








-
Rosaceae is an important plant family within the angiosperm class, possessing high economic value. It includes approximately 124 genera and more than 3,300 species[1]. There are many types of plants in the Rosaceae family that can be used as ornamental plants for landscaping, or as fruits to be enjoyed at people's dining tables. The primary objective in breeding Rosaceae plants is to improve their tolerance to abiotic stress and develop more ornamental varieties to increase their utilization in landscape ecology and construction. One of the important traits that determines the ornamental value of Rosaceae plants is their flower color. Petals are eye-catching and cherished for their beautiful patterns and colors. Some plants have petals with cells of the same color, creating a solid color visual effect, while others have petals with cells of multiple colors, resulting in stripes, spots, or other irregular patterns. The vibrant colors enhance consumers' desire to purchase and add depth to landscape design, capturing attention. The diversity of flower colors is attributed to the variety of pigment types, concentrations, and distribution in different organs and tissues[2,3]. Structural genes regulate the synthesis pathway of pigments, as well as various transcription factors[4−6]. Understanding the regulatory mechanisms of flower color formation is crucial for breeding. The fruit of Rosaceae plants contributes significantly to their commercial value, as the quality of the fruit directly influences consumer purchasing decisions and market value. Fruit quality is determined by physical characteristics such as fruit size, hardness, and color, as well as chemical characteristics like soluble solid content and pH value. Additionally, sensory characteristics such as appearance, taste, and aroma, as well as nutritional parameters like phenolic content and antioxidant capacity, also play a role in determining fruit quality[7,8]. Consumers initially select fruits based on physical characteristics such as color and firmness, while subsequent purchases are primarily influenced by the taste, aroma, and nutritional value of the fruits. The flavor of fruits is primarily sweet and sour, with sugar and acidity being the main influencing factors. The main types of fruit aroma are fruity and light, with volatile aromatic substances being the primary influencing factors. Aroma is a crucial indicator for assessing fruit quality, as it can reflect the flavor characteristics of the fruit. Fruits with a strong aroma are more appealing to consumers. Many fruits, such as Malus pumila, Pyrus spp, Fragaria × ananassa, and Prunus persica, belong to the Rosaceae plant family. Consumers adore them for their unique taste and exceptional nutritional benefits[9−12]. Fruits of the Rosaceae plants exhibit a rich and diverse range of colors due to their high anthocyanin content[4]. Anthocyanins are significant antioxidant phenolic chemicals that have anti-inflammatory and anticancer effects, which makes them useful for promoting health. They also help circumvent cardiovascular illnesses[13,14]. Enhancing the fruit quality of Rosaceae plants is a long-term research topic that requires further investigation. Utilizing high-throughput sequencing technology to investigate the alterations in gene expression levels during the accumulation of fruit sugars, organic acids, and various nutrients, and integrating multi-omics techniques, the functions and regulatory mechanisms of key genes involved in their synthesis, transportation, and accumulation processes will be examined, ultimately constructing a comprehensive regulatory network. The current research thoroughly investigates and leverages omics data from Rosaceae plants to identify genes involved in the production and breakdown of distinctive aroma compounds. The aroma of fruits is synthesized by the combination of multiple volatile substances.
Utilizing omics techniques explores how volatile substances from multiple synthesis pathways cause diversity of fruit aroma types through synergistic/antagonistic effects, and clarifies the reasons why fruits exhibit different aroma types. In addition, due to global climate change, the intensity and frequency of extreme weather events have increased[15]. As phenological stages advance, the flowering time of Rosaceae plants has been progressively earlier, which increases the risk of cold and frost damage[16]. At the same time, factors such as soil salinization and soil drought can also affect plant growth[17]. Rosaceae plants adapt to various stressful environments through molecular-level responses and regulatory mechanisms, thereby ensuring their normal growth and development. When plants in the Rosaceae family encounter stress, they exhibit high expression and synthesis of stress-responsive proteins. These proteins play a crucial role in responding to stress, including enzyme proteins involved in metabolism, regulatory proteins, and resistant proteins that participate in stress responses to fend off attacks. At the same time, transcription factors are also crucial participants in the stress response, regulating the expression of stress-resistant genes through transcription. Omics technology has become a reliable method for studying the molecular mechanisms of stress response in Rosaceae plants.
In recent years, utilizing omics technologies, such as transcriptome, comparative genomics, and genomics, has grown in importance as a means of exploring plant life. The genome serves as the foundation for studies on molecular mechanisms. With genomic data, it is possible to identify and validate new genes or gene clusters related to the synthesis of bioactive compounds through transcriptome, proteomics, metabolomics, and other omics technologies. By utilizing these technologies, the stress-adaptive mechanisms and response mechanisms of Rosaceae plants can be investigated, along with the regulatory mechanisms affecting fruit quality, flower color, and fragrance. This article reviews applications and developments of omics technologies in Rosaceae plant research and provides prospects for future directions in genomics research.
-
The field of genomics analyzes genomes' DNA sequences or the intermediate products of their expression to interpret genetic information. Every analysis starts with a complete, high-quality, and highly accurate genome. Maxam & Gilbert[18] reported a method for determining DNA sequences through chemical degradation, and Sanger et al.[19] invented the dideoxy chain termination method. The use of these methods marked the beginning of first-generation sequencing technology. Although it had high precision, some limitations hindered its broader application.
Subsequently, high-throughput and low-cost second-generation sequencing emerged[20]. Among second-generation sequencing technologies, the Illumina sequencing platform is widely used for genomic sequencing of animals and plants. It has greatly advanced the genome sequencing of Rosaceae plants, including nuclear genome and chloroplast genome sequencing. The Illumina sequencing platform has also been widely applied in the chloroplast genome sequencing in Rosaceae plants. For example, chloroplast genome sequencing has been completed in various plant species such as Prunus mume[21], Prunus salicina[21], Prunus dielsiana[22], Malus domestica[23], Malus toringoides[24], Pyrus pyrifolia[25], Rubus sachalinensis[26], Potentilla parvifolia[27], Crataegus pinnatifida[28], Fragaria pentaphylla[29], Prunus avium[30], and Rosa chinensis var. spontanea[31]. Phylogenetic trees representing the evolutionary relationships of representative Rosaceae plants, based on their chloroplast genomes, have been constructed (Fig. 1a). The chloroplast genome is relatively small in size and contains relatively independent genetic material, including chloroplast DNA and cytoplasmic inheritance. It also has a highly conserved genomic structure. The chloroplast genome can effectively enhance species identification. The whole sequencing of the chloroplast genome offers important insights into the evolutionary trends and phylogenetic connections within the Rosaceae plants. Consequently, it is thought that the chloroplast genome is the perfect system for phylogenetic analysis. However, due to its unique single-parent genetic characteristics, it can only reflect the lineage history of a single parent. The nuclear genome plays a dominant role, containing a vast amount of genetic information that determines most of the physiological, biochemical functions, and morphological characteristics of plants, and it also contains rich genetic variation. Therefore, the nuclear genome, which contains parental genetic information, holds greater value in phylogenetic research. However, the technical requirements for sequencing and assembling the nuclear genome are high, and the data from the nuclear genome is substantial. Some Rosaceae plants that have undergone chloroplast sequencing have not yet completed nuclear genome sequencing and assembly. Phylogenetic analysis of chloroplast genomes showed that F. pentaphylla is closely related to P. parvifolia, while P. mume and P. salicina are closely related. M. toringoides is also closely related to M. domestica (Fig. 1a). Phylogenetic analysis of the nuclear genome revealed that Rosa chinensis and Fragaria vesca are closely related, P. mume and P. salicina are closely related, and M. domestica and P. pyrifolia are closely related (Fig. 1b). The phylogenetic evolution results of the two genomes are consistent. The chloroplast genome is relatively smaller in sequencing difficulty compared to the nuclear genome. This can aid in understanding the phylogenetic relationships between different species in the absence of a nuclear genome.
