









-
Hemerocallis middendorffii (H. middendorffii) belongs to Hemerocallis of Aphoriaceae, which is an excellent open-field perennial flower with high economic value[1,2]. In recent years, with the promotion of eco-friendly and economical landscaping concepts, perennial flowers have gained widespread recognition in garden landscaping, and there has been an increasing demand for new cultivars of Hemerocallis. Therefore, breeding work for Hemerocallis has broad application prospects and high economic value. Currently, traditional field hybrid breeding is the main mode of obtaining excellent new cultivars of Hemerocallis. However, this method is time-consuming and has a long breeding cycle[3]. The use of transgenic technology can overcome the shortcomings of traditional breeding and has significant implications for improving Hemerocallis cultivars[4]. In monocotyledonous plants, only some undifferentiated tissues such as immature embryos can be induced to form calli[5]. H. middendorffii is a typical monocotyledonous plant, and there existed some problems of callus induction difficulty and low proliferation efficiency in genetic transformation in our previous study. It is an important trend to explore and improve the differentiation and transformation efficiency of H. middendorffii to speed up the breeding of new cultivars.
Remarkable regeneration capacity is an important mean for plants to survive in complex environments. Plant regeneration can be divided into three mechanisms, tissue repair, de novo organogenesis, and somatic embryogenesis[6]. De novo organogenesis refers to the process of regenerating adventitious roots or shoots from detached or wounded organs[7,8]. Shoot regeneration is an essential process involving massive cell fate transition in callus cells and spatial reorganization of cell identities[9]. Experimental evidence has shown that callus proliferation is required for successful shoot outgrowth. Shoot regeneration is a two-step process: Firstly, callus formation was promoted by culturing on auxin-rich callus-inducing medium (CIM), and then the callus was transferred to cytokinin-rich shoot induction medium (SIM) to produce adventitious shoots (AS)[10].
It is well known that plant hormones are one of the key factors affecting shoot regeneration, and the balance and interaction between auxin and cytokinin (CK) determine cell fate transitions during this process[11]. Auxin response factors (ARFs), the key mediators of auxin signaling, are mainly involved in plant regeneration[12,13]. IAA-related genes are also involved in AS regeneration, such as AUX1, SAUR, and GH3[14−17]. YUC-mediated auxin biosynthesis is required for efficient shoot regeneration[11]. CK is another major hormone affecting de novo shoot organogenesis. A high CK concentration can make cells lose the characteristics of roots, destroy root primordia, and promote the regeneration of plant shoots[18]. Consistently, many genes related to CK biosynthesis and signal pathways are considered to be key drivers of shoot regeneration. The type A-ARR and B-ARR genes play negative and positive regulatory roles in the CK signaling pathway, respectively[19]. Overexpression of type A-ARR (ARR7 and ARR15) results in the suppression of shoot regeneration, while high-order mutants of A-ARRs display enhanced shoot regeneration[20]. Additionally, other phytohormones, including abscisic acid (ABA), ethylene (ETH), brassinolide (BR), jasmonic acid (JA), and salicylic acid (SA), which also regulate the process of shoot regeneration[21−23]. Previous studies mainly focused on the effects of different hormones and their concentrations on callus induction, proliferation, and adventitious shoots of Hemerocallis[24−26]. However, there is no research to reveal the mechanism of plant hormones and their pathways on the regeneration of H. middendorffii at the molecular level.
Many studies have explored the molecular mechanisms related to shoot regeneration and identified several regeneration-promoting key genes, thereby significantly boosting the regeneration efficiency and genetic transformation of plants. For instance, AINTEGUMENTA-LIKE 5 (AIL5) overexpression increases adventitious shoot regeneration efficiency through activating the expression of shoot development-related genes[13]. Overexpression of the Baby Boom (BBM) gene can enhance bud regeneration ability and genetic transformation efficiency[22,27−29]. It has been documented that lateral organ boundary regulator gene CUP-SHAPED COTYLEDON CUC2 (CUC2) plays a major role in de novo shoot regeneration[30]. Ectopic overexpression of CUC1 or CUC2 can enhance de novo shoot formation and the corresponding double mutant cuc1;cuc2 displays reduced shoot regeneration[31,32].
The WUSCHEL-related homeobox (WOX) gene family is one kind of unique transcription factor in plants, many studies have showed that the WOX gene family members have crucial functions in plant regeneration[33]. Overexpression of the TaWOX5 gene dramatically increases transformation efficiency with less genotype dependency in wheat (Triticum aestivum L.)[34]. In Arabidopsis (Arabidopsis thaliana) and rice (Oryza sativa L.), WOX11 has been identified as the key gene involved in promotion of adventitious rooting. Arabidopsis WOX13 gene facilitates efficient callus formation and organ reconnection by modifying cell wall properties[35]. AtWOX14 can enhance the regeneration of adventitious shoots by affecting the pluripotency of callus cells[36], and its putative rice ortholog OsWOX13 significantly promote shoot regeneration capacity[37]. To date, the function of WOX8 gene has only been reported in a few plant species, including Arabidopsis, Picea abies, Ornithogalum thyrsoides, and Triticum aestivum[38−41]. In Arabidopsis, STIMPY-LIKE (STPL)/WOX8 positively regulates early embryonic growth[38]. In addition, it has been shown that WOX8 and its close homolog WOX9 regulate the development of the basal embryo lineage and also of the apical embryo lineage via noncell autonomous activation of WOX2[42]. PaWOX2 and PaWOX8/9 of Picea abies are expressed at high levels in the early growth stages of zygotic and somatic embryos[39]. TaWOX8 genes could promote immature callus proliferation in Triticum aestivum embryos[41]. Although the roles of the WOX8 gene have been studied, the molecular mechanism of the WOX8 gene and cooperative network with other key genes in regulating callus proliferation and shoot regeneration are largely unknown.
In the present study, the HmWOX8 gene from H. middendorffii was separated using RACE. Through overexpression in Arabidopsis, rice and gene silencing in H. middendorffii, the positive effects of HmWOX8 on callus proliferation and shoot regeneration were identified. In addition, to explore the key candidate genes associated with callus proliferation and shoot regeneration, transcriptome analyses was performed between HmWOX8-overexpression lines (HmWOX8-OE) and wild-type (WT) in rice. Many genes related to hormone signaling pathways and shoot development were significantly and differentially expressed in HmWOX8-OE, and WT. Yeast two-hybrid assays and Bimolecular fluorescence complementation assay confirmed that HmWOX8 could interact with HmCUC2 to promote AS formation. The present findings clarify the positive roles of the WOX8 gene in callus proliferation and shoot regeneration, and provide an important theoretical foundation for further perfecting the TF transcriptional regulation of plant regeneration.
