










-
Tea plant (Camellia sinensis (L.) O. Kuntze) originated from southwest China and subsequently expanded to India, Sri Lanka, and many other countries[1]. A host of people drink tea because of its distinct flavor and health benefits[2]. With market expansion, traditional natural selection and purposeless artificial breeding approaches for selecting high-quality cultivars are falling short of satisfying the demands of the market economy[3,4].
To date, the advancement of PCR and modern sequencing technology has enabled the generation of genotype-based marker-assisted selection (MAS). A combination of crossbreeding and MAS in breeding work can quickly screen for superior varieties with target traits. Furthermore, the construction of genetic maps which can reflect the sequence of functional genes on chromosomes and quantitative trait loci (QTL) are crucial tools for implementing MAS[5]. The higher the density, the more conducive to the subsequent accurate positioning of QTL, which lays a solid genetic foundation for the study of important traits and provides a method for improving the breeding of new varieties of high-quality tea plants[6,7].
In this review, we comprehensively present the current state of the genetic map creation in tea plants, as well as the QTL mapping of significant quality characteristics. We also discussed the application of genome-wide association study (GWAS) tools in genetic tea breeding, which is one of the potential future directions for tea genetics and breeding research, and will promote germplasm utilization for breeding improved tea cultivars.
-
While sequencing technology is evolving, high-quality genome sketches and high-density genetic maps develop uninterrupted, which have greatly accelerated the process of molecular breeding in tea plants. Molecular markers are genetic markers based on genomic sequence differences between organisms and directly reflect the genetic polymorphism at the DNA level. Genetic segregation population, both controlled crossing and natural open crossing are requested for QTL mapping.
Genetic materials for QTLs mapping
-
Due to unfavorable characteristics such as cross-pollination, self-incompatibility, low fruit set, and a long juvenile period in tea plants, traditional natural selection takes at least 20−30 years, which is both time-consuming and labor-intensive[8]. What's more, the highly heterozygous nature of its F1 population limits the range of trait loci position. The conservation and sharing of mapping materials can be achieved through asexual multiplication[9]. That's why a pseudo-testcross strategy can be applied to construct genetic map.
Firstly, parents should be chosen with with large disparities in term of the target trait segregation and a sufficient individuals population size based on the actual scenario, such as population type and mapping aim. Too little genetic difference between parents will reduce parental DNA polymorphism, and too much of a difference will prevent recombination of the chain loci, and even serious bias segregation[10]. The populations often used for mapping in tea plants are mainly hybrid F1 and backcross populations, etc[8,11]. For F1 populations with extreme phenotypic differences, the bulk segregant analysis (BSA) method can be used to quickly identify genomic loci that regulate the quality traits of tea plants. Secondly, it should also be determined that the progeny of the cross bred to obtain a population of offspring segregating for the target traits. After selecting the appropriate mapping parents, the molecular makers can be identified via re-sequencing, simplified genome sequencing, transcriptome sequencing, etc. The transcriptome sequencing data are obtained and then assembled, subsequently the differentially expressed genes can be screened for subsequent QTL mapping. Then, the appropriate mapping software, e.g., MapMaker[12], JionMap[13], HighMap[14] are used for marker linkage analysis. Finally, QTL mapping can be carried out in combination with biochemical data after successful genetic map construction (Fig. 1).
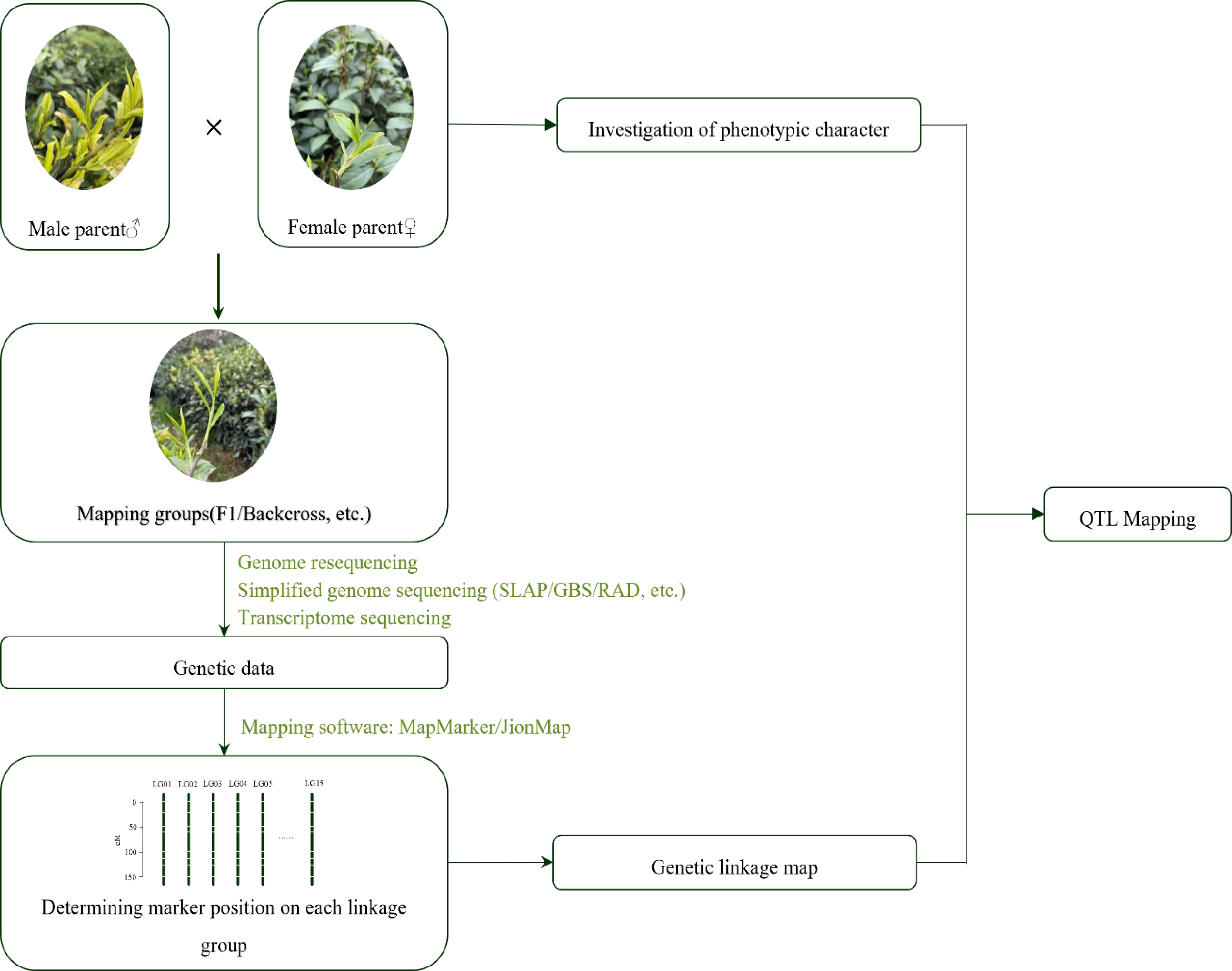
Figure 1.