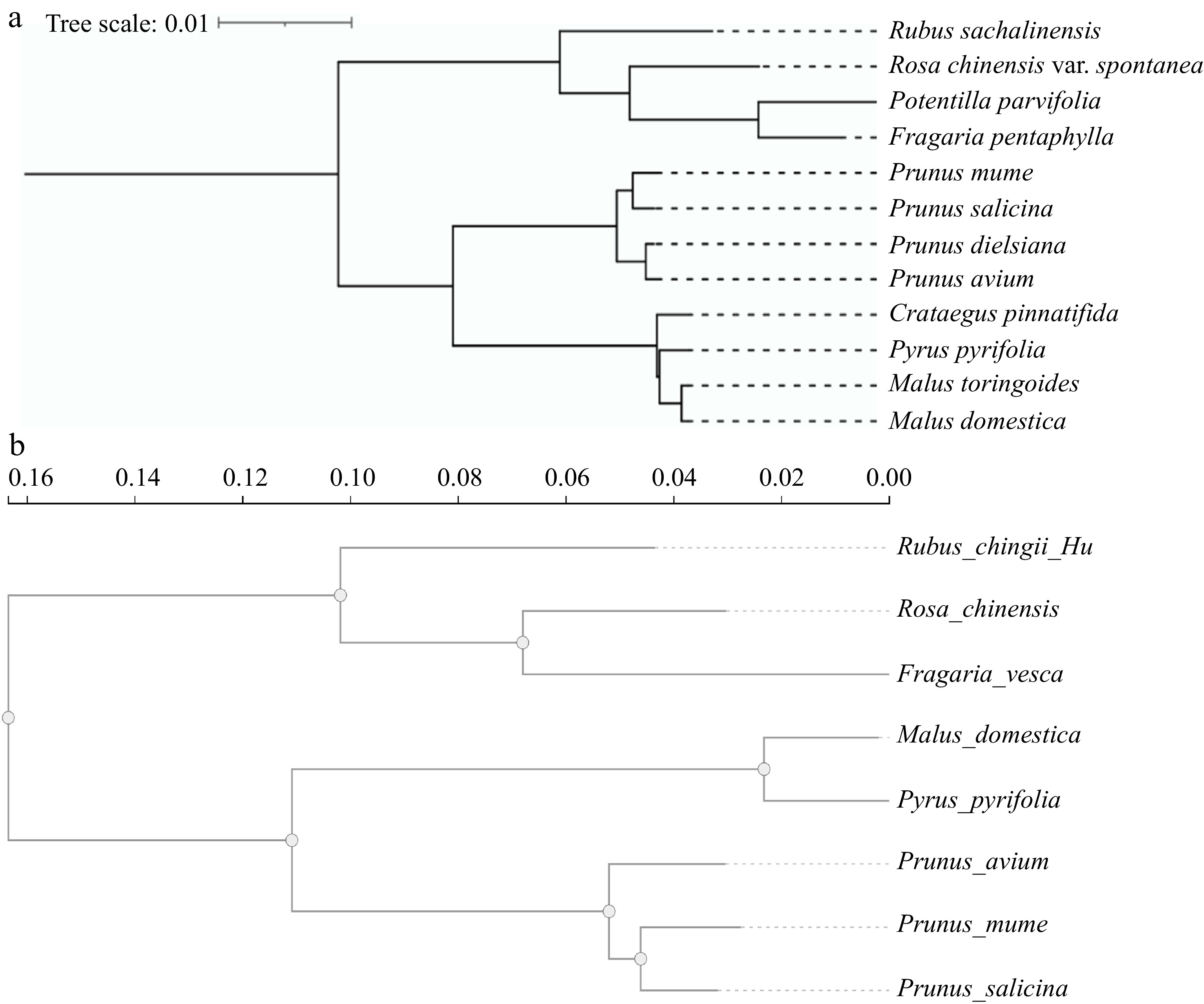
Figure 1.
Phylogenetic evolution tree of Rosaceae plants. (a) The Rosaceae represents the plant chloroplast genome evolutionary tree. (b) The Rosaceae represents the plant nuclear genome evolutionary tree.
Numerous horticultural plants with high economic value are members of the Rosaceae plants. The genomes of these plants show high heterozygosity and complex ploidy as a result of long-term natural variation and artificial domestication. Because of the shorter read length of second-generation sequencing, it is often difficult to differentiate between a high number of repetitive sequences, which can result in unclear or incomplete genome assemblies. For further bioinformatic analysis, this presents substantial obstacles[32]. In contrast, third-generation sequencing technologies offer advantages such as longer read lengths, higher accuracy, greater sensitivity, no PCR amplification bias, and minimal GC bias[33]. These characteristics make third-generation sequencing technologies more suitable for overcoming the limitations of previous sequencing methods in studying the complex genomes of Rosaceae plants.
Oxford Nanopore Technologies' single-molecule nanopore DNA sequencing technology and Pacific Biosciences' single-molecule real-time (SMRT) sequencing technology are two widely used third-generation sequencing technologies. The nanopore sequencer (MinION) which Oxford Nanopore Technologies unveiled in 2014, allows real-time data acquisition from individual molecules, greatly reducing the amount of time needed between sample collection and data processing. Moreover, direct RNA sequencing can be performed without the need for antecedent reverse transcription or amplification[34,35]. In 2022, Pacific Biosciences launched the Revio long-read sequencing system. This system not only achieves higher HiFi data throughput and improved accuracy of HiFi reads but also significantly reduces costs. Many horticultural plants have undergone genome resequencing thanks to the quick development of third-generation sequencing technology. For example, the genome contiguity, accuracy, and completeness of various Rosaceae plants, such as P. persica and Rubus occidentalis, have been significantly improved[36−39].
Because plant genomes are complex and contain many repetitive sequences, it is challenging to assemble a complete genome based solely on sequence reads[32]. To enhance assembly quality, numerous researchers have utilized high-throughput chromosome conformation capture (Hi-C) technology. Hi-C is a high-throughput sequencing-based technique combined with bioinformatics analysis used to determine the spatial relationships of chromatin within the entire genome and the interactions between chromatin on a larger scale, including interchromosomal interactions[40]. Genome assembly has entered a new era with the advancement of third-generation sequencing technologies[41]. Telomere-to-telomere (T2T) genome refers to a 0-gap genome that combines multiple sequencing technologies to achieve high accuracy, high continuity, and high integrity of the telomere-to-telomere level assembly. The assembly of the T2T genome enables the exploration of previously unknown regions, such as telomeres and centromeres, providing a wider research direction for the study of animals and plants. Recently, T2T genome assemblies have been completed for F. vesca[42], Chaenomeles speciosa[41], and P. pyrifolia[43]. Among them, the T2T genome assembly of P. pyrifolia (501.2 Mb) has a contig N50 of 29.26 Mb, which significantly improves the completeness compared to the previous P. pyrifolia genomes ('Cuiguan'[44] and 'Nijisseiki'[45]).
Currently, most Rosaceae plants utilize second-generation sequencing or a combination of second-generation and third-generation sequencing to sequence the genome and assemble the genome at the chromosome level. The genome assembly of Rosaceae plants has become more complete thanks to the extensive application of technologies like Illumina, Nanopore, PacBio, and Hi-C. The scaffold N50 values for Crataegus. pinnatifida var. major and Rosa rugosa reached 44.94 and 56.6 Mb, respectively. The assembled genome sizes of Rosaceae plants range from 214.9 to 1,399.32 Mb, according to earlier research, and the scaffold N50 values range from 2.4 to 56.6 Mb (Fig. 2, Table 1). Prunus domestica is hexaploid, resulting in an abnormally large genome, which reflects its hexaploid nature and the high heterozygosity of its genes.
