








-
Vegetables are one of the basic food sources for human beings, providing essential vitamins, dietary fiber and minerals for human health. Meanwhile, the economic importance of vegetables is increasingly recognized for meeting the diverse dietary needs of a growing population. Polyploids, defined as having three or more sets of chromosomes, often show larger organs and enhanced adaptation to adverse environments. Therefore, polyploidy has been used as an approach for crop breeding, including vegetables. Indeed, polyploids are prevalent in nature, many important crops are polyploids, such as wheat (Triticum aestivum L.), cotton (Gossypium barbadense L.), rapeseed (Brassica napus L.), etc. Polyploid vegetables often possess unique flavors, novel traits, and wider ecological adaptation than their diploid ancestors, providing breeders with greater phenotypic diversity. Comparative transcriptomics analysis demonstrated the importance of polyploidy for the domestication and species, genetic, and physiological diversity in the Brassicaceae family[1,2]. Depending on their genomic composition, polyploids can be classified into autopolyploids, produced by whole genome duplication (WGD) within the same species, and allopolyploids, formed by combining different species. Compared to autopolyploids, allopolyploids are known to be more common and an effective route to hybrid speciation[3].
Sequencing technology facilitated the study of genomics, epigenomics, transcriptomics and other omics. Sanger dideoxynucleotide sequencing pioneered the research into the new world of parsing genetic code. The publication of the first vegetable genome sequence of cucumber (Cucumis sativus L.) in 2009 represents the start of the post-genomic era in vegetable research[4]. After that, integrative analysis using multi-omics data on model plants and crops enabled the investigation of various biological characteristics in a systematic way. To date, the genomes of many vegetable species have been sequenced, a platform (TVIR, http://tvir.bio2db.com) containing 59 vegetable genomes has also been constructed for comparative and functional genomic studies[5].
The development of plant genomics facilitated by sequencing technology opens a new window for studying the evolutionary history of species, which have revealed the deep roots of polyploidy throughout plant evolution[6−8]. Research has shown that all green plants have undergone one or more WGD events during the evolution, pinpointing the significance of polyploidy in plant evolution[9−11]. According to the formation time, polyploids can also be divided into neopolyploids and paleopolyploids[12]. Ploidy is one of the main drivers of diversity in angiosperms[13]. WGD also provides adaptive advantages to cells and organisms, especially in unstable, stressful environments, for example, polyploidy enhances plant salt tolerance[14−16], which improves the natural competitiveness of polyploid plants, making them more likely to survive. In all, the prevalence of natural polyploids documented their evolutionary success[17].
Although there have been examples of successful assembling of different copies of genes within the genome of the same organism from short-reading data[18,19], genomic research in polyploids is still lagging behind due to the short-read length limitations of the first two generations of sequencing and complexity of polyploid genomes. In recent years, the development of long and ultra-long read sequencing provided an unprecedented opportunity for precise genomic analysis in polyploids. For instance, the employment of long read sequencing with Next Generation Sequencing (NGS) as a complement could identify genomic structural variants (SVs) in polyploid crop species like B. napus, which used to be challenging to detect using only NGS data[20].
So far, the genome sequencing and assembly of many polyploid vegetable species have been accomplished, providing vital resources for the research and breeding of vegetables (Fig. 1). For example, the difficulty of assembling the genomes of potatoes is recognized, and recent studies have not only resolved the genome sequences of diploid potatoes[21], but also completed a high-quality chromosome-scale reference genome sequence of a tetraploid variety[22]. Whole-genomic analysis of diploid and polyploid Brassica species revealed highly conserved genomes since the formation of hexaploid ancestor of Brassica[23]. The effects of whole genome triplication (WGT) on intraspecific diversification of B. rapa were reported through pan-genomic analysis using 16 de novo assembled genomes and two reported genomes[24]. This information provides prerequisites for the evolutionary understanding and genetic improvement of vegetables.