QTL mapping construction flow chart.
QTL mapping in tea plants
-
In the early progress of the genetic map in tea plants, the main markers used were randomly amplified polymorph DNA (RAPD), simple sequence repeat (SSR), a few amplified fragment length polymorphisms (AFLP), and cleaved amplified polymorphic sequences (CAPS). The first two generations of molecular marker technology have the advantages of high stability, good repeatability, low cost, and simple operation, but they also have the disadvantages of difficult genotyping, high development difficulty, and a heavy workload. Currently, single nucleotide polymorphism (SNP) molecular marker technology is more widely used in tea plants[15]. The first successful genetic map of the tea plant dates back to 1996. When Tanaka[16] constructed a genetic map of the 'Yabukita' and 'Shizu-Inzaysu131' populations, demonstrated that the pseudo-testcross strategy could be applied to tea plants, but the number of markers were sparse, containing only six linkage groups.
After the construction of the molecular genetic map, and the resolution of-complex quantitative traits, the individual QTLs that locate on specific segments of chromosomes, and their genetic effects, as well as the interaction effects with environment, are estimated, which is called QTL mapping. So far, scholars have been building a more refined genetic map with reference to the pseudo-testcross strategy and excavating a large number of QTLs for quality traits, agronomic traits, and stress resistance in the tea plants. For example, there are QTL loci related to catechins[17], anthocyanins[18,19], caffeine[17,19], free amino acids[20], terpenes aroma[21], as well as bud flush[22,23] , young shoot color, mature leaf size[22], leaf area[24], anthrax disease[19], drought tolerance[25], and short winter-dormancy[26] etc. (Table 1).
Table 1. Summarized papers of genetic map information in tea plants.
No. Mapping population
(♂ × ♀)Population
sizeMarker type Markers Linkage
groupsTotal length
(cM)Average distance
(cM)Ref. 1 Yabukita 46 RAPD 23 6 − − [16] Shizu-Inzaysu131 36 6 − − 2 Sayamakaori 54 RAPD 126 14 1550 11.7 [27] Kana-CK17 140 17 1640 3 SFS150 90 RAPD/AFLP 126 15 1349.7 11.7 [28] TN14/3 − 4 Qimen 4 69 AFLP 208 17 2457.7 11.9 [29] Chaoan Dawuye 200 16 2545.3 5 Fuding Dabaicha 94 RAPD/ISSR 62 6 1180.9 15.7 [30] 6 TRI2043 141 SSR 139 15 1018 2.9 [31] TRI2023 173 15 1192.9 7 Sayamakari 64 SSR/RAPD/CAPS/STS 571 17 3091 1.93 [32] Kana-CK17 632 15 3314 8 TRFCA SFS150 42 RAPD/AFLP/SSR 69 15 1012 14.1 [33] AHP S15/10 31 19 399.5 9 Sayamakaori 54 SSR/RAPD/CAPS/STS 701 15 1305 1.93 [34] Kana-CK17 701 15 1298 10 Longjing43 170 SSR 237 15 1156.9 5.2 [35] Baihaozao 11 Yingshuang 183 SSR 406 15 1143.5 3.0 [17] Beiyue Danzhu 12 Yingshuang 148 SNP/SSR 6448 15 3965 1.0 [36] Beiyue Danzhu 13 Longjing 43 170 SSR 483 15 1226.2 2.5 [22] Baihaozao 14 Wuniuzao 174 SSR 175 16 1156.9 7.4 [37] Longjing 43 15 Fushun 79 RAPD/AFLP/SSR 678 − 1441.6 4.7 [38] Kemsull 16 TRFK 303/577 109 DArT-seq 187 15 1028.1 1.1 [25] GW Ejulu GW Ejulu 152 190 15 1026.6 TRFK 303/577 17 Longjing 43 327 SNP 417 15 1678.52 0.4 [19] Baihaoza 18 Ziyan 176 SNP 131 15 1062.7 9.48 [18] 19 Jinxuan 96 SNP 8956 15 1490.81 0.18 [24] Yuncha1 20 Shuchazao 327 SNP 5325 15 2107.21 0.39 [39] Longjing 43 21 Emei Wenchun 294 SNP 4244 15 1449.19 0.34 [23] 22 Longjing 43 198 SNP 2688 15 1846.32 0.69 [20] Baijiguan 23 Huangdan 148 SNP 3770 15 1754.57 0.47 [21] Jinxuan On the other hand, genome-wide association study (GWAS) is another powerful analysis tool based on the principle of linkage disequilibrium, which is mainly used to mine genetic loci associated with target traits. The basic rationale behind GWAS is to compute the relationship between each marker and a trait of interest that has been scored across unrelated lines or individuals from a broad collection[40]. GWAS does not need to construct a specific hybrid population, using a natural population. The two essential prerequisites in GWAS are genotypes (often based on SNPs) and phenotypes (generally those of distinct attributes). One of the first steps of GWAS establishment is linkage disequilibrium (LD) assessment in the association panel. The LD measure should be used to assess the markers density that is necessary to identify all haplotypes across the genome. GWAS requires selecting tea plant resources with complete phenotypes and abundant genetic variation according to the research needs. In addition, population structure can impact the accuracy of GWAS analysis, so ensuring the accuracy of population structure is necessary to minimize spurious associations[41]. Compared with QTL analysis, GWAS contains a larger number of alleles, which means that GWAS can obtain more gene exchange results[42]. This is also constrained by various conditions, such as the poor correlation between association loci and trait variants detected by GWAS, and the existence of a large number of undetected variant loci. Table 2 outlines the characteristics and differences between GWAS mapping and QTL mapping approach. QTL mapping is carried out using a biparental mapping population.
Table 2. The main characteristics of GWAS mapping and QTL mapping.
QTL mapping GWAS mapping Artificial populations with trait separation Natural populations with rich variation Bi-parental crosses de-novo crossing and selfing Less prone to false positives High possibility of false positives Fewer markers required A larger number of individuals is required Limited number of genotypes and
low allele richnessAbundant alleles but low allele frequency Lower resolution based on the number of recombination High possibility of phenotypic variation More robust in heterogeneity but single genetic basis High map density but low heritability Although the difficulties in GWAS development of tea plants, it is still an important method to detect candidate loci. In addition to the study of candidate genes related to tea plant's growth environment, for example, altitude and region[43]. There is also further exploration of candidate genes related to phenotypic traits such as mature leaf and spring bud germination time[44−47]. A diverse array of work on candidate genes related to important secondary metabolites of tea plants is also underway.