-
H. middendorffii was field grown at the Horticulture Experimental Station of Northeast Agricultural University (Harbin, China; 126.7° E, 45.7° N). The growth conditions in the greenhouse were as follows: relative humidity of 65%−75%, temperature of 20−28 °C, and 12 h of illumination (700 mmol·m−2·s−1) per day. Arabidopsis ecotype Columbia (Col) and transgenic Arabidopsis plants were grown in a climate incubator (4000 lx) at 24 °C under long-day conditions (16 h-light/8 h-dark). WT rice plants (Oryza sativa L. ssp. japonica cv. Nipponbare) and transgenic rice were cultured in climate chamber under a 16-h light (28 °C) : 8-h dark (26 °C) photoperiod.
Gene cloning and vector construction
-
RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) from H. middendorffii leaves, and reverse transcription was performed to obtain cDNA. The full-length cDNA of HmWOX8 were cloned by applying rapid amplification of cDNA ends (RACE) technology. The primers for 5'- and 3'-RACE cDNA were designed based on the transcriptome of H. middendorfii. Following the manufacturer's instructions for the SMARTer RACE 5'/3' Kit (Clontech, Shiga, Japan), the full-length cDNAs were obtained (GenBank ON303271.2). The target fragment was ligated to the pMD19-T cloning vector (Takara Bio, Japan) and subjected to blue-white spot screening. The positive bacteria were sent to Kumei Biotech (Jilin, China) for sequencing. For construction of the overexpression plasmids HmWOX8. After XbaI enzyme digestion, the full-length coding sequences (without a stop codon) were inserted into pCAMBIA1300-35S::GFP vector using ClonExpress II One Step Cloning Kit (Vazyme Biotech Co., Ltd., Nanjing, China).
Sequence alignment and phylogenetic analysis
-
Sequence alignments were performed using the clustalw (
www.genome.jp/tools-bin/clustalw ), and the figure of multiple sequence alignment was plotted by SnapGene software. The conserved domain of HmWOX8 and WOX8 proteins from other plant species were identified using MEME (https://meme-suite.org/meme/tools/meme ). A phylogenetic analysis was constructed using the Neighbor-Joining method with 1,000 bootstrap replicates in MEGA 7 software.Subcellular localization of HmWOX8
-
The empty plasmid without HmWOX8 (35S::GFP) was used as a control. The empty plasmid and pCAMBIA1300-35S::HmWOX8-GFP (pC1300-HmWOX8-GFP) fusion plasmid were transformed into rice protoplasts with nuclear marker, respectively. After 48 h of dark culture, the expression of HmWOX8 in rice protoplasts was observed under a laser copolymerization microscope[43].
Generation of transgenic Arabidopsis plants and callus formation assay
-
The pC1300-HmWOX8-GFP fusion plasmid was transformed into Agrobacterium GV3101 competent cells using a freeze-thaw technique. The wild-type and wox8 mutant of Arabidopsis thaliana were used for transformation by the floral dipping method[44]. The resistant seedlings were screened using 50 mg·L−1 kanamycin-labeled and identified by PCR of HmWOX8 specific primers. Total RNA of from T1 leaves was extracted using TRIzol reagent and reverse-transcribed with the PrimeScript RT reagent kit with gDNA Eraser (TaKaRa). qPCR was performed with the SYBR Premix Ex Taq kit (TaKaRa) using the QuantStudio 1 (Thermo Fisher Scientific Inc, USA). The Arabidopsis thaliana Actin1 gene was used as an internal control to normalize the different samples. Three biological and three technical replicates were performed for each sample. The primers used here are listed in Supplemental Table S1. The transgenic plants were transplanted into sterilized soil until harvest of T3 generation seeds.
The wild A. thaliana and T3-generation HmWOX8 gene overexpressed and recovered A. thaliana stems were used as explants. After cutting them, they were inoculated on Murashige and Skoog (MS) medium containing 0.50 mg·L−1 6-BA + 0.10 mg·L−1 NAA + 30 g·L−1 sucrose to induce callus. Callus images were taken at 20 d after cutting and callus was photographed using a Stereotype microscope (1.25×). A ruler was used to measure the size of the callus and calculate their area. Semi-thin sections were observed using the digital slice scanning system (Thermo Fisher Scientific Inc, USA).
Generation of transgenic rice and callus induction
-
PROKII-ALCR-HmWOX8-GUS recombinant plasmid was inserted into WT rice plants. Transgenic rice seedlings were cultivated, after which total RNA was isolated and cDNA was synthesized. Transgenic rice plants were verified by RT-qPCR. GUS staining was used to verify the establishment of ethanol-induced startup subsystem in rice.
Rice seeds of WT and HmWOX8-OE were sterilized with 75% ethanol for 1 min and 10% sodium hypochlorite for 20 min in sequence, followed by five rinses with sterile distilled water. Then the seeds were placed on MS medium with 0.3 g·L−1 proline + 0.6 g·L−1 hydrolyzed casein + 2 mg·L−1 2,4-D + 30 g·L−1 sucrose + 3 g·L−1 phytagel (pH = 5.9). The growth conditions were 26 °C dark (24 h) with 60%−70% relative humidity. On the second day, rice seeds were sprayed with 2% ethanol. Callus were observed, sampled, and photographed 5, 15, and 25 d after callus induction. Materials at 25 d were photographed with a scanning electron microscope (S3400) for morphological observation. The proliferation-induced callus of WT and HmWOX8-OE were cut into 0.5 cm3 pieces and placed in shoot regeneration medium (N6 + 2 mg·L−1 6-BA + 0.2 mg·L−1 NAA + 30 g·L−1 sucrose + 3 g·L−1 phytagel). After 15 d of culture, the shoot regeneration was observed.