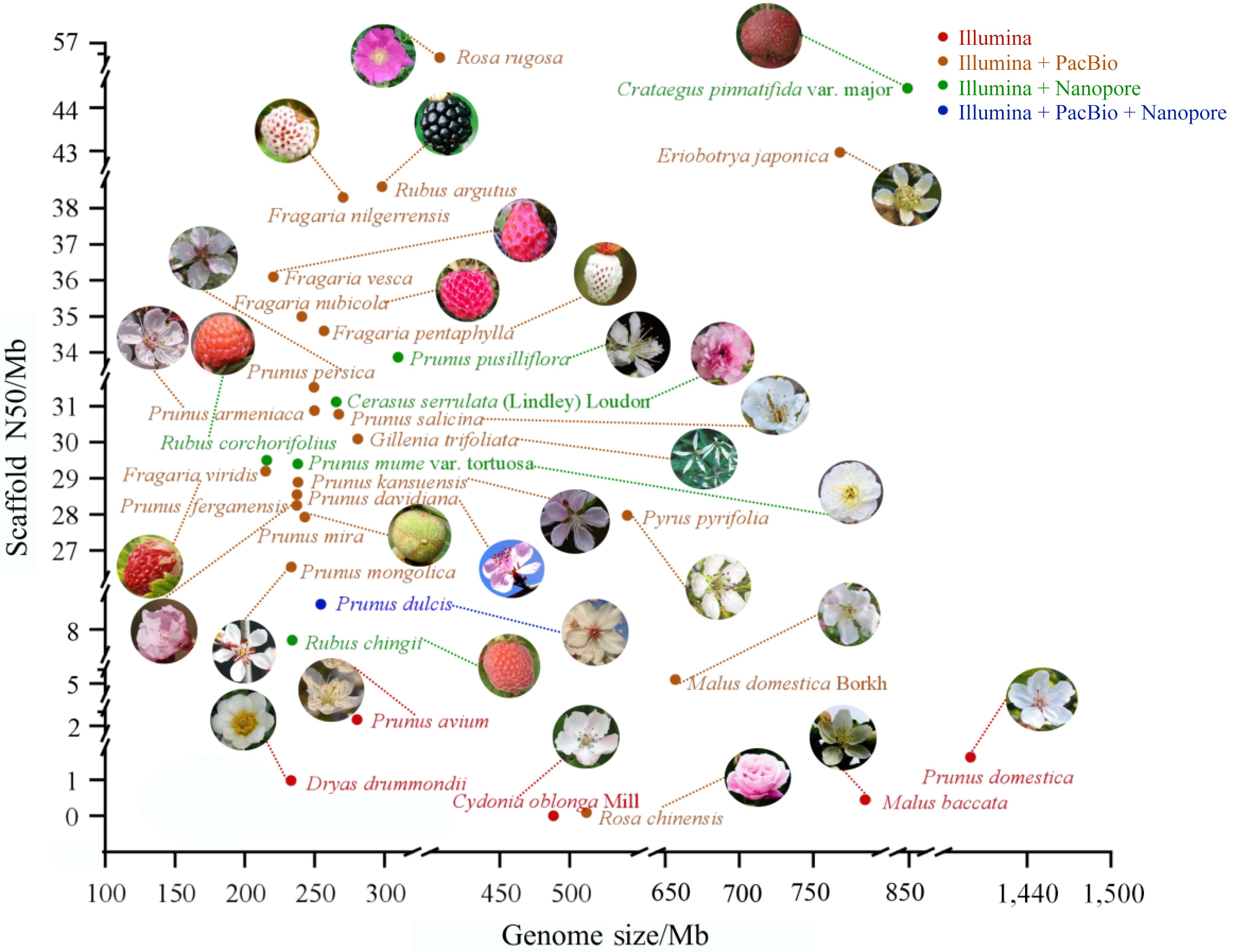
Figure 2.
Summary of plant genome sequencing for Rosaceae. Each plant's genome size is shown by the x-axis, and the scaffold N50 of the genome assembly is represented by the y-axis. Different colors correspond to different sequencing platforms.
Table 1. Progression genome sequencing of Rosaceae.
Species Sequencing method Genome size (Mb) Scaffold N50 (Mb) Ref. Cerasus serrulata (Lindley) Loudon Illumina + Nanopore 265.40 31.12 [46] Prunus avium Illumina 280.33 2.48 [47] Prunus pusilliflora Illumina + Nanopore 309.62 33.87 [48] Dryas drummondii Illumina 233.00 0.98 [49] Eriobotrya japonica Illumina + Nanopore 760.98 43.16 [50] Gillenia trifoliata PacBio 280.76 30.09 [50] Fragaria vesca Illumina + PacBio 220.40 36.1 [51] Fragaria nilgerrensis Illumina + PacBio 270.30 38.3 [52] Fragaria viridis Illumina + PacBio 214.90 29.2 [53] Fragaria nubicola Illumina + PacBio 240.75 35 [53] Fragaria pentaphylla Illumina + PacBio 256.74 34.6 [54] Prunus armeniaca Illumina + PacBio 249.73 30.88 [37] Prunus persica Illumina + PacBio 249.41 31.53 [37] Prunus davidiana Illumina + PacBio 237.29 28.55 [55] Prunus kansuensis Illumina + PacBio 238.06 28.89 [55] Prunus ferganensis Illumina + PacBio 237.24 28.25 [55] Prunus mume var. tortuosa Illumina + Nanopore 237.80 29.4 [56] Pyrus pyrifolia Illumina + PacBio 541.30 27.97 [44] Prunus salicina Illumina + PacBio 267.18 30.78 [37] Prunus domestica Illumina 1,399.32 1.63 [57] Malus domestica Borkh. Illumina + PacBio 643.2 5.5 [58] Malus baccata Illumina 779.00 0.45 [59] Prunus mira Illumina + PacBio 242.94 27.93 [55] Prunus dulcis Illumina + PacBio + Nanopore 254.45 9.2 [60] Prunus mongolica Illumina + PacBio 233.17 26.54 [61] Cydonia oblonga Mill. Illumina 488.40 0.0024 [62] Crataegus pinnatifida var. major Illumina + Nanopore 856.88 44.94 [63] Rosa chinensis Illumina + PacBio 512.00 0.09 [64] Rosa rugosa Illumina + PacBio 407.00 56.6 [65] Rubus chingii Hu Illumina + Nanopore 219.80 8.2 [66] Rubus corchorifolius Illumina + Nanopore 215.74 29.5 [67] Rubus argutus Illumina + PacBio 298.24 38.6 [68] After the sequencing of species is completed, the genomes need to be archived for subsequent data analysis and sharing. Currently, the primary purpose of a database has changed from data storage to online analysis[69]. Genomic databases provide significant convenience for studying horticultural plants. Over the past decade, the Rosaceae Genomics Database (GDR) (
www.rosaceae.org ), established in 2003, has undergone significant expansion in both data and functionality[70]. GDR has collected complete genome assemblies and annotation data for multiple Rosaceae plants, including P. persica, Prunus sibirica, M. domestica, and F. × ananassa. In addition, GDR provides rich data on transcriptomes, genetic maps, genetic markers, quantitative trait locus (QTL) loci, phenotypes, and genotypes, laying an important foundation for genomic research and molecular breeding of Rosaceae plants.Summary
-
As research on Rosaceae plants advances, more Rosaceae plants will be sequenced. Due to the continuous advancement of sequencing technology and splicing algorithms, the selection of plants for sequencing will no longer be constrained by cost or complexity, and the quality of reference genomes will gradually improve. However, genomics still faces significant challenges due to the rapid development of various fields such as information technology and instrumentation, leading to a continuous increase in the total amount of data. With the inclusion of various new indicators and parameters, data is also becoming increasingly complex. In light of the extensive sequencing results, the significant challenges in thorough data mining and interpretation are becoming increasingly apparent. These challenges often present opportunities, and a large amount of unexplained data also brings the potential for endless innovation.
-
Comparative genomics is a field that utilizes advancements in genome sequencing technologies and employs genome maps and various sequencing techniques to perform comparative analysis of genes obtained through sequencing. Its main objective is to study the evolutionary relationships between species by analyzing the functional and structural aspects of genes[71].
