









-
Ethylene is an important plant hormone that plays a vital role in many aspects of plant growth, including fruit development and ripening[1−3]. Climacteric fruit like tomato, apple, pear, kiwifruit and peach display enhanced ethylene production and a peak in respiration at the onset of ripening. It is thought that ethylene regulates the initiation of various ripening-related changes like fruit color, cell wall metabolism, sugar content, flavor and aroma compounds in climacteric fruit[3−5]. In addition, ethylene has been reported to participate in fruit set and early fruit development[6−8]. The inhibition of ethylene production or ethylene-induced responses by external treatments with chemical inhibitors such as ethylene biosynthesis inhibitor aminoethoxyvinylglycine, perception inhibitor silver thiosulphate and 1-methylcyclopropene (1-MCP), induces fruit set efficiency and the growth of set fruits in pear[8], and tomato[6].
Ethylene is synthesized from S-adenosyl-L-methionine (SAM), which is converted to 1-aminocyclopropane carboxylic acid (ACC) under the catalyzation of the enzyme ACC synthase (ACS). Then, ACC is oxidized into ethylene by ACC oxidase (ACO)[9,10]. In the ethylene synthesis pathway, the formation of ACC is generally thought to be the rate-limiting step and therefore, ACS proteins are rate-limiting enzymes in ethylene synthesis[11]. ACS belongs to the family of pyridoxal-5'-phosphate dependent aminotransferases, which require vitamin B6 as a co-factor for activity[9]. According to the presence of particular sequences at the C-terminus, ACS genes can be divided into three major groups: Type I genes encode target sites for mitogen-activated and calcium-dependent protein kinases (MAPK and CDPK, respectively), Type II genes encode target sites for CDPKs and E3 ligases, and Type III genes encode no signaling target sites[12].
Early work has discovered several genes in the ACS family in different species and their differential expression during plant development. There are 12 ACS genes (ACS1 to ACS12) in Arabidopsis, and all of them display different expression patterns throughout growth and development[1, 13]. In tomato, nine genes encoding ACS (LeACS1A, LeACS1B, and LeACS2−8) have been reported[1], with LeACS1A, LeACS2, LeACS4, and LeACS6 are expressed in different stage of fruit development[14, 15]. Three of the genes, LeACS1A, LeACS2, and LeACS4 showed a ripening-related increase in expression, while LeACS6 transcripts were present in mature green fruit but declined as ripening was initiated[15]. A total of 19 ACS genes were identified in apple, with six expressed in fruit[16]. Among them, MdACS3a is expressed lower and before fruit ripening, while MdACS1 is expressed abundantly after fruit ripening[17]. These results indicated that fruit development relies on the expression of different ACS genes to form ethylene at distinct developmental stages.
There are two systems of ethylene biosynthesis in higher plants: System 1 is auto-inhibitory and responsible for producing basal levels of ethylene in all tissues, including non-ripening fruit; System 2 is auto-stimulatory and produces the burst of ethylene during the ripening of climacteric fruit[15, 18]. The ethylene production in Systems 1 and 2 is regulated by differential expression of ACS genes. In tomato, System 1 is regulated by the expression of LeACS1A and LeACS6. As fruit transitions to the ripening stage, the expression of LeACS1A and LeACS4 is induced, dependent on the RIN MADS-box transcription factor, and System-2 ethylene synthesis is subsequently initiated and maintained by LeACS2[15]. In apple, MdACS1 is well-known to control the climacteric ethylene burst during fruit ripening, while MdACS6 is responsible for ethylene production in System-1 and MdACS3a is a main regulator of ethylene production through the transition from System 1 to System 2[17, 19].
Six PpACSs were identified in nectarine, and three of them are expressed during nectarine fruit development and ripening[20]. Among them, PpACS1 is expressed abundantly at the ripening stage and plays important roles in regulating fruit softening[20, 21]. However, peach fruit development and ripening consists of several growth phases: the first exponential growth phase (S1), the onset of pit hardening (S2), the second exponential growth phase (S3), and ripening (S4). Aside from the ripening stage, the regulation of PpACS genes to control ethylene synthesis during the other development phases especially the transition between different growth phases, remains unclear.
In this study, we identified eight PpACS genes in peach and analyzed their gene structures, conserved domains, motifs and promoters. Samples were taken from additional, intervals before and after full bloom to perform detailed examinations of ACS activity and transcript levels of the PpACS genes during fruit development and ripening. The PpACS transcript expression patterns were also examined under ethylene and 1-MCP treatments. These results provide valuable information for the study of the PpACS gene functions during peach fruit development.
-
For PpACS expression profiles and ACS activity analysis, peach fruit at different developmental stages were collected from the cultivar 'Jinqiuhongmi' about 7 days' before and after full bloom (DAFB). For ethylene and 1-MCP treatment, the variety 'Huangshuimi' peach fruit at 70 DAFB were collected and treated with 100 g·L−1 of ethephon, 10 μL·L−1 of 1-methylcyclopropene (1-MCP) or distilled water according to Cheng et al[22]. Fruit samples were collected at 0, 2 and 4 d after treatment. All collected materials were immediately frozen in liquid nitrogen and stored at −80 °C until use. The peach cultivars used in this study were seven-year-old trees and maintained at the Fruit Tree Germplasm Repository of Henan Agricultural University (Henan Province, China).