RNA extraction and transcriptome analysis
-
Total RNA was extracted using TRIzol reagent from the 15 d of WT and HmWOX8-OE lines rice in shoot culture medium. The quality and purity of the RNA were checked using a NanoDrop ND-8000 spectrophotometer (Thermo Fisher Scientific, Pittsburgh, PA, USA), and RNA sequencing was performed in Meiji (Shanghai, China). A total of six libraries (three biological replicates) were sequenced on the Illumina Novaseq 6000 platform. Raw reads were cleaned and filtered. Gene expression levels were then estimated with FPKM[45]. DESeq was used to detect DEGs between the OE and WT lines with the following criteria: p-value < 0.05 and |log2(FoldChange)| > 1. All identified DEGs were mapped to gene ontology (GO) and Tokyo Encyclopedia of Genes and Genomes (KEGG) databases. The significantly enriched biochemical pathways were obtained using KOBAS with corrected p-value < 0.05.
For the expression validation, 14 DEGs were randomly selected from RNA-seq data to analyze the relative gene expression levels. Total RNA from the callus was isolated according to the manufacturer's protocol. The qRT-PCR was performed according to a previously described method. All the experiments were repeated three times. The relative expression of genes was calculated using the 2−ΔΔCᴛ method[46]. The primers used for qRT-PCR are shown in Supplemental Table S2.
Virus-induced gene silencing (VIGS) in H. middendorffii
-
The pTRV2-HmWOX8 vector was constructed using a 205 bp fragment of HmWOX8 with BamH I restriction sites. After BamH I enzyme digestion, the 205 bp fragment of HmWOX8 was inserted into the pTRV2 vector using ClonExpress II One Step Cloning Kit, and then transformed into E. coli DH5α. pTRV2-HmWOX8 was introduced into the Agrobacterium EHA105 competent state using the freeze–thaw method. Single colonies were cultured in the corresponding liquid LB media until the cells reached an OD600 of 1.0−1.3. The Agrobacterium cells were centrifuged at 6,000 rpm for 6 min. The bacterial fluid was collected and suspended in 4.74 g·L−1 of MS, 400 μmol·L−1 of acetosyringone, 10 mmol·L−1 of MgCl2, 10 mmol·L−1 MES, 400 mg·L−1 cysteine, and 5 ml·L−1 Twain 20, (pH = 5.6). The OD600 was adjusted to 1.2. pTRV1 and mixed with pTRV2-HmWOX8 bacterial liquid 1:1, and the two bacterial liquids were fully mixed at room temperature. Callus induced by proliferation were selected (differentiated and etiolated tissues were removed), and cut into small pieces of 0.3 cm3. After 7 d, infection liquid was injected into the callus with a 1 mL sterile syringe with a needle. Once every 7 d, this was repeated twice. The infected callus and wild-type callus were treated in the dark for 24 h, and then grown at 22 °C in a greenhouse with a 16 h/8 h light/dark cycle. AS regeneration ability was quantified by estimating the AS increment coefficient, number of ASs per explant, and AS regenerative efficiency. The results of three experiments were analyzed, with each experiment conducted using 32 explants. Quantification parameters were as follows:
AS increment coefficient = number of ASs/number of explants; number of ASs per explant = number of ASs/number of explants that regenerated ASs; and AS regenerative efficiency (%) = (number of explants that regenerated ASs/number of explants) × 100% (for the calculation, only shoots longer than 1 cm were considered).
Yeast two-hybrid (Y2H) assay and Bimolecular fluorescence complementation (BiFC) assay
-
The full-length coding region of HmWOX8 was cloned into the pGADT7 vector as prey, and HmCUC2 was inserted into the pGBKT7 vector as bait, respectively. These vectors were transformed into the Y2H gold strain. First, transformants were inoculated on synthetic defined plates SD/-Leu/-Trp (DDO) using the daubing method and incubated at 28 °C for 3−5 d. When colonies on the plates grew to 2−3 mm, the co-transformed colonies were transferred to SD/-Leu/-Trp/-His/-Ade/X-a-gal plates and incubated at 28 °C for 3−5 d to observe their growth. The interaction between two fusion proteins were identified by detecting the blue color generated by yeast cells on plates.
For the BiFC assay, the full-length coding regions of HmWOX8 without stop codons was inserted into pCAMBIA1300-35S-N-GFPN vector, HmCUC2 without stop codons was inserted into pCAMBIA1300-35S-C-GFPC vector, respectively. For transient expression, Agrobacterium tumefaciens strain GV3101 carrying those constructs was infiltrated with p19 (1:1:1 ratio; OD600 = 0.5) into the abaxial side of leaves from tobacco. After 3 d of agroinfiltration, fluorescent signals were analyzed with a confocal microscope.
Statistical analyses
-
Data were analyzed using one-way analyses of variance with SPSS v10.0 software (SPSS, Inc., Chicago, IL, USA). The mean values were compared via the least significant difference test at the 0.05 probability level. The GraphPad Prism v8 (Graphpad, USA) was used for plotting.
-
The open reading frame of HmWOX8 consisted of 777 bp and was predicted to encode a protein of 258 amino acids (GenBank: ON303271.2). The theoretical isoelectric point (pI) of HmWOX8 was 4.80. The calculated molecular weight of HmWOX8 was 29.25 kD. Sequence alignment analysis showed HmWOX8 and WOX8 proteins from other plant species contained a homeodomain (HD) conserved domain (Fig. 1a, b). The neighbor-joining method (NJ) was used for multiple sequence alignment analysis of the amino acid sequence of HmWOX8 to build a phylogenetic tree. Results showed that the HmWOX8 was highly homologous with AoWOX8 (Fig. 1c).