Comparative genomics analysis has revealed that the population of Rubus corchorifolius in Hunan province (China) exhibits the highest genetic diversity, indicating a larger ancestral population. In contrast, the population of R. corchorifolius in Yunnan province has undergone strong natural selection, particularly in genes associated with flavonoid synthesis and plant hormone signal transduction. This indicates that Yunnan R. corchorifolius exhibits strong adaptability to the local environment[67]. High-quality genomes of Prunus davidiana, Prunus mira, Prunus kansuensis, and Prunus ferganensis have been assembled by researchers. When compared to cultivated peaches, these varieties exhibit a wide range of genetic variations, as indicated by comparative genomics studies. Among these, the estimated divergence time between P. mira and wild peaches is 1.15 million years ago (Mya), coinciding with a period of intense tectonic movement in the Qinghai-Tibet Plateau[55]. Chromosome doubling, rearrangements, and deletion are common alterations in the genome structure that occur during a species' evolutionary process. Functional evolution and species divergence are closely associated with these phenomena. Studies have shown that large-scale duplication events are widespread in plants[72,73]. In a comparative genomic analysis by Jiu et al.[48], it was discovered that Prunus pusilliflora shares a closer phylogenetic relationship with Prunus serrulata and Prunus yedoensis. The divergence between P. pusilliflora and the two cherry species mentioned above occurred before 4.18 Mya. All three cherry species experienced a shared whole-genome triplication event, known as the γ-WGT.
Summary
-
The whole-genome sequencing data of multiple Rosaceae plants has been made publicly available, providing abundant research materials for comparative analysis among multiple genomes and laying a solid foundation for addressing biological questions. Additionally, utilizing whole-genome sequencing data, researchers can selectively identify high-quality genes to support fundamental biological studies on plant biosynthesis, physiology, and biochemistry. This can lead to the improvement of existing economic plants, enhancement of plant stress resistance, and ultimately reduce production costs while providing high-quality materials for genetic breeding.
-
Genome-wide association studies (GWAS) are commonly used to explore genetic structure in plant populations. GWAS identifies allelic variant loci that are substantially correlated with target traits and analyzes the genetic effects of these allelic genotypes on phenotypes. This technique has been extensively used to identify genes and variant loci linked to significant agronomic traits such as flower color, quality, and resistance.
GWAS on stress resistance in Rosaceae plants
-
Fusarium wilt poses a threat to the growth and development of strawberries, significantly impacting their yield and quality. Through GWAS analysis, candidate genes related to disease resistance and defense in strawberries have been identified[74]. Flesh browning is a physiological disorder in apples that significantly affects their storage and marketability. Tazawa et al. conducted a study on the genetic characteristics of flesh browning in apples[75], focusing on the variety 'Aori27', which is known to be less susceptible to flesh browning. Subsequently, Kunihisa et al.[76] discovered through GWAS that flesh browning is primarily regulated by polyphenol content and the activity of polyphenol oxidase (PPO). This research has provided new insights for improving flesh browning in apples and has contributed to the development of non-browning apple varieties. The tissue-specific expression of DNA binding with one finger (Dof) in R. chinensis, as well as their differential expression in response to salt and drought stress, has been elucidated through the application of GWAS in ornamental gardening. This finding paves the way for genetically enhancing stress tolerance features in different types of roses and further advances the investigation of the gene function of potential Dofs[77]. Through GWAS, it has been discovered that P. mume's dormancy induction and freezing tolerance are significantly influenced by the expression of two ethylene-related genes, ethylene-insensitive3-like (EIL) and ethylene-responsive factor (ERF)[78].
GWAS on flower color in Rosaceae plants
-
Flowers, as the reproductive organs of higher plants, exhibit vibrant colors that attract pollinators and facilitate the reproduction of the next generation. One of the most significant biological characteristics and morphological markers of ornamental plants is flower color, which is frequently used as a key factor in the classification of plants. Through GWAS, QTLs and candidate gene regions associated with significant ornamental traits in P. mume have been identified. For example, MYB108, which encodes an R2R3 MYB transcription factor, is positively correlated with the colors of petals, stigmas, calyxes, and flower buds. These discoveries establish the groundwork for unraveling the genetic mechanisms underlying important traits in P. mume and conducting molecular breeding[79]. Prunus persica f. versicolor, having high ornamental value, can produce bicolored and chimera flowers. The candidate gene dicer-like 2 (DCL2), which controls flower color variegation in P. persica f. versicolor, has been identified through GWAS and other analytical methods[80]. GWAS was conducted using 312 ornamental cherry blossoms as materials, and seven QTLs related to flower color were identified. Among them, is the glycosyltransferase encoded by evm. model. LG02. 1464 which is one of the important enzymes in the biosynthesis of glucosides in anthocyanins. evm. model. LG02. 1464 is a significant candidate gene, which may play a role in controlling the color of cherry blossom flowers[81].
GWAS on fruit quality in Rosaceae plants
-
The accurate deciphering of the genetic regulatory mechanisms underlying significant agricultural traits, such as fruit shape, development, and volatile compounds in Rosaceae plants, is made possible through GWAS. Whole-genome sequencing was performed on five plants, namely P. persica, P. salicina, Prunus armeniaca, P. mira, and P. davidiana[37]. Through GWAS analysis, the researchers conducted a structural variation (usually refers to the large-length sequence changes and positional relationship changes on the genome) analysis on crucial agricultural traits. The results of this investigation demonstrated the important functions of OVATE transcription factor and microRNA172d, NAC (NAM, ATAF1/2, and CUC1/2) transcription factors, in fruit morphology, fruit development time, and fruit shape. To unravel the genetic mechanism behind the unique aroma of peaches, GWAS was utilized to identify QTLs associated with volatile compounds in peaches, and a candidate gene, Pp4G000350, related to these compounds was identified[82]. Using genomic data from 149 species of the Malus genus, a GWAS analysis was conducted on seven fatty acids. It was found that the transient overexpression of SUPERMAN-like (SUP) increased the levels of linoleic acid, and linolenic acid, as well as three aromatic compounds: hexanal, 2-hexanal, and 1-hexanol in the fruit. This provides a theoretical basis for improving the quality of apples[83]. In the study of pear fruit quality, GWAS analysis identified candidate genes associated with sucrose and glucose accumulation. This offers a wealth of genetic resources for further research aimed at enhancing the quality of pear fruit[84].
Summary
-
GWAS is an effective research strategy for identifying genes/loci associated with complex traits in Rosaceae plants. It provides a novel approach to systematically unravel the molecular genetic mechanisms underlying complex traits in Rosaceae plants and establishes a strong foundation for transitioning from traditional breeding to efficient and targeted molecular design breeding in Rosaceae plants. In addition, GWAS analysis needs to overcome the limitations of identifying low-frequency rare allele variations. Finally, the loci associated with GWAS need to be fine-mapped using various omics technologies to accurately pinpoint functional genes and elucidate the impact of allele variations on the target traits. This will lead to a more thorough comprehension of the molecular genetic mechanisms behind complex traits in Rosaceae plants.
-
Transcriptome research examines patterns of transcriptional regulation and gene expression. By utilizing transcriptome techniques to identify variations in gene expression levels, this study elucidates the genetic regulatory mechanisms that underlie significant traits, including stress resistance, flower color, floral fragrance, and fruit quality. This is an important approach for studying the stress resistance and ornamental traits of horticultural plants. With the advancement of transcriptome sequencing in Rosaceae plants in response to different environmental stresses and ornamental traits, many stress-resistant response genes and ornamental trait-related genes have been identified. This lays the groundwork for understanding the mechanisms of plant adaptation to stress and the development of ornamental characteristics.
