









-
Downy mildews are notoriously destructive diseases causing devastating losses on both agronomic and ornamental crops worldwide. With reports on wild impatiens dating back to 1877[1], the first report of impatiens downy mildew (IDM) on cultivated I. walleriana in the United States did not occur until 2004[2]. The fatal consequences of this disease on impatiens production have gained worldwide news coverage, as impatiens was previously the top annual bedding plant in the floriculture industry. Specifically, production in Florida averaged USD
${\$} $ Because this pathogen is a newly emerging one, there is little known about P. obducens. There are few genomic resources available in regard to both I. walleriana and P. obducens. Recently, the first de novo assembly of P. obducens resulted in a 202-Mb genome[3]. Polymorphic SSR marker candidates were discovered and will hopefully serve as an exciting new source as the ongoing molecular studies of this pathogen continues. While there is much to learn about the genetics of P. obducens, recent research available regarding I. walleriana was published identifying disease resistance genes as well as SSRs and SNPs sites which may aid in breeding impatiens for resistance to IDM[4]. There is strong consumer demand for IDM-resistant I. walleriana cultivars. In addition to studying the host and its pathogen separately, understanding the plant defenses in the host-pathogen interaction during infection is critical to understand IDM disease and introduce disease resistance in susceptible host cultivars.
Plant pathogens contain specific structures or molecules known as pathogen associated molecular patterns (PAMP), which are recognized by Pattern recognition receptors (PRRs) to active plant defense. This is known as PAMP-Triggered Immunity (PTI)[5]. PTI is a non-specific defense mechanism allowing for recognition of both non-pathogenic and pathogen organisms, and thus combatting pathogen invasion and further colonization[6]. A second layer of immunity, effector-triggered immunity (ETI) is activated if a pathogen survives PTI. ETI depends on pathogen effectors, secreted in host cells by the pathogen, resulting in programmed cell death at the site of infection, or a hypersensitive response (HR) in an incompatible reaction[5]. HR is prompted by host resistance (R) genes, which are divided into five classes: (i) the CNL class comprising of R genes encoding a protein with at least one N-terminal coiled-coil, a nucleotide binding site and a leucine-rich repeat (CC-NB-LRR), (ii) TNL class containing a Toll-interleukin receptor-like domain, a nucleotide binding site and a leucine-rich repeat (TIR-NB-LRR), (iii) RLP class which is a receptor-like protein containing receptor serine-threonine kinase-like domain and an extracellular leucine-rich repeat (ser/thr-LRR), (iv) RLK which are receptor-like kinase containing an extracellular leucine-rich repeat in addition to the kinase domain (Kin-LRR), (v) 'Others' class includes any other genes described as using other molecular mechanisms to confer resistance[7]. In addition to the genes that positively regulate plant defense, numerous susceptibility (S) genes that negatively regulate defense responses have been discovered during the last two decades. These S genes promote infection by facilitating pathogen penetration or fulfilling their structural or metabolic needs[8]. Currently, the best studied S gene is the MLO gene that confers susceptibility to powdery mildew pathogens. Genome editing using CRISPR/Cas-9 provides an excellent tool to knock out the S genes to enhance disease resistance in plants. A number of host S genes have been identified in case of Phytophthora (an oomycete genus) spp.[9] and silencing of some of these S genes by using CRISPR/Cas-9 based genome editing in potato resulted in enhanced resistance against late blight pathogen P. infestans[10]. Recently, Ma et al. reported a WRKY40 transcription factor (TF) as a susceptibility factor that promotes downy mildew of grapes caused by Plasmopara viticola[11].
Our research focused on transcriptome analysis of I. walleriana during compatible interaction with P. obducens. The main focus was to identify candidate genes that may be involved in impatiens susceptibility to P. obducens. These results could provide insights and lay the groundwork for future studies in understanding both host-pathogen interactions in IDM. Here, we present findings from the first analysis of differential gene expression during the infection of I. walleriana by the IDM causal agent P. obducens.
-
Impatiens walleriana cv. 'Super Elfin White' plants were grown in growth chambers maintained at 24 °C with a 12 h light/dark photoperiod. The P. obducens isolate was maintained on impatiens plants in the growth chamber. Fully expanded leaves of eight impatiens plants were inoculated by pipetting 50 μL sporangial suspension (1 × 108 sporangia/mL) of P. obducens on the abaxial leaf surface. Infection was verified under microscopic observations by staining leaves with tryphan-blue at each time point. Leaves were collected at five different time points (0, 12, 48, 120, and 240 h), flash frozen in liquid nitrogen, and stored at −80 °C until use. Each treatment (time point) consisted of two biological replicates, resulting in a total of 10 leaf tissue RNA samples. P. obducens sporangia for pathogen baseline samples were carefully scraped off the abaxial surface of impatiens leaves showing sporulation and were also flash frozen and stored before RNA isolation.
Library preparation and sequencing
-
Total RNA was isolated from P. obducens sporangia and inoculated impatiens leaves using the RNeasy Plant Mini Kit (Qiagen, Germantown, MD, USA) according to manufacturer's protocols. RNA concentration and quality were determined using Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). Library sample preparation was done using a stranded mRNA-seq kit (KAPA Biosystems, Inc., Wilmington, MA, USA) and sequencing was performed using an Illumina HiSeq 2500 v4 platform (Illumina, Inc., San Diego, CA, USA) at the Center for Genomic and Computational Biology (GCB) at Duke University. Each library was sequenced using the pair-end (2 × 125 bp) protocol. A total of 24 libraries were made (Table 1). The raw data is submitted to NCBI SRA database under the accession number PRJNA1032710.
Table 1. Statistical summary of RNA sequencing data.
