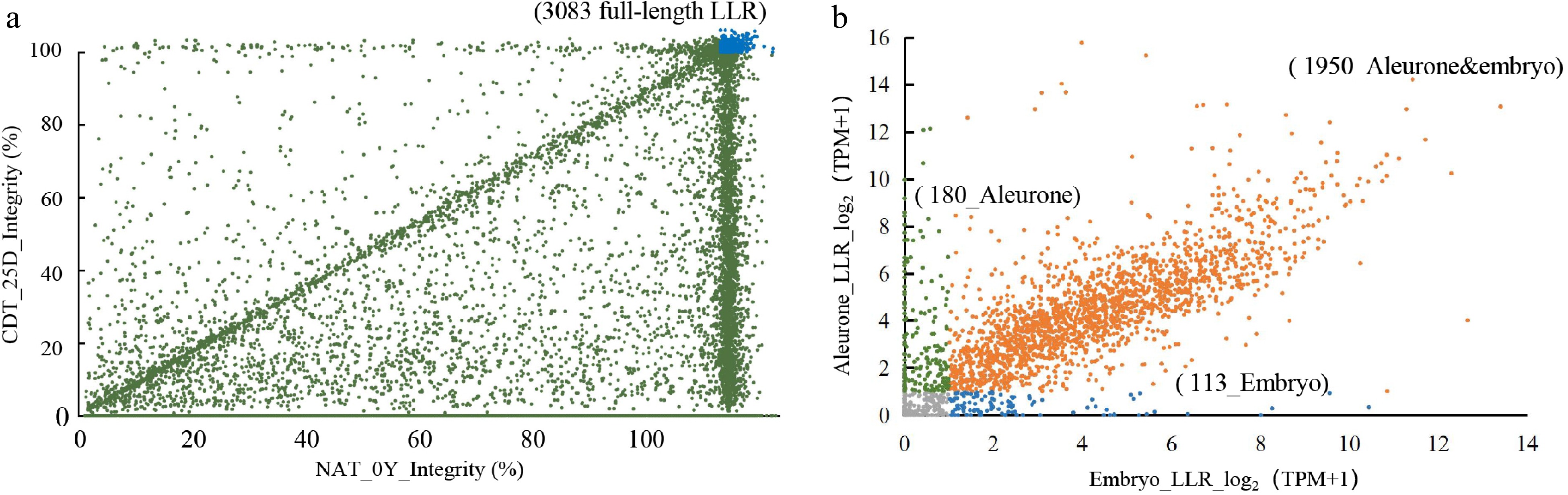
Figure 3.
Identification of full-length long-lived mRNA (LLRs) and validation of transcript levels by Nanopore sequencing and Illumina RNA-seq. (a) Comparative analysis of full-length mRNA in seeds of NAT_0Y and CDT_25D. The blue portion represents 3083 LLRs identified in both samples with at least one full-length transcript. (b) The transcriptomic profile of 3083 full-length LLRs identified by Nanopore sequencing was analyzed. LLRs with a TPM ≥ 1 were considered to have detectable transcript levels. These LLRs were then selected by identifying overlapping genes in all eight aging gradient samples. Aleurone-specific LLRs were annotated with green, embryo-specific LLRs with blue, and LLRs in both tissues were annotated with yellow.
Transcriptome profile of wheat embryo and aleurone layer after CDT and NAT
-
To ensure the purity of tissue separation, we selected four genes with distinct expression patterns between embryos and aleurone layers based on the public transcriptome data from Wheatomics[45]. We performed qPCR analysis to confirm a significant difference in relative transcription levels between these two tissues (Supplemental Fig. S1c). We conducted RNA-Seq using the embryo and aleurone layer of Chinese Spring seeds after CDT and NAT to further investigate the transcriptome changes during seed aging. We generated 3.1 billion high-quality clean reads, with Q30 values ranging from 87% to 94%, and the mean GC% content was 55%~61% (Supplemental Table S5). Principal component analysis (PCA) and correlation heatmap showed that these RNA-seq samples could be separated into four categories based on aging treatments and tissues (Supplemental Fig. S1d, S1e). The results showed that gene transcription levels gradually decreased in both the embryo and the aleurone layer during seed aging (Supplemental Fig. S3a, S3b). We identified 19,736 stored mRNAs in the embryo and 21,433 in the aleurone layer with TPM ≥ 1 in both NAT and CDT seeds (Supplemental Fig. S3c; Supplemental Table S6). The transcription of 3083 LLRs detected by Nanopore sequencing was supported by Illumina RNA-seq. We identified 2,130 and 2,064 LLRs in the aleurone layer and embryo, respectively, and 1,950 LLRs were shared in both tissues (Fig. 3b; Supplemental Table S7).
