









-
Panax ginseng is a perennial herb, which is widely used in medicine in Asia and Europe. It has always been one of the most valuable traditional medicinal herbs in China though mostly cultivated for medicinal demand in the commercial market for the past two decades(Jin et al., 2022). However, the cultivation of ginseng in farmlands is often challenged by soil borne fungal diseases, including ginseng root rot disease, ginseng rusty root rot and ginseng black spot disease, seriously threating root yield and quality (Eo et al., 2013; Liu et al., 2022). Causing a root yield loss of up to 80%, the ginseng root rot disease appears with high incidence, is contagious and hard to detect (Lee, 2004; Kim, 2012; Ryu et al., 2014; Deng et al., 2023). This disease is caused by infection of fungal pathogens such as Fusarium solani, Cylindrocarpon destructans, Phytophthora cactorum and Pythium ultimum (Lee, 2004; Eo et al., 2013; Wang et al., 2016), frequently identified in infected ginseng root or the rhizosphere, particularly in replanted fields (Lee, 2004). While much attention has been given to the origin and infection in cultivated ginseng production, developing quantitative detection and assessment of the pathogenic microbes is becoming urgent for safeguarding ginseng production.
So far, a number of laboratory protocols have been reported such as the plate count method (Deng et al., 2023), machine vision (Harakannanavar et al., 2022), infrared thermal imaging technology (Zhu et al., 2018), spectroscopy method (Shin et al., 2023) and molecular biological identification method (Li et al., 2020; Almario et al., 2013; Zhang et al., 2023). Among these, the plate count method is used widely for classical direct and quantitative identification of the pathogenic fungal species. However, the quantification was impacted by the experiment proficiency of the lab operators and the variability of the soil and field conditions (Li et al., 2022). Moreover, plate cultivation with artificial medium often fails to recognize specific soil microorganisms potentially associated with the disease (Chen, 2013). To overcome such shortcomings, polymerase chain reaction (PCR) could detect nucleic acids of target genes without cultivation of pathogens and thus allow reliable quantification of the pathogenic microbes (Renvoisé et al., 2013; Kreitmann et al., 2023). Accordingly, novel real-time quantitative polymerase-chain-reaction (RT-qPCR) has been widely used in pathogen detection and biomedical diagnostics (Pabinger et al., 2014). As a new generation of absolute quantitative PCR technique, droplet-based digital PCR (ddPCR), independent of standard curve, has been proven to be of high sensitivity, precision, and reproducibility (Hindson et al., 2013; Hou et al., 2023). Compared with RT-qPCR, ddPCR decreases interference from non-target nucleic acid fragments, and the results are less affected by amplification efficiency and PCR inhibitors (Hou et al. 2023). From the perspective of practical application, ddPCR has more application value for early diagnosis and ongoing monitoring of diseases.
In this study, we developed a specific ddPCR method to detect and quantify the gene abundance of F. solani. By testing a number of samples, we compare the sensitivity and accuracy with that of the classic RT-qPCR method. In addition, we assess the applicability of the method, by statistical correlation of the root rot disease incidence of ginseng plants with the abundance of the pathogens quantified for the root-zone soil. The objectivity is to provide a reliable and feasible tool for the detection of F. solani in ginseng cultivation, so as to provide a quick and precise diagnosis of pathogen infection for early warning of ginseng diseases.
-
The positive strain used was Fusarium solani while the negative strains included Sclerotinia ginseng, Ilyonectria robusta, Alternaria panax, Botrytis cinerea, Fusarium oxysporum, Fusarium graminearum and Fusarium moniliforme.
Ginseng seedlings (3-year old) and soil
-
The ginseng seedlings (3-year old) were provided by the ginseng seedling breeding base with Jilin Shenwang Plant Protection Technology Co. Ltd (Jilin, China). Used for pot experiment was ginseng-harvested topsoil (brown loam) collected from the Experiment Base of ginseng planting of Jilin Agricultural University, located in Choushui Village, Fusong County, Jilin Province, China (127°5′12.08″ N, 42°23′48.16″ W). After the harvest of ginseng, the soil was plowed twice at a depth of 30 to 40 cm, each interval was 7−10 d, and the missing ginseng roots and weeds were removed during the ploughing process. The soil was left for a winter, then taken for experiment after thawing in April of the following year. Its basic properties were as follows: soil pH of 5.57, available nitrogen of 29.05 mg·kg−1, Olsen-phosphorus of 107.53 mg·kg−1 and available potassium of 170.56 mg·kg−1 as well as organic carbon content of 3.17%.
Primers and probe
-
The ITS sequence was used as the target genetic location for detecting only in F. solani. Primers and probe sequences were generated by Primer Express® Software (Ver 2.0, PE Applied Biosystems). The amplify fragments was consistent with the conserved sequences from GenBank when compared by Vector NTI software. Theoretical specificity of the primers was examined by Primer-BLAST on the National Center for Biotechnology Information (NCBI) website (
www.ncbi.nlm.nih.gov/tools/primer-blast/ ), the comparison results showed that the primers in this study could only amplify F. solani, and the probe was labeled with Fluorescein (FAM). Primers and probes were synthetized by Sangon Biotech, Shanghai China (Table 1). The same primers and probe were used in both ddPCR and RT-qPCR reactions.Table 1. Detailed information of primers and probes used in this study.