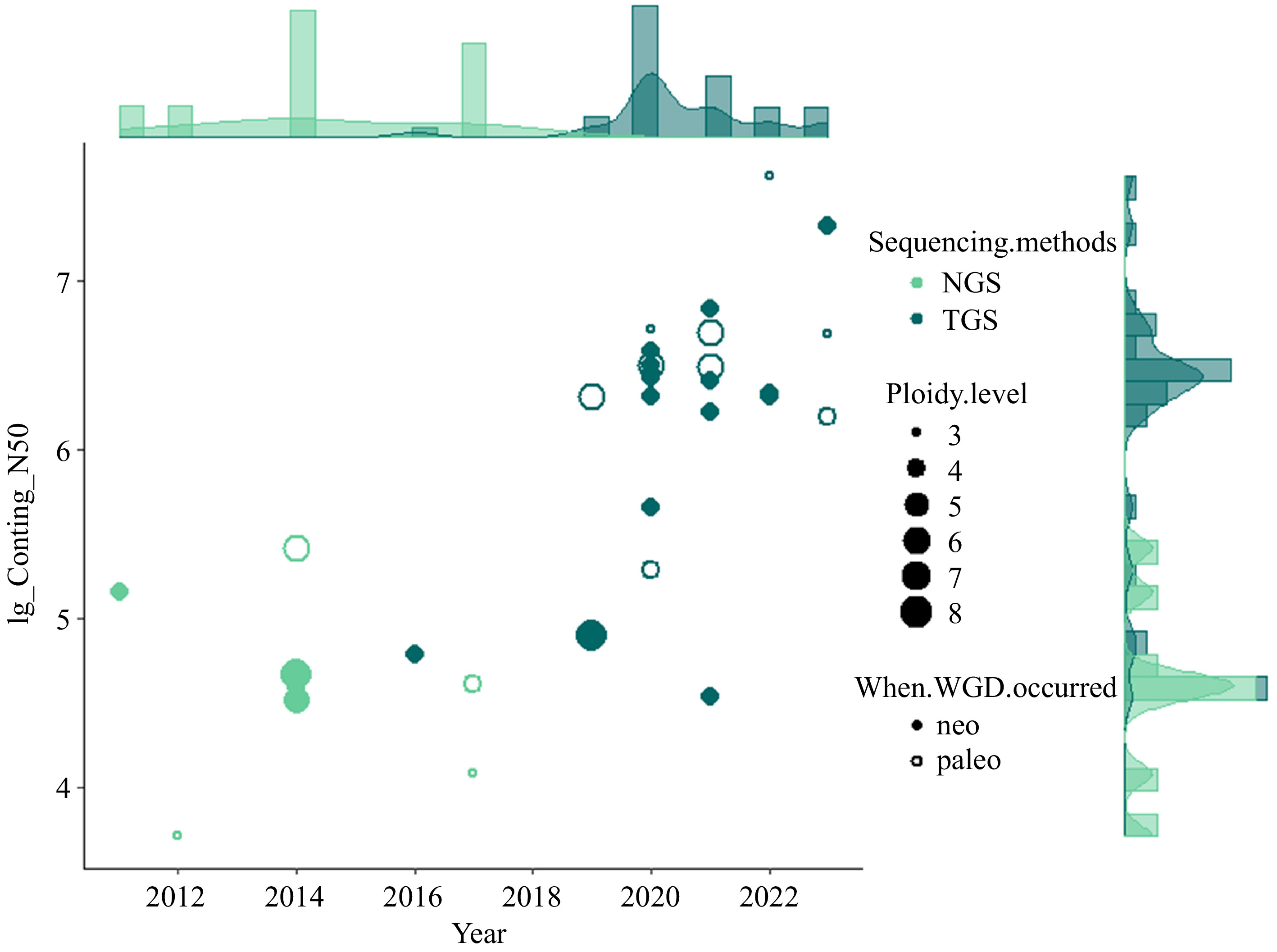
Figure 1.
Overview of currently sequenced polyploid vegetables with the contig N50 (log10) of assemblies and publication year as Y and X axis, respectively. The type of sequencing technology (Next-generation sequencing, NGS and Third-generation sequencing, TGS) are indicated with different colors. The size of data points is scaled by the ploidy level. Neo- and paleo- polyploids are differentiated with closed and open data points.
