









-
In plants, Ca2+ is an integral element in several stress related signallings. During environmental stresses, the cytosolic Ca2+ concentration tends to rise rapidly, which is further co-ordinate with EF-hand domain-containing proteins[1]. Low temperature stress is an environmental challenge that severely limits the geographical distribution and survival rate of certain perennial plants[2]. Nowadays, some plants have evolved specific and effective molecular mechanisms to resist cold damage, and many functional genes involved in cold response have been identified in plants, with some of these genes being closely associated with Ca2+. Stomatal immunity is regulated by pathogen-associated molecular patterns (PAMPs)- and abscisic acid (ABA)-triggered signalling in different ways. Cytoplasmic Ca2+ signature in the guard cells plays a vital function in stomatal immunity, but the mechanism of Ca2+ import is unknown. In the study of the mechanism of PAMPs triggering stomatal closure, it was found that four types of CNGCs (CNGC2, 5, 6, 9, and 12) were enriched in Arabidopsis thaliana guard cells and involved in ABA induced cytoplasmic Ca2+ oscillations. However, only some inactive stomatal movements indicate that other Ca2+ channels (possibly OSCAs) are actively involved in Ca2+ mediated stomatal immunity. In addition, CDPKs mediated phosphorylation of CNGC6 also indicates the existence of alternative pathways. However, the interrelationships between various Ca2+ channels and their mechanisms of simultaneous activation during stomatal defense need to be validated in future research. In addition, ROS induced Ca2+ signaling is not affected in cells, indicating the function of these channels downstream of the ROS pathway[3]. Ca2+ signaling represents a universal transduction signal in plants, mediated by a complex network of Ca2+ binding proteins, many of which function as Ca2+ sensors and act on downstream responses[4]. Calcium-dependent protein kinases (CDPKs) form functional complexes with CBL-interacting protein kinases (CIPKs), enabling plants to respond to various environmental signals and regulate ion fluxes[5]. The CBL-CIPK complex plays a crucial role in signal transduction pathways, where Ca2+ serves as a second messenger, particularly in the regulation of ion transporter activities in response to non-biological signals[6]. In poplar trees, the PeCBL/PeCIPK complex has been identified and shown to play a role in Na+/K+ homeostasis[7].
Over the past few decades, numerous CBL-CIPK complexes have been shown to be involved in signal transduction under non-biological stress. The CBL-CIPK signaling network system, composed of CBLs and their target proteins CIPKs (CBL-interacting protein kinases), plays a significant role in plant responses to various abiotic stresses such as drought, salinity, and low temperature[8−11]. The plant CBL gene family typically consists of 10 members and was initially discovered in A. thaliana, but has since been successfully isolated from various crops such as Zea mays, Populus alba, Gossypium sp, and Oryza sativa[11−14]. Numerous studies have shown that calcineurin B like proteins (CBL) play an important role in plant stress response. Recent studies on Setaria italica have found that SiCBL3 is widely involved in the response of S. italica seedlings to various abiotic stress conditions, such as PEG, salt, high temperature, low temperature, and ABA; SiCBL3 is highly expressed during normal heading and filling stages, and is extensively induced under drought stress during the jointing, heading, and filling stages of S. italica[15]. Studying the gene structure, distribution, and expression characteristics of CBL genes in plant genomes is of great significance in gaining a deeper understanding of their roles in growth, development, and stress responses. The members of the CBL gene family show a strong conservation in their structural features, with most CBL family members containing 7~8 introns, and the positions of these introns within the coding region are relatively fixed[14,16]. The differences in domain structures among different CBL proteins may contribute to their varying abilities to bind calcium ions, providing a foundation for CBLs to sense changes in Ca2+ concentration triggered by various stimulus signals[17]. In recent years, CBL genes from multiple species have been successfully isolated and functionally validated[18−24]. In A. thaliana, AtCBL1 and AtCBL9 are localized to the plasma membrane, and both genes show expression throughout development, although their expression levels are relatively weak in roots. AtCBL5 is also a calcium signaling component located on the plasma membrane, and it exhibits high-level expression in leaves[18,19]. Numerous studies have demonstrated that CBLs exhibit different expression patterns in response to various stressors and during growth and development processes. For example, AtCBL1 is strongly induced by low temperature but is not influenced by exogenous ABA stress[18,20], while AtCBL9 is induced by ABA stress[19]. Although significant progress has been made in the study of CBL genes, most research has been limited to a few plant species, and the majority of CBL genes remain unisolated and functionally characterized.
P. mume is an important ornamental woody plant. It is native to the Sichuan-Yunnan-Tibet region and mainly distributed in the Yangtze River Basin (China). To date, research on freezing resistance in P. mume has primarily focused on the ICE-CBF signaling pathway, while studies on the upstream Ca2+ signaling pathway have been relatively limited. Building upon previous studies on the response of CIPK gene family to low temperature stress, this research focused on identifying and investigating the response to low temperature stress of CBL protein family members that interact with CIPK proteins. The use of bioinformatics analysis methods, that is, based on existing bioinformation databases and resources, the use of mature bioinformatics tools to solve bioinformatics problems. In this study, all CBL gene family members were identified based on the whole genome sequence information of P. mume. The number of CBL genes, gene structure, evolution, and expression patterns under low temperature stress were analyzed at the genome level, providing insights into the biological functions of these family members. Additionally, this study provides an important theoretical basis for the breeding of cold resistant P. mume varieties.
-
The P. mume cultivar 'Zao Lve' was used as a plant material. The 'Zao Lve' variety, after years of cold domestication, could survive and bloom when grown in open fields in North China. For the experiment, two-year-old grafted seedlings of the 'Zao Lve' variety were selected and subjected to low-temperature treatment.
Identification of the P. mume CBL gene family
-
The data for the P. mume gene family in this study were obtained from the P. mume database, including the P. mume gff annotation file and P. mume CDS encoding protein sequences. The CBL gene family A. thaliana was derived from the Uniprot gene database (
www.uniprot.org )[25]. The CBL domain files for each gene member of the obtained A. thaliana gene family were determined and downloaded using PFAM (https://precisionflange.com/ )[26]. To identify the members of the P. mume CBL gene family, the ten published A. thaliana CBL gene family members were used as query genes. The hmmsearch plugin in the hmmer software was utilized for the search (e-value set at 10−5, with other parameters set to default values) to screen for CBL homologous sequences from the P. mume gene family members. The results were then searched against the A. thaliana family in the String database, and members of the P. mume CBL gene family with a similarity greater than 50% were selected.Gene characteristic analysis and protein physicochemical property prediction
-
Based on the identified sequence IDs, the GXF Select tool in TBtools was used to extract the annotation information of the identified sequences. This allowed us to obtain the chromosome numbers and location information of the P. mume CBL gene family members. The six CBL family members were then named based on their respective location information. The ExPASy-Protparam online tool (
https://web.expasy.org/protparam ) was used to analyze the physicochemical properties of the gene family members. It involved amino acid count statistics and predicted protein properties such as molecular weight (MW), theoretical isoelectric point (pI), aliphatic index, instability index, and grand average of hydropathicity (GRAVY). The SignalP v6.0 online tool was used to predict signal peptides, while the TMHMM v2.0 online tool was utilized to predict transmembrane helices. The Cell-PLoc v2.0 online tool (www.csbio.sjtu.edu.cn/bioinf/Cell-PLoc-2 ) was employed for subcellular localization prediction.Phylogenetic analysis of the P. mume CBL gene
-
In this study, the phylogenetic tree of P. mume CBL gene family was analyzed by Neighbor-Joining Algorithm method[27]. The MEGA 7.0 (7170509-X86_64) software was used for phylogenetic analysis of the gene family. The protein sequences of CBLs from A. thaliana, O. sativa, Vitis vinifera and Nicotiana tabacum were obtained from the Uniprot database (
www.uniprot.org )[25]. The MUSCLE program in the software was used for multiple sequence alignment of the selected CBL protein sequences with their corresponding A. thaliana homologous protein sequences (using default parameters). The aligned results were subjected to phylogenetic analysis using the Neighbor-Joining (NJ) method, with the number of differences as the computational model and pairwise deletion as the gap handling option. Bootstrap support was set to 500 for tree reliability assessment. The resulting phylogenetic tree was visualized and beautified using the Evolview online tool (www.evolgenius.info/evolview/#/ )[28].Chromosomal localization of P. mume CBL genes
-
The identified CBL genes were assigned to their corresponding positions on the P. mume genome. The location information of each gene on the chromosomes was obtained using the online tool MG2C (
http://mg2c.iask.in/mg2c_v2.1/ ) to generate a chromosomal localization map.Protein alignment and conserved motif analysis of P. mume CBL proteins
-
Multiple sequence alignment of the selected CBL protein sequences was performed using the Clustal W program in MEGA software (with default parameters). The MEME online tool (
https://meme-suite.org/meme/ ) was utilized for motif analysis to predict conserved motifs and domains present in the protein sequences. Based on the identified P. mume CBL gene family with six gene members, the Gene Structure View (Advanced) tool in TBtools software was used along with the downloaded P. mume annotation information gff file from the P. mume database to analyze the gene structure of the P. mume CBL gene family.Analysis of cis-acting elements in the P. mume CBL gene family
-
In order to analyze the potential cis-acting elements in the promoter sequences of the P. mume CBL gene family, the upstream 700 bp promoter sequence regions were downloaded in bulk from the ATG start codon. The obtained promoter sequences were then analyzed for cis-acting elements using the PlantCARE online tool (
http://bioinformatics.psb.ugent.be/webtools/plantcare/html )[29]. The resulting Tab file was opened in an Excel spreadsheet, and the desired cis-acting elements were filtered. Finally, the Simple BioSequence Viewer function in TBtools software was used to visualize and plot the identified cis-acting elements.Expression analysis of PmCBLs genes
-
In order to further investigate the expression patterns of PmCBL genes, We collected transcriptomic expression profile data from different tissues (roots, stems, leaves, buds, and fruits) and during the natural overwintering process (November, December, January, and February) in P. mume[30].