Gene primer/
probeSequence (5′-3′) Product
size (bp)ITS forward GGAACAGACGGCCCTGTAA 149 reverse TTTCGCTGCGTTCTTCATCG probe CCGCCAGAGGACCCCTAACTCTGTT Optimization of the ddPCR method
-
The ddPCR method was performed in accordance with the manufacturer's instructions (Solarbio, China),in a 20 μL reaction volume included 10 μL of QX200TMddPCRTMEvGreen Supermix, 0.1 μL of each forward and reverse primer (10 μM), 0.1 μL of probe (10 μM), 9.2 μL of ddH2O and 0.5 μL of DNA template. The reactions were optimized for probe concentration (1,000, 750, 500 and 250 nM). The reaction mixture (20 μL) for each sample was loaded into a well of a disposable DG8™ cartridge (Bio-Rad, USA) and 70 μL of Droplet Generation Oil (Bio-Rad, USA) were placed into each of the adjacent oil wells in the cartridge (Bio-Rad, USA). Droplets were produced in each well using a QX200™ droplet generator (Bio-Rad, USA). The droplets were then transferred to a 96-well PCR plate (Bio-Rad, USA). To differentiate the amplitude between the negative and positive droplets and to reduce the background of the negative droplets, we performed a temperature gradient in the annealing step. The ddPCR amplifications were performed with an initial step of 94 °C for 5 min, followed by 50 cycles of 95 °C for 30 s, 64 °C for 60 s and 1 cycle of 98 °C for 10 min, with a final hold at 4 °C. To achieve the best results for the method, a range of annealing temperatures (57 to 64 °C) was tested.
After reaction, the microdroplets from each well were read individually using a QX200 Droplet Reader (Bio-Rad, CA, USA). A threshold was set between the positive and negative microdroplet clusters and the copy number of each well was evaluated with QuantaSoft™ version 1.7.
Optimization of the RT-qPCR method
-
The RT-qPCR was performed using 2× Taqman PCR MasterMix Kit (Solarbio, China) in a volume of 20 μL containing 10 μL of Fast Probe Mixture, 0.1 μL of each forward and reverse primer (10 μM), 0.1 μL of probe (10 μM) and 9.2 μL ddH2O and 0.5 μL of DNA template. The reactions were set up for the ABI7500 (Thermofisher), and amplification programmes were as follows: an initial heating 94 °C for 5 min; followed by 40 cycles of denaturation at 95 °C for 30 s, primer annealing at 64 °C and elongation at 64 °C for 1 min. To achieve the best results for the method, a range of annealing temperatures (57 to 64 °C) were tested and optimized for probe concentration (1,000, 750, 500 and 250 nM).
Analytical assessment of the method proficiency
-
To determine the specificity for F. solani and the cross-reactivity with other microbes, DNA from F. solani, S. ginseng, I. robusta, A. panax, B. cinerea, F. oxysporum, F. graminearum and F. moniliforme were tested. Double-distilled water was used as the negative control.
To evaluate the sensitivity, a 10-fold serial dilution of the DNA (10 to 10−5 ng·μL−1) from the positive sample was used with the assays performed using the optimized conditions as described above. The detection limit was determined as the last serial dilution that gave a positive result.
For evaluating inter-assay reproducibility, each dilution was tested in triplicate and in three independent runs with the RT-qPCR method, and in octuplicates and in eight independent runs with the ddPCR method.
Quantitative analysis in artificially infected samples
Preparation the spore suspension of F. solani
-
The strain of F. solani was stored at −80 °C. After rejuvenescence of the strain, its mycelium was inoculated separately in PDA culture-medium under aseptic conditions. The plates were incubated in artificial climatic chamber at 25 ± 3 °C until the plate was covered with strain. Mycelium were collected and transferred to bottle with a sodium carboxymethylcellulose solution (6 g·L−1), kept shaking for 20 min. The solution was filtered and diluted to a concentration of 3 × 104 spores mL−1 with sterile deionized water.
Quantitative analysis
-
To draw the correlation of the disease incidence of cultivated ginseng with the quantified abundance of F. solani in the soil, a pot experiment with the artificially infected pathogens was conducted in the shade greenhouse within the campus of the Jilin Agricultural University. Three randomly selected ginseng seedlings (3-year old) were transplanted in a pot (23 cm in diameter and 21 cm in height each, with a total of 150 pots) filled with the collected ginseng soil on May 3, 2020. Following growth for 60 d, 90 pots with similar sized ginseng plants were selected and divided into three groups (30 pots each group) subject to further treatments. For Group A with disease induction only, the spore suspension above mentioned was added in the pot soil once at a dosage of 50 ml per plant; for Group B with pesticide control on induced pathogens, a suspension of carbendazim diluted 250 times was further added at a dosage of 50 ml per plant, 3 d following the pathogen induction as for Group A. In addition, Group C was set up as the control, where neither pathogens nor pesticides added. The pot experiment was consistently managed with the standard protocol (GB/T 34789-2017) for ginseng cultivation (Jilin Ginseng and Pilose Antler Management Office et al., 2017) for weed control, irrigation and shading.
A soil sample (3−5 g) of the ginseng root zone in a pot was collected respectively at 3 d, 5 d, 10 d, 20 d, 30 d and 40 d following the treatment on July 15, 2020. These samples were sealed in ice boxes and stored at −80 °C prior to the molecular assays. While soil sampling, the incidence of ginseng disease was observed and recorded based on the symptoms of yellowing and wilting above-ground parts, and the dark brown, hollow and rot roots. The incidence status of ginseng is generally expressed with morbidity, which is the percentage of the number of plants with the symptoms to the total number of plants in a pot.