The highly complex genome structures of neo-polyploids pose great challenges to genome assembly. Not only are advances in sequencing technologies important for the acquisition of high-quality and long-continuity of polyploid genomes, but also the development of assembly algorithms significantly enhance the precision and reliability of assembly and analysis. The phenotypic advantages of polyploids, especially allopolyploids that merge two divergents, but similar genomes, also add difficulties for polyploid genome sequencing, which further hinder the study of its molecular mechanism[25−27]. Hi-C, a new technology created by combining chromosome conformation capture with high-throughput sequencing, can locate genome sequences to chromosomes, which is compatible with NGS sequencing and third-generation sequencing (TGS), is increasingly applied for chromosome-level genome assembly[28,29]. However, in polyploids, the sequence-similar allele fragments can be mistakenly linked together, forming many chimeric assemblies, limiting the assembly accuracy of polyploid genomes[30]. To solve this problem, many algorithms have been developed for the complex sequence structure of polyploid genomes[31], such as canu[32], hifiasm[33], ALLHiC[30,34] and PolyGembler[35]. Of these, ALLHiC is specifically designed to solve the Hi-C assembly puzzle of polyploid species and highly heterozygous genomes by correcting this mismatch of similar fragments in polyploid assembly. For example, the first autopolyploid sugarcane (Saccharum spontaneum L.) genome was deciphered by the ALLHiC algorithm. With an integrative strategy of ALLHiC and canu using HiFi and Hi-C sequencing data, accurate and chromosome-level assembly of polyploid genomes is becoming a routine procedure[36,37]. In this review, the analysis tools used for vegetable genome assemblies were summarized in Table 1.
Table 1. Analysis tools used for vegetable polyploid genome assembly.
Species Ref Analysis tools Year of publication Solanum tuberosum [38] SOAPdenovo; EMBOSS; SOAPdenovoalign 2011 Solanum lycopersicum [39] Newbler; CABOG; BAC FISH; EuGene; ABySS 2012 Brassica napus [40] SOAPdenovo; Newbler; GapCloser; 2014 Fragaria × ananassa [25] SOAPdenovo; Newbler; GapCloser; 2014 Camelina sativa [41] SOAPdenovo; Bambus; GapCloser; NUCmer 2014 Brassica oleracea [42] SOAPdenovo; Bambus; GapCloser; 2014 Brassica juncea var. tumida [43] ALLPATHS-LG; PBjelly; IrysView 2016 Cucurbita maxima Duch. [44] ShortRead; QuorUM; SOAPdenovo; Newbler; GapCloser; Pilon 2017 Lactuca sativa [45] SOAPdenovo; HiRis 2017 Fragaria × ananassa [46] NRGene; HiRise; PBJelly; Pilon; CoGe 2019 Brassica oleracea [47] FALCON; Quiver; BWA-MEM 2019 Brassica napus [48] Falcon; CANU; pbalign; pilon; 3D-DNA; Juicebox; Juicer 2020 Medicago sativa [49] CANU; MECAT; HERA; BWA-MEM; Redundans; Purge Haplotigs; BWA-MEM; Pilon 2020 Medicago sativa [36] CANU; ALLHiC; Jcvi; Juicebox 2020 Brassica oleracea [50] PBcR wgs8.3rc1; FALCON; Quiver; Pilon; BWA-MEM; SALSA; Mummer 2020 Solanum melongena [51] wtdbg2; Racon; Pilon; fragScaff; LACHESIS 2020 Allium sativum [52] FALCON; Quiver; Pilon; Purge Haplotigs; FragScaff; Burrows-Wheeler Aligner; ALLHiC 2020 Brassica napus [53] CANU; Pilon; 3D-DNA; ALLMAPS; ALLHiC 2021 Brassica juncea [54] CANU; BWA mem; MaSuRcA; RefAligner; IrysSolve; Pilon 2021 Brassica juncea [55] Jellyfish; FALCON; Sspace-longread; Quiver; BioNano Solve; BioNano Access; Juicebox; HiCPlotter 2021 Cucumis × hytivus [26] CANU; Pilon; SOAPdenovo; SSPACE; RefAligner; cutadapt; LACHESIS; HiC-Pro 2021 Brassica oleracea [56] Falcon; Arrow; PBJelly 2021 Solanum tuberosum [22] pbccs; hifiasm; BWA-MEM; ALLHiC; jcvi 2022 Solanum tuberosum [57] hifiasm; pilon; 2022 Capsicum annuum [58] MECAT2; Pilon; CANU; Juicer; Juicerbox; 3D-DNA; Phred; CA59 2022 Raphanobrassica [59] JELLYFISH; CANU; Arrow; Pilon; JUICER; 2023 Vicia faba [60] hifiasm; Sniffles; findGSE; purge_haplotigs; Merqury; RagTag; GSAlign; Liftoff 2023 Lactuca sativa [61] CANU; WTDBG2; Arrow; Pilon; 3D-DNA; Juicer; Falcon 2023 Driven by the abundance of data in the genomics era, researchers have an increasing number of research options (e.g., gene editing and multiple omics), and the study of polyploid evolution has recently made significant progress. This review summarizes the research progress in vegetable polyploids driven by sequencing technology and the subsequent studies underpinning important traits and genes, which will further promote germplasm innovation and breeding utilization via polyploidy in vegetables (Fig. 2).