-
Brewed tea quality is basically decided by three kinds of chemical components, i.e., tea catechins, free amino acids (mainly theanine), and purine alkaloids (mainly caffeine), as well as volatile organic compounds. So far, the QTLs mining and candidate genes isolation related to the above main quality chemical components have achieved great progress.
Catechins
-
One of the essentially functional elements influencing tea quality, flavor and the healthy function is catechins, which belong to flavonoids[48], including catechin (C), epicatechin (EC), gallocatechin (GC), epigallocatechin (EGC), epicatechin gallate (ECG), epigallocatechin gallate (EGCG), catechin gallate (CG), and gallocatechin gallate (GCG). The most prevalent of these, EGCG, makes up to 70% of total catechin content (Fig. 2)[49].
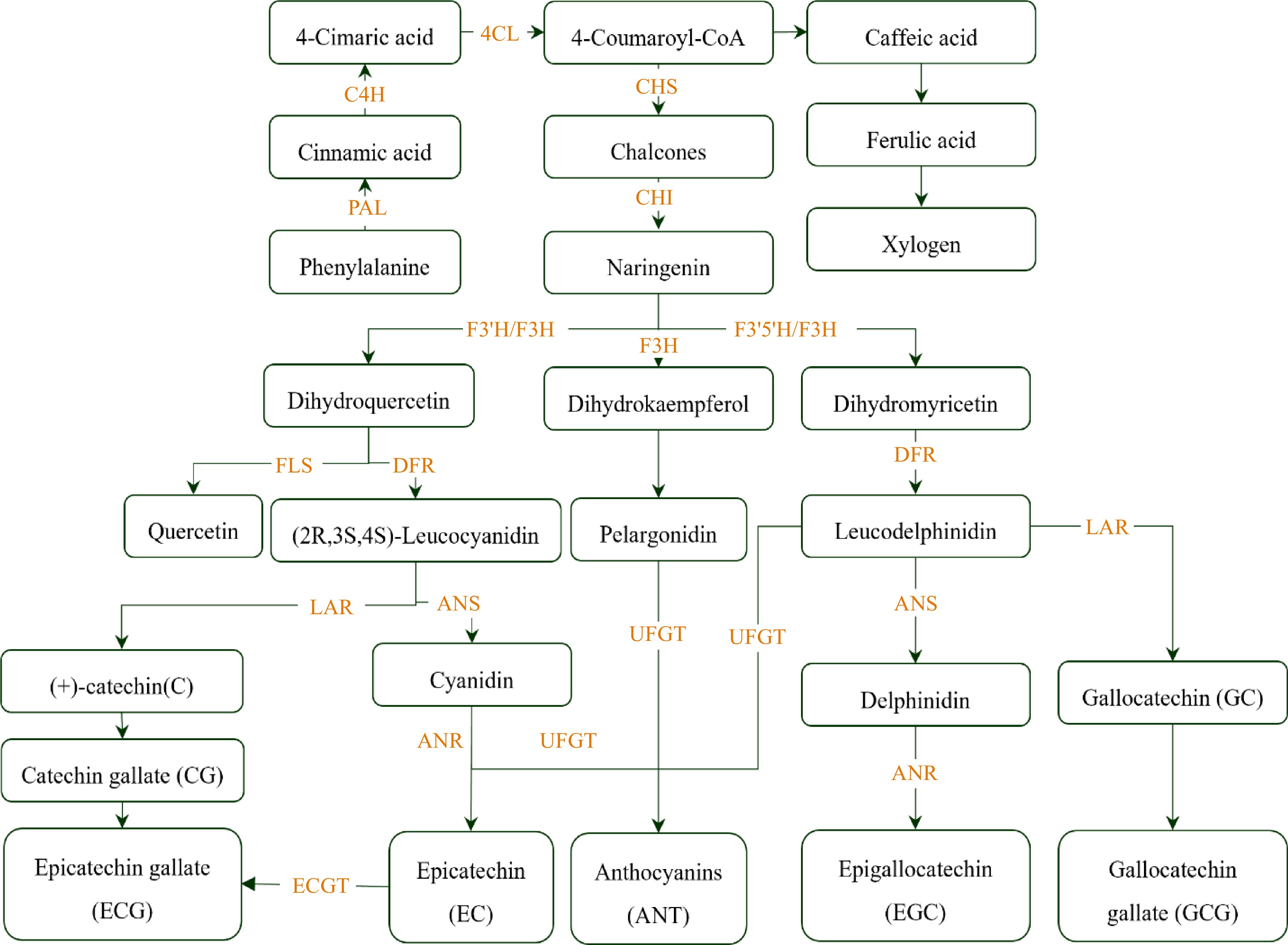
Figure 2.
Schematic presentation of catechin metabolism. PAL, phenylalanine ammonia-lyase; C4H, cinnamate 4-hydroxylase; 4CL, 4-coumarate-CoA ligase; CHS, chalcone synthase; CHI, chalcone isomerase; F3'H, flavonoid 3'-monooxygenase; F3H, flavanone 3-hydroxylase; F3'5'H, flavonoid 3', 5'-hydroxylase; FLS, flavonol synthase; DFR, dihydroflavonol-4-reductase; ANS, anthocyanidin synthase; ANR, anthocyanidin reductase; ECGT, 1-Ogalloyl-b-D-glucose O-galloyltransferase; UFGT, UDP-glucoseflavonoid-3-O-glucosyltransferase; LAR, leucoanthocyanidin reductase; DFR, dihydroflavonol reductase.
Jin et al.[50] discovered four accessions in tea genetic resources with specific total catechins content from 403 local representative tea varieties in China, including one with low total catechins (TC) content (< 60 mg/g) and three with high TC contents(> 200 mg/g), which provided genetic resources for the subsequent exploration of the catechins synthesis pathway. Ma et al.[51] developed 25 EST-SSR makers from 561 microsatellites identified and analyzed their applicability for cross-species/genera amplification and genetic mapping. Later, a total of 25 catechins-related QTLs were found using a moderately saturated genetic map that was created using 406 SSR markers[17]. Among these, nine QTLs on the linkage groups 3, 11, 12, and 15 chromosomal regions were found to be quite stable after years of verification. The map covered a genome length of 1,632.8 cM, with an adjacent marker spacing of 0.1~19.6 cM. An average map distance of 1.5 cM and a linkage group length of 80.2~184.8 cM, laid the groundwork for future investigation of the genes involved in the catechins production pathway. Wang et al.[52] used 'Yingshuang'(YS) and 'Beiyue Danzhu' (BD) as parents to artificially hybridize the F1 population for genotyping. They also constructed a genetic map of the tea plant and QTL analysis was performed subsequently on the catechins components. A total of nine QTLs were detected, mainly distributed among the linkage groups 1, 7, 9, and 12. The genome-wide chromosome-scale assembly was carried out using the 'Shuchazao' genome database, and 64 previously published QTLs papers related to catechins and caffeine were integrated into the chromosome genome[53]. Catechins-related QTLs were concentrated in Chr3, Chr6, Chr7, Chr10, Chr11, and Chr15. Some QTLs were not stable between the two populations. The findings indicated that QTLs in Chr11 may play a significant role in the diversification of Chinese tea genetic resources. The tea plant genome will serve as a reference and a starting point for investigating related gene functions.