Low temperature treatment of plant materials and detection of relative expression levels
-
Seedlings cultivated under long-day conditions (16 h light/8 h dark) at 24 °C were used to investigate the effect of PmCBL genes on cold response. The seedlings were incubated at 4 °C, mimicking a cold environment. One-year-old branches that underwent 4 °C treatment were selected for sampling at various time points including 0, 1, 4, 6, 12, and 24 h. Total RNA was extracted from the samples using a suitable method. First-strand cDNA synthesis was performed using the TIANScript First Strand cDNA Synthesis Kit (Tiangen, China). qRT-PCR was performed using the PikoReal Real-Time PCR System (Thermo Fisher Scientific, CA, USA) and SYBR Premix ExTaq TM (TaKaRa, Dalian, China). The reaction was carried out in a 10 μL system, which included 5 μL of SYBR Premix ExTaqII, 0.25 μL each of forward and reverse primers, 0.5 μL of cDNA, and 3 μL of ddH2O. The PCR program consisted of 40 cycles at 95 °C for 30 s, 95 °C for 5 s, and 60 °C for 40 s, followed by a final extension at 60 °C for 30 s. Three replicates were performed for each sample. The internal reference gene used was PP2A from P. mume. The relative expression levels of PmCBL genes were calculated using the 2−ΔΔCᴛ method. Statistical analysis with standard deviation was performed on the final data.
-
Through bioinformatics analysis methods, a total of six CBL gene family members were identified (Fig. 1a) and named as PmCBL1~PmCBL6 (Table 1). These six CBL genes were unevenly distributed on the chromosomes. PmCBL1 was located on chromosome 1, PmCBL2 and PmCBL3 were located on chromosome 5, and PmCBL4, PmCBL5, and PmCBL6 were located on chromosome 7. In the PmCBL family, the coding sequence (CDS) of PmCBL6 was the longest, comprising 2,475 bp. PmCBL1 had the second-longest CDS with 2,418 bp, followed by PmCBL3 with 1,893 bp, PmCBL2 with 1,805 bp, PmCBL5 with 1,564 bp, and PmCBL4 with the shortest CDS of 1,252 bp. The subcellular localization prediction of these six genes was all on the cell membrane. Through analysis of conserved protein domains, it was found that PmCBLs contain six conserved motifs (Motif 1~6) (Fig. 1b). Specifically, PmCBL2, PmCBL3, PmCBL5, and PmCBL6 contained all six motifs (Motif 1~6), while PmCBL1 and PmCBL4 contained Motif 1~4 and Motif 6 but did not contain Motif 5. From the results of protein multiple sequence alignment among P. mume, A. thaliana, V. vinifera, and O. sativa, it could be observed that P. mume showed higher homology with A. thaliana and V. vinifera, while the homology with O. sativa was relatively lower (Fig. 2).
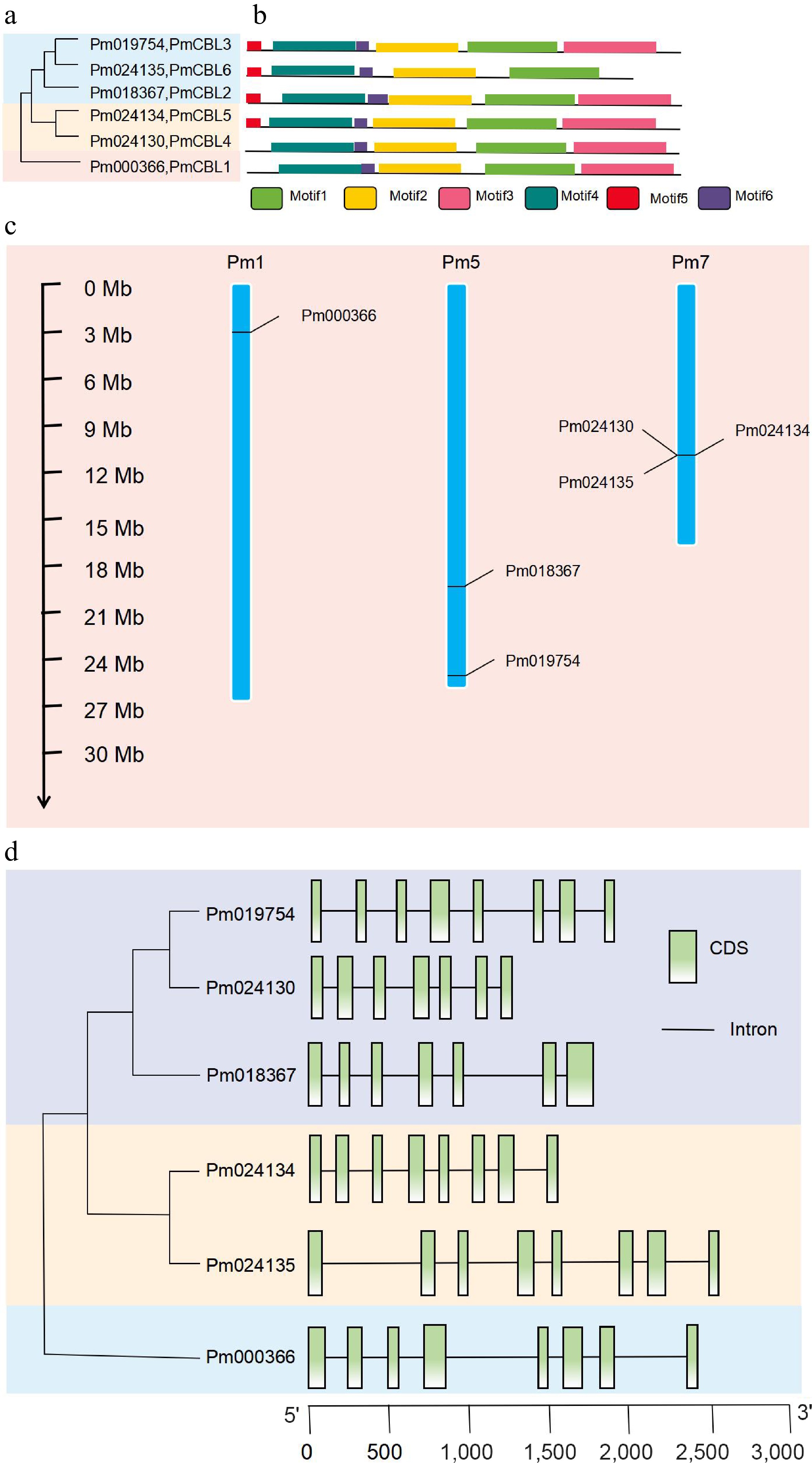
Figure 1.