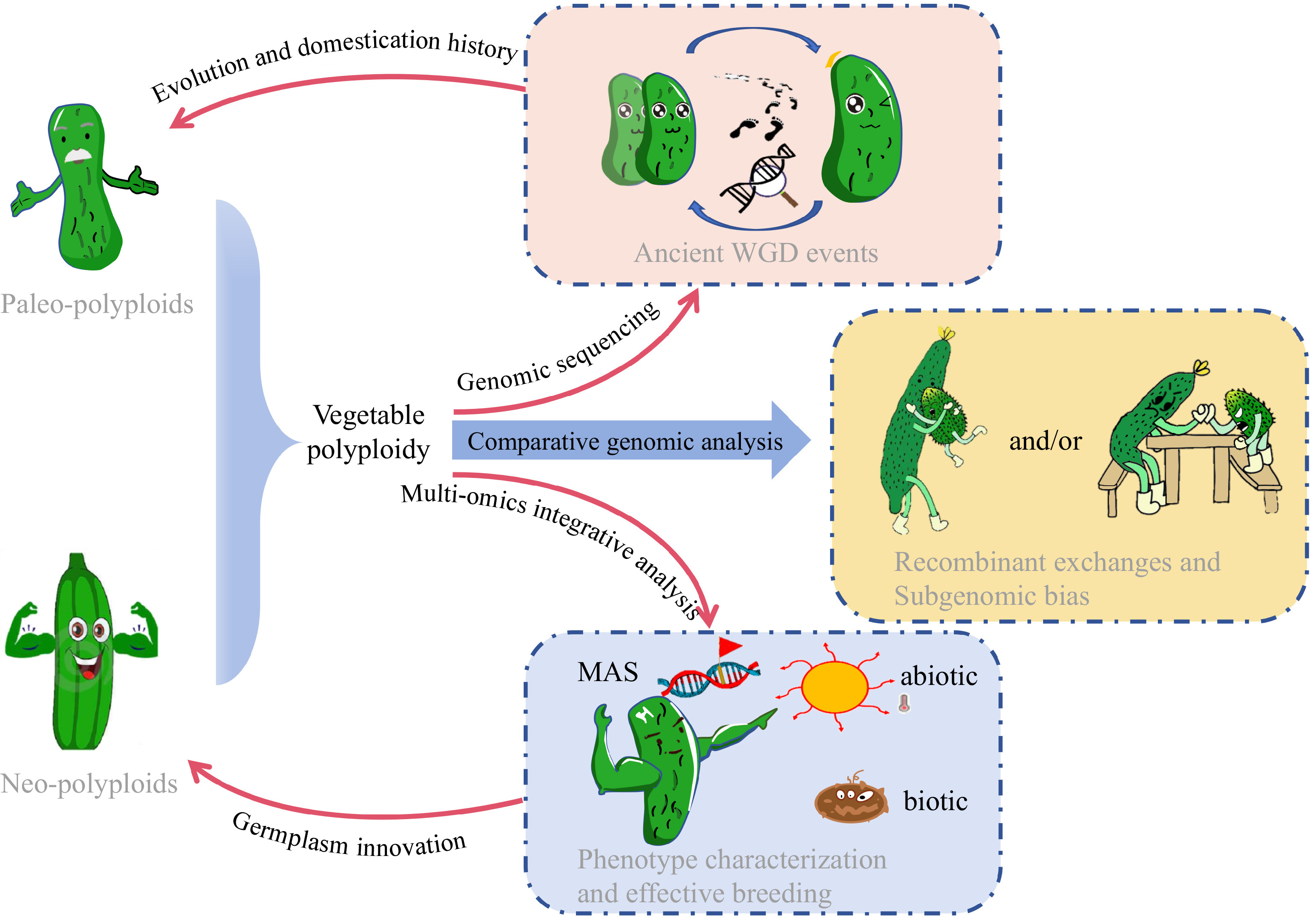
Figure 2.