Quantitative analysis in field samples
-
For evaluating efficacy of the developed method of ddPCR, 40 rhizosphere samples under varying incidence conditions were collected, as per the method described by Liu (Liu et al., 2022), from the ginseng planting base of the Choushui Township, Fusong County, Jilin Province, China. The rhizosphere samples covered healthy ginseng plant and infected plant with root rot, rust rot, black spot or gray mold with a disease severity index above 3 (Fang, 1998; Jilin Ginseng and Pilose Antler Management Office et al., 2017). In addition, four independent topsoil samples for each category of disease infection were collected respectively in August and September when ginseng is actively growing. All samples stored in sampling kits were shipped to the laboratory and tested with the ddPCR protocol described above within 24 hr following sampling. Each sample was analyzed in triplicate.
Statistical analysis
-
All data were expressed as the means plus/minus one standard deviation of a treatment plot and processed with DPS 9.50 and Origin 2018 software. Statistical analysis of variance was performed with ANOVA, LSD methord using SPSS software (Version 20.0). A difference among treatments or a correlation between analyzed parameters was defined as significant at p < 0.05.
-
Specific primers and probes were used in the RT-qPCR protocol. For the first step, different probe concentrations (250, 500, 750 and 1,000 nM) were assessed using the RT-qPCR method. The results showed that the optimal concentration was 500 nM. The optimal annealing temperature for the RT-qPCR method was identified by testing temperatures of 64, 63, 62, 61, 60, 59, 58, and 57 °C. As shown by the results, an annealing temperature of 64 °C gave the best amplification effect.
Finally, an optimal probe concentration of 500 nM and an annealing temperature of 64 °C were the operation conditions for the RT-qPCR method established for F. solani, and the condition was applied to the ddPCR method.
To evaluate the specificity of the ddPCR and RT-qPCR method, DNA from F. solani, S. ginseng, I. robusta, A. panax, B. cinerea, F. oxysporum, F. graminearum and F. moniliforme were tested. As shown in Fig. 1a, the DNA primers were tested for amplification with repeated cycles up to 40 with the RT-qPCR method. Whereby, only F. solani DNA samples appeared positive with the fluorescence amplification (dR) whilst the negative control and samples containing all the other pathogens tested negative response with amplification. The amplified products were sequenced by Sangon Biotech, Shanghai China, and compared with the known species sequences in NCBI database, the results showed, that this 149 bp length gene sequence appears only in F. solani and is 100% homologous to the target gene sequence. In Fig. 1b of the ddPCR method, F. solani DNA samples exhibited concentrated Cha amplitude signals across A04-C04 while those of negative strains displayed few but scattered signals across D10~H11. Clearly, both the ddPCR and qPCR methods were shown to be specific for the detection of F. solani.
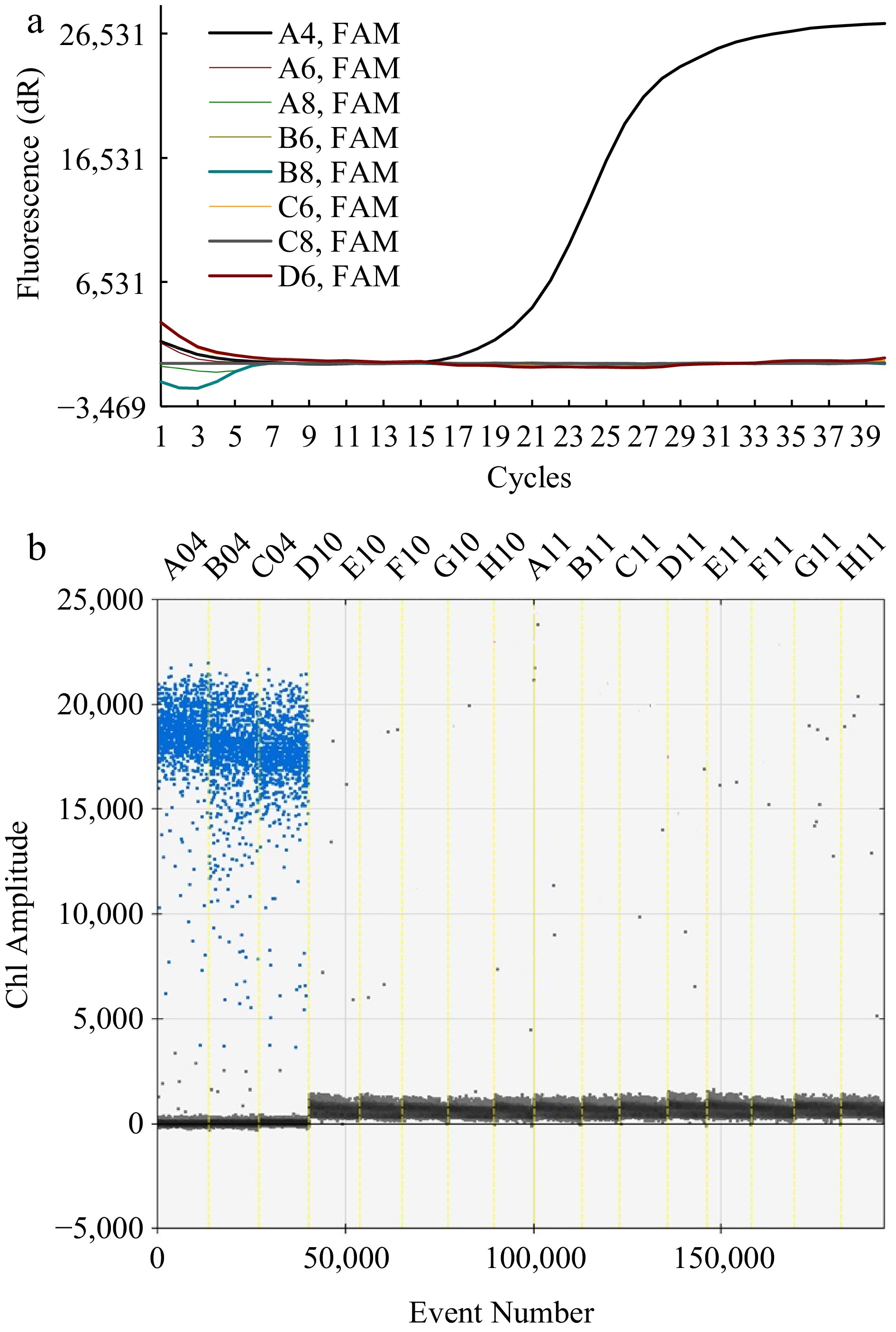
Figure 1.