Transcriptome sequencing on stress resistance of Rosaceae plants
-
Plants inevitably encounter abiotic stresses during their growth, and transcriptome research helps to understand the plant's response mechanisms to environmental stress at the transcriptional level. Transcriptome sequencing has been widely used in studies related to plant responses to environmental stress due to the rapid advancement of high-throughput sequencing technologies. The transcriptome map of Rosa hybrida under low temperature and dark treatment was studied. A total of eight up-regulated genes and one down-regulated gene were identified[85]. Identifying genes related to the molecular regulation of cold acclimation is beneficial for cultivating rose varieties with strong cold tolerance. The ability of loquat leaves to withstand low temperatures has been linked to genes that encode phenylalanine ammonia-lyase, anthocyanin synthase, and D-sorbitol-6-phosphate dehydrogenase, as revealed by transcriptome sequencing conducted on the leaves subjected to a 4 °C treatment[86]. Transcriptome analysis of Rosa xanthina f. spontanea subjected to 4 and −20 °C low-temperature stress elucidated its response mechanisms to cold stress[87]. The researchers assembled the high-quality genome of the 'Cuiguan' pear and used transcriptome sequencing to identify several novel genes that may be involved in regulating dormancy release[44]. This study provides deeper insights into the regulatory mechanisms of bud dormancy release and chilling requirements. Key transcription factor families, including basic-helix-loop-helix (bHLH), WRKY, ERF, and MYB, involved in the response to salt stress, were identified using P. mume as the material[88]. The full transcriptome of R. chinensis was constructed, and several differentially expressed genes (DEGs) were identified. The previously unidentified splicing sites and new genes in the R. chinensis transcriptome have been identified, and the complex regulatory networks of drought stress response genes have been elucidated[89]. Heat stress restricts the growth of roses and adversely affects the yield and quality of cut roses. The transcriptome profiles of R. chinensis under heat stress at different time points were compared, and 6175 DGEs were identified. This provides evidence for the early regulation of R. chinensis' heat stress response[90]. RNA-Seq technology was utilized to analyze the petal transcriptome of R. hybrida, and differentially transcribed genes (DTGs) were identified. Among them, the abundance of brassinosteroid (BR)-related genes was the highest. The application of exogenous BR to rose petals enhanced their defense response to Botrytis cinerea, further demonstrating the role of BR in petal resistance to B. cinerea[91]. Single-cell RNA sequencing (scRNA-seq) has significant advantages over traditional transcriptome sequencing for conducting unbiased studies with minimal sample input. Using F. vesca as the material, a single-cell gene expression map of strawberries was constructed using scRNA-seq. The study revealed the process of signal transformation from normal function to defense response in epidermal and mesophyll cells of strawberries after B. cinerea infection by integrating data from different stages[92]. Due to the variation in plant tolerance to different environmental stresses and the severity of stress, plants exhibit diverse responses. Analyzing the transcriptome profiles of plants under various stress conditions enables the identification of DEGs that are involved in plant responses to these stresses. By annotating and functionally classifying these DEGs, as well as analyzing their expression patterns, it is possible to identify candidate genes that are responsive to various stresses. This process lays the groundwork for further studies on the molecular mechanisms of plant stress responses and the development of new stress-tolerant varieties.
Transcriptome sequencing on flower color of Rosaceae plants
-
At present, candidate genes controlling flower color formation have been identified in multiple Rosaceae plants through transcriptome sequencing. Transcriptome analysis of petals at different developmental stages of the pink-flowered strawberry variety 'Siji Hong' revealed genes associated with anthocyanin or other flavonoid biosynthesis, providing insights into the mechanism of flower coloring in pink-flowered strawberries[93]. Transcriptome sequencing was conducted during key stages of flower development in Malus halliana to analyze the mechanism of flower senescence. The results showed that the downregulation of MhMYB10 expression during this process resulted in petal fading[94]. Transcriptome sequencing has been conducted on the R. chinensis variety 'Old Blush' and R. rugosa to investigate the regulatory pathways involved in anthocyanin biosynthesis. This work provides a theoretical framework for investigating the genetic regulation mechanism of flower color in Rosa plants[95,96]. Through transcriptome sequencing analysis of the red rose and its white flower mutant, it was confirmed that the insertion of the Rosa1 transposon into the promoter region of the RcMYB114 gene resulted in high-level gene expression, leading to the appearance of red roses. Conversely, the absence of this transposon resulted in a white phenotype in roses[97].
Transcriptome sequencing on fruit quality of Rosaceae plants
-
Anthocyanins are a family of flavonoid chemicals that are helpful to human diets and have a role in pigment deposition in plant tissues[13]. To modify the anthocyanin content of fruits and enhance fruit quality, transcriptome studies have been conducted to investigate the biosynthetic pathways and composition of anthocyanins in fruits. It was discovered through transcriptome analysis of peel and flesh tissues that the expression of MYB10 is the primary factor influencing the variations in fruit color in light pink, red, and dark red octoploid strawberries[98]. PacbHLH13 and PacbHLH74 were identified as crucial regulatory factors in the control of anthocyanin synthesis in sweet cherry fruits, as determined by transcriptome co-expression network analysis[99]. Transcriptome sequencing of slices from apples of different colors, such as the 'May' variety, revealed nine genes that might be implicated in the metabolism of anthocyanins in flesh. This study explores the potential regulatory mechanisms underlying anthocyanin accumulation in apple fruits[100]. Transcriptome analysis of C. pinnatifida revealed multiple genes related to red fruit flesh formation and elucidated the mechanism of anthocyanin synthesis in C. pinnatifida fruit flesh[101]. Extending the duration of light exposure for fruit crops can enhance sugar accumulation, promote fruit coloring, and improve the flavor and appearance of the fruit. Mei et al.[102] revealed that MdWRKY40 may attach to the glutathione S-transferase (GSTF) promoter, which facilitates the anthocyanin glycosides' transportation and storage in the peel. By attaching itself to the phosphofructokinase (PFK) promoter, MdMYB108 encourages the buildup of soluble sugars in the flesh. These findings provide theoretical support for the impact of regulating the light environment on the quality of apple fruit.
Summary
-
The continuous advancement of sequencing technologies has enabled the discovery of a greater number of reliable transcripts, as well as the ability to conduct more comprehensive sequencing of the transcriptomes of Rosaceae plants. However, transcriptome in Rosaceae plants also faces a series of new challenges. The massive amount of data poses challenges in information processing. For example, how to align and identify homologous genes among multiple similar sequences to obtain high-quality transcriptomes. Transcriptome of Rosaceae plants is a highly promising area of research. In the future, the application of new technologies such as scRNA-seq and spatial transcriptome in Rosaceae plants will enable more in-depth analysis of the complex characteristics of the transcriptome, thereby advancing the research and development of Rosaceae plant germplasm resources.
-
The field of proteomics investigates the composition and diverse functional characteristics of cellular proteins[103], intending to study protein functions and interactions. The economic and resistance traits of Rosaceae plants involve numerous proteins. Quantitative analysis using proteomic techniques can help solve the biological problems associated with complex traits and establish correlations between genotypes and phenotypes. This can reveal the genetic regulation mechanisms of important traits in Rosaceae plants.
Proteomics study on the stress resistance of Rosaceae plants
-
Proteomics aims to study the overall protein levels and has been widely applied in researching the molecular mechanisms underlying plant resistance to abiotic stress. It focuses on analyzing the structure, function, and expression abundance of proteins in specific tissues or at specific time points[104]. Protein profiles of rose petals at various developmental stages were created using two-dimensional polyacrylamide gel electrophoresis. The analysis revealed that the developing petals contained a significant amount of stress-related proteins[105]. Two-dimensional electrophoresis and label-free quantitative proteomics were used to identify proteins associated with cold tolerance in strawberries. These proteins include molecular chaperones, antioxidant/detoxification enzymes, and metabolic enzymes[106]. In Rosa beggeriana, it was found that proteins related to cold stress were significantly up-regulated during winter dormancy[107]. This discovery lays the foundation for elucidating the regulatory mechanisms of cold tolerance in R. beggeriana. By analyzing the variations in protein response to salt stress in the roots of Prunus cerasus using proteomics, researchers discovered that fructose 1,6-bisphosphate aldolase isoforms undergo mutual conversion. This finding provides further insight into the regulatory mechanisms of P. cerasus in response to salt stress[108]. Researchers used quantitative proteomic sequencing on the F1 hybrid population of 'Golden Delicious' and 'Fuji Nagafu No. 2' to investigate the molecular mechanisms underlying apple resistance to apple ring rot. Their findings demonstrated the important role of sorbitol, a major photosynthetic product in apples, in the resistance of apple trees to apple ring rot. Apple ring rot resistance can be enhanced by elevating the sorbitol content in callus tissue through the overexpression of sorbitol-6-phosphate dehydrogenase (S6PDH), a crucial enzyme in the sorbitol synthesis pathway. On the other hand, callus tissue that has had the MdS6PDH1 gene silenced displays the opposite phenotype[109]. By identifying specific proteins and analyzing their expression characteristics at various stages and in different organs of plant growth and development, proteomics research methods can shed light on the mechanisms underlying plants' responses to stress and adversity.