Schematic diagram illustrating the application of high-throughput sequencing for evolutionary genomics, promoting germplasm innovation and effective breeding in neo- and paleo- polyploid vegetables with cucumber as an example.
-
Whole genome sequencing methods, including survey sequence analysis of RNA or DNA whole genome assembly and so on, provide data for the inference of ancient WGD events in plant evolutionary history[62]. High-quality chromosome-scale genome assembly enables orthologous genes identification and revealed conserved homologous regions in the homology studies of Cucumis diploid and polyploid[26]. These are essential evidence for understanding the evolution and domestication of crops.
Through the analysis of high-quality genomes that have been sequenced and assembled, genome-wide comparative genomic analysis of Cucurbitaceae species identified an overlooked ancient Cucurbit-Common Tetraploidization (CCT) event shortly after the Core-Eudicotium-Ordinary Hexaploid (ECH), which might contribute to the origination and divergence of Cucurbitaceae plants. But by analysis of their sequences, Cucurbitaceae crops also largely retain the genetic information of their diploid progenitor cells, a strong genetic stability after paleopolyploidy that is not common in studied cases of polyploid evolution.[44,63]. This feature provides advantages for studying the evolution of Cucurbitaceae. Genomic analysis of Chinese cabbage (B. rapa ssp. pekinensis) revealed whole-genome triplication (WGT) events during evolution and domestication. Compared with Arabidopsis thaliana, this WGT and subgenome dominance increased the total number of genes of Chinese cabbage by about three times, and promoted the expansion of multiple morphotypes[64]. Ancestors of Allium sativum have lived through two WGD events except for the ancient WGD shared by monocotyledons, suggesting that WGD promotes genome expansion and the proliferation of transposable elements (TEs) in A. sativum[52]. However, WGD is certainly not the only factor that affects genome size. No evidence of recent WGD were found in faba bean (Vicia faba), many genes were duplicated in tandem, which may contribute to its giant genome[60]. The study of stem lettuce (Lactuca sativa var. angustana) also showed that the expansion of gene families of ADS, TAD and KAS was due to both WGD and tandem duplication[61].
Extensive research has shown that allopolyploidy triggers genome-wide gene structural and expressional changes in the first few generations due to the merger of more than two divergent genomes within a single nucleus[65], followed by gradually reduced genetic changes with the process of diploidization. This eventually contributed to the successfully establishment as a new species by returning to stable reproductive process and adapted to new environmental niches[66]. It is found that diploidization and fertility recovery are faster in synthetic allopolyploids than naturally occurring allopolyploids[26], suggesting that breeding via polyploidy is an efficient and feasible way to improve crops.
During the process of introduction, the divergent environments may drive the changes of polyploids to adapt local environment, which could increase the genetic diversity. For example, the genome of rapeseed (B. napus) differentiated rapidly through regional hybridization and intensive selection after the introduction from Europe to Asia, largely expanded the gene pool[67].