Specific detection of primers used for the RT-qPCR and ddPCR methods. (a) Fluorescence intensity in response to amplification cycles with RT-qPCR methods. The curve with amplification was for A4 (F. solani) and that without amplification was for the negative strains (A6~D6). (b) Ch1 amplitude in response to event number using the ddPCR method. The blue signals concentrated across A04, B04, and C04 reflected the DNA of F. solani while those scattered across D10~H11 represented the DNA of the negative strain, including S. ginseng (D10, E10), I. robusta (F10, G10), A. Panax (H10, A11), B. cinerea (B11, C11), F. oxysporum (D11, E11), F. graminearum (F11, G11), and F. moniliforme (H11).
Based on the data shown in Fig. 2a, the relative standard deviation (RSD) reached 0.31% for the tests among the three independent runs in the RT-qPCR method. Comparably, a RSD of 0.35% was obtained for the tests among the eight runs with the ddPCR method (Fig. 2b). These results indicated a high precision of the determination of both assays, being mutually comparable in quantitative determination of F. solani in rhizosphere.
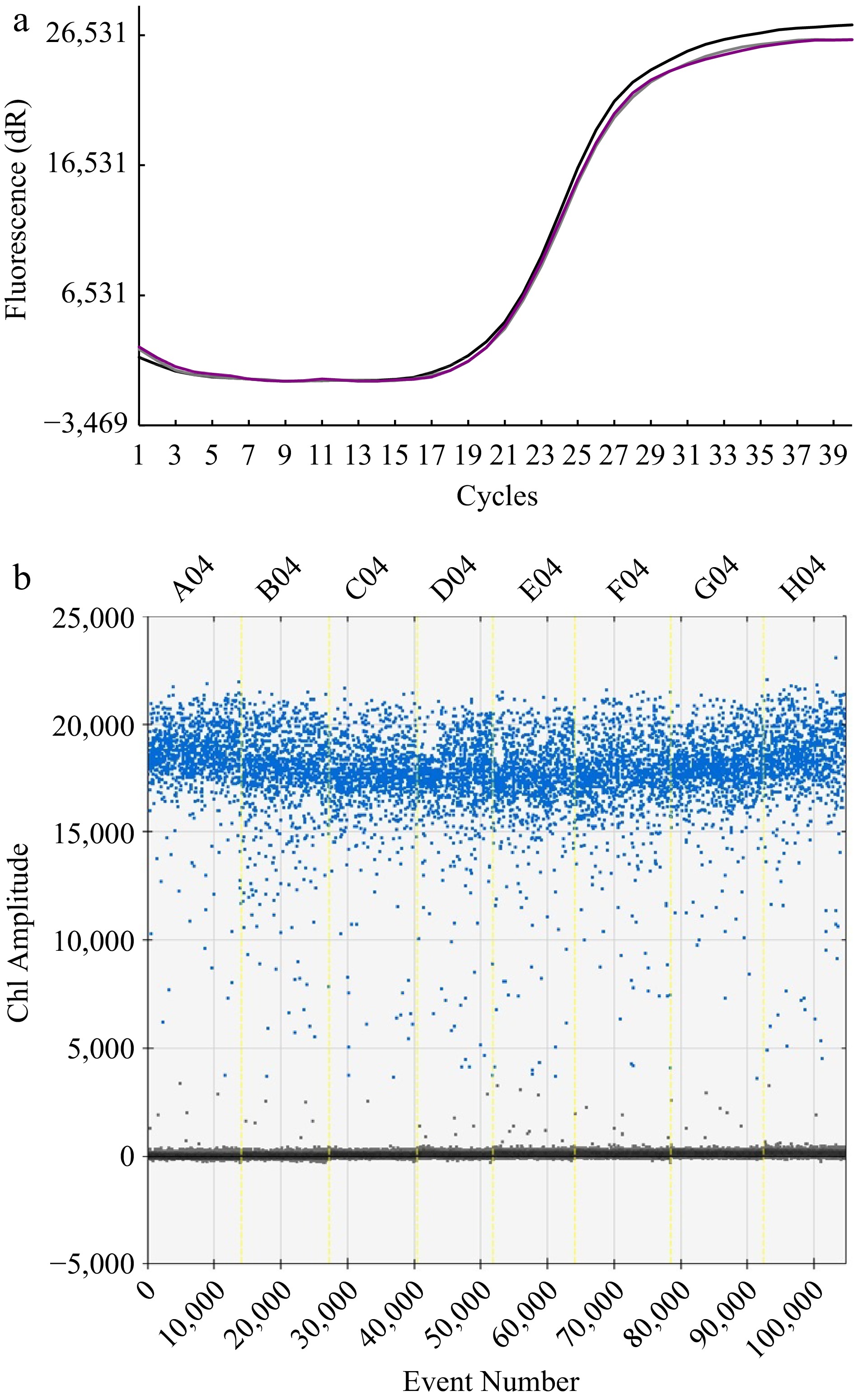
Figure 2.