Proteomics study on flower color of Rosaceae plants
-
The objective of utilizing proteomics methodologies and techniques to identify proteins (enzymes) associated with flower color variations in Rosaceae plants is to uncover the molecular mechanisms that underlie flower color variation at the protein level. Wu et al. using proteomics analysis[110], speculated that auxin-binding proteins and allene oxide cyclase (AOC) could be key factors contributing to the color variation in P. mume 'Fuban Tiaozhi' flowers. Leucoanthocyanidin dioxygenase (LDOX), WD40 (activates the transcription of flavonoid biosynthetic genes and facilitates protein–protein interactions), acetyl-CoA carboxylase (ACC), and PPO II have been linked to pigment biosynthesis pathways and may play a significant role in the post-translational modifications of peach flowers, as indicated by proteomic studies conducted on different peach bud varieties[111]. Lu et al.[112] found that the protein degradation and autophagy pathways, especially those associated with peptidases, are abundant in the protein profile of R. hybrida. These results indicate that autophagy and protein degradation are important causes of senescence of alfalfa petals. Using P. persica f. versicolor as the material, differentially expressed proteins (DEPs) were identified in red and white flowers at various developmental phases. The results showed that the activity of anthocyanin synthase and GST proteins involved in anthocyanin transport were significantly upregulated in red flowers, highlighting their crucial roles in the process of anthocyanin variation in P. persica f. versicolor[113].
Proteomics study on fruit quality of Rosaceae plants
-
Plants undergo aging as an essential part of their life cycle, and this process is often accompanied by various modifications to their biological and biochemical functions. Leaf senescence strongly inhibits carbohydrate accumulation and delays fruit development, thereby affecting its flavor. Wang et al. investigated the effects of melatonin treatment on leaf senescence in Malus hupehensis[114]. The study also analyzed the protein profiles of the plant's leaves in response to different treatments. They discovered that melatonin treatment caused the downregulation of proteins that are typically upregulated during leaf senescence, providing insight into the mechanism through which exogenous melatonin in M. hupehensis delays leaf senescence. One of the primary methods extensively used in fruit production and transportation for postharvest fruit preservation is low-temperature storage. Based on isobaric tags for relative and absolute quantitation (iTRAQ) analysis, we investigated the changes in protein expression during peach fruit ripening and senescence under room temperature and low-temperature conditions. This analysis led to the identification of 325 DEPs. The findings showed that low-temperature storage prolonged the shelf life of peach fruits by regulating antioxidant capacity, inhibiting Ca2+ signaling transduction, and significantly affecting the conversion of starch to fructose and glucose[115]. Proteomic analysis was used to identify DEPs that are associated with the aroma of peach fruits under cold stress[116]. This study revealed that key proteins such as Lipoxygenase (LOX), aldehyde dehydrogenase (AD), alcohol acyl transferase (AAT), and chorismate synthase (CR) are involved in this process.
Summary
-
The research and application of proteomics in Rosaceae plants have made significant progress, but there are still some challenges that need to be addressed in future research. For instance, the intricate nature and dispersion of proteins necessitate researchers to continually enhance the methods for collecting and analyzing proteomic data. In addition to having a much broader range of amino acid residues compared to nucleotide residues, proteins also undergo complex post-translational modifications such as glycosylation and phosphorylation. These modifications make the separation and analysis of proteins extremely challenging. However, research and application of proteomics in Rosaceae plants still hold great potential for elucidating life processes such as growth, development, and metabolic regulation.
-
Metabolomics is a field that involves the qualitative and quantitative analysis of metabolites present in a biological organism during a specific period. It aims to elucidate the metabolic processes and dynamic changes within the organism. Metabolomics techniques are important for investigating traits related to stress resistance and fruit quality in Rosaceae plants.
Metabolomics study on the stress resistance of Rosaceae plants
-
Rosaceae plants often grow in challenging environments with various and changing stressors. Even though these stressors cause visible morphological changes in plants, such as wilting due to water deficiency, plants respond to them through a variety of mechanisms. These mechanisms include the release of metabolites and the regulation of gene expression. The plant responds to stress by releasing organic acids, removing reactive oxygen species, and expressing heat shock proteins, among other mechanisms. The transcription factor families, such as MYB and WRKY, also play an important role in this process. By employing multi-platform untargeted metabolomics techniques, we studied the metabolic changes in 'Camarosa' strawberry fruits grown under drought and salt stress conditions. The findings showed that metabolites linked to the suppression of reactive oxygen species, cell wall, and membrane lipid production are involved in the metabolic disturbance that occurs in plants under stress. These metabolites could function as osmotic stress indicators[117]. Metabolomics analysis of apple seedlings has provided a theoretical basis for improving their nitrogen and phosphorus absorption efficiency by identifying significantly different metabolites[118,119]. Using P. mume 'Meiren' as the material, a comprehensive transcriptome and metabolomic analysis was conducted to study the genes and metabolites associated with low temperatures. It was found that gene families such as MYB, WRKY, and NAC were upregulated after low-temperature treatment. Additionally, most metabolites exhibited higher metabolic levels under low-temperature conditions, indicating a significant impact of low temperature on the metabolite content of P. mume 'Meiren'[120]. The regulatory mechanisms of apple varieties with different levels of cold tolerance under freezing stress were investigated using both cold-tolerant and cold-sensitive varieties. The results showed that variations in metabolites, such as raffinose, stachyose, 4-aminobutyric acid, spermidine, and ascorbic acid, were the primary factors influencing the differences in cold tolerance among various apple varieties[121]. This study deepens our understanding of the regulatory mechanisms employed by apple trees to cope with freeze injury during the dormant period.
Metabolomics study on floral fragrance and flower color of Rosaceae plants
-
The types and levels of pigments that influence flower color exhibit significant individual variation, and their composition and abundance also show diversity as the flowering time progresses. Genes that are differentially expressed in the two flower colors, such as chalcone synthase (CHS), dihydroflavonol 4-reductase (DFR), and anthocyanidin 5,3-O-glucosyltransferase (GT1), were identified through transcriptome sequencing and metabolomic analysis of the petals of the miniature rose variety 'Neptune King' and its natural mutant, the deep pink miniature rose 'Queen'. This laid the groundwork for research into the biosynthetic metabolic pathways of anthocyanins in miniature roses[122]. The study of flavonoid metabolites at various stages of petal development in roses revealed that anthocyanins are responsible for turning roses red under sunlight. This finding facilitates the investigation of how light influences flower color[123]. Fragrance is one of the important commercial traits of ornamental plants and often captivates people more than flower color. Several aromatic varieties of R. rugosa were subjected to metabolic analysis to examine the patterns of volatile organic compound (VOC) content in the biosynthesis pathways of phenylalanine, tyrosine, tryptophan, and monoterpenes. R. rugosa's genetic regulatory mechanisms for fragrance were clarified by this analysis[124]. Different varieties of P. mume were examined for their volatile metabolites using headspace-solid phase microextraction-gas chromatography-mass spectrometer (HS-SPME-GC-MS) technology, and the primary metabolites that influence fragrance were identified. The mechanisms underlying the variations in fragrance among different varieties of P. mume have been clarified[125].
Metabolomics study on fruit quality of Rosaceae plants
-
The taste of fruits is a crucial factor that inpacts consumer preferences. Zou et al.[126] studied the potential metabolic factors contributing to the different textures of loquat and discovered that they were associated with the composition and abundance of carbohydrates, organic acids, amino acids, and phenolic compounds. This research provided new insights for enhancing the quality of loquat. The analysis of the metabolites of ten different strawberry varieties revealed that the main factors influencing the nutritional differences between these varieties are amino acid metabolism, anthocyanin synthesis, and flavonoid synthesis pathways. The theoretical foundation for determining the nutritional composition of different strawberry varieties was provided by Wang et al.[127]. Spatial metabolomics can directly measure the spatial distribution of metabolites in plant samples, providing a deeper understanding of the internal regulatory mechanisms of plant components. The distribution of soluble carbohydrates, such as fructose, glucose, and sorbitol, in apples, was analyzed using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry imaging (MALDI-TOF MSI). The study confirmed a notable increase in sucrose content in the central region of the flesh compared to the periphery[128]. By examining the changes and distribution patterns of chemical substances during the growth process of strawberries using spatial metabolomics, the study found that citric acid and sugar were uniformly distributed throughout the entire fruit at each stage of maturity, whereas anthocyanins were primarily concentrated at the edges of the fruit[129]. Many Rosaceae plants have medicinal and edible value, and metabolomics can be used to screen for active metabolites within them. A non-targeted analysis of the chemical composition of loquat roots using ultra-high-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry (UHPLC-QTOF-MS) technology showed that the extract is rich in health-promoting triterpenoids and phenolic compounds[130]. The leaf extract of Rubus idaeus L. was found to have the ability to protect fibroblast cells from ultraviolet radiation (UVB) damage, as demonstrated by liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) spectroscopy. This discovery provides theoretical support for investigating the examination of antioxidant metabolites in raspberry leaves[131]. Metabolomic studies have shown that phenolic extracts from apples induce metabolic changes in human THP-1 macrophages and contain natural compounds with anti-inflammatory properties[132].