Phylogenetic analysis, protein motif structure, chromosome localization, and gene structure analysis of PmCBL genes. (a) Phylogenetic analysis of PmCBL genes. (b) Protein motif structure of PmCBL genes. (c) Chromosome localization analysis of PmCBL genes. (d) Gene structure analysis of PmCBL genes.
Table 1. Members of the P. mume PmCBL gene family and their main molecular characteristics and information.
Gene name Gene ID Chromosome Position Subcelllar localization CDS (bp) Intron PmCBL1 Pm000366 Chr01 2287536~2289774 Cell membrane 2,418 5 PmCBL2 Pm018367 Chr05 17870110~17871915 Cell membrane 1,805 4 PmCBL3 Pm019754 Chr05 25555186~25557079 Cell membrane 1,893 5 PmCBL4 Pm024130 Chr07 10584318~10585570 Cell membrane 1,252 4 PmCBL5 Pm024134 Chr07 10621981~10623545 Cell membrane 1,564 5 PmCBL6 Pm024135 Chr07 10622516~10628186 Cell membrane 2,474 5 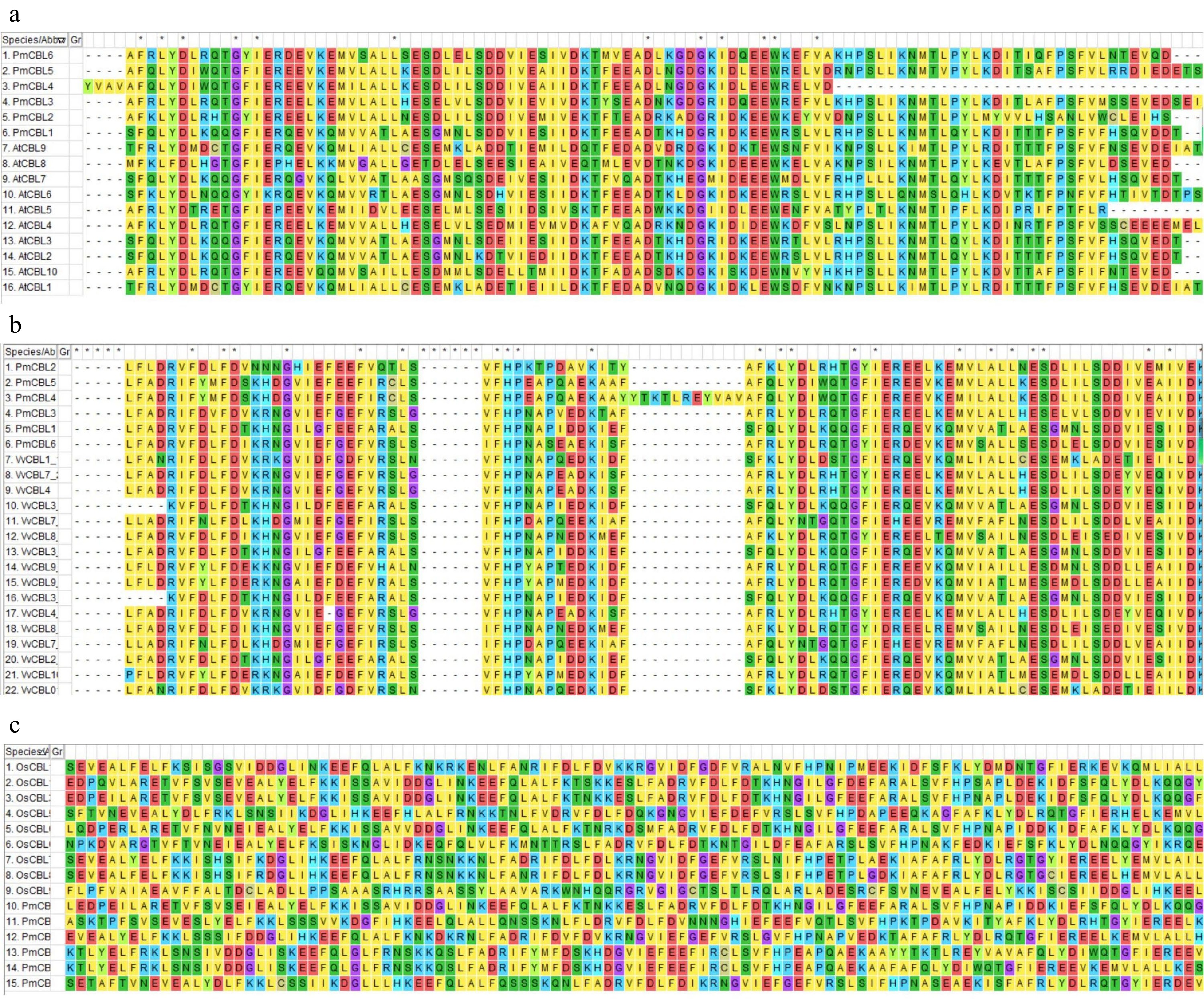
Figure 2.
Protein amino acid sequence comparison diagram. (a) Protein amino acid sequence comparison diagram between P. mume and A. thaliana. (b) Protein amino acid sequence comparison diagram between P. mume and V. vinifera. (c) Protein amino acid sequence comparison diagram between P. mume and O. sativa.
Physicochemical property analysis of the P. mume CBL gene family
-
The physicochemical properties of the six CBL homologous sequences were analyzed (Table 2). The results showed that the amino acid (aa) numbers of the PmCBL protein sequences ranged from 186 (PmCBL4) to 226 (PmCBL1), with molecular weights (MV) ranging from 21.66 kDa (PmCBL4) to 26.05 kDa (PmCBL1). The theoretical isoelectric points (pI) ranged from 4.57 to 5.14. The instability index values ranged from 35.97 to 49.06, with only PmCBL1 having an instability index below 40, indicating high stability of CBL protein. The grand average of hydropathicity (GRAVY) values for PmCBL1 ~ PmCBL6 were all negative, indicating that these six proteins were hydrophilic. The aliphatic index values ranged from 90.09~96.96, indicating that all six proteins were lipophilic. None of the six gene family members contained signal peptides or transmembrane helices.
Table 2. Physicochemical properties of P. mume CBL gene family members.
Protein name Gene ID Number of amino acids Molecular weight Thereotical pI Instability index Signal peptide PmCBL1 Pm000366 226 26052.71 4.82 49.06 NO PmCBL2 Pm018367 217 24903.52 5.14 35.97 NO PmCBL3 Pm019754 212 24429.82 4.73 46.65 NO PmCBL4 Pm024130 186 21665.75 4.6 40.14 NO PmCBL5 Pm024134 213 24720.17 4.57 40.98 NO PmCBL6 Pm024135 218 25144.71 4.86 46.47 NO Evolutionary relationship analysis of the CBL gene family in P. mume
-
To understand the evolutionary relationship between members of the P. mume CBL gene family, a phylogenetic tree was constructed using the selected six PmCBL genes, 17 OsCBLs genes from O. sativa, 16 VvCBLs genes from V. vinifera, 20 NtCBLs genes from N. tabacum and 10 AtCBLs genes from the model plant A. thaliana (Fig. 3). The results of the evolutionary analysis showed that the CBL gene family could be divided into three groups, each containing among them, P. mume Group I included PmCBL2, PmCBL3, PmCBL4, PmCBL5, and PmCBL6. Group III consisted of PmCBL1. In Group I, PmCBL4 and PmCBL5 were paralogous gene pairs. This suggested that the CBL gene in P. mume underwent expansion, expansion and replication. Some genes in AtCBLs, NtCBLs, VvCBLs and PmCBLs could be considered as orthologous gene pairs, such as AtCBL4 and PmCBL3, NtCBL14 and PmCBL2, VvCBL8 and PmCBL6. The discovery of orthologous gene pairs suggests the existence of ancient CBL genes before P. mume and A. thaliana, N. tabacum, V. vinifera were also similar.