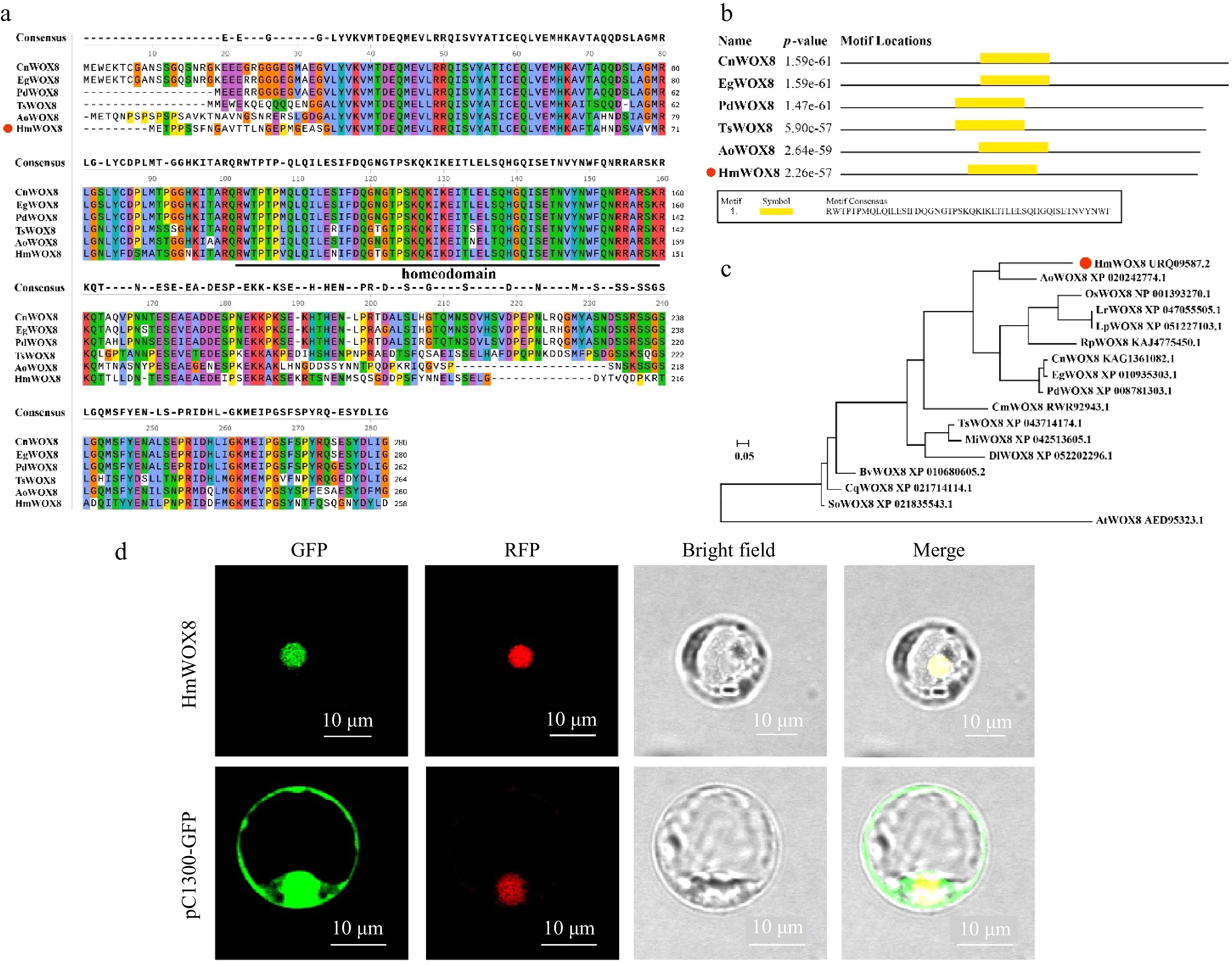
Figure 1.
Multiple sequence alignment, phylogenetic tree analysis and subcellular localization. (a) Multiple sequence alignment. (b) Conserved domain of HmWOX8 and five orthologous WOX8 proteins. The orthologous proteins were Cocos nucifera (CnWOX8), Elaeis guineensis (EgWOX8), Phoenix dactylifera (PdWOX8), Telopea speciosissima (TsWOX8), Asparagus officinalis (AoWOX8). (c) The Neighbor-Joining phylogenetic tree analysis of the amino acid sequence alignment of the HmWOX8 protein and WOX8 proteins from other plant species. The N-J phylogenetic tree was constructed using MEGA11. AoWOX8 (Asparagus officinalis, XP_0202427), OsWOX8 (Oryza sativa, NP_001393270.1), LrWOX8 (Lolium rigidum, XP_047055505.1), LpWOX8 (Lolium perenne, XP_051227103.1), RpWOX8 (Rhynchospora pubera, KAJ4775450.1), CnWOX8 (Cocos nucifera, KAG1361082.1), EgWOX8 (Elaeis guineensis, XP_010935303.1), PdWOX8 (Phoenix dactylifera, XP_008781303.1), CmWOX8 (Cinnamomum micranthum f. kanehirae, RWR92943.1), TsWOX8 (Telopea speciosissima, XP_043714174.1), MiWOX8 (Macadamia integrifolia, XP_042513605.1), DlWOX8 (Diospyros lotus, XP_052202296.1), BvWOX8 (Beta vulgaris subsp. Vulgaris, XP_010680605.2), CqWOX8 (Chenopodium quinoa, XP_021714114.1), SoWOX8 (Spinacia oleracea, XP_021835543.1), AtWOX8 (Arabidopsis thaliana, AED95323.1). (d) Subcellular localization of HmWOX8 protein in rice protoplasts. Scale bars = 10 μm.
To determine the subcellular localization of HmWOX8, the pCAMBIA1300-35S::HmWOX8-GFP (pC1300-HmWOX8-GFP) fusion plasmid were introduced into rice protoplasts with the nuclear marker. Under a laser confocal microscope, the fluorescence of HmWOX8 coincided with the fluorescence of a nuclear marker located in the nucleus (Fig. 1d).
Overexpression of HmWOX8 improves callus proliferation efficiency in Arabidopsis
-
To confirm the functions of the HmWOX8 gene in callus proliferation, expression vector pCAMBIA1300-HmWOX8-GFP was constructed and transformed into wild-type Arabidopsis and wox8 mutant. Six independent T3 generation overexpressed lines and five independent T3 generation recovery lines of the HmWOX8 gene were examined for kanamycin resistance, and the result was subsequently confirmed with PCR identification (Supplemental Fig. S1a, b). These lines were screened using the selection method described above until T3 generation overexpressed and recovery seeds were obtained. The expression level of HmWOX8 significantly increased in the transformed lines according to qRT-PCR assays (Fig. 2a, b). While WT explants form well-developed calli from the petiole cut end, HmWOX8 overexpression explants generate compact calli with a significant increase in the projection area. Phenotype analysis revealed that callus areas were bigger in HmWOX8 overexpression lines compared to the wild-type (Fig. 2c, d, i). Relative to WT, the areas of callus were increased by restoring HmWOX8 to wox8 mutant (Fig. 2c, e, i). To further identify the developmental function of HmWOX8 in callus proliferation, whether the reduction of callus areas in WT was due to the reduction in cell number or cell size was next asked. Therefore, the semi-thin sections at the median plane of the calli were performed. All the cells contained in the sections were calculated manually, found that WT calli have 22 large cells (n = 3, big cells > 2,500 μm2) (Fig. 2f), whereas HmWOX8 overexpression calli have 128 large cells (n = 3, big cells > 2,500 μm2) for each section on average (Fig. 2g), implicating that cell proliferation was increased in the HmWOX8 overexpression calli. The wox8 mutant calli have 17 big cells (n = 3, big cells > 2,500 μm2). Although there are only 17 large cells in the wox8 mutant callus (the number of large cells is less than that of WT, the callus area was significantly larger than that of WT (Fig. 2i). This is because there are more intermediate cells in the callus of wox8 mutation than WT (400 μm2 < intermediate cells < 2,500 μm2) (Fig. 2g).