Degradation percentage of LLRs
-
To assess the stability of LLRs under NAT (0, 1, 3, 5, and 8 years) and CDT (5, 15, and 25 d) conditions, we measured the gene fold change (FC) using Illumina RNA-seq. In the aleurone layer, there were more LLRs with log2FC ≥ 0 than LLRs with log2FC < 0 under both NAT and CDT conditions (Fig. 4a). However, in the embryo, the number of LLRs with log2FC ≥ 0 decreased during seed aging and was less than that of LLRs with log2FC < 0 in CDT_25D seeds (Fig. 4b). These results suggested that the stability of LLRs differs between the aleurone layer and embryo and that LLRs in the aleurone layer were generally more stable than those in the embryo under aging conditions.
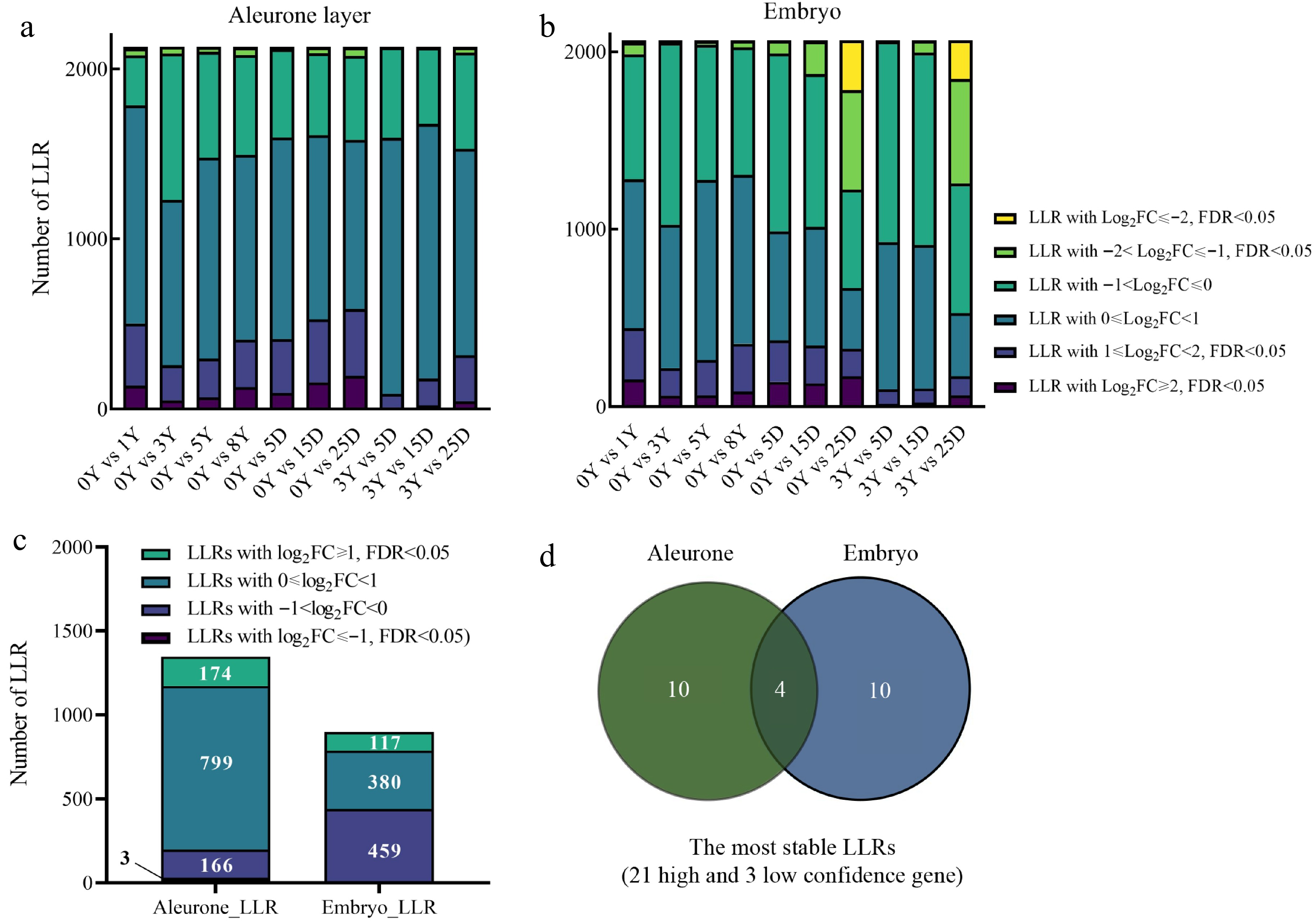
Figure 4.