-
DNA sequence recombinant exchanges often occurs shortly after polyploidy in plants, which has been studied in many polyploids[68−71]. The rearrangement may have led to gene loss such as the loss of PII in the ancestors of Compositae[61]. Neo-allopolyploids are challenged to maintain meiotic stability for being able to reproduce after merging two or more divergent genomes. However, there is a possibility of crossover between chromosomes from different ancestors, known as inter-homoeologue crossover, which will lead to aneuploidy gamete production, hampering normal meiotic pairing. Therefore, the key to maintain stable allopolyploid meiosis is to prevent the formation of inter-homoeologue crossovers[72]. Studies have shown that changes in MSH4 expression may have specific effects on homoeologous crossover, which suggests that MSH4 proteins may be 'major players' in allopolyploid meiotic adaptation[69].
Homoeologs are a pair of genes or chromosomes from one ancestor which separated until the allopolyploidy event brought it together again[73]. Homoeologous rearrangements between homoeologous chromosomes have different levels and degrees of influence on genotype, genetic stability, and seed yield of offspring after meiosis according to parental inheritance and positive and reverse crossing[74]. For example, the tendency of homoeologous recombination in B. napus is mainly associated with TEs in the diploid ancestors[75]. This phenomenon produces new transcripts that can further result in new protein variants and thus contribute to the evolution of allopolyploid plant species[68], which occurred frequently in the process of polyploidy. This exchange played an important role in the evolution and diversity of B. napus[70]. Exchanges of large chromosomal segments among homoeologous chromosomes may affect the expression of many genes, but many homoeologous exchanges are tolerated with hardly any changes. However, for those dose-sensitive genes, biased expression changes may occur to favor the re-establishment under certain selection pressures[71]. The identification and manipulation of related genes to control changes between homoeologous chromosomes is receiving increased attention, which is crucial for polyploidy research and utilization. The development of sequencing technology provides a feasible approach to detect the changes and its regulating genes in polyploids. It has been demonstrated that recombination-related genes like Fanconi Anemia Complementation Group M (FANCM) is related to meiotic stability, so that homoeologous chromosome recombination, chromosomal deletion and secondary meiosis can be regulated via manipulation of FANCM[59]. By quantification of homoeologous recombination levels in B. napus, three quantitative trait loci (QTLs) were identified for the contribution of the control of homoeologous exchanges, with a major QTL on chromosome A9, BnaPh1, contributing between 32% and 58% to the observed variation[76]. Integrative strategy including sequencing technology can accelerate plant breeding via allopolyploidization with extensive genetic exchanges and creation of new genes.
-
During the process of hybridization and whole genome replication, drastic genome-wide changes happened in the newly formed allopolyploids and their offspring[77]. These changes can be asymmetric between subgenomes. Subgenomes with less sequence loss and higher gene expression levels are termed dominant, while the other subgenomes are termed submissive, this phenomenon is collectively known as subgenome dominance[78,79]. Nevertheless, from the perspective of individual genes, the expression pattern can vary, with genes from submissive subgenomes expressing higher than those homoeologous from dominant subgenomes[80].
By studying genomic data from strawberry and epigenomics data from rapeseeds. Previous studies have shown that the bias in subgenomic gene expression levels is closely related to the genetic background of the subgenome, usually immediately after the first hybridization event[46,81]. Although the number of genes with this expression bias increases with generation, the difference between biased genes towards parents is narrowing and continues to have profound effects on plant evolution. In Brassica polyploids, the subgenomic expression bias of ancestral genomes has a persistent impact on the intraspecific diversity of existing Brassica crops[24,82]. However, in Cucurbita species, no significant subgenome dominance were detected[44], which may be due to the similar genetic background between the ancestral species. Despite such few cases, subgenome dominance were more often seen and played essential roles in contributing to their agricultural value, such as B. napus[83,84], Fragaria × ananassa[46] and Cucumis × hytivus[26].
The combination of multi-omics provides an effective way to interpretate the nature of subgenome dominance from different angles. In addition to changes in genomic structure and expression, subgenome dominance is associated with many epigenetic changes, including DNA methylation, small RNA (sRNA) regulation and chromatin modifications. Changes of sRNA ensures the initial genomic stability of neo-allopolyploid formation by controlling for factors such as various noncoding elements and stress-related gene expression[85]. Previous studies of synthetic allopolyploid wheat have found that small inferring RNAs and chromatin modifications have different effects on different subgenomes, which may be closely related to differences in expression between subgenomic genes[86].