Sample ID Raw reads (PE) Clean reads 0H_Control_R1 23,520,868 18,287,675 0H_Control_R2 27,793,058 24,193,950 12H_Control_R1 24,320,452 20,299,572 12H_Control_R2 19,672,888 15,319,055 24H_Control_R1 24,320,452 21,024,513 24H_Control_R2 19,672,888 13,922,270 48H_Control_R1 24,162,182 17,057,095 48H_Control_R2 17,226,072 19,581,661 120H_Control_R1 20,526,634 23,639,470 120H_Control_R2 23,321,216 18,259,179 240H_Control_R1 27,654,810 14,085,034 240H_Control_R2 21,765,004 21,712,471 0H_Infected_R1 37,971,712 29,005,241 0H_Infected_R2 37,778,302 29,248,606 12H_Infected_R1 33,006,920 24,968,942 12H_Infected_R2 31,223,426 24,440,783 24H_Infected_R1 34,379,442 26,500,567 24H_Infected_R2 36,662,544 27,860,148 48H_Infected_R1 31,490,402 23,140,744 48H_Infected_R2 33,238,084 24,795,526 120H_Infected_R1 37,946,602 29,078,825 120H_Infected_R2 34,596,692 25,346,127 240H_Infected_R1 33,746,888 23,867,301 240H_Infected_R2 33,041,348 24,552,405 Total 528,964,774 RNA-Seq analysis
-
Raw reads were imported into CLC Genomics Workbench R9 (Qiagen) and assessed for quality. Sequencing adaptors and low-quality reads were trimmed by 15 and 5 bp from the 5' and 3' ends, respectively. A trimming quality score of 0.005 was used. Cleaned high quality reads from samples were assembled de novo using the default parameters in the CLC Genomics Workbench 9.5.3. Because of the complexity of transcriptome data, a word size 25 and bubble size 1,000 were used for de novo assembly.
Assembly optimization
-
Contigs were subsequently fed to the program cd-hi-est for removal of any redundancy in the sequence, using a sequence identity threshold of 0.95[12]. After cd-hit clustering analysis, selected contigs were subjected to comprehensive functional annotation through BLASTx to remove P. obducens contigs. Filtered pathogen free contigs were again subjected to comprehensive functional annotation using Blast2GO 5 PRO[13]. Cloud BLAST tool was used to run BLASTX on transcripts against the non-redundant plant genome NCBI database using an error cutoff value of 1 × 10−5. Only those contigs were kept that had high homology with the plant genes.
Differential expression analysis, functional annotation and enrichment analysis
-
Differentially expressed transcripts (DETs) were determined by using the R package DESeq2 using default parameters. Differential expression analysis was done using the transcript counts table generated in CLC genomics. The DETs were filtered at a threshold of log2fold > |2| and p adj < 0.001. Comprehensive functional annotation of DETs was done using Blast2GO 5 PRO and gene ontology (GO) mapping of the DETs was conducted to produce GO term distribution. A Venn diagram analysis on DETs at all timepoints was done using the online tool provided by VIB and Ghent University (
http://bioinformatics.psb.ugent.be/webtools/Venn/ ). Singular Enrichment Analysis (SEA) AgriGO tools were used for gene set enrichment analysis, their comparisons and PAGE[14]. Finally, functions of the DETs, specifically in common plant pathways, were determined using MapMan software[15,16]. Since MapMan and AgriGO annotations are only available for few plant species, we used the Arabidopsis homologs for our de novo constructed impatiens transcripts. -
In total, 689,038,886 reads were generated from all 24 samples (libraries). After quality assessment and trimming, a total of 528,964,774 clean reads were used to construct a de novo assembly in the CLC Genomics workbench which resulted in 91,164 contigs with an N50 value of 1,650 bp (Table 1). These contigs were fed into the cd-hit-est cluster analysis based on similarity, producing 89,987 contigs. Filtering through BLASTx to remove P. obducens contigs resulted in 73,022 contigs. This optimized assembly was used as a reference and reads from all the samples were mapped back to this optimized pooled reference assembly and a read count table was generated.
Differential expression analysis was done on this table using DEseq2. Only those transcripts were considered as differentially expressed genes that have log2fold > |2| and p adj < 0.001. According to this criterion, the number of DETs between infected and control samples ranged from around 3,000 to 4,500 across all six time points (Table 2) with the highest number of DETs (4,549) at 0 h and the lowest at 120 h. The number of upregulated genes was greater than the number of downregulated genes at all time points with the most upregulation happening at 240 h (2,672).
Table 2. Pairwise comparisons between infected vs control samples at all six time points.
Comparison
(infected vs
control)Total
DETsUpregulated
DETsDownregulated
DETsUnique to
timepoint
DETs0 h 4,549 2,470 2,068 298 12 h 4,098 2,132 1,966 140 24 h 4,369 2,235 2,133 201 48 h 3,619 2,006 1,613 112 120 h 3,607 2,066 1,541 97 240 h 4,491 2,672 1,819 502 Functional annotation and gene set enrichment analysis
-
Comprehensive functional annotation of DETs was conducted using Blast2GO software. Species distribution showed little homology with other plant species. There is a lack of impatiens genes available within the NCBI database, therefore the species available with the largest number of BLAST hits was Vitis vinifera (Supplemental Fig. S1). Gene ontology (GO) terms were assigned to DETs among all three GO categories i.e., biological process (BP), molecular function (MF) and cellular component (CC) (Supplemental Fig. S2). Most BP hits were placed in the categories of catalytic and binding and transport activity. The DETs in the MF category were mostly placed within metabolic and cellular processes, response to stimulus, and biological regulation. In CC, most DETs fell under the membrane category (Fig. 1). In order to find out enriched GO terms at each time point, Singular Enrichment Analysis (SEA) was performed and cross comparison of SEA results across all time points was done by using SEACOMPARE (Cross comparison of SEA) tool of AgriGO (Supplemental Figs S3 & S4). Response to chitin (GO:0010200) and response to carbohydrate stimulus (GO:0009743) were the top two significantly enriched GO terms across all six time points. Regulation of biosynthetic process (GO:0009889), regulation of transcription (GO:0045449), response to stimulus (GO:0050896), response to fungus (GO:0009620) and many other daughter terms of response to stress, cellular and metabolic processes were significantly enriched at all time points. To find out DETs unique to each time point, venn diagram analysis was done followed by their SEA analysis (Fig. 2). At 0 h, 298 unique DETs were found that were enriched in catalytic activity (GO:0003824), cell part (GO:0044464) and plasma membrane (GO:0005886) GO terms. Cell part and plasma membrane GO terms were also enriched among unique DETs at 24, 120 and 240 h. Various responses to stress and stimulus related GO terms were enriched in DETs unique to 12 h time point. The highest number of unique DETs (502) was found at 240 h and the enriched GO terms among these DETs were mostly related to metabolic processes (GO:008152), phosphorylation related metabolic processes (GO:0016310, GO:0006796), macromolecule modification and localization (GO:0043412, GO:0033036) and intracellular organelle like: cytoplasm (GO:0005737), chloroplast (GO:0009507) and plastid (GO:0009536). Detailed SEA comparison analysis of DETs unique at each time point can be found in Supplemental Figs S5 & S6.