The degradation pattern of long-lived mRNA (LLRs). (a), (b) LLRs with different fold change (FC) among groups of comparison. (c) This refers to the count of genes that overlap with the same FC across different aging samples from the same tissue. Specifically, the samples include natural aging treatment (NAT) for 0, 1, 3, 5, and 8 years and controlled deterioration treatment (CDT) for 5, 15, and 25 d seeds. (d) Venn diagram comparisons of the most stable LLRs in the embryo and aleurone layer. The most stable LLRs were the overlapping LLRs with log2FC ≥ 1 and FDR < 0.05 between NAT_3Y and CDT (5, 15, and 25 d), which were selected from the overlapping LLRs with log2FC ≥ 1 and FDR < 0.05 (compared to NAT_0Y) as mentioned in Fig. 4c.
Further analysis demonstrated that the LLRs had different FCs across various seed aging samples and fresh NAT_0Y seeds, likely due to differences in their Transcripts Per Million (TPMs) (Supplemental Table S6, S7). We examined the overlapping LLRs among all aged samples to identify LLRs with similar FC features in aleurone layers or embryos. We found that a significant portion of genes (14.80% from the aleurone layer and 49.00% from the embryo) were LLRs with log2FC < 0. Several LLRs log2FC ≤ 1 and FDR < 0.05 were identified in all overlapping LLR sets (Fig. 4c; Supplemental Table S8). These LLRs with log2FC < 0 were associated with translation and transportation, as shown by Go enrichment analysis (Supplemental Fig. S4a, S4b; Supplemental Table S9). In contrast, LLRs with log2FC ≥ 1, FDR < 0.05 were associated with salt stress, heat, protein folding, reactive oxygen species, protein complex oligomerization, and abscisic acid metabolism (Supplemental Fig. S4c, S4d; Supplemental Table S9). LLRs with log2FC ≥ 1 and FDR < 0.05 may be more stable (Supplemental Table S8) since elevated read counts were obtained in the degraded samples. In addition, the TPMs of LLRs with log2FC ≥ 1 and FDR < 0.05 were relatively higher in NAT_3Y seeds than in NAT_0Y (Supplemental Table S7). Therefore, these LLRs with significantly increased transcripts level that still meets the criteria of log2FC ≥ 1 and FDR < 0.05 when compared to CDT_5D, CDT_15D, and CDT_25D samples with NAT_3Y may have the highest stability during aging. Finally, we identified 24 LLRs with the highest stability during aging (Fig. 4d), among which three were low-confidence genes. So only 21 LLRs were subjected to further analysis. Analysis of these 21 transcripts in embryo and aleurone layer tissues of fresh (NAT_0Y), NAT_3Y, and CDT_25D seeds showed a uniform distribution rather than a gradual increase from 5' to 3' end, eliminating the sequencing bias of Illumina RNA-seq. This result also demonstrated the full-length characteristic of these most stable LLRs (Supplemental Fig. S5).
Characterization of the most stable LLRs
-
The length of the coding sequence for high-confidence genes varied from 54 to 16,080 bp[45], whereas the 21 most stable LLRs identified in this study had coding sequence lengths of 336−720 bp in the aleurone layer and 195−591 bp in the embryo (Fig. 5a−c). Of all high-confidence genes, 74,181 had coding sequence lengths greater than 720 bp. In contrast, the length of 33,711 genes is not longer than 720 bp. Although the number of transcripts with length longer than 720 bp was 2.2 times greater than those shorter than 720 bp, none of them were identified as the most stable LLR, indicating that shorter transcripts may be more stable during seed aging in wheat (Fig. 5a−c). Furthermore, the average GC% contents, which is a reason for the stability of transcript, of the LLRs in the aleurone layer and embryo (approximately 75%) was higher than that of all high-confidence genes (approximately 55%), suggesting that the gene with higher GC% content may be more stable during seed aging (Fig. 5d).