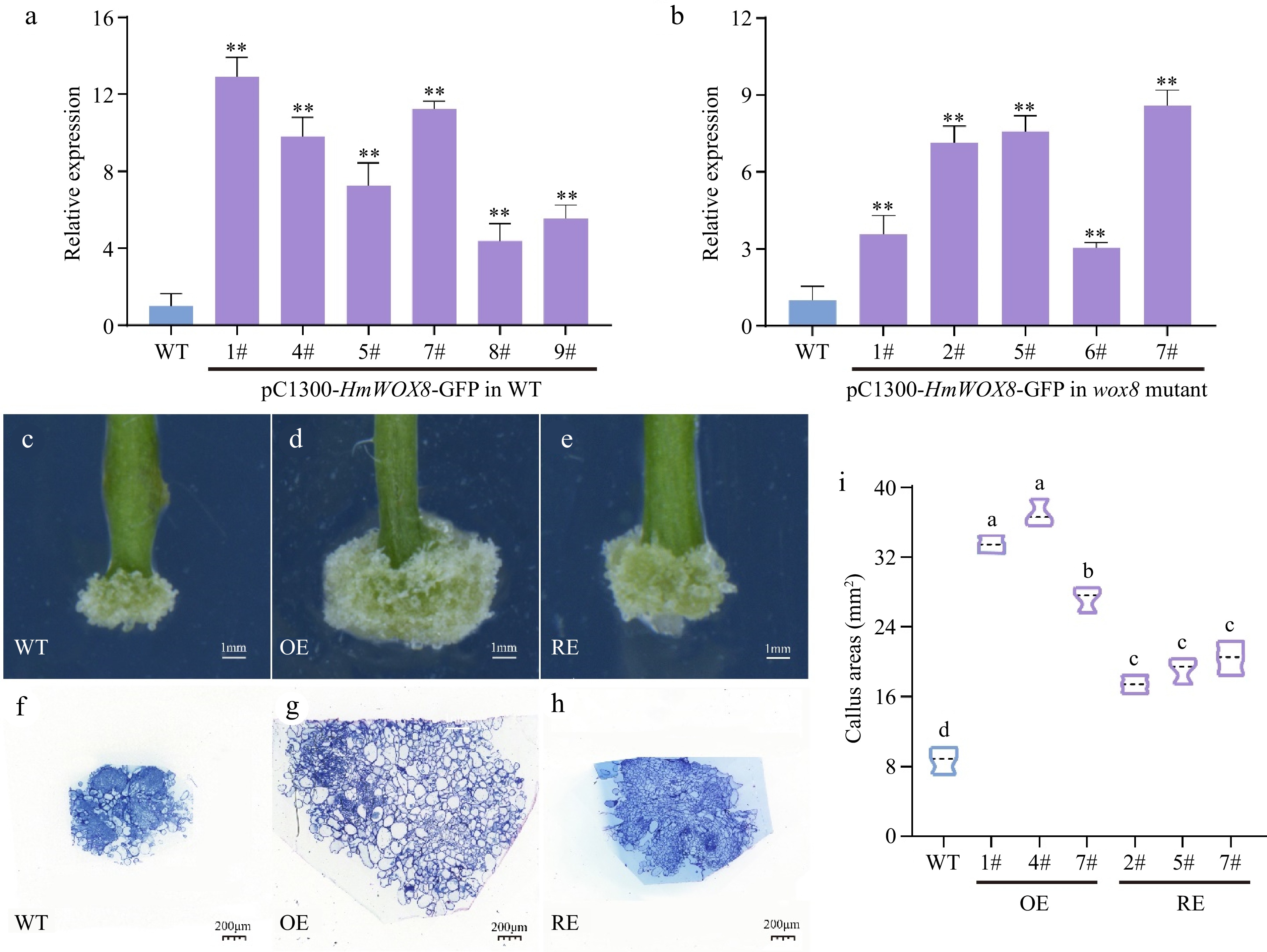
Figure 2.
Functional analysis of HmWOX8 transgenic A. thaliana. (a), (b) Identification of HmWOX8 gene expression in transformed Arabidopsis positive plants and wox8 mutant positive plants. (c)−(e) Callus phenotype of wild-type, HmWOX8-overexpression and recovery T3 transgenic plants. Scale bars = 1 mm. (f)−(h) Semi-thin cross sections of wild-type, HmWOX8-overexpression and recovery T3 transgenic plants, Scale bars = 200 μm. (i) Comparison of Wild-type (WT), HmWOX8 overexpression (OE) and recovery (RE) of callus areas in Arabidopsis. ** indicate significance at p < 0.01. The different lowercase letters indicate significant differences (p < 0.05).
Effect of HmWOX8 on the callus proliferation efficiency of rice
-
The exact role and underlying mechanism of HmWOX8 in plant regeneration are still unclear. To verify whether the overexpression of HmWOX8 (HmWOX8-OE) affects callus proliferation, PROKII-ALCR-HmWOX8-GUS recombinant plasmid was inserted into wild-type rice. The GUS staining method was used to verify the establishment of ethanol-induced startup subsystem in transgenic rice (Supplemental Fig. S2a−d). The expression level of the HmWOX8 gene in overexpressing transgenic callus was significantly higher than that in wild-type (WT) callus (Supplemental Fig. S2e). Considering the same genetic background and cultivation environment, all phenotypic differences between HmWOX8-OE and WT callus were due to the overexpression of HmWOX8. The callus-associated phenotypes of HmWOX8-OE transgenic and WT callus were observed. The callus phenotype after continuous culture for 5, 15, 25 d are shown in Fig. 3a, transgenic callus areas were significantly higher than that of WT callus areas. Compared with WT, the callus area of HmWOX8-OE has exhibited an increase by 2.52-fold at early stages of callus formation (5d) (Fig. 3b). After continuous culture for 15 d, overexpressing HmWOX8 callus areas reached 39.62 mm2, which was 1.83-fold that of WT (Fig. 3b). Consistent with 5 and 15 d, the 25th day of callus induction also showed an increase of callus density in overexpressing HmWOX8 lines, compared to that in the WT (1.71-fold) (Fig. 3b). Collectively, these results indicated that the overexpression of HmWOX8 could significantly improve the proliferation efficiency of callus. The HmWOX8-OE and WT rice calli cultured for 25 d were observed by scanning electron microscope. The results showed that the surface and length-cutting section of WT were loose and irregular, while HmWOX8-OE have a compact and regular calli with relatively large volume (Fig. 3c). In addition, cell clusters of HmWOX8-OE calli had formed protrusions (Fig. 3c).