Yamashita et al.[54] evaluated the magnitude of the potential of genomic predictions (GP) and GWAS tools in studying secondary metabolites of tea genetic breeding, which provided novel clues for the subsequent wide application of GP and GWAS in genomic-assisted breeding of tea plants. In addition, researchers identified 13 candidate genes that are closely linked to EGCG and caffeine content, which can help to breed new high-quality tea varieties. In view of the relatively few studies on the combination of phenotypic traits, quality components and genome-wide association analysis of tea plants. Hazra et al.[55] used 23 groups of Darjeeling tea for GWAS of agronomic traits and quality components, and finally detected 12 SNPs were closely related to EGCG content. Fang et al.[56] conducted an analysis of the content levels of important secondary metabolites of tea plants in three seasons using GWAS, and the study identified a total of 307 SNP markers. Of these, 209 SNPs were found in all three seasons. After further analysis, 60 SNPs were highly correlated with catechin content.
Amino acids
-
Free amino acids, most importantly theanine, are prominent secondary metabolites that play a vital role in the formation of umami and sweet flavors in tea fusion[57]. Generally, theanine synthesis occurs predominantly in the roots of tea plants and is transferred to new shoots[58]. Theanine is also biosynthesized in leaves, though less so. Its content declines along with the leaf aging[59]. As shown in Fig. 3, the dynamic accumulation of theanine is infected by various external conditions, which include light[45,60], season, temperature, growth cycle, etc[61].
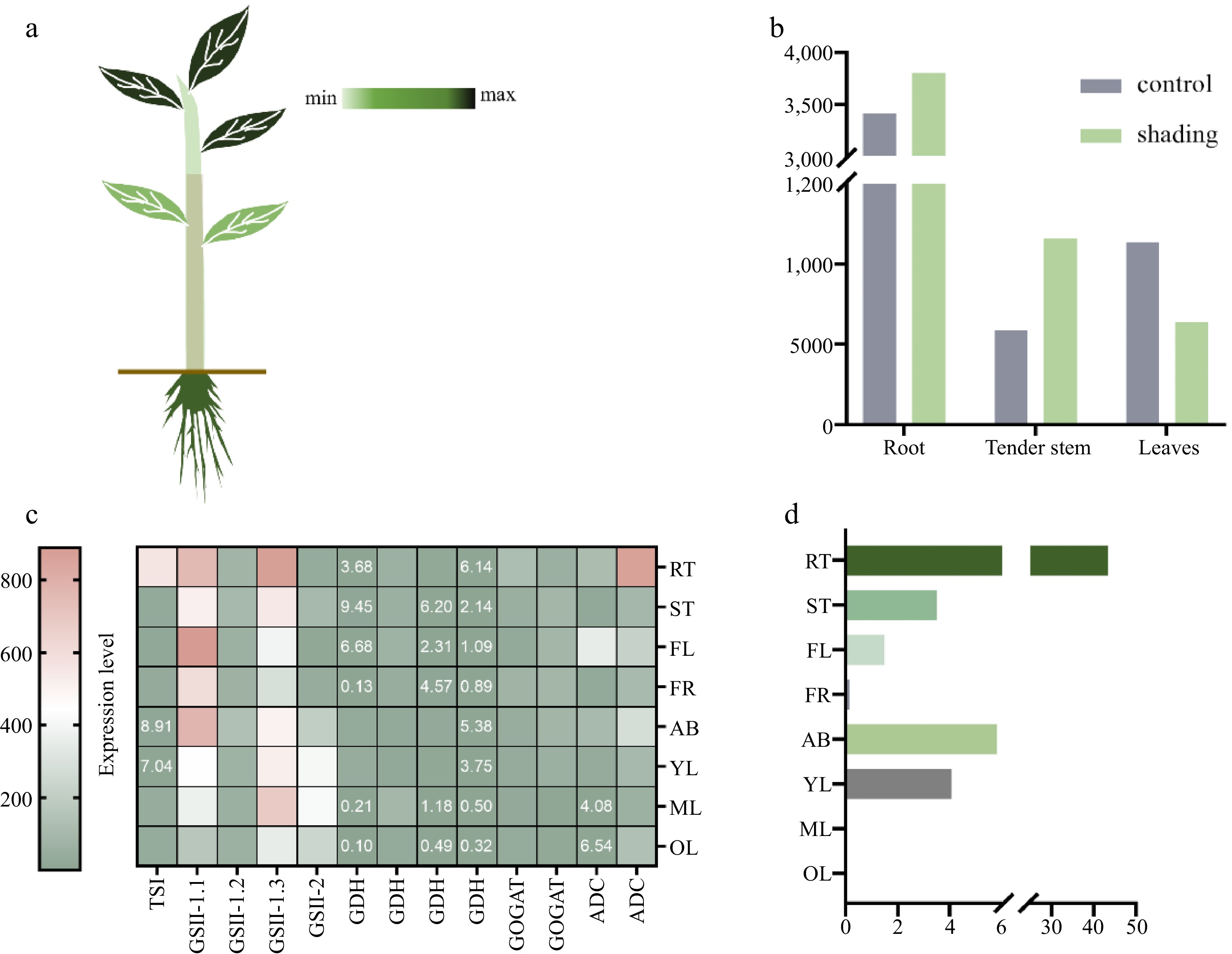
Figure 3.
Effect of different factors on theanine accumulation. (a) Theanine content in different tissues: young leaves > roots > old leaves > stems. (b) Theanine content in different tissues under shading (Data source is from Yang et al.[60]). (c) Expression patterns of candidate genes in different tissues in theanine biosynthesis of cultivar 'Shuchazao': apical buds (AB), young leaves (YL), mature leaves (ML), old leaves (OL), young stems (ST), flowers (FL), young fruits (FR) and tender roots (RT) (Date source is from Wei et al.[1]). (d) The content of theanine in different tissues of tea cultivar 'Shuchazao': The dry weight contents of theanine were detected by HPLC analysis in eight different tissues of tea cultivar 'Shuchazao' (Date source is from Wei et al.[1]).