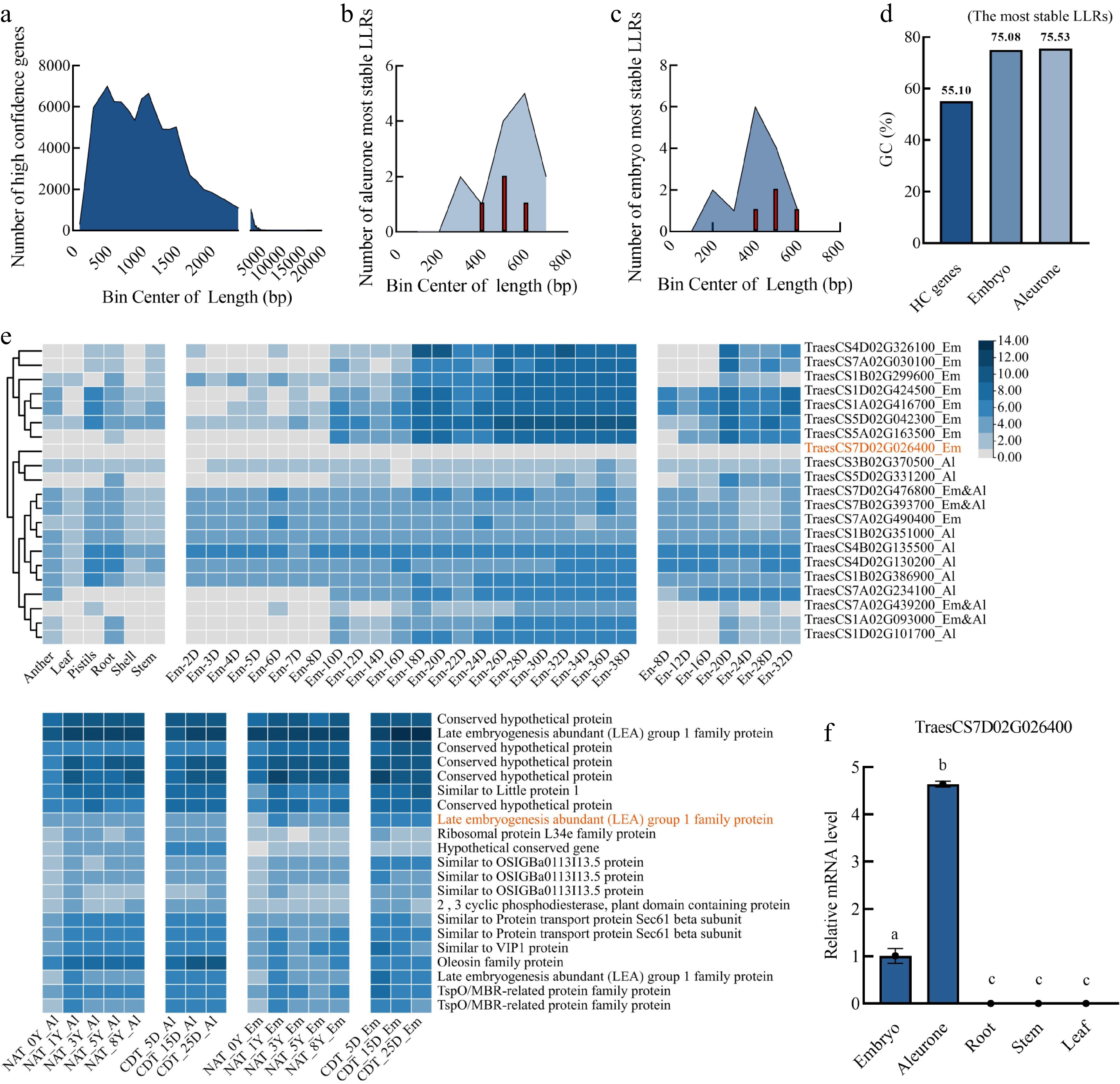
Figure 5.
Characterization of the most stable long-lived mRNAs (LLRs) in the embryo and aleurone layers. (a), (b), (c) Frequency analysis of the coding sequence length (x-axis) and gene numbers (y-axis). (a) Total high confidence genes. (b) The most stable LLRs in the aleurone layer. (c) The most stable LLRs in the embryo. (d) The average GC (%) content of all the high confidence genes in wheat seeds and the most stable LLRs in the wheat embryo and aleurone layer. (e) Heatmap visualization of RNA-seq data (Chinese Spring) from anther, leaf, pistils, root, shell, stem, the developing embryo (2−38 d), developing endosperm (8−32 d), NAT (0, 1, 3, 5, and 8 years) seeds, and CDT (5, 15, and 25 d) seeds. Embryo, EM; Aleurone layer, Al; Endosperm, En. LLRs in both embryo and aleurone layer,m Em&AL. (f) The relative expression levels of TraesCS7D02G026400 in root, stem, and leaf were determined by qPCR using actin as an internal reference gene. Two independent experiments were performed with triplicate samples in each experiment. The relative expression levels were calculated using the TBtools (Simple q-PCR summary) software. Using different lowercase letters to represent significant differences (p < 0.01, one-way ANOVA with Tukey's test).
The promoters of the 21 most stable LLRs were found to contain motifs associated with various plant hormone responses, including abscisic acid response (ABRE), light response (G-box), and hormone response (Supplemental Fig. S6, Supplemental Table S10). The promoter of 17 genes contains motifs related to cis-acting regulatory elements involved in methyl jasmonate (MeJA) response (CGTCA motif and TGACG motif) (Supplemental Fig. S6). Additionally, motifs related to gibberellin response (P-BOX) and auxin response (TGA elements) were identified in three of the four LLRs that were detected in both the aleurone layer and embryo (Supplemental Fig. S6).
The four most stable LLRs in the embryo and aleurone layer were annotated as members of the LEA 1 protein family (TraesCS7A02G439200), TSPO/MBR-related protein family (TraesCS1A02G093000), and OSIGBa0113113.5 protein (TraesCS7D02G476800, TraesCS7B02G393700) (Fig. 5e ). The expression analysis in anther, leaf, pistils, root, shell, stem, embryo, endosperm, NAT, and CDT wheat seeds indicated that TraesCS7D02G026400 was a seed-specific gene as well as an embryo-specific stable LLR in RNA seq data (Fig. 5e; Supplemental Table S11). The qPCR results showed a significantly higher relative mRNA level of TraesCS7D02G026400 in the embryo and aleurone layer compared with the root, stem, and leaf (Fig. 5f, Supplemental Table S2). None of the other 20 most stable LLRs was found to be seed-specific (Fig. 5e; Supplemental Table S11).