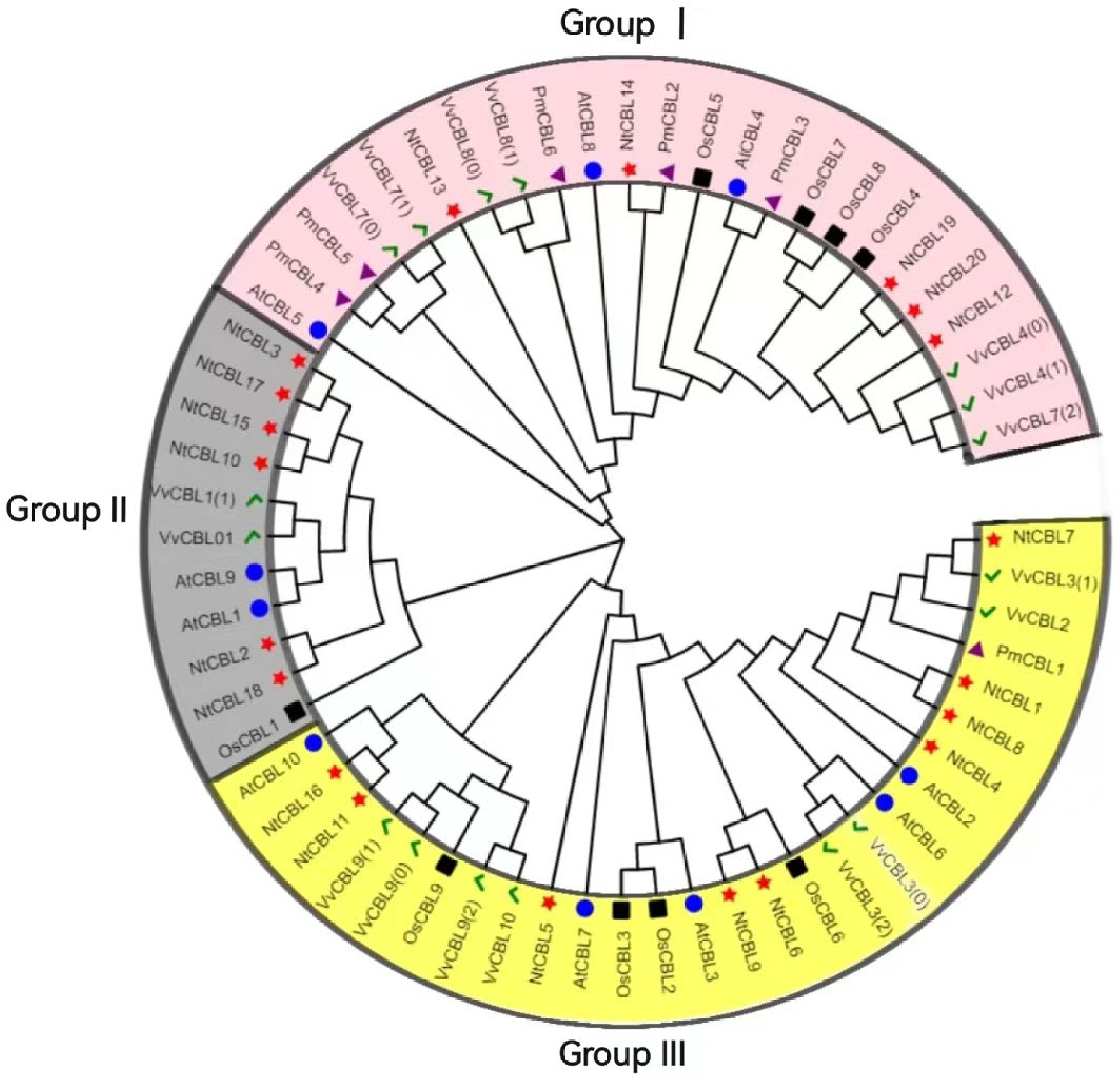
Figure 3.
Phylogenetic tree of P. mume, A. thaliana, O. sativa, N. tabacum and V. vinifera. The green checkmark represents V. vinifera, the black square represents O. sativa, the blue circle represents A. thaliana, the red star represents N. tabacum, and the purple triangle represents P. mume.
Analysis of gene structure and chromosome localization of the P. mume CBL gene family
-
Further analysis of the gene structure of the P. mume CBL gene family revealed that the six PmCBL genes share a similar overall gene structure (Fig. 1d). Specifically, PmCBL3 had eight exons separated by introns. PmCBL4 had seven exons with approximately equal sizes separated by introns. PmCBL2 had six exons separated by introns, with two exons being relatively far apart. PmCBL5 had eight exons separated by introns, and the sizes of these eight exons were roughly similar with a similar distance between each pair of exons. PmCBL6 had eight exons separated by introns, with some exons having larger inter-exon distances. PmCBL1 also had eight exons separated by introns, and similarly, some exons had larger inter-exon distances.
Based on the results from the MG2C online tool (Fig. 1c), the six identified P. mume CBL genes were found to be located on three chromosomes. PmCBL2 and PmCBL3 were located on one chromosome, PmCBL4, PmCBL5, and PmCBL6 were located on another chromosome, and PmCBL1 was located on a separate chromosome.
Analysis of cis-acting elements in the P. mume CBL genes
-
In order to further investigate the potential functional roles of PmCBL genes, the 700 bp sequence upstream of the start codon of each PmCBL gene was extracted as its promoter region. Cis-acting element analysis was performed on this region, focusing on important elements that had been extensively studied and were associated with plant growth and development, as well as stress responses. (Table 3, Fig. 4) The results showed that a total of 17 cis-acting element types responsive to plant hormones and stress were identified, and there were differences in the types and quantities of elements among different genes. Among them, all genes contained common cis-acting elements (CAAT-box) in their promoter and enhancer regions, with slight differences in the number of elements ranging from 2~6. PmCBL3 had the highest number of six (CAAT-box elements) , while PmCBL4 had the lowest two. Two cis-acting regulatory elements involved in MeJA response (CGTCA-motif and TGACG-motif) were found, and five genes contained both of these elements, with an equal number in each gene. Five genes contained the core promoter element (TATA-box) located around the transcription start site −30, namely PmCBL1 ~ PmCBL5, but there was a large variation in the number of elements among the members, ranging from 4~11. PmCBL1 had one light-responsive element (GT1-motif), while the other genes did not contain it. There were four light-responsive elements identified: PmCBL1, PmCBL4, and PmCBL5 each contained one GATA-motif, PmCBL2 contained one I-box, PmCBL1 contained one TCCC-motif, and PmCBL3 contained one LAMP-element. Two cis-acting regulatory elements involved in zein protein metabolism regulation were found: O2-site and MBS. PmCBL3, PmCBL4, and PmCBL5 each contained one O2-site, PmCBL6 contained two O2-sites, and PmCBL4 and PmCBL6 each contained one MBS. One cis-acting regulatory element involved in light response (G-box) was identified, with PmCBL4 and PmCBL5 each containing one. One cis-acting regulatory element required for anaerobic induction ARE was found, with PmCBL2 and PmCBL5 each containing one. One cis-acting regulatory element (A-box) involved in cis-element regulation was found, with PmCBL5 and PmCBL6 each containing one. One cis-acting element involved in abscisic acid response ABRE was found, with PmCBL4 and PmCBL5 each containing one. One cis-acting element involved in auxin response (TGA-element) was found in PmCBL6. One cis-acting element involved in salicylic acid response (TCA-element) was found in PmCBL4.