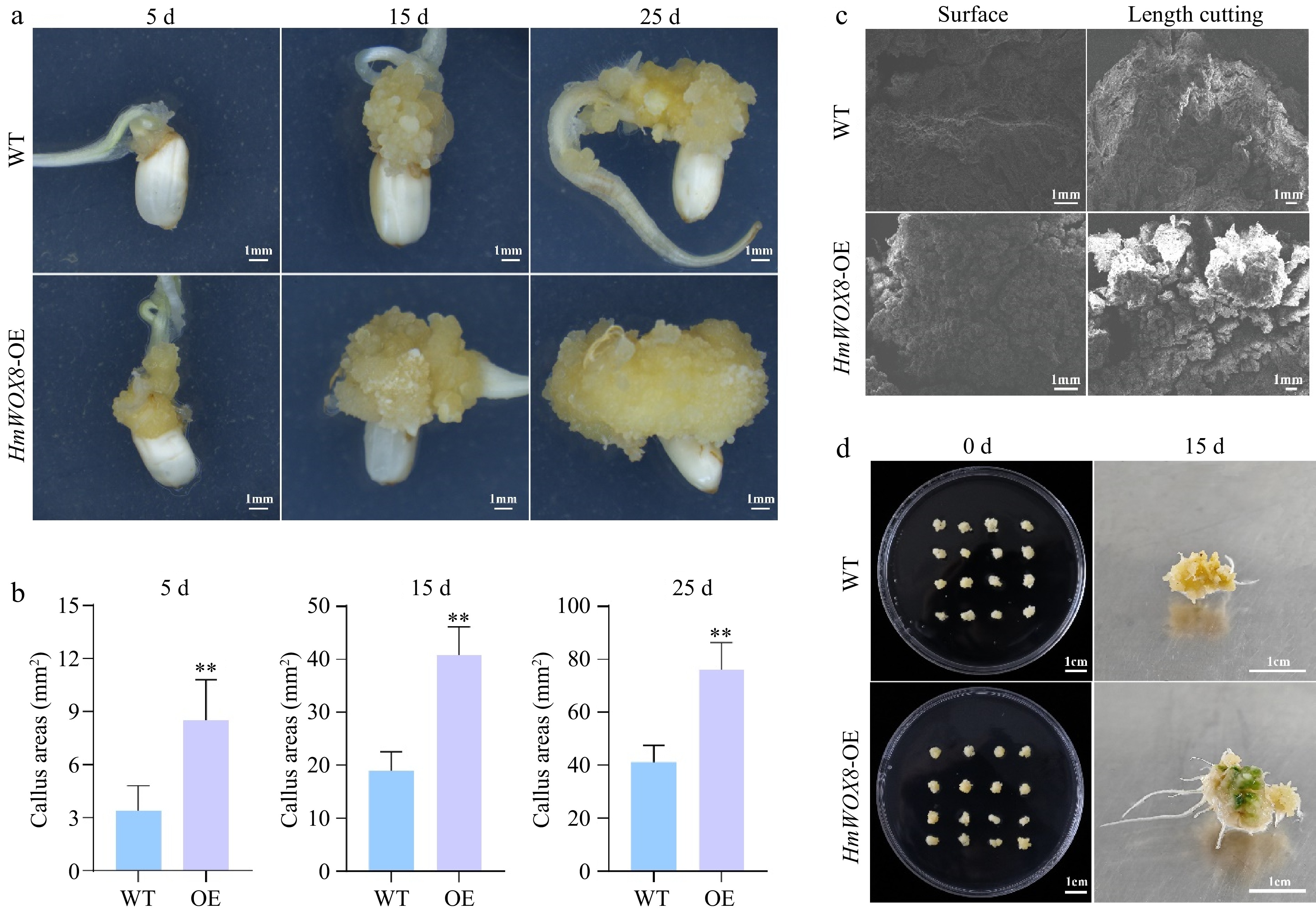
Figure 3.
Effects of HmWOX8 overexpression on callus proliferation and shoot regeneration in rice. (a) The callus phenotype of WT and HmWOX8-OE after continuous culture for 5 d, 15 d, and 25 d. Scale bars = 1 cm. (b) Comparison of callus area between WT and HmWOX8-OE after continuous culture for 5 d, 15 d, and 25 d. (c) The scanning electron microscope of WT and HmWOX8-OE. Scale bars = 1 mm. (d) The shoot phenotype of WT and HmWOX8-OE after 15 d in regeneration medium. Scale bars = 1 cm. ** indicate significance at p < 0.01.
To test the potential role of HmWOX8 in adventitious shoot regeneration, the HmWOX8-OE and WT rice callus were cut into 0.5 cm3 pieces and placed in the shoot regeneration medium. After 15 d, the callus of WT rice proliferated normally, and there were no regenerated green buds (Fig. 3d). Nevertheless, the callus area of over-expressed HmWOX8 increased, and green buds appeared (Fig. 3d). The visual evaluation indicated that the AS regeneration ability of transgenic lines was significantly improved. Therefore, HmWOX8 was speculated to play a crucial role in AS regeneration.
Comparative transcriptome analysis between WT and HmWOX8-OE in rice
Volcano map and KEGG enrichment analysis
-
To identify the regulatory genes in HmWOX8-overexpression lines, transcriptome sequencing of HmWOX8-OE and WT were performed. Compared to WT, a total of 2,373 DEGs were identified, including 1,037 upregulated and 1,336 downregulated genes in HmWOX8-OE (Fig. 4a). In the KEGG analysis, these DEGs were enriched in the pathways of 'Plant hormone signal transduction' (map04075; p-value = 3.9e-03), 'Phenylpropanoid biosynthesis' (map00940; p-value = 1.9e-03), and 'Starch and sucrose metabolism' (map00500; p-value = 2.1e-02) in HmWOX8-OE (Fig. 4b). Furthermore, GO enrichment patterns were generally consistent with KEGG analysis. The pathway 'carbohydrate metabolic process' in the biological process category (GO:0005975; p-value = 8.18e-06) was significantly enriched in HmWOX8-OE compared to WT (Supplemental Fig. S3). In the molecular function category, 'glycosyltransferase activity' (GO:0016757; p-value = 2.67e-06) was the most abundant pathway (Supplemental Fig. S3). In addition, the dominant subcategory was 'extracellular region' (GO:0005576; p-value =8.90e-06) in the cellular component (Supplemental Fig. S3).