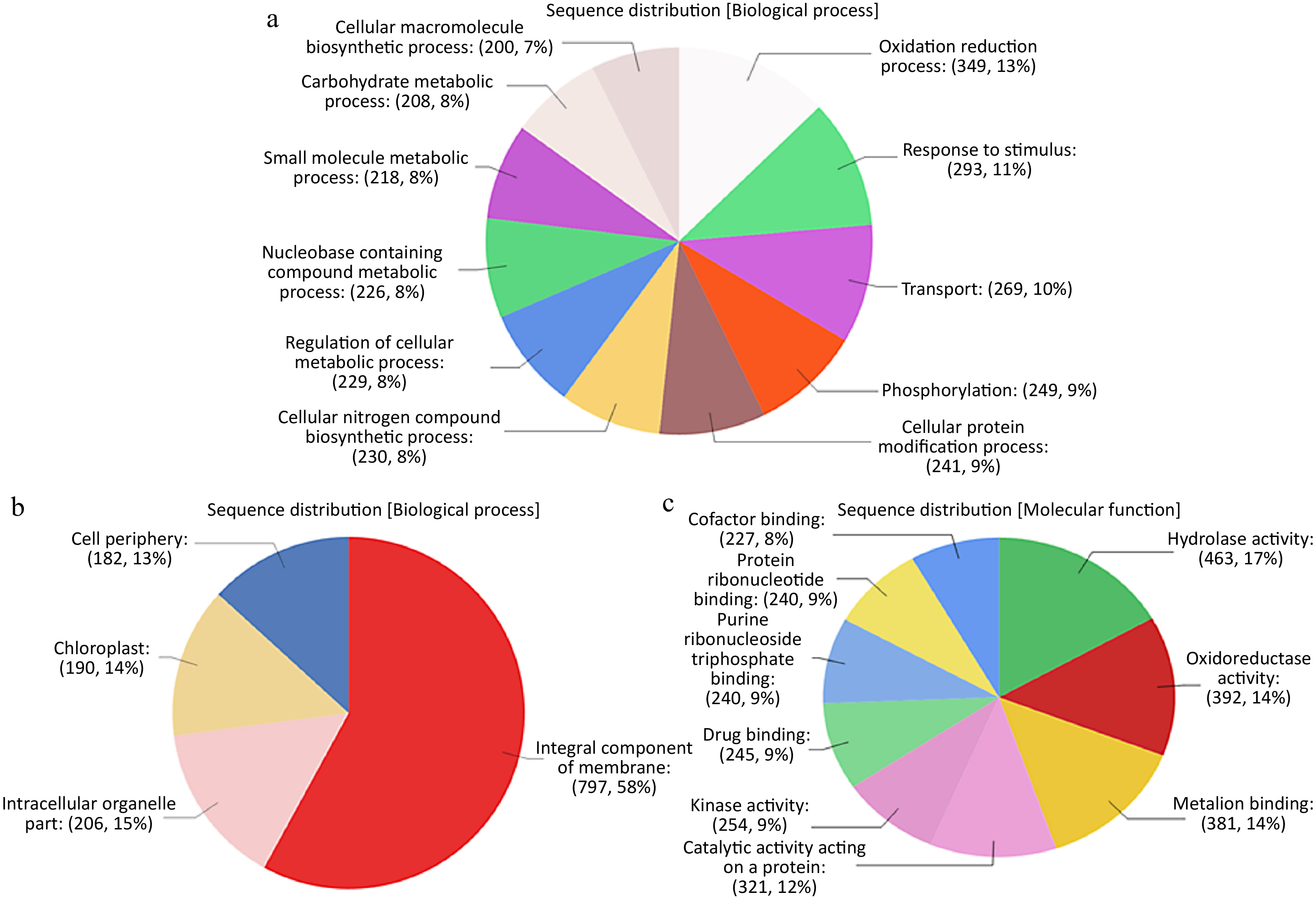
Figure 1.
(a) Sequence distribution of DETs among Biological Processes (BP) GO terms. (b) Sequence distribution of DETs among Cellular Components (CC) GO terms. (c) Sequence distribution of DETs among Molecular Functions(MF) GO terms.
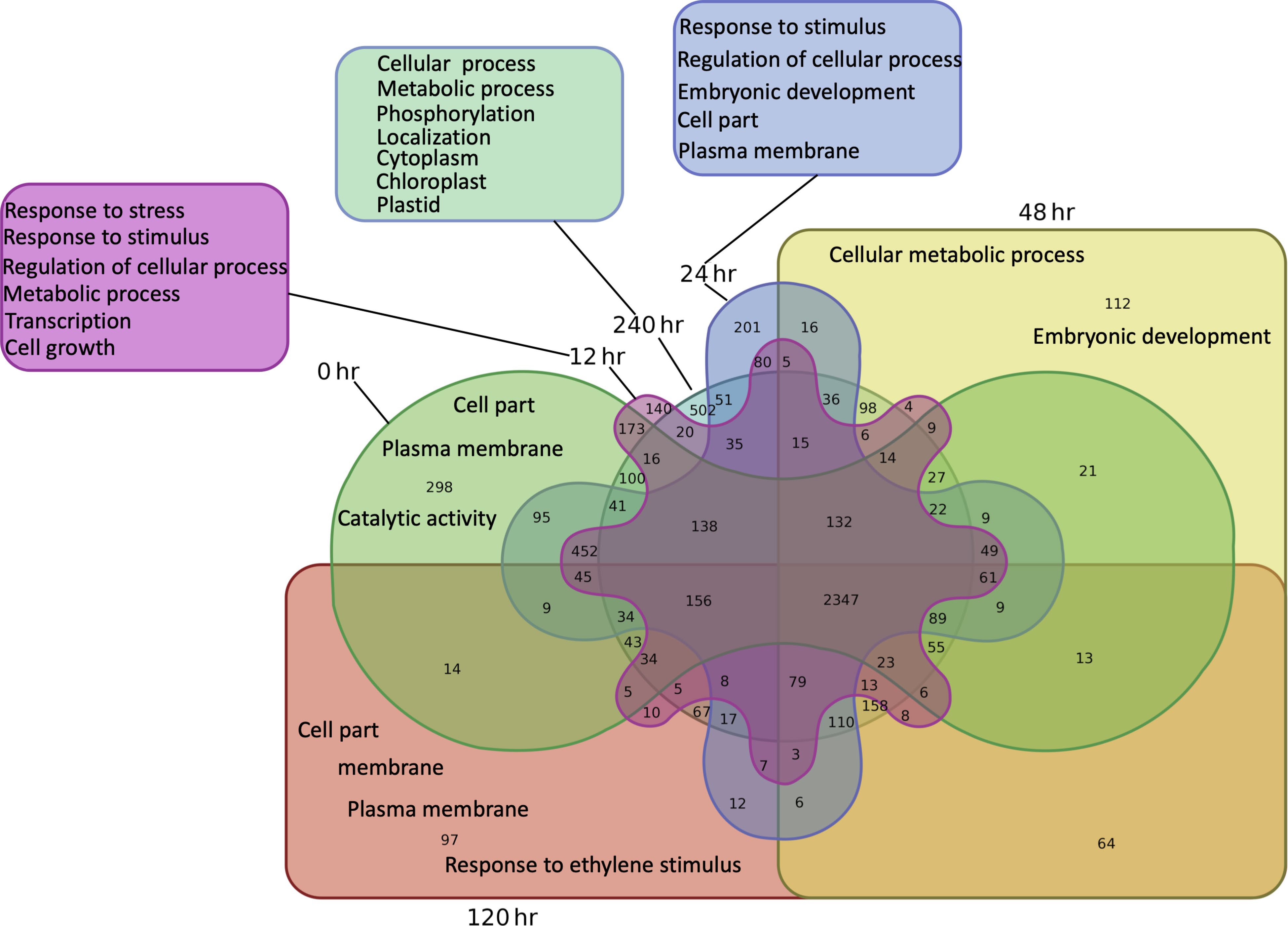
Figure 2.