Table 3. Analysis of cis-acting elements in the P. mume CBL gene family members.
Gene PmCBL1 PmCBL2 PmCBL3 PmCBL4 PmCBL5 PmCBL6 Gene ID Pm000366 Pm018367 Pm019754 Pm024130 Pm024134 Pm024135 CAAT-box 3 4 6 2 4 4 CGTCA-motif 2 1 1 2 1 TGACG-motif 2 1 1 2 1 TATA-box 4 4 4 11 8 GT1-motif 1 TCCC-motif 1 GATA-motif 1 1 1 ARE 1 1 G-box 1 1 MBS 1 1 TGA-element 1 A-box 1 1 I-box 1 O2-site 1 1 1 2 LAMP-element 1 ABRE 1 1 TCA-element 1 CAAT-box was a common cis-acting element in the promoter and enhancer regions. CGTCA-motif/TGACG-motif was cis-acting regulatory elements involved in MeJA response. TATA-box was a core promoter element located around the transcription start site (-30). GT1-motif was a light-responsive element. TCA-element was a cis-acting element involved in salicylic acid response. TGA-element was an element involved in auxin response. ABRE was a cis-acting element involved in abscisic acid response. A-box was a cis-acting regulatory element. ARE was a cis-acting element required for anaerobic induction. G-box was a cis-acting element involved in light response. O2-site/MBS was cis-acting regulatory elements involved in zein protein metabolism regulation. TCCC-motif/GATA-motif/I-box/LAMP-element was part of light-responsive elements. 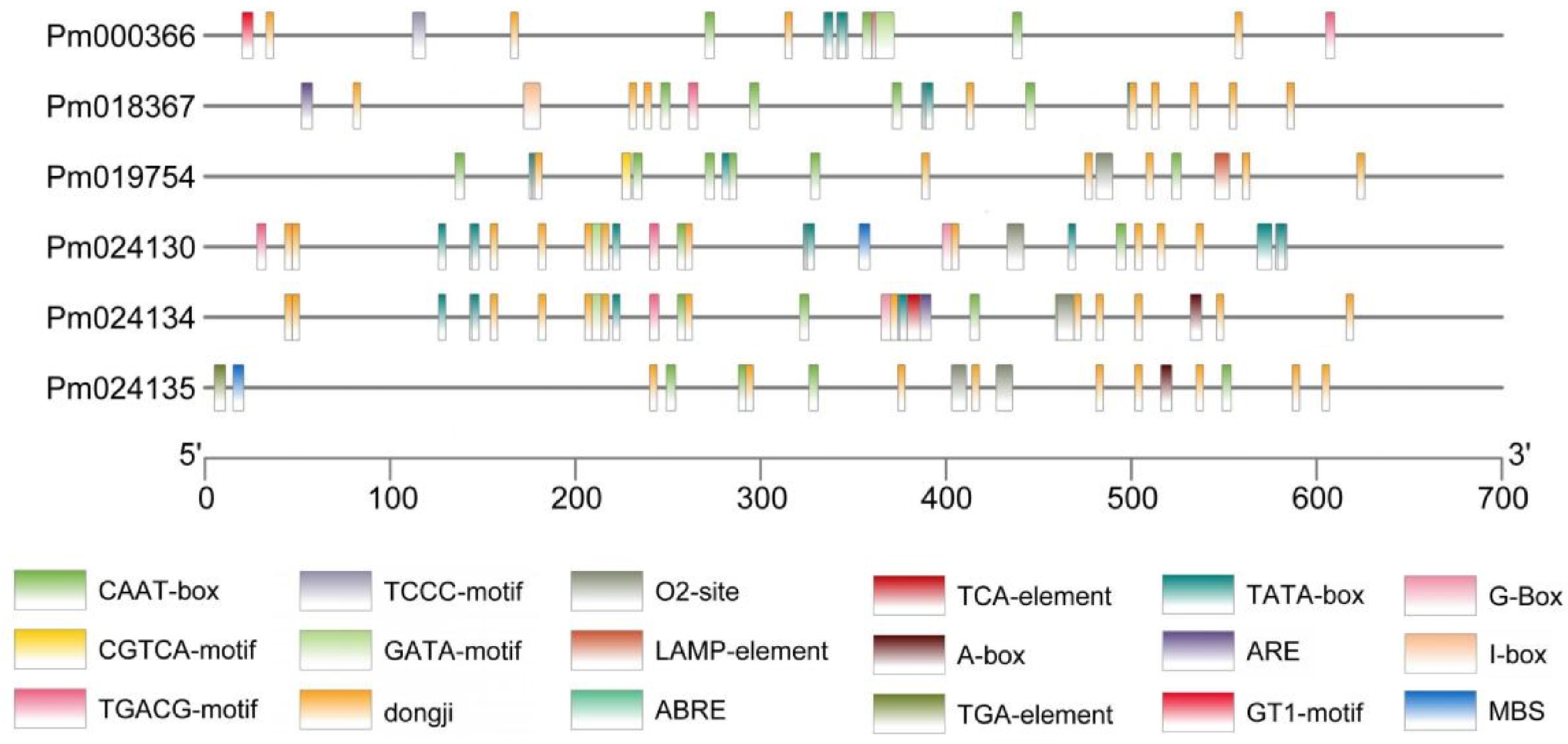
Figure 4.
Analysis of cis-acting elements in PmCBL genes.
Gene expression of the P. mume CBL gene family
-
By analyzing transcriptome data from different parts of the P. mume, tissue-specific expression patterns were observed in different members of the P. mume CBL gene family (Fig. 5). PmCBL1 showed higher expression levels in flower buds, PmCBL2 exhibited higher expression levels in roots, PmCBL3 and PmCBL6 had higher expression levels in leaves, and PmCBL4 and PmCBL5 showed higher expression levels in stems. From this, it could be observed that PmCBL3 and PmCBL6 had similar expression patterns, while PmCBL4 and PmCBL5 had similar expression patterns. This suggested that they belonged to the same Group I subfamily.