Li et al.[62] used an artificial mapping population using 'YS' × 'BD' and performed QTL mapping combined with free amino acids. Nineteen QTLs were discovered and spread across seven linkage groups. Among them, CsTS involved in the theanine biosynthesis pathway was mapped to linkage group 03 following cDNA sequence analysis, and the SNPs sites were converted into dCAPS markers, which offered vital assurance for the identification and gene cloning. A high-density genetic map was constructed using 166 F1 populations derived from the cross of 'Baijiguan' and 'Longjing 43' as parents[63]. There were found to be 25 QTLs related to free amino acids, dispersed throughout nine linkage groups, including linkage groups 01, 03, 04, 06, 08, 11, 13, 14, and 15. Several attempts have been made to improve the accuracy of the genetic map. Huang et al.[20] made use of 198 individuals obtained from the same parental hybrid population as test subjects for genotyping to create a high-density genetic map. SNP markers (2,688) were collected, with the range being 93.41 to 171.28 cM. Researchers collected relevant biochemical data for two years, and discovered 18 QTLs for free amino acid content, which were located in the linkage groups 03, 06, 11, and 14, respectively. The confidence interval of the two years was 0.18~48.16 cM. Three stable main-effect QTLs related to theanine (Thea), glutamate (Glu), and Aspartic acid (Asp) on linkage group 03 were verified. They could be used as loci for subsequent molecular marker-assisted selection with the aim of selecting offspring resources and improving tea breeding efficiently.
Purine alkaloids
-
Purine alkaloids in tea plants mainly include caffeine, theobromine and theacrine[64]. Caffeine (1,3,7-trimethylxanthine) is the most prevalent purine alkaloid, and it not only contributes to the distinct taste and flavor of tea fusion but has also been shown to have tremendous health-promoting benefits[65]. Caffeine, another prominent characteristic secondary metabolite in the tea plants, is formed by sequentially methylating xanthosine at the 7-, 3-, and 1-N positions[66]. The primary synthesis pathway of caffeine is confirmed to be xanthosine→7-methylxanthosine→7-methylxanthine→theobromine (3,7-dimethylxanthine) →caffeine (Fig. 4)[67,68].
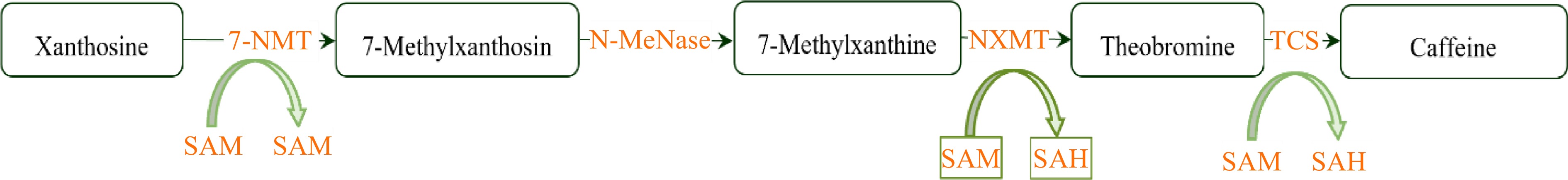
Figure 4.
Schematic representation of caffeine metabolism. 7-NMT, 7-Methylxanthosine synthase; N-MeNase, N-Methylnucleotidase; MXMT, Theobromine synthase; TCS, Tea caffeine synthase; SAM, S-Adenosyl methionine; SAH, S-Adenosyl-L-homocysteine.
Following the continuous determination of caffeine content in 403 tea accessions, six specific genetic resources were successfully screened, including three high-caffeine, one high-theacrine, and two high-theobromine[50]. Ma et al.[36] developed 6,042 valid SNP markers using specific-locus amplified fragment sequencing (SLAF-seq) based on the low-density genetic map. The final genetic map contained 6,448 molecular markers with a total length of 3,965 cM and an average locus spacing of 1.0 cM, which laid a solid foundation for the group's subsequent downstream genetic analysis. The F1 testcross population of 'YS' and 'BD' was used as the experimental material. The quantitative trait QTL mapping of four related traits, including caffeine, theobromine, the sum of caffeine and theobromine, and the ratio of caffeine to theobromine, were performed by Ma et al.[69]. The LOD significance threshold of each trait ranged from 4.8 to 5.5 cM, and a total of 10 major QTLs were detected. Three stable QTLs were verified within two years. Among them, qCAF1 related to caffeine stability was distributed on linkage group 01, with a confidence interval of 39.943~101.543 cM within three years, an overlapping area of 61.459~62.554 cM, and a length of 1.095 cM. Four QTLs related to theobromine (TBR) were also mapped on linkage groups 03, 12 and 13. In addition, Ma also conducted a preliminary study on the relationship between N-methyltransferases (NMT) and QTL. A total of 11 NMTs were found, of which seven distributed among the five linkage groups were detected; the potential location and structure of CsTCS1 (TEA012514) were also preliminarily explored, which provided clues for subsequent function verification.
Theacrine is a special purine alkaloid, which is significantly related to the bitterness of bitter tea, and has various physiological activities such as sedation and hypnosis. The researchers excavated 30 transcription factors related to the regulation of bitter tea metabolism, and speculated that transcription factors belonging to the NAC and HD-ZIP transcription factor families, i,g, were most likely to be key genes involved in the metabolism of theacrine[70].
Volatile components
-
Aroma has a significant impact on the character and quality of tea.Volatile components not only contribute to the aroma of tea, but also play a role in the interaction between tea plants and the external environment. Chen et al.[21] used the constructed high-density genetic linkage map of tea plants to identify 42 QTLs related to volatile components, of which eight were related to the content of monoterpenes and 12 were related to the content of semiterpenes, providing a theoretical basis for the follow-up in-depth study of the molecular mechanism of volatile terpenes.
-
The timing of spring bud flush and young leaf color are important agronomic traits deciding the harvesting time in the year and the tea processing suitability. Mature leaf size and stress resistance are also important traits related to adaptability, tea quality and safety. Their QTLs and related candidates in tea plants have recently made promising progress.
Time of spring bud flush
-
Tea plants are a key commercial crop, and the fresh leaves are its primary production organ. Therefore, the timing of spring bud flush (TBF) which is one of the most important agronomic traits of tea plants, is an essential economic value[47]. Tea plants adapting to low temperatures with early sprouting in early spring can accumulate important secondary metabolites that not only improve its quality but are also beneficial to human health as well as ensuring fewer pests and diseases appreciated by many consumers. Therefore, early TBF varieties have higher market competitiveness and are becoming important breeding targets for tea plants.