It has been hypothesized that the combination of different epigenetic states (such as histone modifications and DNA methylation levels) and the distribution of TEs resulted in the imbalance in subgenome populations[46,80,87,88]. For example, the combination of different degrees of DNA methylation, the different histone modifications, and different TE content between subgenomes in rapeseed could cause subgenomic imbalances[88]. Alger & Edger hypothesized that difference in gene expression levels between subgenomes may be because of methylation of the TEs rather than the TEs themselves[79]. However, this hypothesis is challenged by recent research in synthetic oilseed rape, they found that the difference in TE dose between allopolyploid subgenomes is not sufficient to cause subgenome bias expression[84]. Histone modifications could also contribute to the formation of subgenome dominance[82,88]. In addition, different tissues and cells may also have different subgenome bias. For example, one subgenome of allopolyploid blueberries has higher gene expression at almost all studied tissues and developmental stages, but the other subgenome has higher gene expression during fruit development[89]. While the reasons for the formation of subgenome dominance are complex, it is well acknowledged that this phenomenon plays a very important role in polyploid evolution and requires further study.
-
Polyploidy is a classical and efficient crop breeding approach, which can produce novel characters within a very short time. Indeed, many crops have been domesticated from these polyploid ancestors, suggesting the significant role of polyploidy. Polyploids often show better quality and adaptative characteristics than diploids, but their genomes are also larger and more complex, with a larger proportion of redundant genes and regulators, making the association of mutant alleles in polyploid crops difficult to identify. Most polyploid crops have recessive mutations, which seriously hinders the use of forward genetics, therefore, reverse genetics are more promising in the study of polyploid species[90,91]. In any case, an accurate and contiguous reference assembly provided by the cutting-age sequencing and assembling techniques is important for the study of polyploid crops.
Based on the improvement of throughput and read length of sequencing technology, breakthroughs have been made in marker assisted selection (MAS) of polyploid crops such as Brassicaceae[92] and Solanaceae[21]. Through single-cell transcriptome sequencing, the responses of different cell types of cabbage to high temperature were uncovered, and the cell-specific photosynthetic response and differential genes were revealed[64,93]. Along with the high-quality chromosome-scale assembly of octaploid strawberries (F. × ananassa), metabolomic analysis showed that dominant subgenomes largely control several biological pathways associated with important agronomic traits[46]. Genome-wide association study (GWAS) using single nucleotide polymorphisms (SNPs) generated from resequencing data of a specific population has been successfully applied to detect quantitative trait loci in many crop species, including vegetables[94]. Causal variants were also identified in polyploid species by using modified strategy of GWAS methods like single-dose SNPs[95]. Polyploid tailored software like GWASpoly were also introduced and used for GWAS in polyploid sugarcane[96] and potato[97]. Moreover, multi-omics datasets were also integrated with GWAS to uncover trait-determining genes in allopolyploid oilseed rape (B. napus L.)[98] and octoploid strawberry[99]. The newly developed tools or integrative strategy of multi-omics pave the way for effectively elucidating the underlying mechanism of various phenotypes in polyploid vegetables, and thus can be extremely useful for crop germplasm enhancement.
-
Polyploid vegetable crops are widely grown due to their unique traits for higher yields and better qualities. With the utilization of advanced sequencing technology, sequences of many complex vegetable genomes, especially polyploids, have been successfully characterized. It shows that the contribution of polyploidy to the rapid expansion of angiosperms, known as 'Darwin's abominable mystery'. More importantly, as polyploidy is one of the main approaches for germplasm innovation, these outputs provide fundamentally important resources for crop enhancement. The use of sequencing and related methodologies to reveal its related molecular mechanisms is of specific significance for the better use of polyploidy for vegetable crop improvement. This is especially the case for vegetable crops where the vegetative parts are consumed, thus, organ magnification due to polyploidy bring direct economic benefits.