Venn diagram of pair-wise comparisons of DETs unique to timepoints and most enriched GO 210 terms among unique DETs at each time point.
To gain more understanding about the functional annotations of DETs with respect to their expression levels across all time points separately, parametric analysis of gene set enrichment (PAGE) was performed (Fig. 3). PAGE is an efficient and sensitive tool that ranks annotated gene clusters with respect to their expression levels. PAGE analysis revealed that catalytic activity is the most induced molecular function across all six time points. Signaling, reproduction, reproductive process, multicellular organismal process, development process, cellular process, transcription regulation, enzyme activity and binding were significantly induced throughout the infection cycle. Biological processes, like cellular component organization and biogenesis, localization and establishment of localization were all repressed across all time points. Most of the enriched cellular components like cell part, organelle and extracellular region were downregulated at all six time points, whereas macromolecular complex was induced at 0 h, slightly induced at 12 h and repressed across the rest of the time points. Immune system process and response to stimulus were induced throughout the infection with highest induction at 0 h (Fig. 3). Cross comparison of daughter GO terms in PAGE analysis revealed that GO terms related to photosynthesis are downregulated across all timepoints, but their number is increased from 48 h post inoculation to 240 h post inoculation (Supplemental Figs S7 & S8).
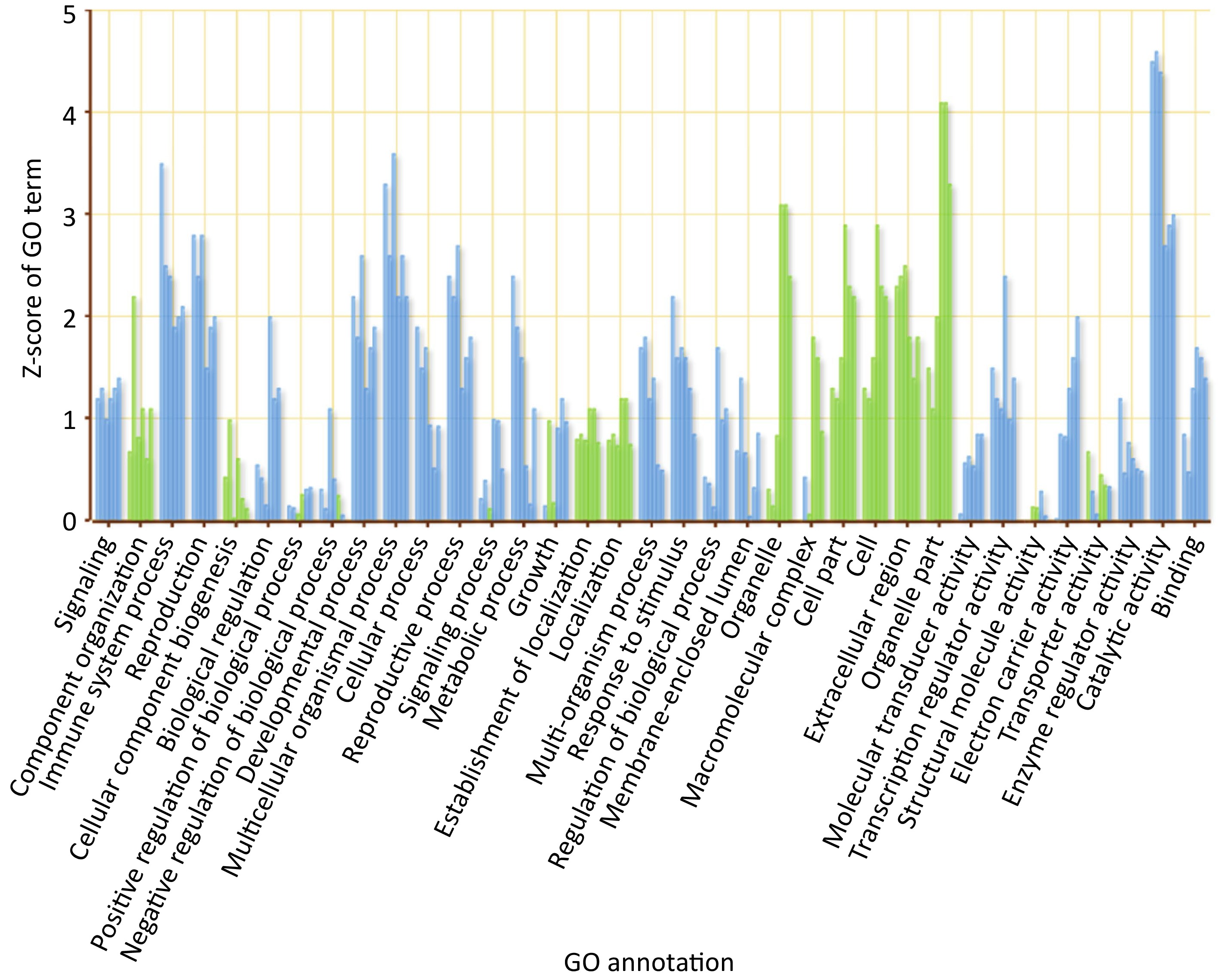
Figure 3.