Identification of the PpACS genes in peach
-
The genome sequences (Version 2.0) used in this study were downloaded from GDR (
www.rosaceae.org/gb/gbrowse/prunus_persica_v2.0/ ). All PpACS genes in peach were identified by BlastP using the ACS genes of Arabidopsis thaliana as the query sequence[12] downloaded from the TAIR database (www.arabidopsis.org/ ). The potential PpACS genes were reconfirmed through comparison with the Swiss-Port database in NCBI-BLASTP (https://blast.ncbi.nlm.nih.gov/Blast.cgi ).The exon-intron structures and the conserved motifs of the PpACS genes were analyzed and visualized using TBtools[23] with the parameters of zero or one repetition and 10 maximum number of motifs.
The 2,000 bp upstream of each PpACS coding sequence (CDS) was extracted as the promoter by TBtools[23]. The promoter cis-acting elements were predicted through the PlantCARE (
http://bioinformatics.psb.ugent.be/webtools/plantcare/html/ )[24] and visualized using TBtools[23].Multiple sequence alignment, phylogenetic analysis, and protein structure prediction of PpACSs
-
The predicted CDS length, predicted isoelectric point (pI) and molecular weights (MWs) of all confirmed PpACSs were predicted by ExPASy-ProtParam (
https://web.expasy.org/protparam/ ), and their subcellular localization was predicted by Plant-mPLo (www.csbio.sjtu.edu.cn/bioinf/plant-multi/ ).The multiple sequence alignment was generated by the BioEdit Sequence Alignment Editor[25]. A total number of 23 ACS proteins from Arabidopsis, tomato and apple were downloaded from the Phytozome database (
https://phytozome-next.jgi.doe.gov/ )[26] and were used for phylogenetic analysis together with the PpACSs. Maximum likelihood and neighbor-joining trees of ACS proteins were generated using MEGA 11.0 with 1,000 replicates (Jukes-Cantor distance matrix).Quantitative real-time PCR analysis
-
Total RNA was extracted using the Spin Column Plant total RNA Purification Kit (Sangon Biotech, Shanghai, China), and the first-strand cDNA was synthesized using the PrimeScriptTM RT reagent Kit (TaKaRa, Dalian, China). qRT-PCR was conducted in a total reaction volume of 20 μL containing 10 μL of SYBR Green I Master Mix (Takara, Dalian, China), 0.2 μM of each primer, and 100 ng of template cDNA. The amplification was performed on an ABI Prism 7500 FAST Sequence Detection System (Applied Biosystems, Madrid, CA, USA) with the following program: pre-denaturation at 95 °C for 30 s, followed by 40 cycles of 95 °C for 30 s, 60 °C for 34 s, and 72 °C for 1 min. The peach PpTEF2 (Prupe.4G138900) gene was selected as constitutive controls[27]. According to the Ct value, the gene expression level was calculated by the 2−ΔΔCT method[28]. Primers used for qRT-PCR were designed using Primer-BLAST[29] and are listed in Supplemental Table S1. Three biological replicates were performed for each analysis.
Analysis of ACS activity during fruit development
-
The peach fruits collected at different developmental stages after full bloom were also used to detect ACS activity. Firstly, fruit samples were ground with liquid nitrogen, and 0.1 g samples were homogenized by hand with 2 mL phosphate buffer (pH 7.4), and then centrifuged at 1,000 g for 20 min. The supernatant was collected to detect ACS activity. The Plant ACC synthase (ACS) ELISA Kit (Ruishou, Shanghai, China) was used to perform a chromogenic reaction according to the instructions. Briefly, a total reaction volume of 50 μL samples or standard, 100 μL of enzyme conjugate were added to a microtiter plate, covered with an adhesive strip and incubated for 60 min at 37 °C, the microtiter plate was washed four times. Then, 50 μL Substrate A and Substrate B were added to each well respectively, gently mixed and incubated for 15 min at 37 °C. Lastly, 50 μL Stop Solution were added and the reaction solution wwas measured at a wavelength of 450 nm using a microplate reader within 15 min. A standard curve was generated and used to calculate the ACS activity. Three biological replicates were performed.
-
An early study identified six PpACS genes in the nectarine genome[20]. Here, we have found eight PpACS genes in the peach genome, including the six reported PpACSs and two other genes named PpACS7 and PpACS8 (Supplemental Table S2). According to previous studies, the ACS proteins can be classified into four types based on their C-terminal sequence characteristics[12, 30]. Here, a phylogenetic tree was constructed, and sequence alignment of the C-terminal amino acids of the ACS proteins was performed.
The phylogenetic analysis showed that the eight PpACS proteins from peach were classified into four types (Type I, Type II, Type III and AT, or aminotransferase) together with the ACS proteins from Arabidopsis, tomato and apple (Fig. 1). For instance, PpACS1 and PpACS5 clustered with Type I ACS proteins, PpACS4 and PpACS6 with Type II, and PpACS2 and PpACS3 with Type III (Fig. 1). The proteins that cluster in the AT type, namely PpACS7 and PpACS8 from peach, AtACS10 and AtACS12 from Arabidopsis, and MdACS6 from apple, are likely aminotransferases (AT) that lack ACS activities (Fig. 1).