Degradation of LLRs among different wheat varieties
-
We tried to design primers for the 21 most stable LLRs based on the Chinese Spring genome sequence, but their high GC content brought challenges for primer design and PCR amplification. Therefore, we selected 7 LLRs (LLR 2, 3, 10, 11, 13, 15, and 20) to measure their stability in 18 wheat varieties after aging (Fig. 6a). To evaluate the stability of LLRs, we used three genes previously shown to degrade gradually as wheat seeds aged, and they belonged to the short-lived mRNA (SLR) set in our study[20]. After natural aging for 3 years, SLRs could be amplified in more than ten varieties. However, after CDT for 25 days, all three SLRs were no longer detectable in any of the 18 wheat varieties (Fig. 6b). In contrast, all seven LLRs could be successfully amplified in more than 11 varieties after both aging treatments (Supplemental Fig. S7). These results suggest that LLRs are relatively stable during seed aging, even in different wheat varieties.
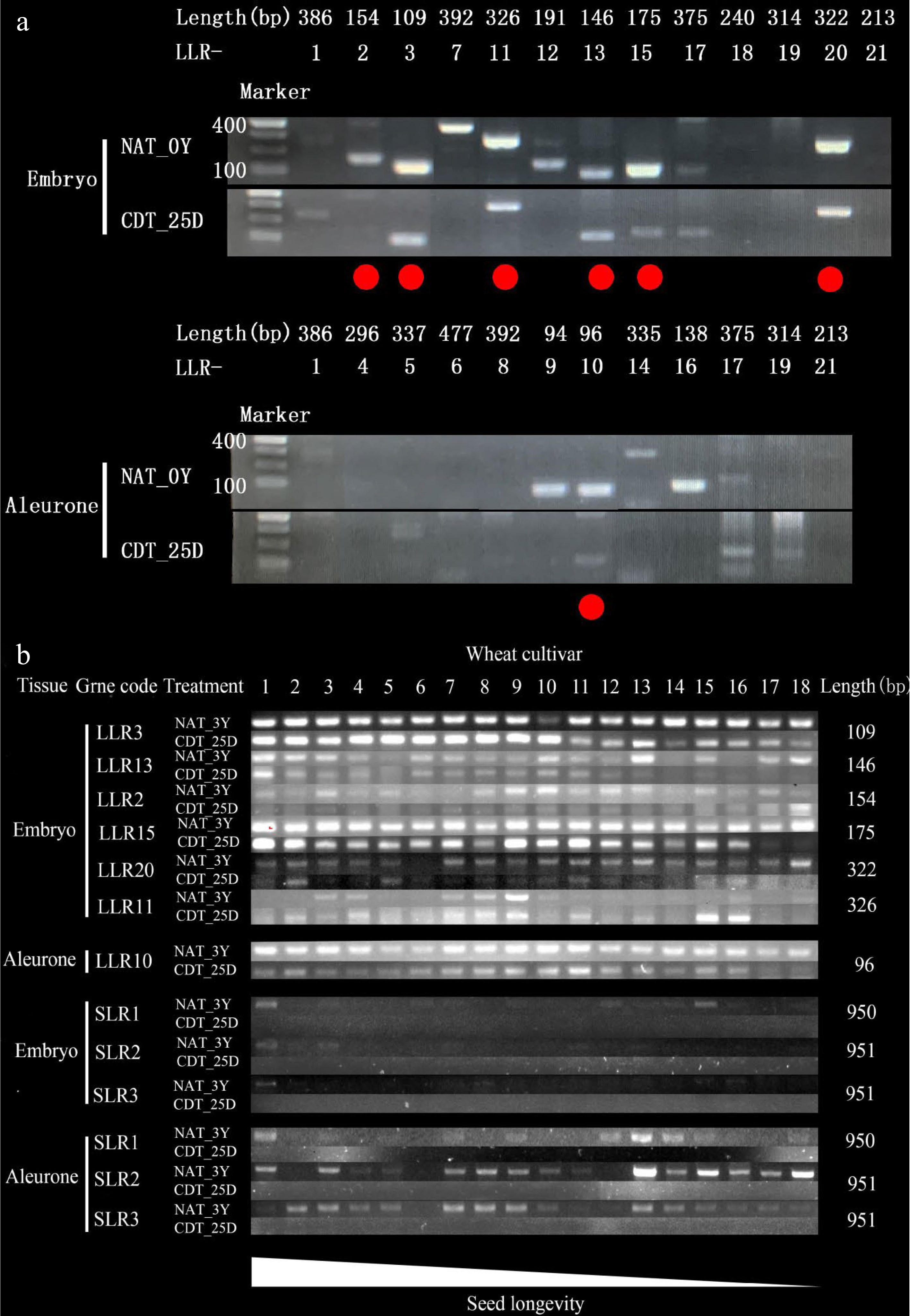
Figure 6.