It has been proposed that TBF is controlled by multiple genes in perennial trees. Research related to TFB traits in tea plants has also progressed. Tan et al.[22] identified two major QTLs related to TFB, qTBF1-1 and qTBF1-2 on linkage group 01. And in 2018, further time, location, and population validation were conducted on QTLs related to TBF, with testing conduction, and it was found that only 'Longjing 43' experienced QTL separation phenomenon[71]. In 2022, Tan et al.[23] utilized the constructed genetic map to identify five QTLs, among which there were two main effect QTLs, distributed in chr03 and chr04. This laid a solid foundation for the subsequent in-depth study of TBF traits in tea plants.
Young leaf color
-
Tea plants with diverse leaf colors, such as purple and yellow, have the potential to improve human health and are very popular around the world. It is necessary to select tea plant varieties with higher nutritional value. Tan et al.[18] detected 13 QTLs according to the LOD threshold, of which the distribution of QTLs related to anthocyanidin and young shoot color was linkage groups 05, 08 and 13.
Mature leaf size
-
Leaves are the main object of tea plant utilization, which is closely related to yield and has high economic value. If the leaf area is too small, it will affect economic returns, while if the leaf area is too large, it will cause deterioration of the population's lighting conditions and following yield. Therefore, the study of QTL associated with leaf area traits is critical to production value.
Tan et al.[23] detected five QTLs for mature leaf length. There was not QTL for mature leaf width. qMLL9 was detected in all datasets but not always significant at the genome-wide level (p < 0.05). An et al.[24] reconstructed the genetic map using genome sequencing technology and identified 25 QTL markers related to leaf area in the second linkage group of 28−101 cM. The above research related to leaf area provides strong data and a map for the separation of tea plant traits and further functional gene mapping.
Biotic and abiotic resistance
-
The growth of tea plants is facing many adverse environmental factors, such as high and low temperatures, drought, strong light, diseases and pests. Improving tea plant resistance and selecting resistant varieties are of great significance for tea production. Koech et al.[72] used the percent relative water content (%RWC) of tea leaves after the five-hour withering method to evaluate the drought resistance ability of tea plants. They identified a total of three QTLs related to drought resistance, distributed in linkage groups 02, 06, and 09. Xu et al.[37] identified eight QTLs controlling the anthrax trait of tea plants, among which there was a major QTL on linkage group 10, with a LOD value of 6.80 and a 13.8% contribution rate of phenotypic variation.
-
Previously, a lot of basic work on forward genetics have been done, including the construction of the tea genetic map and QTL mapping of quality traits, etc. The QTL intervals associated with the main quality traits of tea plants have been preliminary clarified. In contrast, the genetic mechanism of the quantitative trait of the tea plants is not clear. The results of further identification and verification of key candidate genes through genetic transformation are integrated to provide crucial molecular marker genes for tea plant molecular breeding.
Candidate genes mining
-
Jin et al.[73] found 10 SNPs on the flavonoid 3',5'-hydroxylase (F3'5'H) gene locus that were significantly associated with catechins, and studied the function of SNP-711 / SNP-655 in F3'5'H through gene expression analysis. The two SNPs identified had a strong correlation with the ratio of di/tri-hydroxylated catechins and catechin content, which may affect the transcriptional regulation function of catechins in tea plants. In addition, the team also examined some QTLs closely related to catechins-HOS15[74]. It was also suggested that the specific gene SCPL1A (TEA034034.1) in the tea plant was significant for the gallic acidification of catechins. Furthermore, Jiang et al.[75] discovered a link between a SNP between the chalcone synthase (CHS) gene and catechin content.
Three molecular markers related to caffeine variation were initially identified as TM89, TM211, and TM200[76]. Related studies found that the tea caffeine synthase 1 (TCS1) allele had six different types of allelic variations. Three new full-length cDNA sequences of CSTCS 1 alleles were successfully isolated and identified from specific germplasm resources[77]. The researchers identified 87 SNPs and then developed two CAPS markers: SNP4318 and SNP6252. In addition, SNP4318 related to caffeine content was also found. The site-directed mutagenesis experiment also verified this conclusion.
Tan et al.[23] identified 22 genes as major candidate genes for the five QTLs related to the timing of spring bud flush(TBF) trait, namely qSPI3, qSPI4,mqSPI5, mqSPI9 and mqSPI14. These genes are related to plant hormones, some are related to F-box proteins and DNA methylation, and some are transcription factors. This has a basic role in breeding early TBF tea individuals and further functional gene verification.
Furthermore, GWAS also provides a new means for candidate gene mining. Fang et al.[56] detected seven SNPs connected with theanine using association analysis of SNPs and metabolites, the majority of which were related to amino acid production and metabolism.
Candidate gene verification
-
Wang et al.[63] constructed 16 molecular fingerprints based on the allelic genes of different varieties screened in the laboratory and screened three pairs of primers (TM156, TM508, and MSG0380) for identification of different albino and yellowing tea plant varieties. The researchers continued to use specific primers to screen out the rare allelic variation TCS1g of TCS1. The results of protein activity detection stated that TCS1g had similar functions to TCS1b and TCS1c, both of which had only theobromine activity and no caffeine activity. It was speculated that the amino acid residue at position 221 is the key site that caused TCS1g to have only theobromine activity[78]. Recent research discovered a new TCS allele, TCSh, which had both theobromine synthase and caffeine synthase (CS) activities. CsTCS activity was determined by the 225th and 269th amino acid residues. The promoter activity of TCS1e and TCS1f were low. In addition, the team also identified a key cis-acting element, which laid a solid foundation for further study of TCS1. Based on the previous research results, nine TCS1 alleles were found, and three functional models were constructed (Fig. 5)[79]. Fang et al.[56] used a combination of GWAS and metabolite association analysis to uncover 80 SNPs associated with caffeine content and partially validate them in a tea plant population. Eight of the 30 SNP markers used for validation were associated with caffeine.