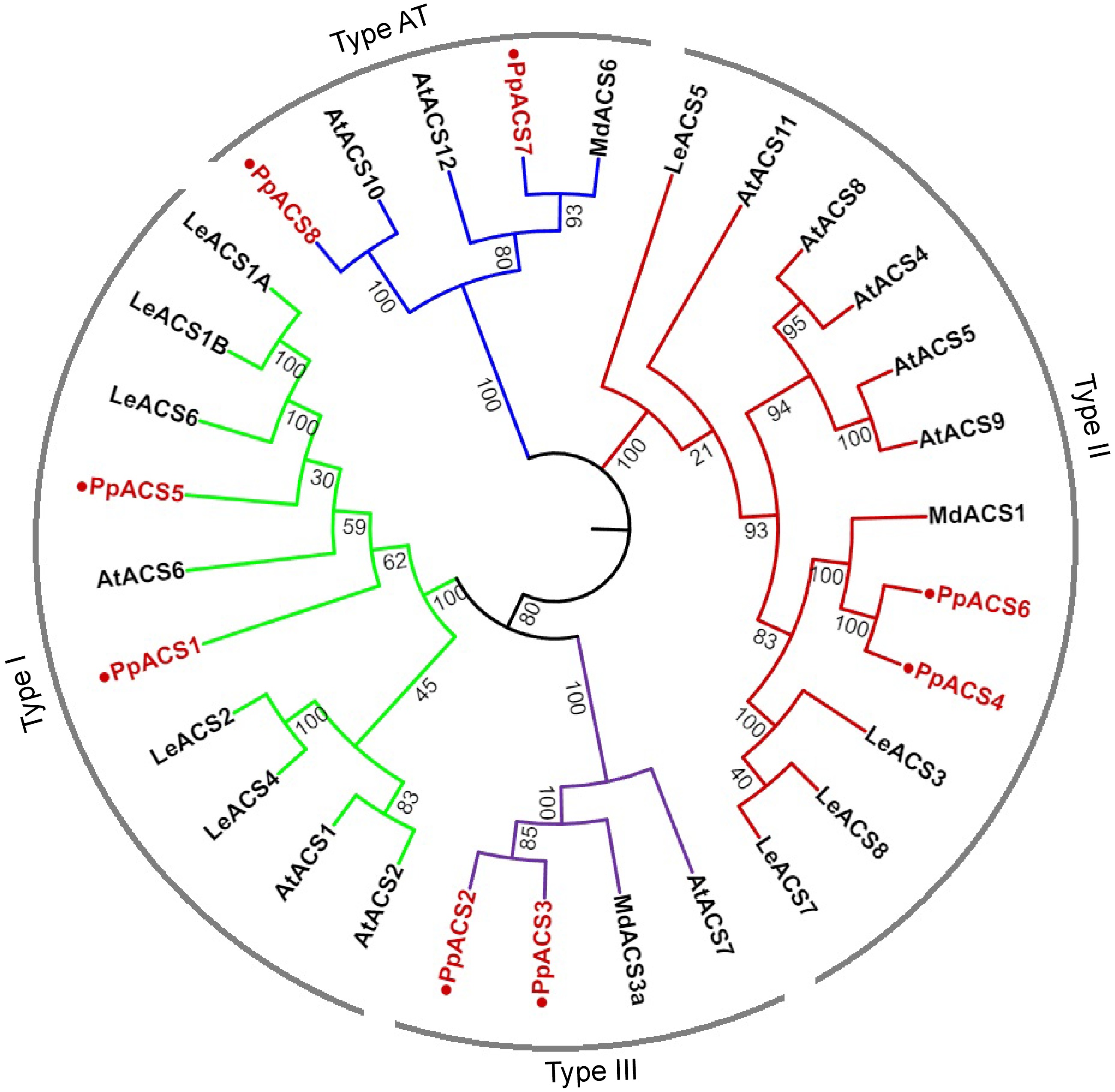
Figure 1.
Phylogenetic analysis of ACS proteins from Arabidopsis, tomato, apple, and peach. A neighbor-joining tree of ACS proteins was generated using the MEGA11.0 with 1000 replicates (Jukes-Cantor distance matrix). ACS proteins of peach are marked with red color. Deduced amino acid sequences from Arabidopsis [AtACS1 (At3g61510), AtACS2 (At1g01480), AtACS4 (At2g22810), AtACS5 (At5g65800), AtACS6 (At4g11280), AtACS7 (At4g26200), AtACS8 (At4g37770), AtACS9 (At3g49700), AtACS10 (At1g62960), AtACS11 (At4g08040), AtACS12 (At5g51690)], apple [MdACS1 (U89156), MdACS3a (AB243060), MdACS6 (MDP0000133334)] and tomato [LeACS1A (U18056), LeACS1B (U18057), LeACS2 (X59139), LeACS3 (U17972), LeACS4 (X59146), LeACS5 (AF167425), LeACS6 (AF167428), LeACS7 (AF043122), LeACS8 AF167427)] were used. AT type ACS proteins are more like aminotransferases.