The transcript levels of the most stable long-lived mRNAs (LLRs) in naturally aged treatment (NAT) and controlled deterioration treatment (CDT) wheat seeds detected by RT-PCR. The number of PCR cycles varied depending on their mRNA abundance in the unaged seeds. PCR products were run in agarose gels. (a) The 21 most stable LLRs in NAT for 0 Y (NAT_0Y) and CDT for 25 d (CDT_25D) Chinese Spring seeds were analyzed, respectively (see Supplemental Table S2 for the gene list). The 10 genes in 18 wheat varieties that NAT for 3 years (NAT_3Y) seeds and CDT_25D seeds were analyzed. (See Supplemental Table S1 for the wheat cultivar code and Supplemental Table S2 for the gene list). The lower wheat cultivar code number indicates higher seed longevity in (b).
Furthermore, to investigate the variation in seed longevity, we examined the stability of LLR13, LLR15, and LLR20 in four wheat varieties (SM691, AK58, ZM366, and XM36) with different degrees of reduction in seed germination percentage (ΔGP) after 25 d of CDT treatment. The results showed that LLR13, LLR15, and LLR20 degraded significantly in SM691 (ΔGP = 97.79%) and AK58 (ΔGP = 97.99%) but persisted in ZM366 (ΔGP = 4.11%) and XM36 (ΔGP = 10.75%) (Supplemental Fig. S7). These findings suggest that the presence or absence of specific combinations of LLRs can serve as molecular markers to estimate seed longevity, and LLR13, LLR15, and LLR20 may be promising candidates for further investigation into their roles in seed aging and longevity.
-
Seed germination refers to the physiological process culminating in the emergence of the embryo from its enclosing coverings, including the endosperm, perisperm, testa, or pericarp. Starch degradation, initiated by GA secreted by the embryo during germination, is considered a post-germinative event[31,32]. The scutellum, rather than the aleurone epithelium, is mainly responsible for the synthesis of α-amylase during the initial stages of germination in wheat, rye, oats, and maize[47]. However, any malfunctioning embryo or aleurone sections can affect seed germination[48]. Seed GP was correlated with the aleurone layer RIN and the embryo RIN (Fig. 2b). The mutagenic substances formed during aging would act early during the germination of seeds. The deleterious effects of aged endosperm on a young embryo[49] might be related to the decreased activity of antioxidant enzymes, such as catalase, peroxidase, dehydrogenase, and amylase[50]. The accumulation of toxic compounds in the aged endosperm or aleurone can induce chromosomal breakage in young embryos[51]. The response of aleurone layers from normal and aged seeds to heat shock has been investigated. Only aleurone layers from normally germinated seeds could recommence substantial α-amylase synthesis during recovery[52]. One of the LLRs identified in the aleurone layer was an oleosin family protein (TraesCS7A02G234100) (Fig. 5e), which may be involved in oil body mobilization during post-germinative seedling growth and may prevent the coalescence of protein storage vacuoles[53−55]. Our study found that the aleurone layer had more stored mRNAs and LLRs in aged seeds than the embryos (Supplemental Fig. S3a, S3b; Fig. 4c). Lipid oxidation has been implicated in seed deterioration, and detailed analyses of the changes in the lipidome during long-term dry storage of a range of genotypes of oilseed rape wheat, barley and Arabidopsis support this claim[56−58]. The lipid content of wheat embryo (8%−15%) is higher than that of other seed tissues (bran, aleurone, and endosperm; 6.8%−7.5%)[59]. Additionally, the embryos and endosperms or aleurone layer have different enzymatic patterns, highlighting that the two seed compartments age independently[6]. These differences between embryos and endosperm (aleurone layer) may cause varying degradation percentages of mRNAs (Fig. 4a, b).
Integrating NGS and full-length sequencing to obtain LLRs accurately
-
Our study observed reduced RNA integrity in the embryo and aleurone layer of aged seeds (Fig. 2a), with the lowest RIN being 5.5 (Fig. 2a). Despite a RIN lower than 3, the length of mRNA is still longer than 8000 bp[35]. Although Illumina's TPM and FC can predict mRNA degradation trends[40,44], mRNA fragmentation errors may exist with short-read sequencing. Therefore, it is difficult to determine whether the interruption of mRNAs is caused by seed aging or by the sequencing technology used, as Illumina technology can interrupt mRNAs before sequencing (Supplemental Table S5). By using the NEBNext Poly (A) mRNA magnetic isolation module and cDNA synthesis, Nanopore full-length sequencing was employed in our study to enrich and identify mRNAs that remain intact during aging[41]. Integrating Nanopore and Illumina sequencing enables the identification of LLRs with at least one full-length transcript and predicts mRNA degradation trends in aged seeds. Thus, our approach can effectively exclude the effects of mRNA fragmentation errors, leading to more accurate identification of LLRs. In conclusion, LLRs can be predicted by the FC determined by short-read sequencing[44], and fragment mRNA errors can be excluded by full-length Nanopore sequencing[36], demonstrating the integration of both sequencing technologies is a powerful tool for identifying stable mRNAs in aged seeds.