Summary
-
Metabolomics is still in its infancy as an emerging discipline and has a lot of unanswered questions. These include improving the sensitivity of detection, developing universal detection methods, conducting unbiased high-throughput quantitative analysis, and providing comprehensive annotation of metabolomics data. Metabolomics technology has advanced from studying a few metabolites to simultaneously studying a large number of metabolites involved in a specific metabolic process. Nevertheless, obtaining a complete plant metabolome is difficult due to the incomplete databases related to Rosaceae plants and the complexity of plant metabolic processes.
-
From a biological perspective, the genome provides the basis for molecular studies, while the transcriptome reveals the current state of gene expression, reflecting mechanisms such as transcriptional regulation and post-transcriptional regulation. In comparison, proteomics directly studies the proteins in organisms, while metabolomics examines the changes in metabolites over time in biological systems following stimulation or perturbation. Metabolomics is the closest link to the phenotype and serves as a bridge connecting genes and phenotypes. However, individual omics approaches can only explain specific biological phenomena within their respective fields. Comprehensive multi-omics analysis, on the other hand, enhances our understanding of the underlying complex mechanisms of biological phenotypes. The use of multi-omics joint analysis techniques, such as transcriptome-bound metabolomics and genome-bound metabolomics, is a result of the rapid advancement of sequencing technology.
Through multi-omics analysis, researchers discovered that the sucrose signaling pathway enhances the salt-alkali stress resistance of M. halliana by inducing the expression of CYP75B1 (flavonoid 3′-monooxygenase) and T31B5-170 (auxin-responsive GH3 family). This induction leads to flavonoid formation and promotes auxin signal transduction[133]. Transcriptome and metabolomic technologies were used to analyze heat-treated loquat fruit to study the effects of heat stress on the fruit[134]. This analysis allowed for a comprehensive understanding of the complex molecular mechanisms underlying the fruit's response to heat stress. Multi-omics analysis revealed that excessive nitrogen fertilizer application in apple cultivation can also lead to a decrease in soluble sugars and flavonoid compounds in fruits, resulting in a decline in fruit quality[135]. Using QTL mapping and GWAS, a closely related gene, Pm024213, was found to help explain the regulatory mechanism of the weeping trait in P. mume. Pm024213 specifically expresses itself in the buds and branches of weeping P. mume flowers and is involved in the synthesis of auxin and lignin[136]. By integrating transcriptome and proteomic analyses, we studied key candidate genes and proteins involved in floral bud differentiation, elongation, and flowering stages in loquat. This research lays the foundation for unraveling the genetic regulatory mechanisms of loquat flower development[137]. The transcriptome sequencing and methylation analysis of petals at different stages of the 'Fenhong Zhusha' variety of P. mume revealed extensive involvement of DNA methylation in the biosynthesis of floral scent. This implies that DNA methylation may be essential for controlling the biosynthesis of floral fragrance[138]. A total of 191 candidate genes related to P. mume blooming period were identified through a combination of GWAS and transcriptome sequencing[139]. By conducting a combined analysis of transcriptome and DNA methylation profiles in mature fruits of apple cultivars 'Honeycrisp' and 'Qinguan', two candidate genes, tonoplast sugar transporter (TST) and MdMa11 (encoding a P3A-ATPase), were identified that may be associated with the soluble sugar content and acidity of the fruits[140]. Transcriptome sequencing was performed on the peel of two different-colored varieties of Cerasus humilis, and combined with anthocyanin metabolomic analysis, candidate genes associated with anthocyanin expression were identified[141]. In the investigation of the nutritional and medicinal properties of the 'Big Five-pointed Star' loquat, scientists have successfully assembled the genome of the loquat. Comparative genomics analysis revealed distinctive genetic and molecular mechanisms of genome recombination and repair in the 'Big Five-pointed Star' loquat. Metabolomic analysis of the roots, leaves, and flowers of the loquat plant led to the discovery of numerous active components and their associated genes involved in biosynthesis[142].
Summary
-
Many important traits in Rosaceae plants are determined by complex regulatory networks constructed through the interaction of multiple genes, protein interactions, and the interplay of metabolites. However, relying on a single omics technology often lacks comprehensiveness. With the emergence of the multi-omics era, it has become essential to integrate genomics, transcriptomics, proteomics, metabolomics, and other omics levels in the investigation of complex traits in Rosaceae plants. By integrating information from different levels, such as genes, proteins, and metabolites, comprehensive analyses can be conducted. This integration will facilitate the construction of a comprehensive genetic regulatory network for complex traits, as well as the interpretation of the growth and development processes at different levels that underlie the complex traits of Rosaceae plants.
-
Considerable progress has been achieved in employing omics techniques to analyze the flower color, fruit quality, and regulatory mechanisms of Rosaceae plants in response to abiotic stress (Table 2). Over time, omics technology is constantly advancing (Fig. 3). Emerging technologies have made it convenient to study the life activities of Rosaceae plants, and the research on Rosaceae plants is no longer limited to traditional omics technology. Researchers have initiated investigations into the transcripts and metabolites of Rosaceae plant tissues at the single-cell level aiming to systematically study the gene expression and metabolism in specific tissues and cell types at particular developmental stages. In recent years, there has been rapid development in the use of scRNA-seq and spatial multi-omics techniques to study cellular heterogeneity within plant tissues and analyze plant life activities.
Table 2. The main findings of omics analysis of Rosaceae plants.