Further amino acid sequence alignment of the C-termini of the ACS proteins from Arabidopsis and peach confirmed the conservative characteristics of the PpACS proteins (Fig. 2). The Type I ACS proteins have four conserved serine residues, three of which are phosphorylation sites of MAPKs and one of which is a phosphorylation site of CDPKs (Fig. 2). The Type II ACS proteins contained only one CDPK phosphorylation sites or conserved serine residues at the C-terminus (Fig. 2). Both the Type III and AT-type showed the shortest C-terminals, lacking both types of conserved serine residues (Fig. 2).

Figure 2.
Amino acid alignment of the C-termini of ACS proteins from Arabidopsis and peach. The conserved serine (S) residues of the ACS proteins are marked with black boxes according to previous research[31].
Gene structure and conserved motif composition of PpACSs
-
The analysis of exon-intron structure is essential to understanding the evolutionary history within gene families[32]. The exons and introns of the ACS genes in peach were analyzed by TBtools. The PpACS members in the same subfamily showed roughly similar exon/intron distribution patterns in terms of the exon length and the number of introns (Fig. 3b). The Type I enzymes PpACS1 and PpACS5, PpACS4 and PpACS6 (Type II), PpACS7 and PpACS8 (AT type) all contained four exons and three introns, while PpACS2 and PpACS3 (Type III) contained three exons and two introns (Fig. 3a, b). The conserved motifs of the PpACS genes were analyzed, and the results showed that all members contained nine motifs (Fig. 3c). Motif 1−8 were common and distributed among all members, while motif 9 was included in members of Type I, Type II and Type III, but not in the AT Type. Only the AT-type genes contained motif 10, suggesting a highly specific function. Sequence logos of the ten conserved motifs included the seven conserved domains of ACS proteins (Fig. 3d).

Figure 3.
Gene structure and conserved protein motifs of the PpACS family in peach. (a) The phylogenetic tree based on all PpACS proteins was constructed using the maximum likelihood method with 1,000 bootstrap. (b) The gene structures of the PpACS genes. The green and yellow boxes represent UTRs and CDSs respectively. (c) The conserved motifs of the PpACS proteins were identified using the MEME program. (d) The sequence logos of the conserved motifs numbered from 1 to 10. The seven conserved domains of ACS are marked with red boxes and named as box 1−7.
Promoter cis-elements analysis of the PpACS genes
-
In order to better understand the potential regulatory function of PpACS genes in peach, the possible cis-regulatory elements (CREs) were identified in the 2,000-bp upstream region from each transcription start site using the PlantCARE online web tool. The predicted CREs of the PpACS genes contained the core promoter elements, environmental stress-related elements, hormone-responsive elements, light-responsive elements, site-binding-related elements, and some others binding sites (Supplemental Table S3). The largest number of elements were the core promoter elements that contained the TATA-box, AT-TATA and CAAT-box (Supplemental Table S3). The light-responsive elements were abundant, including Box 4, the G-box, and the TCT-motif (Supplemental Table S3). There were also various hormone-responsive elements, including the TCA-element involved in salicylic acid (SA) responsiveness, the TATC-box and gibberellin (GA)-response element (GARE) involved in GA responsiveness, the abscisic acid (ABA)-responsive element (ABRE), the AuxRR-core and TGA involved in auxin responsiveness, the CGTCA-motif and TGACG-motif involved in MeJA-responsiveness and the ethylene-responsive element (ERE) (Fig. 4b, c). The most numerous element was the ABRE, which was widely distributed among the PpACSs, except for PpACS3 and PpACS7. The second most common element was ERE (Fig. 4b, c). This analysis indicates that the PpACS genes are regulated by ABA and ethylene signals.
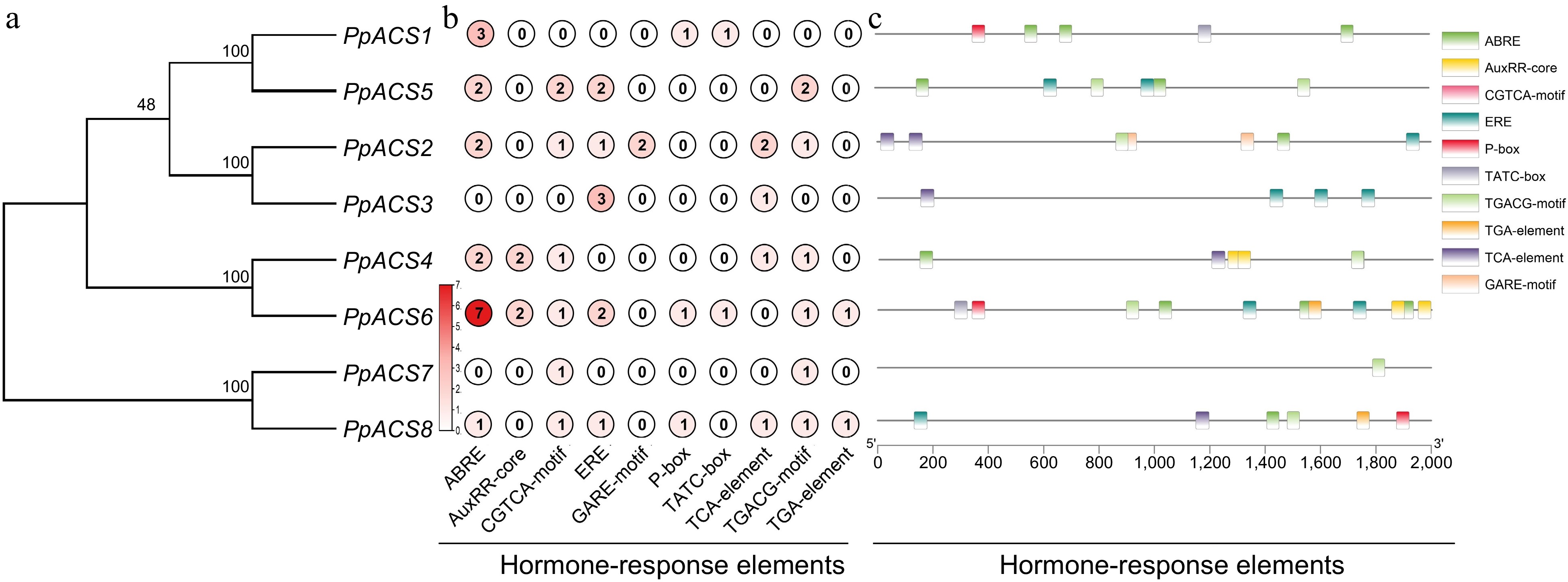
Figure 4.