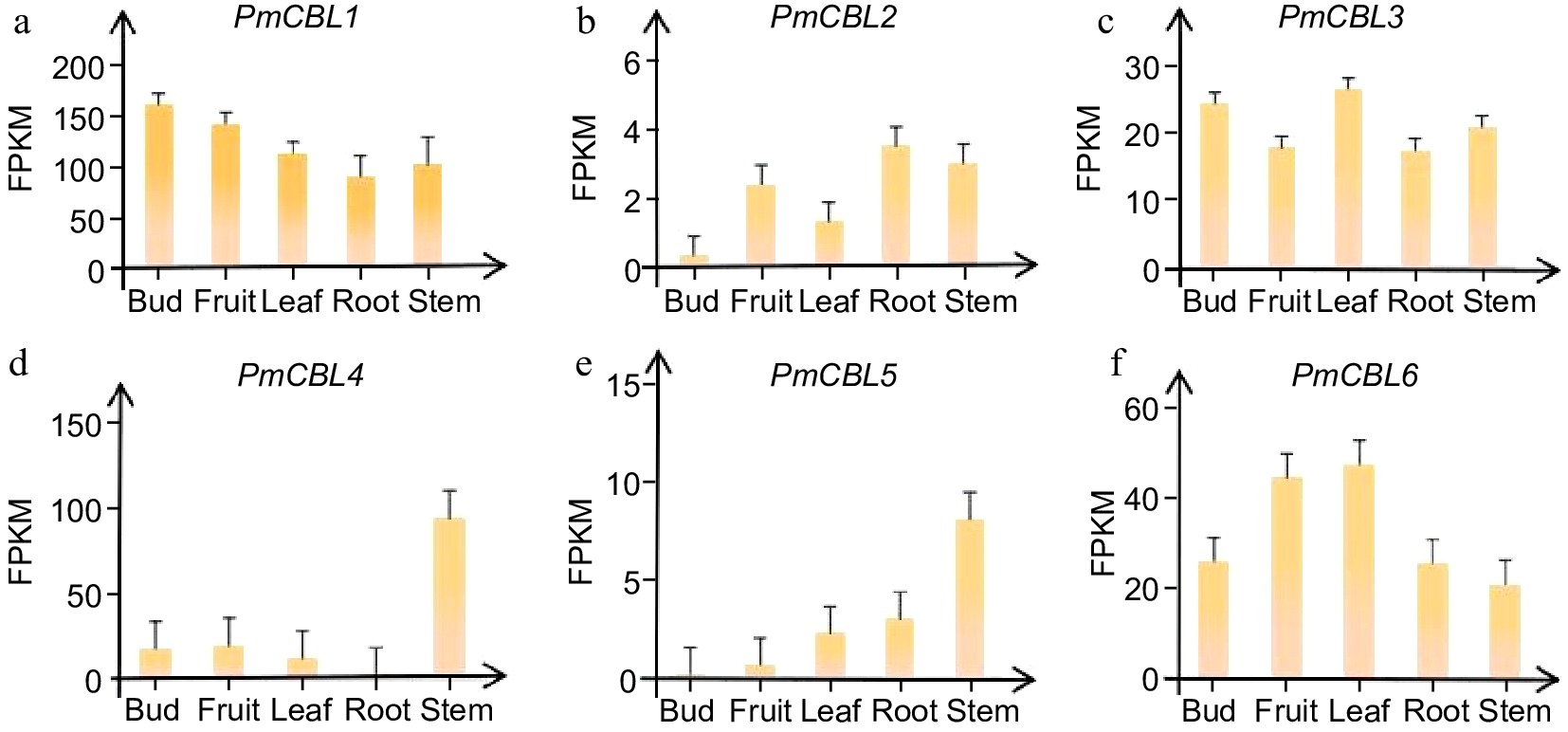
Figure 5.
Expression patterns of PmCBL genes in different tissue parts of P. mume.
By analyzing the transcriptome data of P. mume during wintering, the wintering process of P. mume could be divided into three stages: the early stages of overwintering, which was November (DD); midwinter, which was December (NDD); late overwintering which was January (LD); and naturalness, which was February (NF). The expression patterns of the P. mume CBL gene family could be obtained (see Fig. 6), with different genes showing different expression patterns. PmCBL1 showed an upregulation trend in all three stages compared to the NF stage, reaching its peak in the LD stage, with expression levels 1.23 times higher than the NF stage. PmCBL2 exhibited both upregulation and downregulation trends in the three stages compared to the NF stage. It reached its maximum expression level in the NDD stage, being 1.22 times higher than the NF stage. However, it reached its minimum expression level in the LD stage, showing a downregulation of 1.3 times compared to the NF stage. Overall, PmCBL2 displayed a downregulation trend. PmCBL3 showed an upregulation trend in all three stages compared to the NF stage, with the highest expression level observed in the DD stage, being 1.06 times higher than the NF stage. PmCBL4 showed a downregulation trend in all three stages compared to the NF stage, with the minimum expression level observed in the DD stage, showing a downregulation of 1.20 times compared to the NF stage. PmCBL5 exhibited a downregulation trend relative to the NF stage, reaching its minimum expression level in the NDD stage, showing a downregulation of 8.28 times compared to the NF stage. PmCBL6 showed an upregulation trend relative to the NF stage, with an upregulation of 1.42 times compared to the NF stage. PmCBL4 and PmCBL5 were known to had significant roles in the wintering process of P. mume.
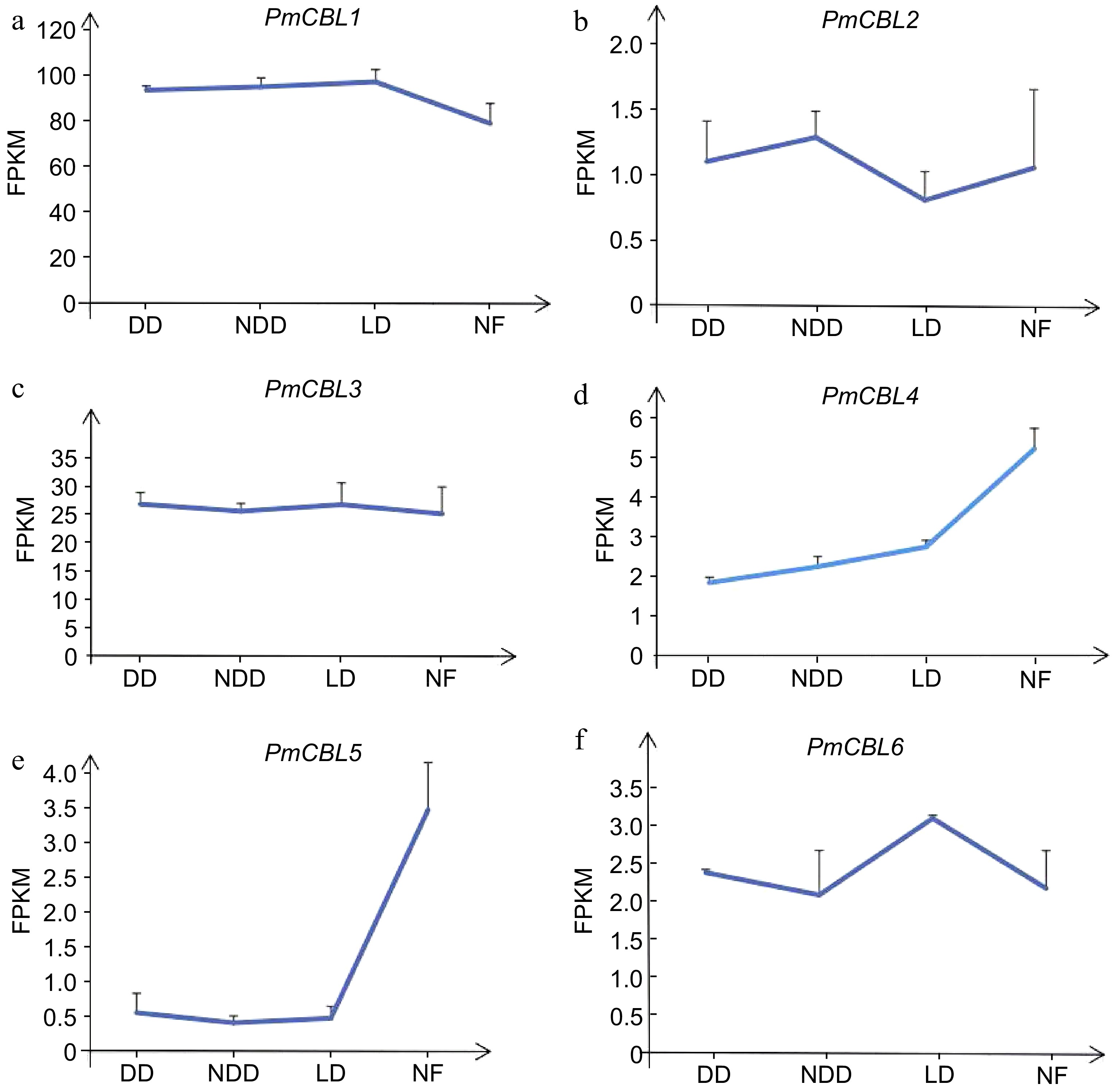
Figure 6.
The expression pattern of PmCBLs genes during overwintering. DD, November; NDD, December; LD, January; NF, February.