LLRs may be related to cell survival and seed longevity
-
Poly(A) polymerase activity decreases with age, and the translational levels decrease in aged wheat embryos[60,61]. Transcript degradation of the elongation factor EF-1 occurs both in the embryo during NAT and CDT but still exists in the embryo and aleurone layer in CDT_25D (Supplemental Table S6). A longevity-related QTL (Q.Lng.ipk.2A.1) contains a candidate gene similar to the translation elongation factor EFG/EF2 protein[62]. Transcripts related to ribosomal functions, particularly translation, are overrepresented in the stable mRNAs group and may indicate the importance of reconstituting the translational machinery during germination[3]. Among the analyzed mRNAs, 21 selected LLRs were more stable (Fig. 4d). The coding sequence of these LLRs was enriched with three repeats of the sequence TCCTCCTCC, which might be related to transcription factor IIIA and ribosomal protein L5[63]. The ribosomal L34e and preprotein translocase family proteins mRNA were detected as the aleurone layer's most stable LLRs (Fig. 5e). The longevity markers 7D (Wpt-0934) and 7A (wPt-0303) also reveal the relationship between ribosomal proteins and seed longevity[64].
In the aleurone layer, VIP1 was identified as the most stable LLR. It plays a role in the osmosensory response by binding to the 5'-AGCTGT/G-3' DNA sequence and is found in the promoters of the hypoosmolarity-responsive genes CYP707A1 and CYP707A3[65]. LEA 1, TSPO, and OSIGBa0113113.5 were identified as the most stable LLRs in both embryos and aleurone layers (Fig. 5e). The seed-specific expressed gene (TraesCS7D02G026400) is annotated as an LEA 1 family protein (Fig. 5e). The LEA 1 proteins, which have evolutionary and functional characteristics of an ancestral plant protein group, are also present in other eukaryotes and the Archaea and Bacteria domains[66]. In Arabidopsis, maize, and Medicago, LEA 1 protein is correlated with seed vigor and longevity[67−69]. Wheat seed longevity markers on 4B (wPt-1272) have identified some genes described as dehydrin-/LEA group proteins[64]. TSPO (Fig. 5e) expression seems to be correlated with LEA4-5 protein (TraesCS7A02G439200) expression in Arabidopsis[70]. TSPO is a stress-induced, posttranslationally regulated, and early secretory pathway-localized plant cell membrane protein involved in transient intracellular ABA-dependent stress signaling and has roles in apoptosis[71,72]. LLR 13, 15, and 20 were more stable in high longevity varieties than short longevity varieties after aging (Supplemental Fig. S7), suggesting that these stable LLRs may contribute to seed survival[40]. In addition to the 21 most stable mRNAs, several LLRs with log2FC ≥ 0 were identified in both the embryos and the aleurone layer, and they may be necessary for seed longevity. For example, the heat shock protein (HSP) and 1-cysteine peroxiredoxin antioxidant (PER1) were identified as LLRs (Fig. 4c, Supplemental Table S8). The heat shock protein OsHSP18.2 improved seed longevity under CDT[73]. A PER1 protein from Nelumbo nucifera enhances seed longevity and stress tolerance in Arabidopsis, and the PER1 protein is stable in high-vigor wheat after aging treatment[74,75]. A multi-omic study revealed a bZIP23-PER1A–mediated detoxification pathway to enhance seed vigor in rice[27]. These mRNAs existed after NAT and CDT, but the molecular mechanisms responsible for their role in wheat seed longevity and germination have not yet been clarified.
Seeds translate stored mRNAs during germination using stored ribosomes, and RNA integrity is closely related to seed vigor[19,21]. The germination of dry wheat seeds correlates with the embryo and the living aleurone cell mRNAs[30]. Our study identified specific LLRs related to longevity by comparing high-vigor and low-vigor varieties, and we examined the degradation rates of mRNA by transcriptome profiling[40]. We verified full-length LLRs using Nanopore sequencing[36,44]. While LLRs have a short and high GC content, the protected manner of mRNAs results in mRNAs having variant degradation percentages[23,76]. However, fission due to free radical attacks at random bases is also evident[20,36]. Further investigation is necessary to uncover the complex roles of these LLRs in seed longevity and the mechanism of seed resurrection. Overall, our study provides valuable insights into the mechanisms of plant cell survival and may contribute to developing more effective seed storage and preservation strategies.