Analysis of the cis-elements in the promoters of the PpACS genes. The cis-elements of the PpACS genes were determined by the PlantCARE online web tool and visualized by the TB-tool. (a) Phylogenetic tree of all PpACS proteins. (b) Number of hormone-responsive elements detected in each PpACS gene promoter. (c) Visualization of hormone-responsive elements in the PpACS promoters, with ten different cis-elements displayed by different colored boxes.
ACS activity and PpACS expression profiles during peach fruit development
-
To further investigate the potential functions of the PpACS genes during peach fruit development and ripening, the ACS activity and PpACS expression profiles were determined. Peach fruit samples were numbered negatively as the days before full bloom, while the days after full bloom were divided into four phases: first exponential growth phase (S1), the onset of pit hardening (S2), the second exponential growth phase (S3), and ripening (S4) (Fig. 5a, b).
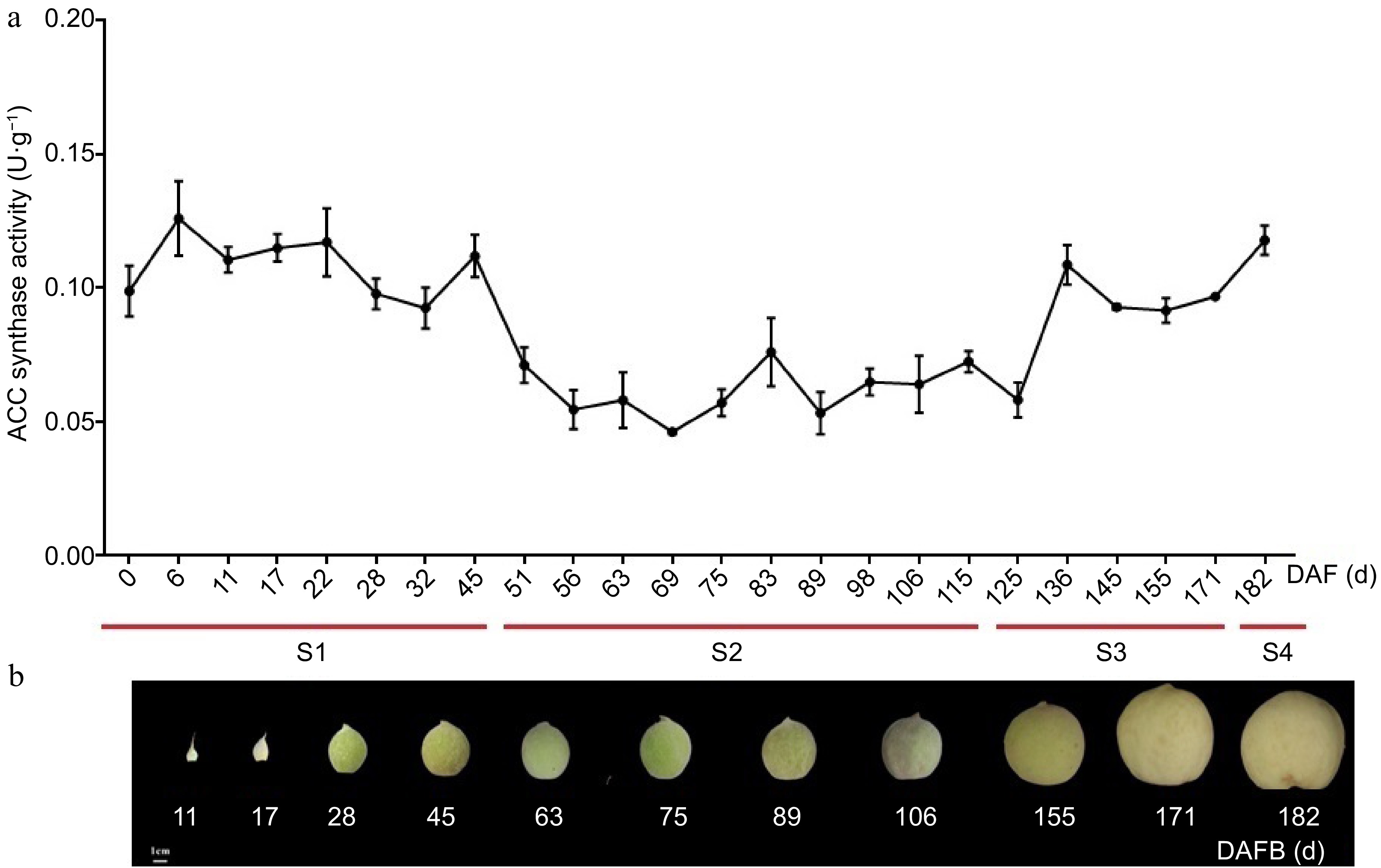
Figure 5.