The test for reproducibility of the methods. (a) Florescence response to repeated cycle of amplification with RT-qPCR method. Note that the curves of three independent runs are almost consistent. (b) Ch1 amplitude response to event number.
Based on the curves in Fig. 3a, sharp fluorescence response was observed at a DNA concentration of 10−2 ng·μL−1 (the third curve on the left), relevant to a gene abundance of 920 copies μL−1. With the ddPCR method, whereas, discernible amplitude signals could appear at a concentration as low as 10−5 ng·μL−1 (relevant to a gene abundance of 0.92 copies μL−1) though very strong at concentrations above 10−3 ng·μL−1. The established F. solani-specific ddPCR method was able to detect very low concentrations of template DNA, being much more sensitive than the RT-qPCR method.
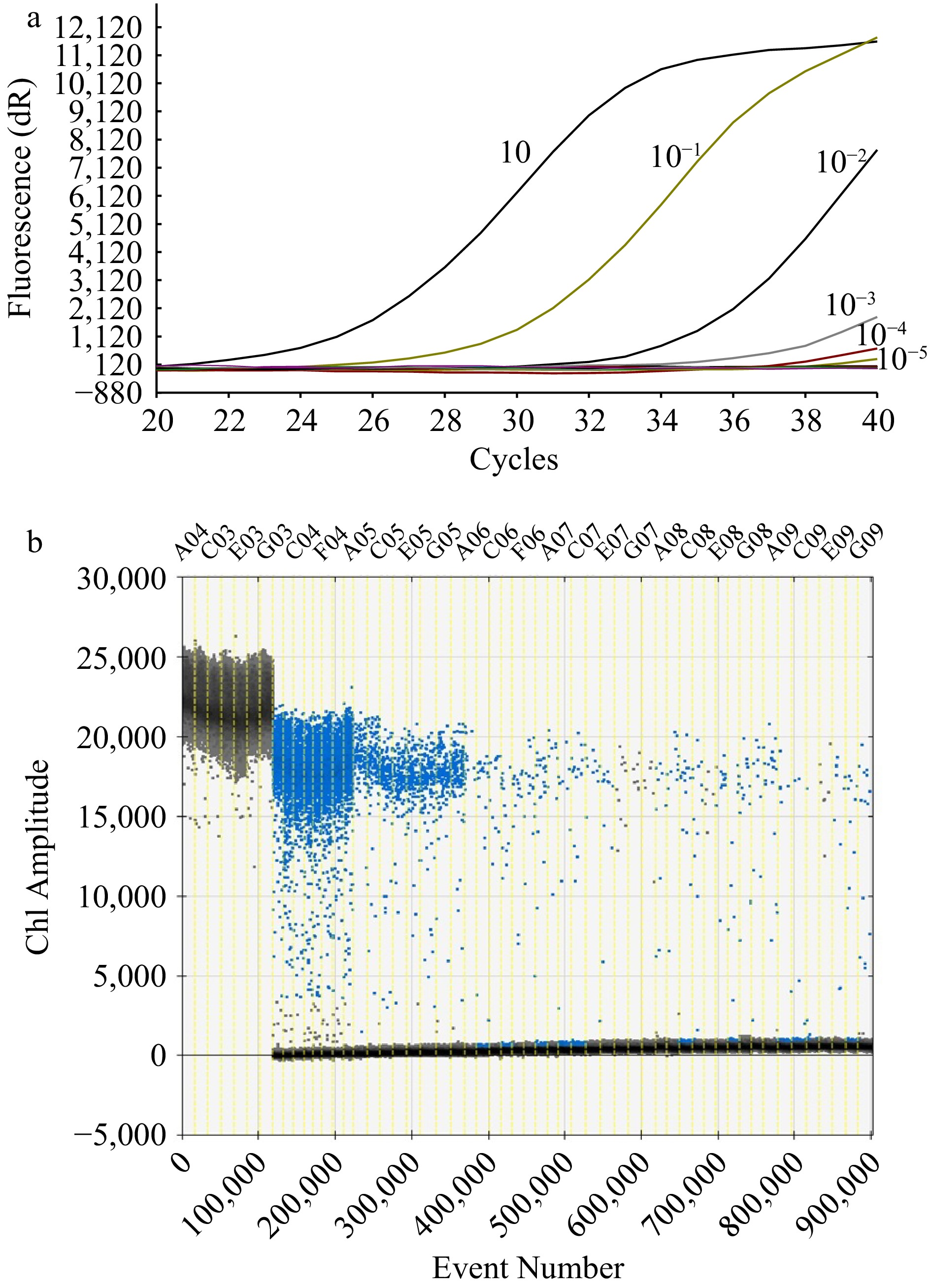
Figure 3.
Sensitivity test of the methods. (a) Amplification curve with RT-qPCR method of a concentration of 10, 10−1, 10−2, 10−3, 10−4, 10−5 ng·μL−1 respectively from left to right. (b) Ch1 amplitude response to event number with the ddPCR method. The black signals represented the positive strain (A03, C03, E03, G03) while the blue signals in the regions represented the concentration respectively at 10 (C04, F04), 10−1 (A05, C05, E05, G05), 10−2 (A06, C06, F06), 10−3 (A07, C07, E07, G07), 10−4 (A08, C08, E08, G08), 10−5 ng Μl−1 (A09, C09, E09, G09).