This research was funded by the Major Program of National Agricultural Science and Technology of China (NK20220607), the National Natural Science Foundation of China (U22A20472), the National Key Research and Development Program of China (2018YFE0112000), the Sichuan Science and Technology Support Project (2021YFH0077; 2021YFYZ0027; 23NSFSC0770), the Science and Technology Support Project of Chengdu (2021-GH03-00002-HZ) and the open research fund of SKL-CGEUSC (SKL-ZD202212).
-
The authors declare that they have no conflict of interest.
- Supplemental Fig. S1 Tissue purity and correlation between biological replicates in Illumina RNA-seq.
- Supplemental Fig. S2 The water content and RNA integrity number (RIN) of different varieties.
- Supplemental Fig. S3 Identification of stored mRNA in aged wheat seeds by Illumina RNA-seq.
- Supplemental Fig. S4 Gene Ontology (GO) enrichment of overlapping long-lived mRNAs (LLRs).
- Supplemental Fig. S5 The sequencing depth from 5' to 3' of the most stable long-lived mRNAs (LLRs) and easily degraded mRNAs.
- Supplemental Fig. S6 Sequence analysis of the most stable long-lived mRNAs (LLRs).
- Supplemental Fig. S7 The correlation between longevity and the most stable long-lived mRNAs (LLRs).
- Supplemental Table S1 Wheat accessions used in this study.
- Supplemental Table S2 Quality of Nanopore-Seq data obtained in this study for NAT_0Y (fresh seeds) and CDT_25D seeds.
- Supplemental Table S3 Full-length long-lived mRNAs (LLRs) identified by Nanopore-seq data.
- Supplemental Table S4 Quality of Illumina RNA-seq data.
- Supplemental Table S5 Identification of stored mRNAs in embryo and aleurone layer using Illumina RNA-seq.
- Supplemental Table S6 Transcripts Per Million (TPM) analysis of full-length long-lived mRNAs (LLRs) in embryo and aleurone layers using Illumina RNA-seq.
- Supplemental Table S7 long-lived mRNAs (LLRs) with different fold changes (FC) in the embryos and aleurone layers compared with NAT_0Y.
- Supplemental Table S8 Gene Ontology (GO) enrichment analysis of overlapping long-lived mRNAs (LLRs) with relatively decreased and significantly increased transcript levels in embryos and aleurone layers.
- Supplemental Table S9 Motifs in the promoter of the most stable long-lived mRNAs.
- Supplemental Table S10 Expression profiles of the most stable long-lived mRNAs in different tissues and treatments.
- Supplemental Table S11 List of genes and primers used for Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and Real-Time Quantitative Polymerase Chain Reaction (qPCR) experiments.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press on behalf of Hainan Yazhou Bay Seed Laboratory. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Liang W, Dong H, Guo X, Rodríguez V, Cheng M, et al. 2023. Identification of long-lived and stable mRNAs in the aged seeds of wheat. Seed Biology 2:14 doi: 10.48130/SeedBio-2023-0014 

Identification of long-lived and stable mRNAs in the aged seeds of wheat
- Received: 28 October 2022
- Accepted: 28 July 2023
- Published online: 07 October 2023
Abstract: Seed germination relies on preserving mRNA integrity in dry seeds. However, the quality of mRNA in aged wheat seeds is not well understood. Here, we investigated 20 wheat varieties for seed longevity using controlled deterioration treatment (CDT) and identified that Chinese Spring seeds exhibit moderate longevity. We observed correlations between seed viability and RNA integrity in the aleurone and embryo cells after aging-treatment. Nanopore sequencing of whole seeds from natural aging treatment (NAT) and CDT for 25 d identified 3,083 full-length transcripts. We performed RNA-seq transcriptome profiling to classify the tissue origin of these transcripts under eight aging treatments, revealing the presence of 2,064 overlapping long-lived mRNAs (LLRs) in the seed embryo and 2,130 in the aleurone layers. These LLRs corresponded to genes with detectable transcription levels and at least one full-length transcript in their coding sequence. Notably, degradation percentages of these mRNAs varied among seeds of different wheat varieties with similar ages. We predicted 21 most stable LLRs with high GC% content and short coding sequence length, among which only one LLR was seed-specifically expressed and belonged to the late-embryogenesis-abundant (LEA) protein family. RT-PCR confirmed the expression of the seven LLR fragments in the aleurone layer and embryo of Chinese Spring seeds. We found three of the most stable LLRs (LLR13, LLR15, and LLR20) identified in Chinese Spring were more stable in high longevity varieties than in short longevity varieties after aging, indicating their potential roles in seed longevity and germination.
-
Key words:
- Stored mRNAs /
- Wheat /
- Aged seeds /
- Long-lived mRNAs /
- Embryos /
- Aleurone layer