Total ACC synthase activity at different stages of fruit development in peach. (a) Total ACC synthase activity in different stages of peach fruit development. The values are means ± SD of three independent biological replicates for ACC synthase activity. (b) Sampling time of peach fruit at different development stages, which was divided into four periods (S1, S2, S3 and S4 identified with red lines) according to the days after full bloom (DAFB).
The total ACS activity fluctuated during fruit development (Fig. 5a). ACS activity was higher during the stage 1 and decreased significantly during the S2 (Fig. 5a). There was a rapid increase in activity within stage-3 at 136 DAFB, and the activity remained high during stage 3 and 4 (Fig. 5a).
The expression profiles of the PpACS genes showed that the eight PpACS genes were differentially expressed during peach fruit development. Among them, the AT-type genes PpACS7 and PpACS8 exhibited higher expression during fruit development (Fig. 6g & h). Transcripts of PpACS1, PpACS5, and PpACS6 also accumulated during fruit development and ripening (Fig. 6a−c), while those for PpACS2, PpACS3, and PpACS4 exhibited minimal expression in peach fruit (Fig. 6d−f). PpACS1, which has been identified as playing important roles in peach fruit ripening[33, 34] showed a burst expression at the S4 stage (Fig. 6a). Interestingly, PpACS5, PpACS6, PpACS7, and PpACS8 showed clear expression peaks at the S1 to S2 transition (Fig. 6b, c & g). The ACS genes were mainly expressed at the early (S1) and ripening stages (S3−S4) during the fruit development and may function for ethylene synthesis during these stages.
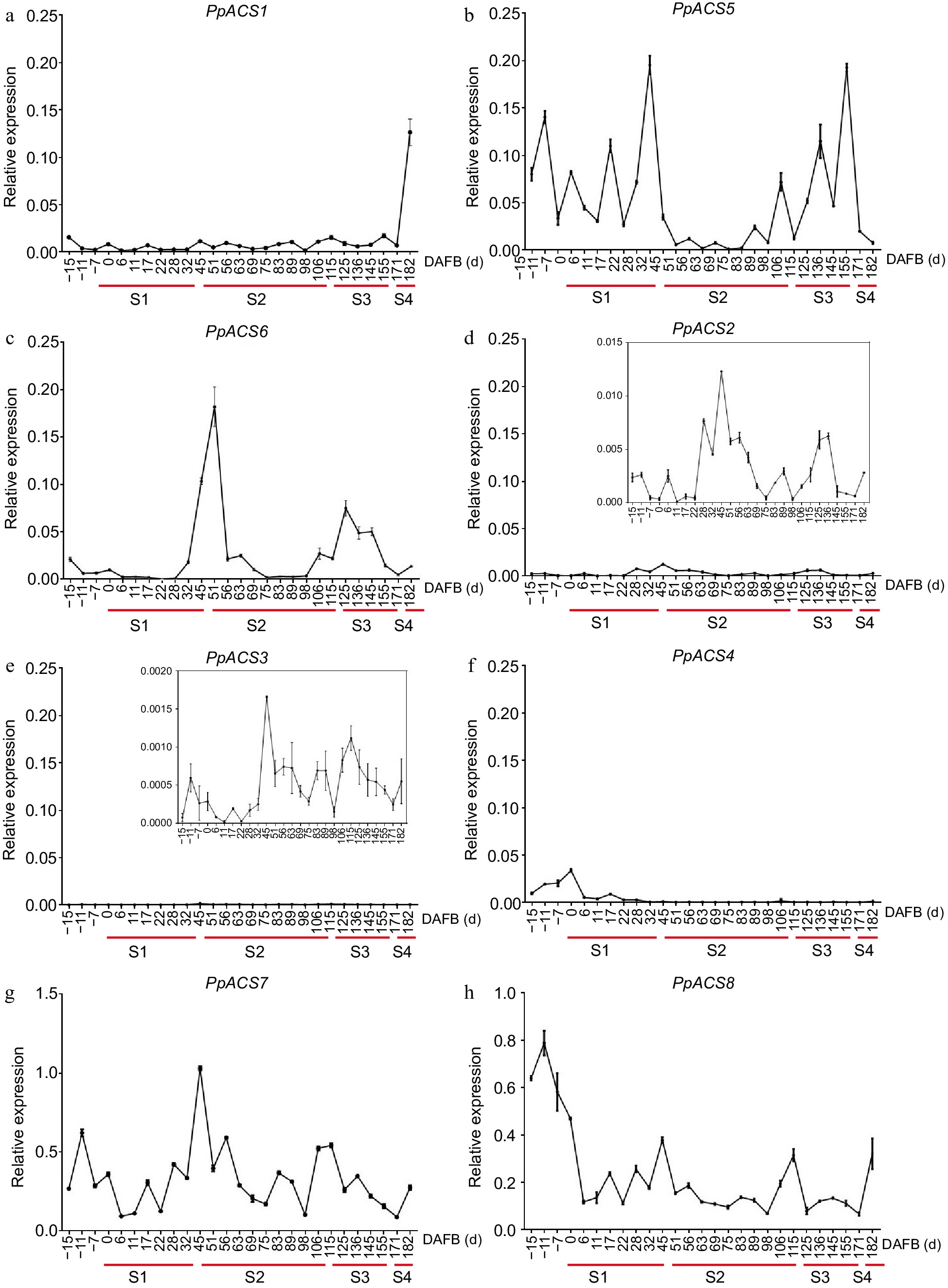
Figure 6.