Quantification of F. solani and disease incidence with Ginseng in pot culture
-
With the established ddPCR protocol, the dynamic abundance of F. solani in the soil of the pot experiment was quantitatively traced (Fig. 4, Supplemental Fig. S1). For the control group (Fig. 4a), the gene abundance of F. solani gradually increased from 927.5 to 8,470 copies g−1 over the whole 60 d growth. Whereas, a bi-model abundance dynamic was found for the disease induction group, with which the first peak was 23,467.5 copies g−1 at 10 d after transplantation while the second peak was 62,765.5 copies g−1 at 40 d after transplantation respectively. With the disease induction plus pesticide treatment, the gene abundance of F. solani was similar to the control though significantly higher at 10 d after transplantation than the control. In other words, the gene abundance of the pathogenic fungus was seen decreased by 38% following the pesticide addition over that before the pesticide addition. The established ddPCR method thus allowed a clear but sharp differentiation in the gene abundance between the disease treatments in the pot experiment.
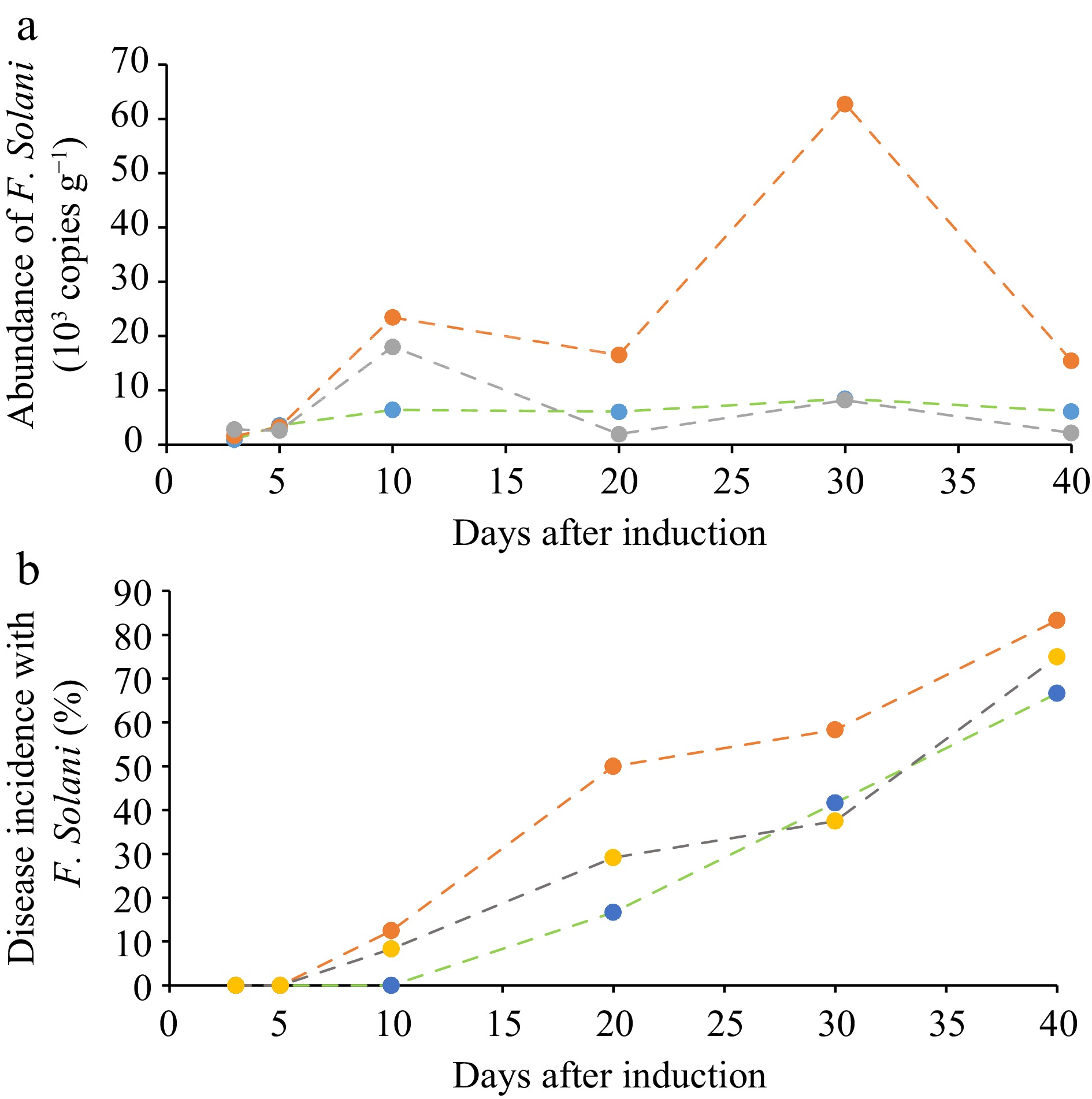
Figure 4.
Quantitative dynamics of F. solani in ginseng root zone. (a) Observed with the developed ddPCR method and (b) the disease incidence rate (%) of ginseng plants in the pot experiment. Green line, control; Red line, the disease induction treatment; Gray line, disease induction plus pesticide treatment.