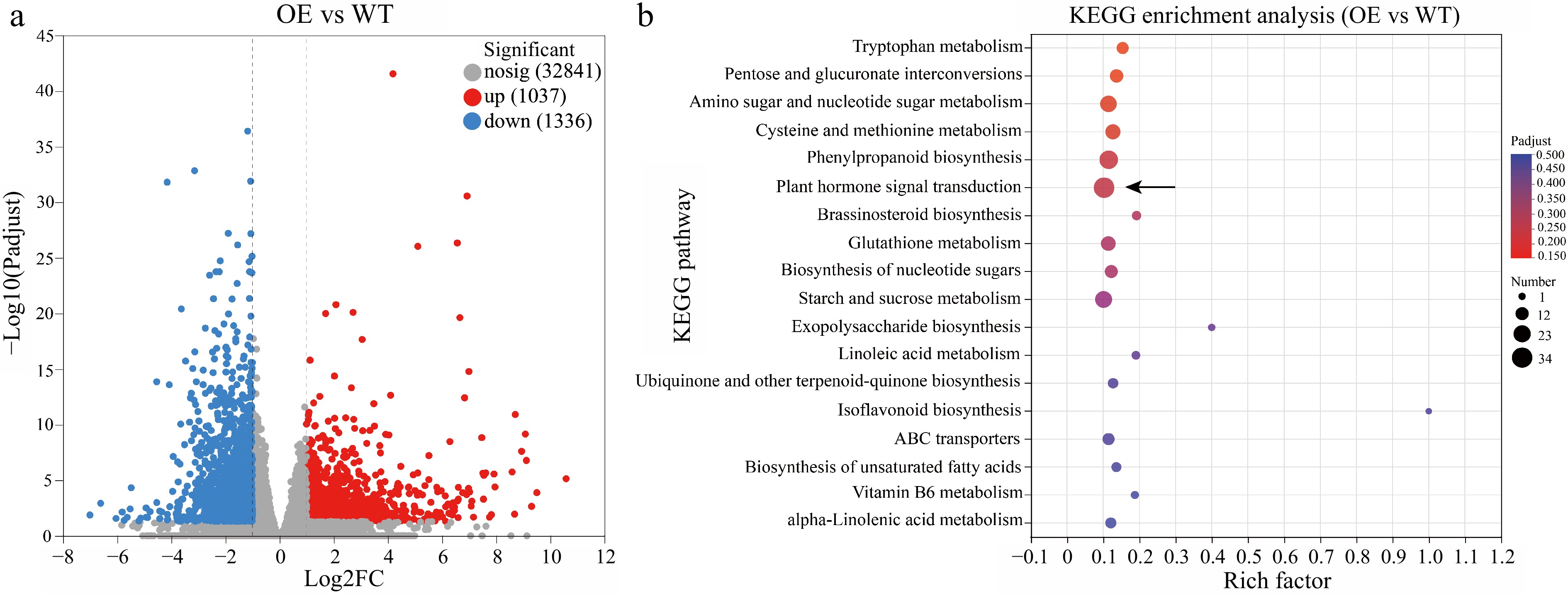
Figure 4.
Volcano map and KEGG enrichment analysis. (a) Volcano map analysis. (b) KEGG enrichment analysis of differentially expressed genes (DEGs) between HmWOX8-OE and WT.
DEGs involved plant hormone signal transduction and shoot regeneration
-
The DEGs related to the plant hormone signal transduction pathway were analyzed based on the KEGG enrichment analysis. A total of 56 enriched DEGs were associated with seven sub-pathways of the plant hormone signal transduction pathway (Fig. 5a). This includes 30 DEGs from the auxin sub-pathway, four from the CK sub-pathway, and nine DEGs from the ABA sub-pathway. It also includes three DEGs from the ETH sub-pathway, two from the BR sub-pathway, two from the JA sub-pathway, and six DEGs from the SA sub-pathway (Fig. 5a). In the auxin sub-pathway, four upregulated ARF genes and six downregulated GH3 genes were detected in HmWOX8-OE vs WT. The expression levels of other auxin signaling-related genes, including AUX1, AUX/IAA, and SAUR, were changed between the HmWOX8-OE lines and WT plants (Fig. 5b). When considering the CK signal transduction pathway, upregulated ARR2 (B-ARR) was detected, whereas three differentially expressed A-ARR (ARR4, ARR5, and ARR9) were downregulated between the HmWOX8-OE lines and WT plants (Fig. 5b). The transcriptome analysis revealed that PYL/PYL and PP2C genes were differentially expressed, with five DEGs downregulated and three upregulated in HmWOX8-OE. One gene was upregulated, which encodes SnRK2 (Fig. 5b). Regarding the ETH signal transduction pathway, CTR1 and EIN2 were downregulated, whereas the ERF1/2 gene was upregulated in HmWOX8-OE lines (Fig. 5b). As for BR signal, BRI1 gene was downregulated, whereas BKI1 gene was upregulated (Fig. 5b). Compared with WT, JAR1 and MYC2 genes were downregulated in HmWOX8-OE of JA sub-pathway (Fig. 5b). Several DEGs involved in the SA sub-pathway were also observed. Three DEGs were annotated as NPR1 genes, one was upregulated and the other two were downregulated in HmWOX8-OE vs WT. One downregulated TGA gene was detected in the HmWOX8-OE lines (Fig. 5b).