Expression profiles of PpACS genes during fruit development of peach. The expression values are means ± SD of three independent biological replicates as analyzed by qRT-PCR. The fruit development stage was divided into four periods (S1, S2, S3 and S4; identified with red lines) according to the days after full bloom (DAFB). (a)−(h) show the expression profiles of PpACS1−PpACS8, respectively.
The effect of ethylene and 1-MCP treatments on PpACS gene expression
-
To further investigate whether PpACS gene expression of is under the control of ethylene in peach fruit, the transcript levels of these PpACS genes were examined after 1-MCP and ethylene treatments. The levels of two transcripts, PpACS1 and PpACS8 were upregulated under ethylene treatment and repressed by 1-MCP treatment (Fig. 7). The expression of PpACS3 was repressed by both ethylene and 1-MCP after treatment for 4 d, while PpACS7 was induced by both ethylene and 1-MCP (Fig. 7). Other PpACS genes, including PpACS2, PpACS4, PpACS5 and PpACS6, were induced by 1-MCP (Fig. 7). All these results indicated that the PpACS genes respond differently to ethylene and 1-MCP.

Figure 7.
Effect of ethylene and 1-MCP treatments on the expressions of PpACS genes. The expression values are means ± SD of three independent biological replicates as determined by qRTPCR. Fruits were treated with ethylene and 1-MCP and then, stored at room temperature until sampling at 2 and 4 d. *p < 0.05, **p < 0.01, ***p < 0.001 (Student's t-test).
-
Genes in the ACS family have been divided into types I, II, III, and AT[12]. In Arabidopsis, AtACS10 and AtACS12 encode aminotransferases and have ATase activity, which have been reported are not likely to be involved in ethylene biosynthesis[12]. Here, PpACS7 and PpACS8 from peach clustered with the AT types together with MdACS6 from apple. It has been reported that MdACS6, a homolog of PpACS7, was expressed continuously during fruit ripening and could regulate ethylene biosynthesis when overexpressed in apple fruit[35]. These results indicate that the AT type ACS genes may have ethylene biosynthesis function during fruit development and need to be further investigated.
Regulation of ethylene biosynthesis is very important in fruit development, and ACS is the key rate-limiting enzyme in the ethylene synthesis pathway. Here we have conducted a detailed examination of ACS gene expression during peach fruit development and ripening. The results indicate that five ACS genes, PpACS1, PpACS5, PpACS6, PpACS7 and PpACS8, are expressed in peach fruit (Fig. 6). Zeng et al.[20] found that PpACS1, PpACS4 and PpACS5 show ripening-related increases in expression during fruit development and ripening in nectarine 'CN13'. PpACS1 showed extremely high expression at in stage 4 fruit, similar to a previous report. PpACS4, which has been reported to show a climacteric pattern from S3-1 to S4-1, exhibited minimal expression in our studies during the entire course of fruit development. Furthermore, the expression of PpACS5 fluctuated during stages 1 and 3, whereas data presented by Zeng et al.[20] showed it decreased in nectarine during fruit development. These discrepancies may be caused by the use of different cultivars or may be due to the greater number of fruit samples at closer intervals that we collected. The increased number of fruit samples offered a greater chance to detect any changes in ACS gene expression during fruit development and ripening.
Ethylene plays an important role in regulating the development and ripening of fruits, especially in climacteric fruits like peach. Peach fruit development follows the double sigmoid curve and is divided into four phases[36]. Fruit ripening in peach was thought to be ethylene dependent. Ethylene production at the ripening stages is mainly regulated by the expression of PpACS1[21, 33]. Here, we also found that total ACS activity and the expression of PpACS1, PpACS5 and PpACS6 were high during S3 and S4 (Fig. 5a & 6). However, it is interesting that there are expression peaks for PpACS5, PpACS6, PpACS7 and PpACS8 at the S1 to S2 transition, with corresponding peaks of total ACS activity. The growth of peach fruit is slowed down at the S1 to S2 transition and is followed by endocarp lignification (onset of pit hardening), which lasts at the end of S2[37]. The duration of S2 stage is highly dependent on the cultivars, being shorter for early ripening varieties and longer for late ripening varieties[37]. The increasing expression of these PpACS genes at early S2 stage indicated the potential function of ethylene for the peach fruit mature date.