GO sequence distribution chart separated by biological process (BP), molecular function (MF) and cellular component (CC).
We generated MapMan figures at each time point in order to provide a visual representation of genes involved in common plant pathways[15,16]. DEGs from all six time points were mapped on the biotic stress pathways. Genes involved in proteolysis were observed to be mainly upregulated across all time points. General signaling genes were about equally downregulated as they were upregulated. The WRKY transcription factors (TFs) were mostly upregulated (Fig. 4).
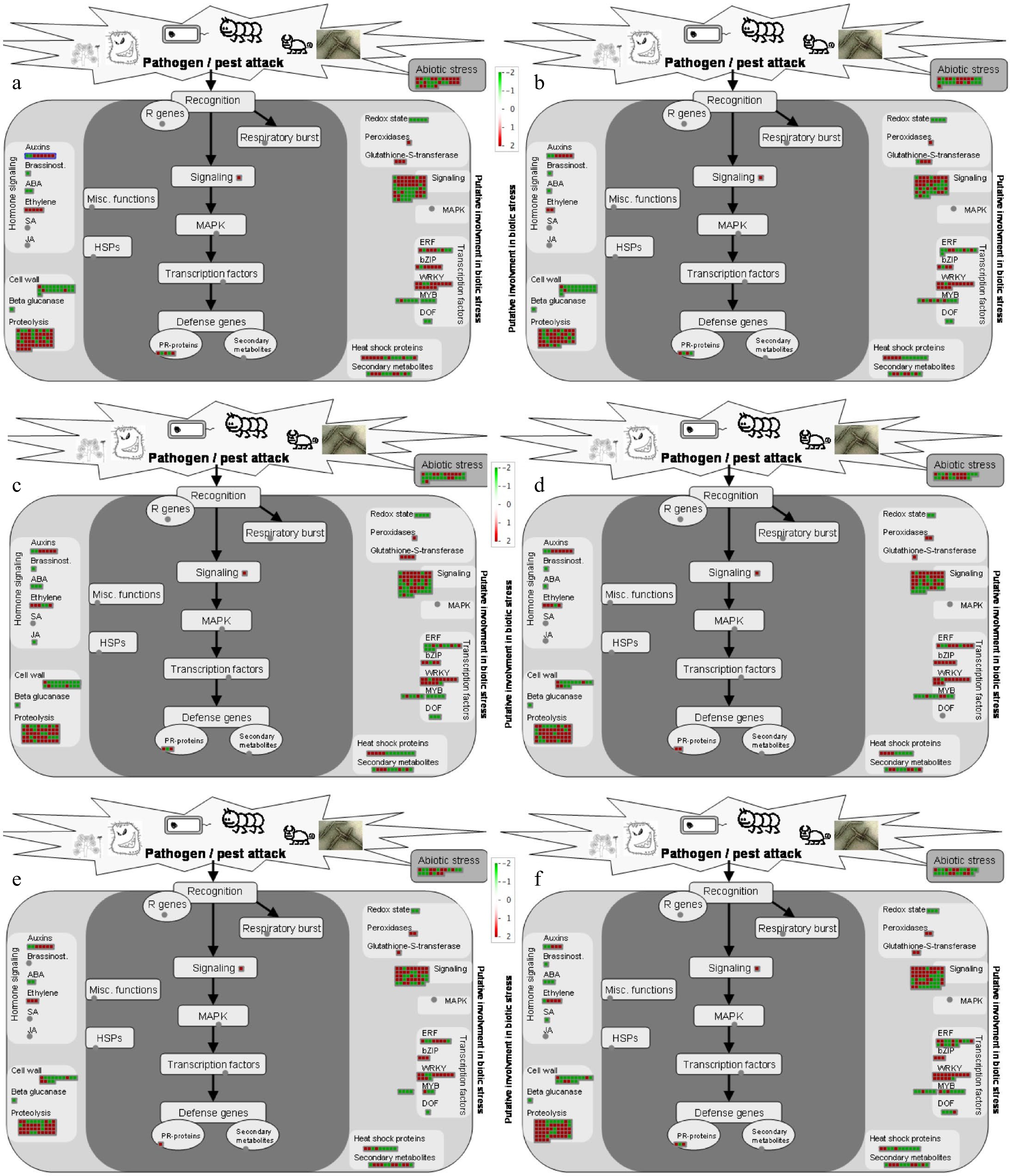
Figure 4.
Visualization of differentially expressed genes involved in biotic stress pathways in response to P. obducens (a) 0 h (hours post-inoculation), (b) 12 h, (c) 24 h, (d) 48 h, (e) 120 h and (f) 240 h. Color gradient represents log2 fold ratios with red representing upregulation and green representing downregulation in treatments over mock roots. Each box represents one transcript.
Genes putatively involved in plant defense
-
Many of the previously reported positive and negative plant defense regulators were found to be differentially expressed in I. walleriana in response to P. obducens. For example, multiple homologs of pathogenesis-related (PR) genes were found among the top differentially expressed genes across all time points. Pathogenesis-related protein 1 (IwPo0054080) was consistently induced at all six time points and presented highest log2FC at 0, 12 and 120 h, second highest log2FC at 24 and 48 and 240 h. Whereas a PR-1 homologue (IwPo0001117) was consistently downregulated (log2FC 11-9) across all time points (Fig. 5 & Supplemental Table S1).