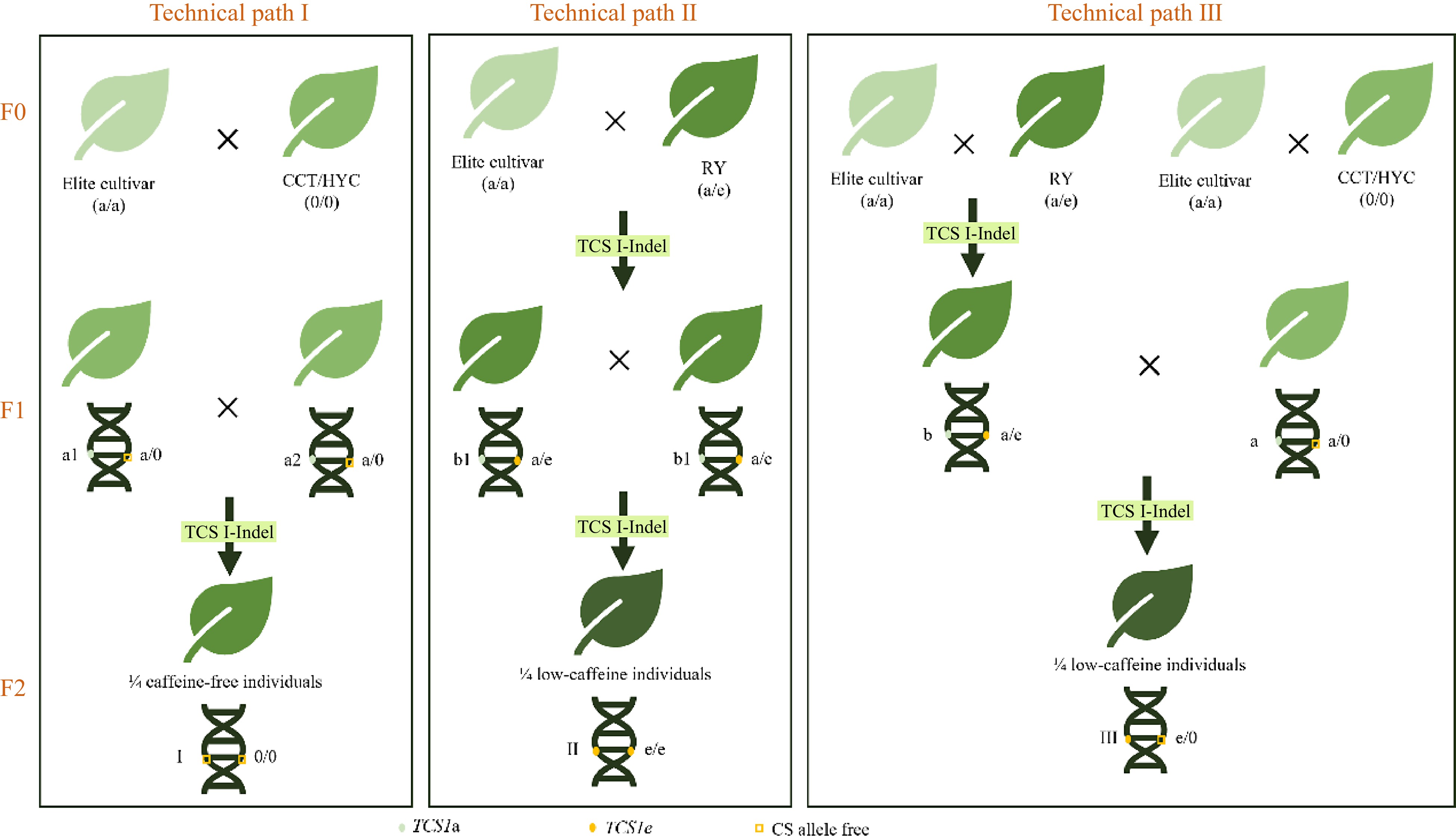
Figure 5.
Route map of three low caffeine tea plant breeding technologies. CCT, 'Cocoa tea', a caffeine-free tea plant from Guangdong, China; HYC, 'Hongyacha', a naturally caffeine-free tea plant from Fujian, China; RY, 'Ruyuan', which contains low caffeine. (Data source is from Wang et al.[81]).
Additionally, functional studies of candidate genes related to agronomic traits have also made some progress. Most recently, Liu et al.[80] discovered a candidate gene, CsDREB17, encoding the AP2/ERF transcription factor of the main QTL qTBF4-1 related to the timing of spring bud flush of tea plants. They demonstrated that it is a negative regulatory factor controlling the TBF of tea plants, providing a reference for further exploring the early growth mechanism of tea plants. Wang et al.[81] used SNP markers to successfully locate CSAN1 on LG08 linkage group. Combined with the results of the team's QTL mapping, it was found that the gene coincided with the main QTL related to the color of tea shoots. In the same year, Wei et al.[82] combined the transcriptome and genome and reported that the QTL mapped as bud leaf color (BLC) : CsMYB75 and CsGSTF1 play a key role in the synergistic effect of anthocyanin hyperaccumulation. Of course, there are still some candidate genes that have not undergone functional verification that need further exploration by researchers (Supplemental Table S2).
-
Although the global tea industry is showing a thriving trend, the lack of basic biological research, for example, the synthesis, metabolism and transport of secondary metabolites in tea plants should not be overlooked. The breeding efficiency is still low and the selection of varieties is difficult. It is well-recognized that the genome of tea plants such as 'Yunkang10'[83], 'Shuchazao'[1,3], 'Biyun'[84], 'DASZ' (wild relative)[85], 'Longjing 43'[9], 'Huangdan'[86], 'Tieguanyin'[87], and 'Duyun Maojian'[88] have been published one after the other. The use of high-quality genomes is becoming increasingly common, therefore choosing a suitable genome is a vital and wiser choice. Choosing a genome that meets quality control and genotyping requirements is more cost-effective. TMDB[89], TPIA[48], TeaCoN[90], TeaMid[91], TeaGPIN[92], TeaPGDB[93], TeaAS[94], TeaGVD[95] and TeaPVs[96] were established. Meanwhile, multi-omics comprehensive databases-Teabase, which provide a mass of open-access data for Camellia, were also published[97] (Fig. 6). In comparison to other crops, such as rice[98,99], the quality and integrity of the tea plant genome still remain severely limited. Further research into the pan-genomes of diverse tea plants types, as well as systematic and in-depth studies on tea plant genomes, are required.

Figure 6.
Timeline of research on tea plant genome databases and platforms.
Clearly, genetic resources are the key to tea genetic improvement. Tea genetics research is significant for both practical and economic reasons. Although tea plant biotechnology breeding emerged relatively late, it has achieved stages of vigorous development resulting in molecular assisted breeding technology, such as constructing increasingly high-quality genetic maps, accurately locating QTL loci closely related to important tea plant traits, and applying GWAS technology based on resequencing.
Because of the biological properties of the tea plants, QTLs mapping is a difficult approach for studying complex traits. Currently, research on the quality traits has achieved considerable success, with the discovery and verification of several molecular markers that are closely connected to the desired traits (Supplemental Table S1). In addition, molecular markers related to agronomic traits and stress tolerance are also of great significance for genetic breeding research in tea plants. However, there are still many markers with relatively low polymorphism. The developed molecular markers rely more on the SSR markers system, resulting in the newly developed QTL being limited to specific hybrid populations. Furthermore, the double-threaded development of molecular markers and breeding efforts limit the practicability of target trait genes to objective factors including environment and genetic background. In the future, the consistency and sharing of resources such as phenotypic data, metabolic data, germplasm resource numbering and resequencing data are very important for the development of future breeding. The good news is that tea genetic breeding work could refer to model crops, such as the rice 3K database, and then the suitable genetic breeding system is gradually developing[96].