Omics analysis Application level Species Omics analysis Application level Species Comparative genomics Species evolution Rubus corchorifolius GWAS Fusarium wil Fragaria × ananassa Prunus davidiana,
Prunus mira,
Prunus kansuensis,
Prunus ferganensisFlesh browning Malus domestica Prunus pusilliflora Salt and drought stress Rosa chinensis Transcriptome Salt stress Prunus mume Cold stress Prunus mume Heat stress Rosa chinensis Flower color Prunus mume Drought stress Rosa chinensis Prunus persica f. versicolor Botrytis. cinerea Rosa hybrida Prunus campanulata 'Plean' Fragaria vesca Fruit morphology Prunus persica, Prunus salicina, Prunus armeniaca, Prunus mira, Prunus davidiana Cold stress Rosa hybrida Fruit aroma Malus domestica Eriobotrya japonica Prunus persica Rosa xanthina f. spontanea Fruit flavor Pyrus spp Prunus pyrifolia Metabolomics Drought and salt stress Fragaria × ananassa Flower color Pink-flowered strawberry Cold stress Prunus mume Malus halliana Malus domestica Rosa chinensis
Rosa rugosaNitrogen and phosphorus absorption Malus domestica Flower color Miniature rose Fruit color Fragaria × ananassa Rosa hybrida Malus domestica Floral fragrance Rosa rugosa Prunus avium Prunus mume Crataegus pinnatifida Fruit flavor Eriobotrya japonica Fruit flavor Malus domestica Fragaria × ananassa Proteomics Abiotic stress Rosa hybrida Malus domestica Cold stress Fragaria × ananassa Fruit color Fragaria × ananassa Rosa beggeriana Active metabolites Rubus idaeus Salt stress Prunus cerasus Eriobotrya japonica Ring rot Malus domestica Malus domestica Flower color Prunus persica Multi-omics Salt stress Malus halliana Rosa hybrida Heat stress Eriobotrya japonica Prunus persica f. versicolor Nitrogen absorption Malus domestica Prunus mume Weeping trait Prunus mume Fruit development Malus halliana Flower development Eriobotrya japonica Fruit storage Prunus persica Floral fragrance Prunus mume Fruit aroma Prunus persica Fruit flavor Malus domestica Fruit color Cerasus humilis Active metabolites Eriobotrya japonica 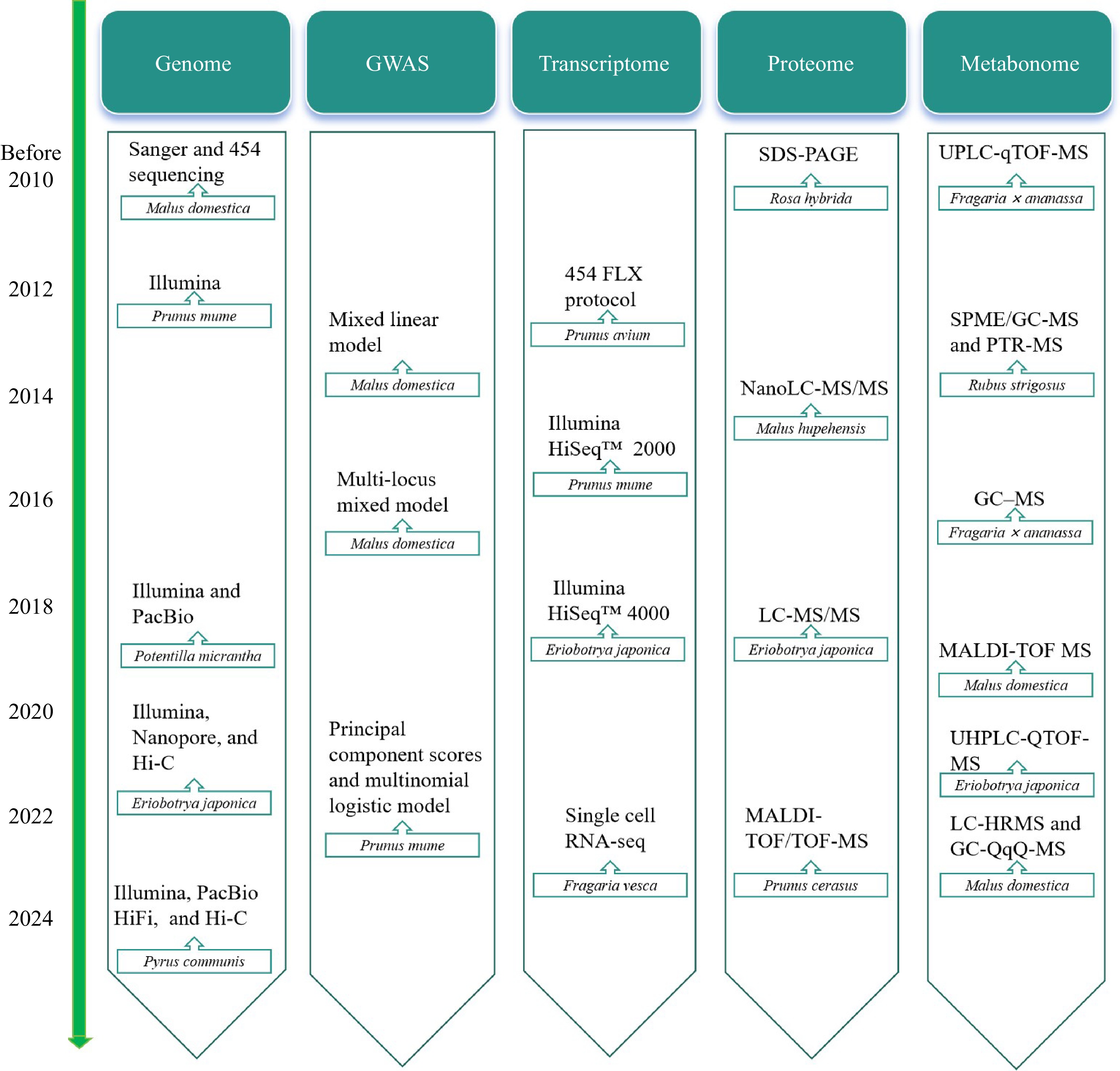
Figure 3.
Timeline for omics research on Rosaceae plants.
Traditional molecular biology methods have limited capabilities in revealing cellular heterogeneity within Rosaceae plant tissues. ScRNA-seq has introduced a new era of cell-based transcriptome analysis, enabling the expedited identification of rare cell types and the discovery of new cell types while characterizing multiple different cell types and states. It provides a more accurate and comprehensive understanding of their roles in life processes. ScRNA-seq technology has been widely used in medical research and is gradually being applied in animal and plant research. A growing number of researchers in plant science have been leveraging scRNA-seq and spatial multi-omics techniques to investigate tissue heterogeneity, development, stress resistance, and breeding, resulting in the resolution of numerous significant scientific challenges in the plant research field. However, the presence of cells presents a primary challenge for the application of spatial multi-omics in plants due to the difficulty in preparing protoplasts. As a result, the use of scRNA-seq and spatial multi-omics techniques in Rosaceae plants is relatively limited. In Rosaceae plants, researchers utilized scRNA-seq to assemble the genomes of two diploid apricot trees and create a gene expression map of strawberries at the single-cell level[92,143]. They also employed spatial metabolomics to investigate the alterations and distribution of chemical compounds during the growth stages of apples and strawberries[128,129].
Future advancements in spatial multi-omics technologies will lead to new research breakthroughs and deepen our understanding of the regulatory mechanisms of various cell and tissue types. Further exploration is required in various aspects of spatial multi-omics technology, including mass spectrometry imaging (MSI) resolution, data analysis software, and instruments. With the advancement of technology, spatial multi-omics will aid in interpreting the growth and development processes of complex traits in Rosaceae plants at various levels and construct a comprehensive genetic regulatory network for complex traits, providing theoretical support for efficient molecular breeding.
-
The authors confirm contribution to the paper as follows: study visualization: Lv W; writing – original draft: Lv W, Miao D, Miao R, Fan D, Meng J, Liu X, Cheng T, Zhang Q; writing – review & editing and formal analysis: Sun L. All authors reviewed the results and approved the final version of the manuscript.
-
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
This study was supported by the Fundamental Research Funds for the Central Universities (QNTD202306); Forestry and Grassland Science and Technology Innovation Youth Top Talent Project of China (No. 2020132608) and Beijing High-Precision Discipline Project, Discipline of Ecological Environment of Urban and Rural Human Settlements.
-
The authors declare that they have no conflict of interest. Lidan Sun is the Editorial Board member of Ornamental Plant Research who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and the research groups.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Lv W, Miao D, Miao R, Fan D, Meng J, et al. 2024. Advances in the omics research of Rosaceae. Ornamental Plant Research 4: e013 doi: 10.48130/opr-0024-0011 

Advances in the omics research of Rosaceae
- Received: 23 November 2023
- Revised: 14 March 2024
- Accepted: 21 March 2024
- Published online: 06 May 2024
Abstract: Rosaceae plants are widely distributed worldwide. These plants, including well-known ornamental plants and those producing temperate fruits, have significant economic value. The utilization of omics technologies has significantly advanced the research progress on Rosaceae plants in various areas, including germplasm resources, molecular breeding, and evolutionary domestication. Here, an overview of the applications of omics technologies in Rosaceae is provided. It includes whole-genome sequencing and assembly of various Rosaceae plants, as well as the utilization of genome-wide association studies, transcriptome, proteomics, metabolomics, and comparative genomics. These techniques are used to understand the genetic regulatory mechanisms underlying important traits in Rosaceae plants, such as flower color, fragrance, stress tolerance, and fruit quality. We outline the prospects of genomics research in Rosaceae plants, aiming to establish a foundation for comprehending the genetic mechanisms of molecular breeding and significant ornamental traits. This research could potentially provide theoretical support for rapidly cultivating new germplasm and varieties.
-
Key words:
- Rosaceae /
- Genomics /
- Genome-wide association study /
- Transcriptome /
- Proteomics /
- Metabolomics /
- Comparative genomics