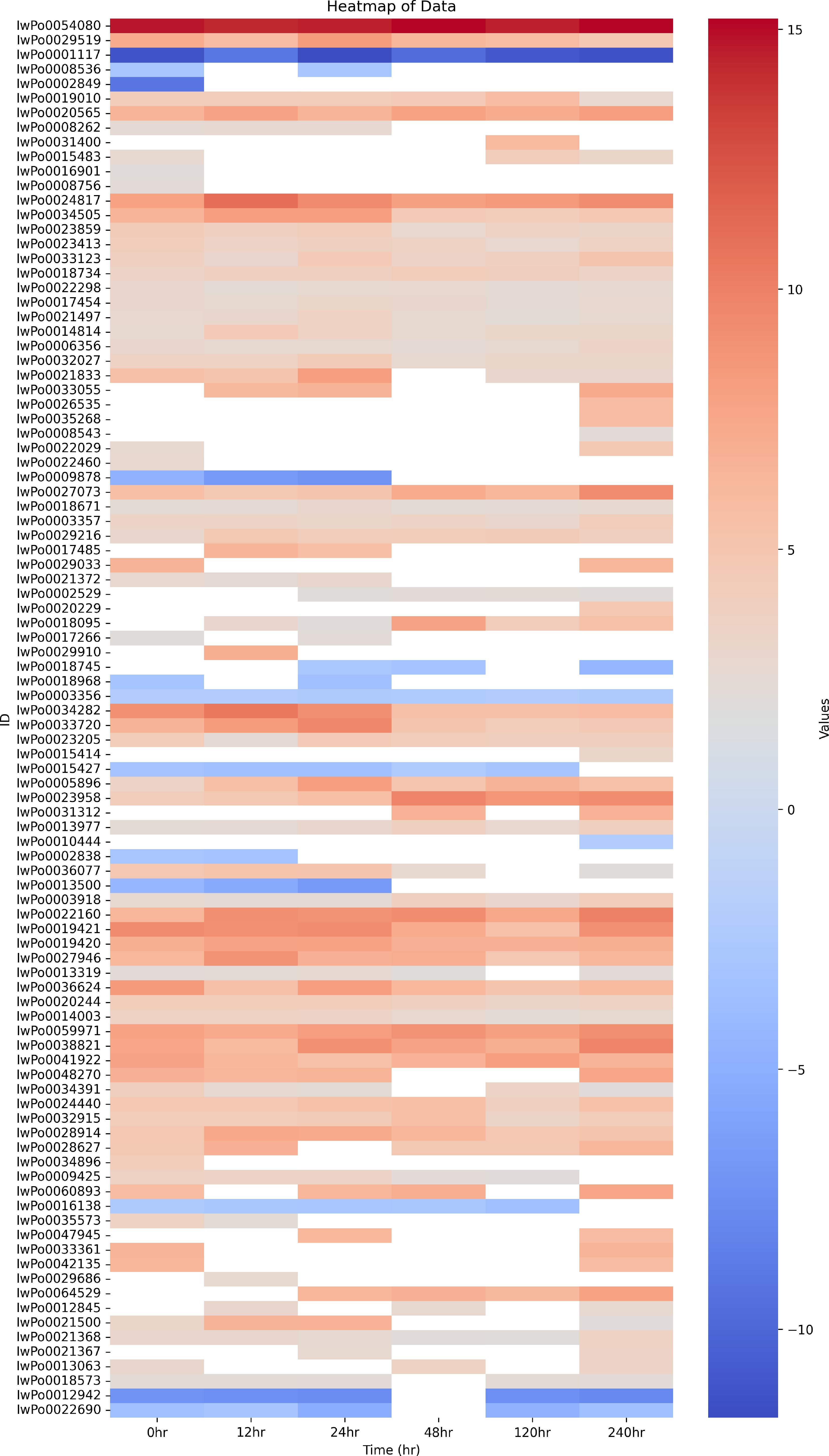
Figure 5.
Heat map of putative plant defense DETs of I. walleriana during infection by P. obducens.
Several R gene classes were also found to be induced in I. walleriana in response to P. obducens infection. A CC-NBS-LRR domain containing transcript (IwPo0019010) annotated as disease resistance protein At1g62630 was found to be consistently upregulated at all six time points. Three DETs code for NB-LRR type disease resistance protein At4g27220. One (IwPo0020565) was induced across all time points, IwPo0008262 was found upregulated at 0, 12 and 24 h and IwPo0031400 was induced only at 120 h. Two DETs encoding RGA3 were found upregulated only at 0 h. Kinase activity (GO:0016301) was found to be significantly upregulated throughout the infection cycle (Supplemental Figs S7 & S8). Many of the receptor-like kinases (RLKs) were found to be differentially expressed across all time points, including 20 leaf rust resistance kinase Lr10-like (LrRLKs) transcripts. One (IwPo0009878) was downregulated at 0, 12 and 24 h and didn't differentially express at later stages of infection. Twelve of them were consistently upregulated at all six time points, three were only induced at 240 h post inoculation, while others were inconsistently differentially expressed across time points. Several other RLKs including G-type lectin S-receptor-like serine/threonine-protein kinase transcripts, wall-associated receptor kinases and stress-induced RLK were found to be mostly upregulated (Fig. 5 & Supplemental Table S1).
In addition to R genes, we also found some previously reported S genes. Six DETs coding for different isoforms of MLO genes were found differentially expressed across all time points. Three of these transcripts were upregulated and three were downregulated. Like MLO enhanced disease resistance 2 (EDR2) is another powdery mildew susceptibility factor. Three DETs were annotated as different EDR isoforms. One EDR2 (IwPo0023958) and one EDR4 (IwPo0013977) were highly upregulated at all six time points, whereas another EDR2 (IwPo0031312) isoform was found highly upregulated at 48 and 240 h. Recently, a TF WRKY40 is reported as S gene in the case of downy mildew of grapes[11]. Along with several other putative plant defense responsive TFs (Supplemental Tables S2−S7), we found 34 different WRKY TFs among top DETs across all time points. Out of these, 32 WRKY TFs were upregulated and two were downregulated during the infection cycle (Fig. 5 & Supplemental Table S1).
In addition to defense genes, we found diverse types of retrotransposons (RTs) among highly differentially expressed transcripts in all infected vs control comparisons. On the basis of differences in their structure and transposition cycle, plant retrotransposons are classified in two major classes: LTR retrotransposons (LTR-RTs) and non-LTR retrotransposons. LTR-RTs are further divided into Ty1-copia group and gypsy group[17]. In total 62 DETs encoding diverse RTs were found across all time points (Fig. 5). Most of these were LTR-RTs and only one was annotated as non-LTR type retrotransposon. Among LTR-RTs, 31 DETs encode Ty1-copia domain containing Retrovirus-related Pol polyprotein from transposon TNT 1-94 (TNTs), 10 DETs code for putative Ty1-copia subclass type RTs and four DETs were annotated as gypsy type RTs. Overall, five DETs of Ty1-copia type and one DET of gypsy type were downregulated and 66 DET RTs (diverse groups) were upregulated. Out of these 66 upregulated RTs, nine were found to be upregulated across all six time points. Only one TNT type RT was found downregulated across all six timepoints (Supplemental Table S8).