The disease incidence dynamics was portrayed in Fig. 4b. The disease incidence of the ginseng plants with disease induction treatment appeared at 10 d following the transplantation, when no incidence occurring in the control treatment. At 110 d after transplantation (40 d after root disease incidence treatment), the incidence of the pathogenic disease reached 83.3%, 75.0% and 66.7% respectively for disease induction treatment, disease induction plus pesticide treatment and the control. This was correlated significantly (r = 0.634, p < 0.05) to gene abundance of the pathogenic fungus in the soil respectively for the treatments. It is worthy to note that there was high gene abundance of F. solani in the cultivated ginseng soil even under the control in this experiment, probably from external sources. Conventional practice using pesticides, for instance carbendazim in this study, had little impact in preventing disease incidence.
Quantification of F. solani and disease incidence in ginseng fields
-
The gene abundance of F. solani in soil under healthy ginseng and unhealthy ginseng associated with four respective categories of pathogenic disease, including root rot, rust rot, black spot or gray mold, are displayed in Table 2 and Supplemental Fig. S2. In the soil with healthy ginseng plants, the fungal species of F. solani was not observed in August but detected in an abundance of 2,100 copies g−1 in September. In the soil with unhealthy plants, however, the pathogenic fungal in the rooted soil was found with gene abundances in a range of 1.7 × 103 copies g−1 to 43.7 × 103 copies g−1 in August and of 0 to 10.6 × 103 copies g−1 in September. The pathogenic fungal abundance was all higher (by 40%−100 %) in August with more active growth than in September, during which ginseng plant tended to wilt as the temperature in the mountainous area became lower. And, the gene abundance of the rooted soil was generally in an order of root rot diseased plants > gray mold diseased plant and rust rot diseased plant > black spot diseased plant, with clear root symptoms observed only on the root rot diseased plants. Apparently, this pathogenic fungus exists widely in ginseng soil even with the healthy plants in September, indicating a potential risk of high disease incidence for ginseng cultivation.
Table 2. Abundance of F. solani in the rooted soil with plants infected by different diseases.
Soil under ginseng Gene abundance of F. solani
(103 copies g−1)August September Healthy plant n.d. d 2.10 ± 0.19b Root rot plants 43.7 ± 1.91a 10.6 ± 0.96a Rust rot plants 4.48 ± 0.97c 2.76 ± 0.29b Gray mold disease plants 29.9 ± 1.33b 1.85 ± 1.28b Black spot disease plants 1.71 ± 0.43d n.d. d n.d., Not detected as below the detection limit of the ddPCR prorocol. Lowercase letters indicate the difference is significant (p < 0.05). -
Molecular detection is the best method for diagnosis of the spread of soilborne plant diseases (Farh et al., 2019). Specific primers are arguably the most critical single components of any PCR assay for their properties control the exquisite specificity and sensitivity that make this method uniquely powerful, and achieve quantitative analysis of the target species. Interspecies discrimination can be achieved with well-designed specific primers (Shi et al., 2023). Plant rhizosphere soils have a high microbial diversity and specific primers are the basis for developing pathogen detection methods for complex soil microbiome. While ITS is one of the genes used for the discrimination of F. solani (Mesapogu et al., 2011), in this study, we developed the method with specific ITS primers along with four strains of pathogenic fungi with ginseng root disease and three strains of related species of F. solani as negative strains. The developed method using the primers allowed a reliable differentiation of F. solani from other Fusarium species and other fungal genera.
Both ddPCR and RT-qPCR methods have been used as a specific and sensitive molecular detection method for Penicillium citrinum, Priestia megaterium, Tomato brown rugose fruit virus and Fusarium asiaticum (Chow et al., 2018; Kaminsky et al., 2022; Vargas-Hernández et al., 2022; Wang et al., 2023). In the study, we developed a ddPCR protocol for detecting F. solani with the specific primers based on the ITS gene. Our tests proved that the ddPCR protocol can detect pathogenic bacteria effectively in soil, with strong specificity, good reproducibility and high sensitivity, similar to or better than the RT-qPCR method in quantitative detection of the fungal species associated with the ginseng diseases. When compared with other molecular detection methods, ddPCR is advantageous as it is sensitive, accurate and does not require an external standard curve (Kiselinova et al., 2014). When compared with the previously reported results, the sensitivity obtained in this study was better than that of PCR (103 pg) (Omori et al., 2018), qRT-PCR (1 pg·μL−1) (Farh et al., 2019) Compared to the RT-qPCR method in this study, ddPCR method exerted a much lower limit of detection (down to 0.92 copies μL−1) , potentially applicable to early diagnosis of ginseng disease with very weak pathogeny (Nishimura et al., 2022). We anticipate that the ddPCR method could be developed into a powerful, accurate, sensitive and precise tool for assessing the surveillance of F. solani in ginseng cultivation.