Quantitative analysis of gene expression
-
To further investigate the expression patterns of PmCBL genes in response to low temperature stress, qRT-PCR experiments were conducted to examine the expression levels of PmCBLs under cold treatment. During the treatment at 4 °C, the expression levels of PmCBL genes showed an upregulation or downregulation trend over a 24-h time course (Fig. 7). PmCBL1, PmCBL2, PmCBL3, PmCBL5, and PmCBL6 showed an upregulation trend in their expression levels. Among them, PmCBL1 and PmCBL5 exhibited the highest expression levels at 24 h, being 12.18-fold and 50.11-fold higher than the pre-treatment levels, respectively. PmCBL2 and PmCBL6 reached their maximum expression levels at 6 h, with a 5.50-fold and 8.90-fold increase, respectively, compared to the pre-treatment levels. PmCBL3 showed the highest expression level at 12 h, which was 11.22-fold higher than the pre-treatment level. PmCBL4 showed a downregulation trend compared to the pre-treatment levels.
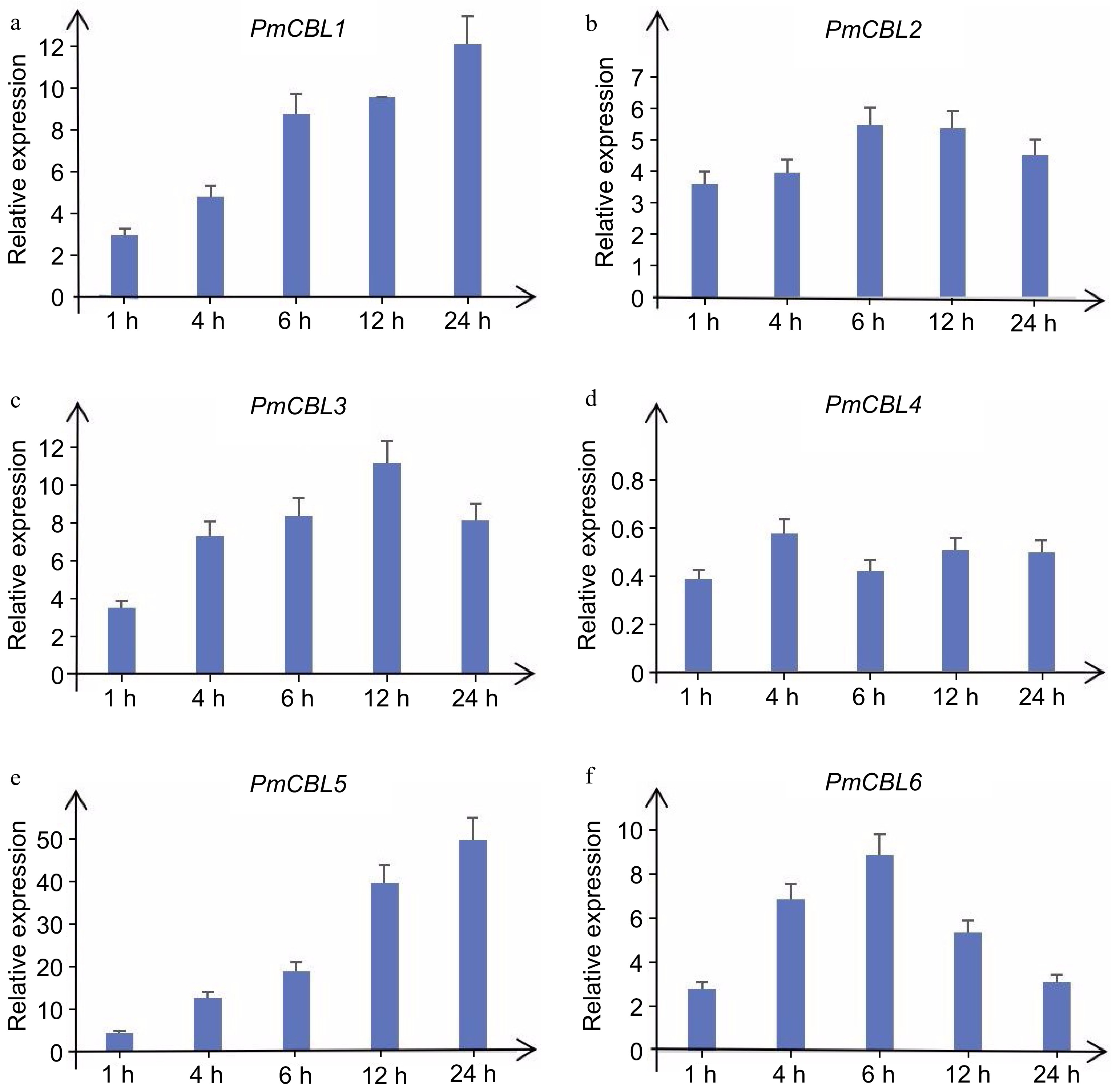
Figure 7.
Expression levels of PmCBL genes under 4 °C treatment.
-
Currently, members of the CBL gene family have been identified in various vegetables, fruits, and cereal crops. The CBL gene family has been extensively studied in various plant species. In V. vinifera, a total of eight members of the CBL gene family were found[31]. Similarly, in O. sativa, 10 members of the CBL gene family were identified[32]. A. thaliana, known for its significance in plant research, has also revealed the presence of 10 CBL gene family members[11]. Solanum lycopersicum, commonly known as tomato, exhibited 13 characterized members of the CBL gene family[33]. Remarkably, Triticum aestivum, or wheat, stands out with the largest number of identified CBL gene family members, totaling 68[34]. Additionally, CBL gene family members have also been identified in Algae, Racomitrium canescens, Pteridophyta, and gourd plants. In this study, a bioinformatics approach was employed to obtain the structural domain files of the CBL gene family and perform an HMMER SEARCH. Through Pfam database, a total of six P. mume CBL gene family members were obtained. These findings shed light on the diversity and complexity of the CBL gene family across different plant species.
The composition of introns and exons can reflect the evolutionary relationships within a gene family. It has been found that CBL genes in dicot model plants such as A. thaliana and poplar contained 6~7 introns. In the six identified P. mume CBL gene family members, all of them contained introns, with a ranged of 7~8 introns and minimal variation in the numbers. This suggested that introns in P. mume CBL genes might be more active during the evolutionary process compared to plants like A. thaliana and poplar. Generally, early-stage plant evolution tends to exhibit a higher enrichment of introns compared to later stages[35], with a higher rate of intron loss than gain[36]. To further explore the evolutionary relationships, phylogenetic trees were constructed using CBL proteins from A. thaliana, V. vinifera, O. sativa, N. tabacum and P. mume. The CBL proteins from these five species can be divided into three subfamilies, which may have evolved from different ancestral sequences. The six CBL genes of P. mume were distributed in Group I and Group III. In Group I, homologous gene pairs of P. mume and homologous gene pairs of P. mume and other species had been found. The identification of homologous gene pairs not only provides insights into the duplication and diversification processes within P. mume's own genome but also sheds light on the phylogenetic relationships between P. mume and other species. This discovery highlights the intricate mechanisms underlying the preservation and replication of genetic information, as well as the evolutionary connections that exist among different organisms.
Low temperature conditions, also known as cold stress, pose a significant threat to plant growth. Freezing stress impedes the growth of most plants and poses a great risk to the cultivation of many perennial woody plants. Previous studies have found that T. aestivuml grown under normal temperature conditions are killed at freezing temperatures of approximately −5 °C. However, if the species undergoes cold acclimation, it can survive at temperatures as low as −20 °C[37]. In previous research on Pyrus, it is found that under low temperature (4 °C) stress, the expression levels of PbCBL2, PbCBL4, and PbCBL8 were upregulated, PbCBL1 and PbCBL3 exhibited downregulation in expression under low-temperature stress[38]. Additionally, in previous studies on V. vinifera, except for VvCBL5, which shows a significant downregulation expression trend under low temperature stress, all seven VvCBL genes show a significant upregulation expression trend[39]. In this study, the P. mume gene database was utilized to obtain the sequences of CBL gene family members. These sequences were used as probes for expression analysis, enabling the investigation of expression characteristics. The results from real-time fluorescence quantitative PCR demonstrated that the expression patterns of the six P. mume PmCBLs varied under low-temperature stress. This variation may be attributed to the involvement of PmCBLs in regulating the signaling pathways associated with low-temperature responses. During the 4 °C treatment, the expression levels of PmCBL genes showed an upregulation or downregulation trend over a 24 h time period. PmCBL1, PmCBL2, PmCBL3, PmCBL5, and PmCBL6 exhibited upregulation in expression, among which PmCBL1, PmCBL3, PmCBL5 and PmCBL6 showed the most significant upregulation, suggesting their crucial roles in regulating P. mume response to low-temperature stress. Through comparative analysis of P. mume, V. vinifera, and Pyrus, it can be seen that they have certain similarities in response to low temperature stress.