Apart from the aforementioned genes, we found multiple calcium signaling genes, aquaporins, transporters, TFs and many other core plant defense genes differentially expressed in I. walleriana in response to infection by P. obducens at all stages of the infection cycle (Supplemental Tables S2−S7).
-
RNA-Seq is an excellent technique to reveal changes in a plant's gene expression in response to pathogens, even when whole genome information is unavailable. With the advent of CRISPR/Cas based genome editing technology, knocking out susceptibility genes to enhance disease resistance has become a promising approach. Transcriptome analysis of a host during disease development provides an excellent way of identifying genes involved in susceptibility. A recent study was published which is the first comparative transcriptome analysis of both a resistant and susceptible impatiens cultivar in order to identify candidate genes for IDM resistance[4]. This study was conducted on non-inoculated, uninfected impatiens leaves. Many putative R genes involved in IDM resistance were identified. To further host-pathogen interaction and identify the candidate S genes involved in promoting IDM, we analyzed transcriptional changes in an inoculated, susceptible cultivar of impatiens at six timepoints. Numerous researchers have reported that many biochemical events take place during early minutes of plant-pathogen interactions[18]. In order to track transcriptional changes during the first contact between plant and pathogen, we collected samples for the first time point immediately following inoculation, designated as 0 h. At 10 d (240 h) post-inoculation, sporulation of P. obducens can be observed on the abaxial leaf surface of susceptible, infected impatiens plants. P. obducens is an obligate biotroph, therefore we wanted to track transcriptional changes throughout the entirety of its infection cycle (0 to 240 h). To our knowledge, this is the first study of the host-pathogen interaction between I. walleriana and P. obducens available.
In 2018, Ball Horticultural Company and KeyGene announced the completion of the entire genome sequencing and assembly of I. walleriana. Because it is not currently available to the public, our study relied on the use of de novo transcriptome assembly of I. walleriana. After assembly, optimization was done in order to preserve biologically meaningful transcripts for further differential expression analysis. Differential gene expressions among infected and control samples were observed immediately after inoculation to the end of infection cycle. Surprisingly, we observed the highest number of differentially expressed genes at 0 h than at any other timepoint, except 240 h. Consistent with other plant-pathogen transcriptome studies, significant enrichment of cellular, physiological and metabolic processes as well as response to stress and stimulus were observed among DETs across all six timepoints. This indicates substantial transcriptional reprogramming in I. walleriana within minutes after inoculation with P. obducens until the end of infection cycle.
Pattern recognition receptors (PRRs) are the first layer of plant defense in the innate immune system of plants[5]. Localized to host cell membranes, they recognize pathogen PAMPs and activate PTI. Plants carry two types of PRRs: receptor-like kinases (RLKs) and receptor-like proteins (RLPs)[19]. The present study showed differential expression of numerous PRRs of both types in impatiens infected with P. obducens during early infection as well as late infection stages. Wall-associated RLKs have been reported as the positive regulators of plant defense against fungi[20]. In our study, we found four wall-associated receptor-like kinases to be upregulated and one to be downregulated throughout the infection cycle of P. obducens in I. walleriana. Another type of RLK, G-type lectin S-receptor-like serine/threonine-protein kinase, was also found to be upregulated across all timepoints, including 0 h. G-type RLKs are reported to be highly induced during a tolerant interaction of a citrus rootstock with a soilborne oomycete pathogen, Phytophthora parasitica[21]. There is no functional evidence about the involvement of G-type RLKs in plant-pathogen interaction. But in rice, the Bph3 locus that contains three G-type RLKs encoding genes (OsLecRK1−OsLecRK3) has been shown to confer broad-spectrum and durable insect resistance for more than 30 years[22]. Further studies may be able to confirm if G-type RLKs play a role in oomycete-plant interactions. We also found several leaf rust disease resistance RLKs (Lr10-like) to be induced during IDM disease development. As the name indicates, Lr10 is reported to confer resistance against leaf rust in wheat[23], but it is not reported in case of oomycete-plant interactions. In addition to several RLKs, we also found seven different NB-LRR type R genes that were mostly induced during IDM disease development. Bhatarrai et al. found most of the IDM resistance candidate genes in impatiens showing high similarity levels to the NB-LRR or LRR gene families and identified two genes as TNL or CNL encoding proteins[4]. These R genes are different from the genes identified in our study.
Recently, the discovery of S genes and knocking them out through genome editing is opening up new horizons for developing disease resistant plants[10]. We have also identified some S genes potentially involved in IDM disease development by P. obducens. Recently, Ma et al. reported a WRKY40 transcription factor as a susceptibility factor that promotes downy mildew of grapes caused by Plasmopara viticola[11]. We found several different WRKY transcription factors being highly induced, including five WRKY40 that were consistently upregulated across all six time points. MLO proteins were initially identified as common powdery mildew susceptibility genes, but recent studies have also found them to be involved in plant-oomycete interactions[24,25]. We found six homologs of MLO genes to be differentially expressed in I. walleriana, three of which were highly upregulated. Enhanced Disease Resistance 2 (EDR2) is another powdery mildew susceptibility gene that is reported to negatively regulate salicylic acid based plant defense[26]. We found three EDR isoforms being differentially expressed in I. walleriana during downy mildew development. One EDR2 and one EDR4 were highly induced throughout the infection. Induction of MLO and EDR genes by P. obducens in I. walleriana could be speculated as another example of MLO and EDR2 genes being involved in host susceptibility to pathogens other than powdery mildew, in this case, an oomycete. Activation and involvement of PR genes have been widely reported during compatible and incompatible oomycete-plant interactions[9,25,27]. We found some PR genes among the top induced DETs throughout the infection cycle.