Despite those progresses achieved by sequencing technology, the availability of multi-omics data and more efficient analysis tools, there are still many problems remaining. Examples are a comprehensive understanding of polyploidization, the significance of subgenome dominance, the genetic determinants of larger organs or not in polyploids, and so on. Polyploid vegetables belong to different families, however, in-depth basic research has been carried out in only few species and results are often contradictory. Another possible reason is that many vegetables are regional, which explain why tomato, cucumber and Brassica crops have in focus. Thus, the research on minor vegetables are way behind major polyploid crops like wheat and cotton, despite the continuous lower costs of sequencing. Research work on 'famous' vegetable crops species are often easier to publish in more influential journals that prefer a wider potential audience, while the work on minor vegetable crops receive less attention and less funding for doing fundamental research, which may affect the motivation and confidence of the corresponding researchers.
Against the background of continuous global population growth, climate change, and deteriorating resource environment, using polyploidy to increase crop diversity and enhance environmental adaptability is essential in crop breeding to secure global food security and food quality. Furthermore, health problems caused by high calorie and high sugar diets in the past makes people pay more attention to the need for healthy dietary fiber, pinpointing the importance for vegetable breeding. The complex genomes and uncertainty of phenotypic changes of polyploids are the main limitations that hamper the development of polyploid breeding. Advanced technologies like ultra-long read and single-cell sequencing, which are essential for polyploid research, are still unaffordable for wide application in vegetable research. We call for more attention and support for research in non-model vegetable crops, which is fundamentally important for diversification in vegetable germplasm and a healthy diet.
Previous evidence has shown the successful application of multi-omics in polyploid crop research. Lower and lower sequencing costs will providing more cost-effective and valuable resources that will provide us with deeper understanding of polyploidy, such as the molecular mechanism of enhanced adaptation to adverse environments. The growing amount of sequencing data puts forward higher requirements for data processing and efficient utilization. How to screen out those key factors from vast amounts of data will be the next question that needs to be addressed. Recent progress on variant prediction using Artificial Intelligence may be a promising direction for polyploidy-based molecular design crop breeding[100]. Meanwhile, even if candidate genes were sifted out, how to verify their function remains a challenge for most non-model vegetable crops. The establishment and optimization of genetic transformation systems in a broader range of vegetables, especially polyploids species, deserves attention with significant resources to secure future proofed crops.
-
The authors confirm contribution to the paper as follows: study conception and design: Yu X; data collection: Du W; draft manuscript preparation: Du W, Wang X, Zhao X, Pei Y, Xia L, Zhao Q, Cheng C, Wang Y, Li J, Qian C, Lou Q, Zhou R, Chen J, Yu X; manuscript revision: Du W, Yu X, Ottosen CO. All authors reviewed the results and approved the final version of the manuscript.
-
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
This research was supported by the National Key Research and Development Program of China (2023YFF1002000), the National Natural Science Foundation of China (32372697) and the Natural Science Foundation of Jiangsu Province (BK2022148).
-
The authors declare that they have no conflict of interest.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Du W, Wang X, Zhao X, Pei Y, Xia L, et al. 2024. How high-throughput sequencing empowers the research of polyploidy in vegetable crops. Vegetable Research 4: e006 doi: 10.48130/vegres-0024-0005 

How high-throughput sequencing empowers the research of polyploidy in vegetable crops
- Received: 29 August 2023
- Revised: 06 December 2023
- Accepted: 09 January 2024
- Published online: 05 February 2024
Abstract: Vegetables are not only economically important, but also essential for a healthy human diet providing fiber, minerals and essential nutrients. All flowering plants, including many vegetable crops, are polyploids, pinpointing the significance of polyploidy in plant evolution and crop breeding. In the last two decades, the fast development of sequencing has facilitated genome wide investigation of genetic and epigenetic changes that has occurred during the polyploidization process. With the achievement of more and more high-quality plant genomes, ancient polyploidization (also known as whole genome duplication, WGD) events have frequently been seen, which is vital for the understanding of domestication and differentiation history of vegetables. Moreover, advanced joint analysis of multi-omics data has been applied for efficient elucidation of underlying molecular mechanisms of complex traits in vegetables. This paper summarizes the status for the research on vegetable polyploids facilitated by high-throughput sequencing, that improve the understanding of plant evolution and support effective vegetable breeding via polyploidy.
-
Key words:
- Polyploid /
- Vegetable /
- Sequencing /
- Evolution /
- Breeding