Cold-responsive genes were cloned, and functional analysis was performed using the whole genome. The exact role of the c-repeat/DRE binding factor (CBF/DRE) in cold tolerance was studied in P. mume[25,40]. After cold treatment, the expression levels of PmCBLs showed either upregulation or downregulation, but these genes exhibited differential expression levels as shown in Fig. 6. These expression patterns, similar to their homologs, suggest that PmCBLs might play important roles in cold response. To date, numerous studies have demonstrated the important role of plant CBL genes in plant stress responses. For instance, AtCBL1 in A. thaliana can be strongly induced by non-biological stresses such as low temperature and injury but is not influenced by exogenous ABA[18,20]. On the other hand, AtCBL9 plays a role in both the ABA signaling pathway and ABA biosynthesis pathway and is primarily involved in the stress response of A. thaliana during the seedling stage[19]. ZmCBL4 in maize can significantly enhance salt tolerance in transgenic A. thaliana[13]. These findings highlight the significance of CBL genes in mediating plant responses to various stressors. Due to the signaling crosstalk among different stresses, multiple Ca2+ signals can be generated even under the same stress conditions. In addition, different CIPK target proteins may bind to the same sensor, and there may be functional redundancy between different CBL genes, making the entire CBL-CIPK signaling pathway complex and diverse. In order to gain a deeper understanding of the CBL-CIPK signaling pathway and how PmCBLs interacts with target proteins to activate downstream responses in response to low temperature stress, it is necessary to further explore other CBL genes.
Through transcriptomic data analysis, the expression patterns of CBL proteins can be obtained. Previous studies have shown that N. tabacum CBL proteins exhibit tissue-specific expression patterns. NtCBL13 and NtCBL14 share similar expression patterns, with low expression levels in all tissues except for roots where they are expressed. On the other hand, NtCBL6, NtCBL8, NtCBL7, NtCBL4, NtCBL5, NtCBL1, and NtCBL9 exhibit similar expression patterns with high expression levels in various tissues, except NtCBL9 which shows lower expression levels in mature roots. The remaining NtCBL proteins have higher expression levels in flower tissues. Overall, NtCBL proteins are abundantly expressed in flowers, leaves, and roots[41]. According to the research conducted on P. mume, the gene expression of PmCBLs varies in different tissue parts of P. mume (Fig. 6). PmCBL1 showed higher expression levels in flower buds, PmCBL2 exhibited higher expression levels in roots, PmCBL3 and PmCBL6 showed higher expression levels in leaves, and PmCBL4 and PmCBL5 display higher expression levels in stems. The gene expression pattern analysis revealed that the expression levels of PmCBL genes were relatively high in stems. The differential expression levels of genes in different tissue parts might be a result of biological evolution. For N. tabacum, NtCBL6, NtCBL8, NtCBL7, NtCBL4, NtCBL5, NtCBL1, and NtCBL9 share similar expression patterns and belong to the Group A subfamily of the evolutionary system, while the remaining NtCBL proteins belong to the Group B subfamily[41]. As for P. mume, PmCBL3, and PmCBL6 exhibit similar expression patterns, and PmCBL4 and PmCBL5 also showed similar expression patterns, indicating that they belong to the Group I subfamily. Therefore, it can be observed that higher homology between genes leads to more similar gene expression patterns.
-
To summarize, we conducted a genome-wide identification of the P. mume CBL gene family for the first time. We identified six CBL genes, among which PmCBL1, PmCBL3, PmCBL5, and PmCBL6 showed the most significant upregulation, suggesting their crucial roles in regulating P. mume response to low-temperature stress. Therefore, this study indicated that PmCBLs might play a key role in enhancing freezing tolerance and winter hardiness in P. mume by modulating responses to low-temperature stress. This study contributed valuable genetic resources that could be utilized for the molecular breeding of cold-resistant P. mume.
-
The authors confirm contribution to the paper as follows: study conception and design: Li P, Zhang Q; data collection and analysis platform: Liu H, Hao L; data analysis and draft manuscript preparation: Liu H; provided help with the experiments: Zhang X, Zhang Y; manuscript revision: Wang H, Wang J, Liu Z, Zhang Q, Li P. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article.
The research was supported by the Science and Technology Project of Hebei Education Department under Grant No. BJK2024033, the Natural Science Foundation of Hebei Province under Grant No. C2021204184 and Hebei Agricultural University College Student Innovation and Entrepreneurship Training Program under Grant No. 202310086003.
-
The authors declare that they have no conflict of interest.
-
Received 22 October 2023; Accepted 27 February 2024; Published online 3 April 2024
-
For the first time, six PmCBL genes had been comprehensively and systematically identified in P. mume.
The PmCBL gene family responded to cold stress, and PmCBL1/3/5/6 were identified as potential key genes involved in regulating cold tolerance in P. mume.
-
# Authors contributed equally: Haolin Liu, Lihong Hao
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Liu H, Hao L, Zhang X, Zhang Y, Wang H, et al. 2024. Identification of the Calcineurin B-like gene family and gene expression patterns in response to low temperature stress in Prunus mume. Tropical Plants 3: e010 doi: 10.48130/tp-0024-0010 

Identification of the Calcineurin B-like gene family and gene expression patterns in response to low temperature stress in Prunus mume
- Received: 22 October 2023
- Revised: 19 February 2024
- Accepted: 27 February 2024
- Published online: 03 April 2024
Abstract: The CBL gene family is an important family in the Ca2+ mediated signal transduction pathway in plants and plays a crucial role in plant stress responses and growth development. However, research on the response of members of the Prunus mume CBL gene family to low temperature stress remains scarce. In this study, we systematically analyzed the protein physicochemical properties, chromosome localization, phylogenetic evolution, gene structure, conserved domains, cis-acting elements, and gene expression patterns in response to low temperature stress of members of the P. mume CBL gene family using bioinformatics tools. Six PmCBL gene family members were identified in the P. mume genome. Phylogenetic trees were constructed, revealing three subfamilies named Group I, Group II, and Group III. In the P. mume gene family, PmCBL4 and PmCBL5 were paralogous genes. The members of the P. mume CBL gene family were unevenly distributed on three chromosomes. The CBL encoding protein, the number of isoelectric points (pI), the number of introns and exons of the six gene families were different. Analysis of the upstream 700 bp promoter sequences of the P. mume CBL gene family revealed the presence of various types of cis-acting elements involved in non-biological stress responses. Among the six identified genes, each gene exhibited different expression patterns in response to low temperature. Among them, the up-regulated expression of PmCBL5 was the largest, and the expression of PmCBL1, PmCBL3 and PmCBL5 showed the up-regulated trend. These results indicated that PmCBL1, PmCBL3, PmCBL5, and PmCBL6 were key genes involved in the response of P. mume to low temperature stress. This study provided comprehensive and systematic analysis of the P. mume CBL gene family members and identified key genes involved in the response to low temperature stress, thereby providing genetic resources for molecular breeding programs aimed at enhancing cold resistance in P. mume.
-
Key words:
- Prunus mume /
- CBL gene family /
- Low temperature stress /
- Gene identification