Retrotransposons (RTs) have been found abundantly in the plant genomes[17]. LTR-RTs represent the most abundant class of transposable elements in plants. Activation of LTR-RTs in plants in response to several biotic and abiotic stresses is reported[28]. In sweet orange, a locus that confers resistance to Citrus tristeza virus is reported to contain five copia-like and three gypsy-like RTs[29]. Tnt1, a copia type LTR-RT of tobacco is reported to be involved in plant defense responses against diverse pathogens[30]. Tnt1 has been reported to be induced in response to elicitins[31], the elicitors of oomycetes[32,33]. Recently, Fu et al. reported differential expression of three Retrovirus-related Pol polyprotein from transposon TNT 1-94 (TNTs) in sweet orange roots affected by citrus blight[34]. One TNT is reported to be upregulated in carrizo citrange roots in response to Phytophthora parasitica at 48 h post inoculation[25]. In this study, we found 31 TNTs, 29 of them were highly induced, whereas three were repressed in I. walleriana during different stages of infection by P. obducens. In rice, six different RTs were found to be unregulated during a susceptible interaction with bacterial leaf streak pathogen Xanthomonas oryzae pv. oryzicola[35]. Since RTs are ubiquitously present among eukaryotes, we have analyzed a dual plant-pathogen transcriptome data. The question arises whether all these RTs are of I. walleriana or include the pathogen's RTs also. As described in our methods, we filtered our data to remove all possible pathogen transcripts before performing the analysis, and DETs were also annotated using cloud BLAST on viridiplantae database. Functional significance behind the transcriptional reprogramming of these RTs in IDM development needs further investigation.
In addition to the aforementioned genes, we found many other core plant defense-related genes to be differentially expressed throughout the infection cycle of IDM. Conclusively, this study revealed the presence of transcriptional changes in common physiological and core plant defense-related pathways in I. walleriana during infection by P. obducens, and many genes identified here are potential candidates to look for IDM disease susceptibility genes.
-
The authors confirm contribution to the paper as follows: study conception and design: Ali GS; analysis and interpretation of results (performed all wet lab experiments): Suarez S; RNA-seq data analysis: Naveed ZA; draft manuscript preparation: Suarez S, Naveed ZA; manuscript revision: Naveed ZA. All authors have read and agreed to the published version of the manuscript.
-
The raw data is submitted to NCBI SRA database under the accession number PRJNA1032710
www.ncbi.nlm.nih.gov/bioproject/?term=(PRJNA1032710)%20AND%20bioproject_sra[filter]%20NOT%20bioproject_gap[filter] . We acknowledge American Floral Endowment for providing funds for this research.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Stephanie Suarez, Zunaira Afzal Naveed
- Supplemental Table S1 Putative plant defense DETs of I. walleriana during infection by P. obducens.
- Supplemental Table S2 Detailed results of differential expression analysis at 0 h ifected vs 0 h control.
- Supplemental Table S3 Detailed results differential expression (infected vs.control) analysis at 12 h.
- Supplemental Table S4 Detailed results differential expression (infected vs.control) analysis at 24 h.
- Supplemental Table S5 Detailed results differential expression (infected vs.control) analysis at 48 h.
- Supplemental Table S6 Detailed results differential expression (infected vs.control) analysis at 120 h.
- Supplemental Table S7 Detailed results differential expression (infected vs.control) analysis at 240 h.
- Supplemental Table S8 DETs encoding retrotransposons in I. walleriana during infection by P. obducens.
- Supplemental Fig. S1 Species distribution chart showing maximum homology with plant species.
- Supplemental Fig. S2 Overall GO term distribution of DETs.
- Supplemental Fig. S3 Snapshot of SEACOMPARE showing cross comparison of Singular Enrichment Analysis (SEA) of total DETs across all 6 time points. Top Panel.
- Supplemental Fig. S4 Snapshot of SEACOMPARE showing cross comparison of Singular Enrichment Analysis (SEA) of total DETs across all 6 time points. Bottom Panel.
- Supplemental Fig. S5 Snapshot of SEACOMPARE showing cross comparison of Singular Enrichment Analysis (SEA) of DETs unique to each time point. Top Panel.
- Supplemental Fig. S6 Snapshot of SEACOMPARE showing cross comparison of Singular Enrichment Analysis (SEA) of DETs unique to each time point. Bottom Panel.
- Supplemental Fig. S7 Snapshot of PAGE SEACOMPARE showing cross comparison of PAGE of DETs across all 6 time points. Top Panel.
- Supplemental Fig. S8 Snapshot of PAGE SEACOMPARE showing cross comparison of PAGE of DETs across all 6 time points. Bottom Panel.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Suarez S, Naveed ZA, Ali GS. 2024. Transcriptional profiling of Impatiens walleriana genes through different stages of downy mildew infection reveals novel genes involved in disease susceptibility. Ornamental Plant Research 4: e008 doi: 10.48130/opr-0024-0006 

Transcriptional profiling of Impatiens walleriana genes through different stages of downy mildew infection reveals novel genes involved in disease susceptibility
- Received: 04 June 2023
- Revised: 26 December 2023
- Accepted: 02 January 2024
- Published online: 27 March 2024
Abstract: Impatiens downy mildew is a highly destructive disease of Impatiens walleriana, an economically important bedding ornamental crop. This disease is caused by a recently emerged pathogen Plasmopara obducens. Since both the host and pathogen are relatively less studied, there are only a few genomic resources available for both I. walleriana and P. obducens. In this study, we have analyzed transcriptional changes in I. walleriana in response to P. obducens infection during different stages of disease development. Our main goal was to identify candidate genes that may be involved in I. walleriana susceptibility to P. obducens. Since the genome of I. walleriana is not available publicly, we constructed and optimized a de novo transcriptome assembly consisting of 73,022 transcripts. Differential expression analysis based on this optimized de novo transcriptome assembly revealed 3,000 to 4,500 differentially expressed transcripts (DETs) at six different time points during infection. Functional annotation of these DETs revealed that numerous plant stress responsive genes are activated and deactivated throughout the infection cycle. Several receptor-like kinases (RLKs), resistance genes, powdery mildew susceptibility genes, transcription factors including a grape downy mildew susceptibility factor, several retrotransposons and many other plant stress related genes were predominantly differentially expressed in I. walleriana in response to P. obducens. Analyses reported here provide molecular insights into the disease susceptibility mechanism of the Impatiens downy mildew and lays out a strong foundation for future studies aimed at improving downy mildew resistance in I. walleriana.