A model for the regulation of PpACS gene expression during nectarine fruit development and ripening was proposed[20]. In immature nectarine fruit, they found that the System 1 ethylene production relies on the expression of PpACS5, while expression of both PpACS1 and PpACS4 increased with fruit ripening and were responsible for the burst in ethylene production in System 2. Our data showed that, besides PpACS5, other PpACS genes, including PpACS6, PpACS7 and PpACS8, which were mainly expressed in other tissues beside fruit, were also involved in System 1 ethylene biosynthesis and were responsible for producing a basal level of ethylene during early fruit development (Supplemental Fig. S1). Furthermore, PpACS4 was not expressed during the whole course of fruit development, suggesting that it may not be involved in System 2 ethylene biosynthesis. These results were also supported by Guo et al.[34]. Additionally, transcription of PpACS1 was inhibited after ethylene treatment, suggesting an ethylene-independent (non-autocatalytic) system in nectarine fruit[20]. In contrast, our data clearly show that the expression of PpACS1 were significantly induced by ethylene and repressed by 1-MCP, which indicated that PpACS1 functions in System 2 ethylene biosynthesis via an ethylene-dependent (autocatalytic) way. The result is inconsistent with previous reports, which may be due to the treatment of the fruit at different developmental stages and/or treatment times. The 'CN13' fruits used for treatment by Zeng et al.[20] were collected at the commercial harvest stage (90 DAFB, S4-1), whereas the fruit used here were harvested at 70 DAFB (S3). Moreover, the increased PpACS1 transcript levels after 2- and 4-d of ethylene treatment in our study suggests that high ethylene levels required for induction of these genes.
-
In the present study, we identified eight PpACS genes from the peach genome. Phylogenetic analysis showed that the eight proteins could be divided into four types (Type I-III and AT), which was also supported by their exon/intron structures, promoter motifs and protein domains. The ACS activity was higher during the early (S1) and ripening (S3 and S4) fruit developmental stages. The PpACSs expression profiles during peach fruit development were investigated, and five PpACS genes (PpACS1, PpACS5, PpACS6, PpACS7 and PpACS8) were expressed in peach fruit. Among them, PpACS1 was highly expressed during peach fruit ripening and induced by ethylene treatment, while PpACS5, PpACS6, PpACS7 and PpACS8 were expressed at the S1 to S2 transition, suggesting their different roles in ethylene biosynthesis systems during peach fruit development.
-
The authors confirm contribution to the paper as follows: study conception and design: Zhang L, Feng J; draft manuscript preparation: Wang X, Zhang L; manuscript revision: Tan B, Zheng X, Feng J; experiments performed: Wang X, Dong K; formal analysis: Ye X, Cheng J, Wang W. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analysed during this study are included in the published article and its supplementary information files.
The work was conducted at the Henan Provincial Key Laboratory of Fruit and Cucurbit Biology and supported by the National Natural Science Foundation of China (32002014), the National Key Research and Development Program of China (2019YFD1000104), and the Special Fund for Henan Agriculture Research System (S2014-11-G02).
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Langlang Zhang, Jiancan Feng
- Supplemental Table S1 The Primer sequences of genes.
- Supplemental Table S2 Molecular characteristics of PpACS genes in peach.
- Supplemental Table S3 Classification statistics of PpACSs promoter cis-elements.
- Supplemental Fig. S1 Expression profiles of PpACS genes in different tissues.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang X, Dong K, Cheng J, Tan B, Zheng X, et al. 2024. Genome-wide identification and expression pattern analysis of the ACS gene family during fruit development in peach. Fruit Research 4: e004 doi: 10.48130/frures-0023-0040 

Genome-wide identification and expression pattern analysis of the ACS gene family during fruit development in peach
- Received: 18 August 2023
- Revised: 21 September 2023
- Accepted: 23 October 2023
- Published online: 05 January 2024
Abstract: Ethylene plays an important role in regulating the development and ripening of fruits, and 1-aminocyclopropane carboxylic acid synthase is the key rate-limiting enzyme in ethylene synthesis pathway. In this study, eight PpACS genes were identified from the peach genome [Prunus persica (L.) Batsch], and their phylogeny, gene structures, promoter motifs and expression patterns were analyzed. The PpACS genes could be divided into four types, and the genes with similar structures and motif distribution clustered together. Identification of the cis-elements in the promoters revealed that the PpACS genes may respond to various hormones. Furthermore, expression analysis showed that five (PpACS1, PpACS5, PpACS6, PpACS7 and PpACS8) of the eight PpACS genes were expressed at different stages during peach fruit development. Among them, PpACS1 was highly expressed at the ripening stage and induced by ethylene. The expression peaks of PpACS5, PpACS6, PpACS7 and PpACS8 during the transition from first exponential growth to pit hardening (S1 to S2) indicated a potential function of ethylene during this important transition. Taken together, these results provide valuable information for future investigation into the functions of the PpACS genes during peach fruit development and ripening.
-
Key words:
- Peach /
- ACS genes /
- Fruit development /
- Ripening /
- Ethylene