Furthermore, the ddPCR method also showed better reproducibility than the RT-qPCR method. Previous studies have shown that there is no established linear relationship between the occurrence of disease and the abundance of pathogens though there was positive correlations between the disease index and the number of pathogens (Lu et al., 2019). The incidence of Fusarium wilt is closely related to the gene abundance of Fusarium oxysporum present in the soil, when the number of F. oxysporum in the soil reached more than 103 cfu g−1, it was likely to result in an outbreak of Fusarium wilt (Liang et al., 2013). In this study, the gene abundance of F. solani was in a range of 1−8.5 thousands copies g−1 in ginseng cultured soil, indicating a wide occurrence of the pathogenic fungal species in ginseng soil, and highly accumulated in mid-August and decreased in September. This phenomenon may be related to the close relationship between the growth of microorganisms such as fungi in the soil and the biological and abiotic factors in the environment. According to the biological characteristics of F. solani (Shen et al., 2012), the optimal growth temperature is 30 °C, and its sporulation quantity and conidium germination rate are 2.70 × 107 / dish,100%. When the ambient temperature is 15 °C, the sporulation quantity and conidium germination rate drop to 0.03 × 107 / dish and 4.4%, with a decrease of 98.9% and 95.6%, respectively. When the ambient temperature was reduced to 10 °C, there was no spore production and germination. The average temperature in September is 5~18 °C of Fusong (China), so the temperature drop may be an important reason for the decrease in the number of F. solani. Second, Yaw (Akosah et al., 2021) found that Fusarium was among the dominant genera of the rhizosphere and rhizoplane during flowering, while at senescence they were replaced by the closely related genus Monographella, FUN Guild analysis confirms the role of the potato plant in assembling fungal taxa and guilds. Dalvinder (Singh et al., 2009) studied the interactions of temperature and water potential in displacement of Fusarium pseudograminearum (Fp) by fungal antagonists, they found that the ability to displace Fp under cool dry conditions appears to be critical. It can be seen that the dominance of different fungal populations in soil is related to the growth stage, and there are dynamic changes in the dominance of different fungal populations under the comprehensive action of environmental factors such as moisture and temperature. However, the population with direct succession relationship with F. solani in rhizosphere soil of ginseng remains to be further studied.
Both with the treated soil in pot experiments and the field ginseng soil, the abundance of the disease-inducing species peaked at the time of active growth of ginseng plant, being consistent with the disease emergence observed in the field in August. This was partly explained with the correlation of root rot disease incidence with the fungal abundance, which was reported with a biochar soil improvement experiment for ginseng disease control (Liu et al., 2022). Such well-established correlation may value its potential application in diagnosis and assessment of ginseng disease spread in ginseng cultivation (Marta et al., 2015). Overall, this method could be used to quantitatively characterize the potential incidence of ginseng root disease in the monitoring and prevention of root rot in ginseng cultivation.
This study was funded partly by Jilin Provincial Science Technology Department under a grant number of 20230204022YY, 20210204184YY. We thank Prof. Dr. Changtian Li and Dr. Zheliang Lv for their kind assistance in field work of sampling and disease observation. The advice and comments for the work by Dr. Cuijin Liu are also acknowledged.
-
The authors declare that they have no conflict of interest.
- Supplemental Fig. S1 Healthy ginseng(a), initial infection ginseng (b), aboveground part (c) and underground part(d) of ginseng infected with F. solani in pot experiment.
- Supplemental Fig. S2 Disease symptoms of Root rot(1a, 2a), Rust rot(1b, 2b), Gray mold(1c, 2c) and Black spot(1d, 2d) on root, leaf and seedling in ginseng field.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yang L, Zhou S, Nie D, Liu C, Yu L, et al. 2023. Developing PCR-based novel molecular assays to quantitatively detect Fusarium solani in ginseng soil for assessing soil health in ginseng cultivation. Soil Science and Environment 2:7 doi: 10.48130/SSE-2023-0007 

Developing PCR-based novel molecular assays to quantitatively detect Fusarium solani in ginseng soil for assessing soil health in ginseng cultivation
- Received: 24 May 2023
- Accepted: 19 August 2023
- Published online: 21 September 2023
Abstract: The fungal species of Fusarium solani (F. solani) is closely associated with the soil borne root rot disease of Panax ginseng, leading to a decline in the quality of the medicinal materials. In this study, we established experimental conditions of two PCR-based microbiological assays, a droplet digital PCR (ddPCR) method and a RT-qPCR method, and tested their efficacy of quick and quantitative detection of F. solani DNA in ginseng soil. These methods were further evaluated in quantifying F. solani for rhizosphere samples of ginseng plants with different status of pathogenic disease infection. The results showed that the two methods were both highly specific and reproducible in detecting and quantifying the gene of F. solani, without any observable cross-reaction with other species. Compared to the RT-qPCR method in this study, ddPCR method exerted a higher sensitivity of detection (0.92 copies μL−1) than RT-qPCR method (920 copies μL−1), and free of reliance on a standard calibration curve. With the ddPCR method, the absolute quantification of nucleic acid samples is made possible by changing the detection standard from fluorescence intensity to the presence or absence of fluorescence, and the accuracy of detection has been significantly improved. The abundance of F. solani were 0−2,100 copies g−1 dws, 927.5−8,470 copies g−1 dws and 10,605−43,697 copies g−1 dws respectively in uncultured ginseng soil, soil from healthy ginseng and the soil from infected ginseng. The quantitative abundance of the pathogenic fungal species infected by the diseases but also track the dynamic change in the gene abundance of F. solani in disease monitoring with ginseng growth. While the gene abundance of F. solani was highly accumulated in mid-August and decreased in September, the pathogenic fungal gene abundance tested was closely correlated to the incidence rate of ginseng root rot. Apparently, this pathogenic fungus is exists widely in ginseng soil even with the healthy plants in September, indicating a potentially great risk of high disease incidence for ginseng cultivation. Therefore, the developed ddPCR assay could be reliable and feasible, potentially applied in early diagnosis of the infection by the pathogenic fungal of F. solani in ginseng cultivation and in routine assessment of the disease occurrence with ginseng growth.
-
Key words:
- Root rot /
- Incidence rate /
- Soil borne disease /
- Fusarium solani /
- Rhizosphere /
- Panax Ginseng