Tea plant GWAS research is thought to have great development potential, which has provided novel clues for enhancing the pace of research on the complex traits of tea plants (Fig. 7)[100]. But there's no denying that most traits in polyploid tea plants are complex, and the loci currently detected by QTL and GWAS explain are only a small fraction of the phenotypic variation that is much smaller than the heritability of the traits. More in-depth studies are needed in the future to minimize the loss of genetic loci. Additional study in the following areas can be performed to solve the aforementioned difficulties.
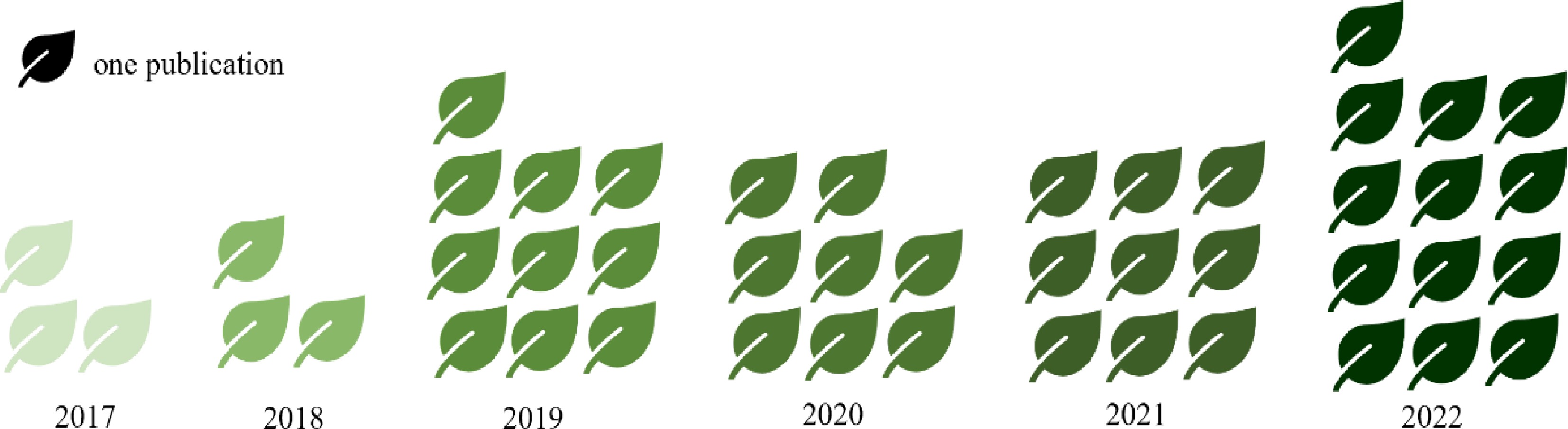
Figure 7.
Growing number of articles on tea plant GWAS. The number of articles is implied by the amount of tea plants. All data were obtained from the web of science (
www.webofscience.com ) by searching the terms 'tea plants' and 'genome-wide association study'.(1) The metabolic mechanisms of target genes need intensive study, including upstream and downstream metabolites and the relationship between product biosynthesis and transport. Furthermore, the sample size should be expanded and the co-expression network of trait coupling should be accelerated. Researchers need to devote attention to studying the relationship between genes and clarifying the regulatory mechanism with the aim of promoting the discovery of candidate genes related to important traits of the tea plants.
(2) To make full and rational use of the different tea genetic resources and improve the diversity of genetic breeding materials. Increasing the number of populations and shortening the interval of population markers can also effectively improve the quality of QTL mapping[101]. In addition, it is indispensable to choose the appropriate QTLs analysis method. In order to comprehend the biochemical pathways underlyingly complex characteristics, emphasis should also be placed on the identification of interacting QTLs. Additionally, reasonable use of various computer systems can significantly increase the effectiveness and accuracy of QTL analysis.
(3) Emphasis on the application of high-throughput phenotyping technology in tea phenotype evaluation to overcome the problems of poor accuracy and unstable data caused by manual measurement[102]. Obtaining high-quality phenotypic data and then combining it with genome sequencing to obtain more valid genetic loci and genes.
(4) Combining multi-omics data including transcriptome and metabolome for metabolite genetic variation and genome-wide association analysis to solve the problem of low genetic potency of tea plants. Zheng et al.[103] established gene co-expression networks (GCN) based on the results of weighted correlation network analysis (WGCNA) and constructed a regulatory network to reveal catechin biosynthesis. They concluded that new uncharacterized TF families such as WRKYs, SBPs, and MADSs have potential regulatory effects on catechin biosynthesis. In the future, multi-omics association analysis can also be established at the RNA level to further reveal the regulatory network of target traits.
(5) On the basis of high-throughput genome sequencing, we are supposed to focus on the development of new breeding techniques, including the establishment of tea plant transformation systems, the application of crisper technology, and molecular design breeding[104]. Additionally, we also need to pay attention to the analysis, processing, management, and access of meaningful data[7].
-
Conceptualization and writing and figure preparation, ZH Wang; review and editing, R Huang, S Ercisli, DG Moon; supervision, L Chen. All authors reviewed, read, and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (U19A2030, 32072631), the China Agricultural Research System of MOF and MARA (CARS-019), and the Chinese Academy of Agricultural Sciences through the Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2021-TRICAAS) to LC.
-
The authors declare that they have no conflict of interest. Liang Chen is the Editorial Board member of Beverage Plant Research who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and his research groups.
- Supplemental Table S1 The genetic and physical details of catechin-, caffeine-, theanine and agronomical- related QTLs.
- Supplemental Table S2 The genetic and physical details of catechin-, caffeine-, theanine and agronomical- related QTLs potential genes.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang Z, Huang R, Moon DG, Ercisli S, Chen L. 2023. Achievements and prospects of QTL mapping and beneficial genes and alleles mining for important quality and agronomic traits in tea plant (Camellia sinensis). Beverage Plant Research 3:22 doi: 10.48130/BPR-2023-0022 

Achievements and prospects of QTL mapping and beneficial genes and alleles mining for important quality and agronomic traits in tea plant (Camellia sinensis)
- Received: 27 July 2023
- Revised: 08 August 2023
- Accepted: 24 August 2023
- Published online: 07 September 2023
Abstract: Tea is one of the most significant non-alcoholic beverages globally due to its unique secondary metabolites. Therefore, it is essential to apply molecular technologies in conjunction with various phenotypes for candidate gene mining and identification, regulating the synthesis and degradation of secondary metabolites contributing to tea quality, in order to enhance effective tea breeding. To date, there are various tea genetic resources and numerous high-density genetic maps owing to the progress and development of the tea plant genome. In this review, we comprehensively reflect the mining and identification of quality-related candidate genes using quantitative trait loci (QTL) mapping and genome-wide association study (GWAS) in tea plants in recent years. Functional verification and promotion of these candidate genes were also discussed.