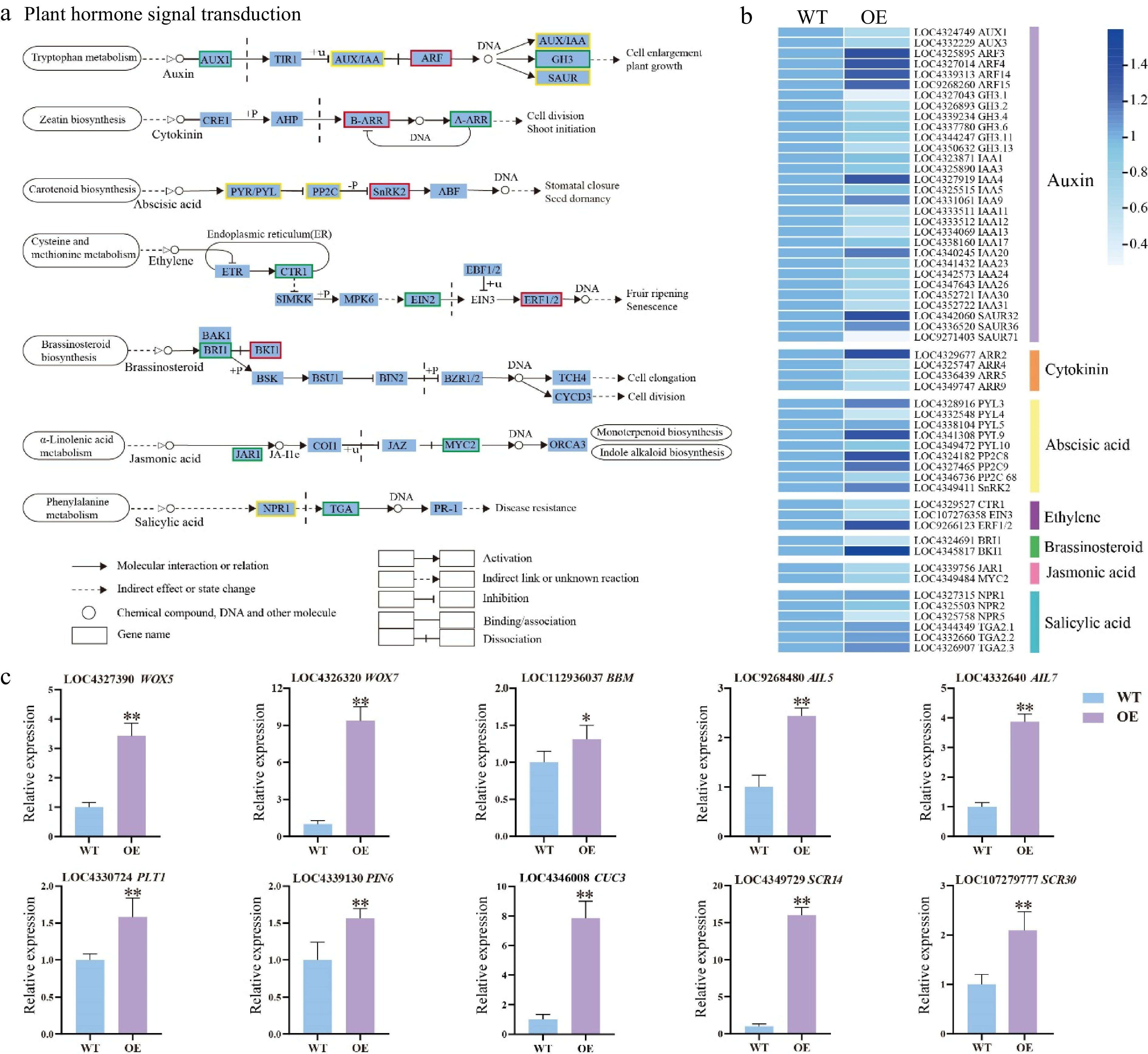
Figure 5.
DEGs related to phytohormone signaling transduction pathway and shoot development-related genes. (a) Expression of the phytohormone signal pathway. The color of the background frame indicates the pattern of gene expression: red for upregulation, green for downregulation, and yellow for both upregulation and downregulation. The bars represent standard deviation. (b) Heatmap of the DEGs related to plant hormone signal transduction pathway. (c) Relative expression of shoot development-related genes. Error bars = standard deviation. * p < 0.05, ** p < 0.01 (Student's t-test).
Based on RNA-seq data, shoot development-related genes were analyzed in the HmWOX8 transgenic plants during AS formation. The expression levels of WOX5, 7, and AIL5, 7 were upregulated in HmWOX8-OE lines. Expression levels of the BBM gene were higher in HmWOX8-OE plants than in WT. Compared with the WT plants, PLT1 PIN6, and CUC3 genes were significantly upregulated in the HmWOX8-OE lines. Moreover, SCR14, and SCR30 genes was upregulated in HmWOX8-OE lines (Fig. 5c).
Validation of transcriptome data by qRT-PCR analysis
-
To verify the accuracy and reproducibility of the transcriptome sequencing data, the transcript abundance of 14 selected DEGs was analyzed by qRT-PCR. The expression patterns of these genes are basically consistent with the transcriptome data (Supplemental Fig. S4). The results showed the high reliability of RNA-seq data.
Functional verification of HmWOX8 in H. middendorffii
-
To further investigate the function of HmWOX8 in H. middendorffii, agrobacterium cultures harboring pTRV2-empty, and pTRV2-HmWOX8 were used to infiltrate the callus of the same size (Fig. 6a). When the callus was infected twice with the recombinant TRV construct (15 d), a morphological difference was observed between the WT, and VIGS-HmWOX8 plants (Fig. 6b). The callus area of the silent HmWOX8 gene was smaller than that of the empty control and WT callus (Fig. 6b). To verify whether HmWOX8 was silent, the expression level of HmWOX8 in the callus infected with pTRV1 + pTRV2-HmWOX8 was measured using the qRT-PCR. Compared with WT and empty, the content of the HmWOX8 gene was significantly reduced in VIGS-HmWOX8 lines (Fig. 6c). After 15 d in vitro culture on shoot regeneration medium, the empty control and WT callus have begun to sprout, while the callus of silent HmWOX8 gene grows slowly (Fig. 6d). Compared to that in the WT and empty, transgenic callus areas were significantly lower. AS increment coefficient and average number of regenerated AS per callus for WT and empty plants were clearly higher than those for VIGS-HmWOX8 (Fig. 6e). After 30 d, WT and empty have differentiated into green leaves, while the callus of silent HmWOX8 has just sprouted. AS regenerative efficiency in WT and empty were significantly higher than that of VIGS-HmWOX8, and no significant difference in AS regenerative efficiency was noted between WT and empty (Fig. 6g). AS increment coefficient and average number of regenerated AS per callus in VIGS-HmWOX8 callus was lower than that in WT and empty plants (Fig. 6g). These results indicated that HmWOX8 promotes AS formation.
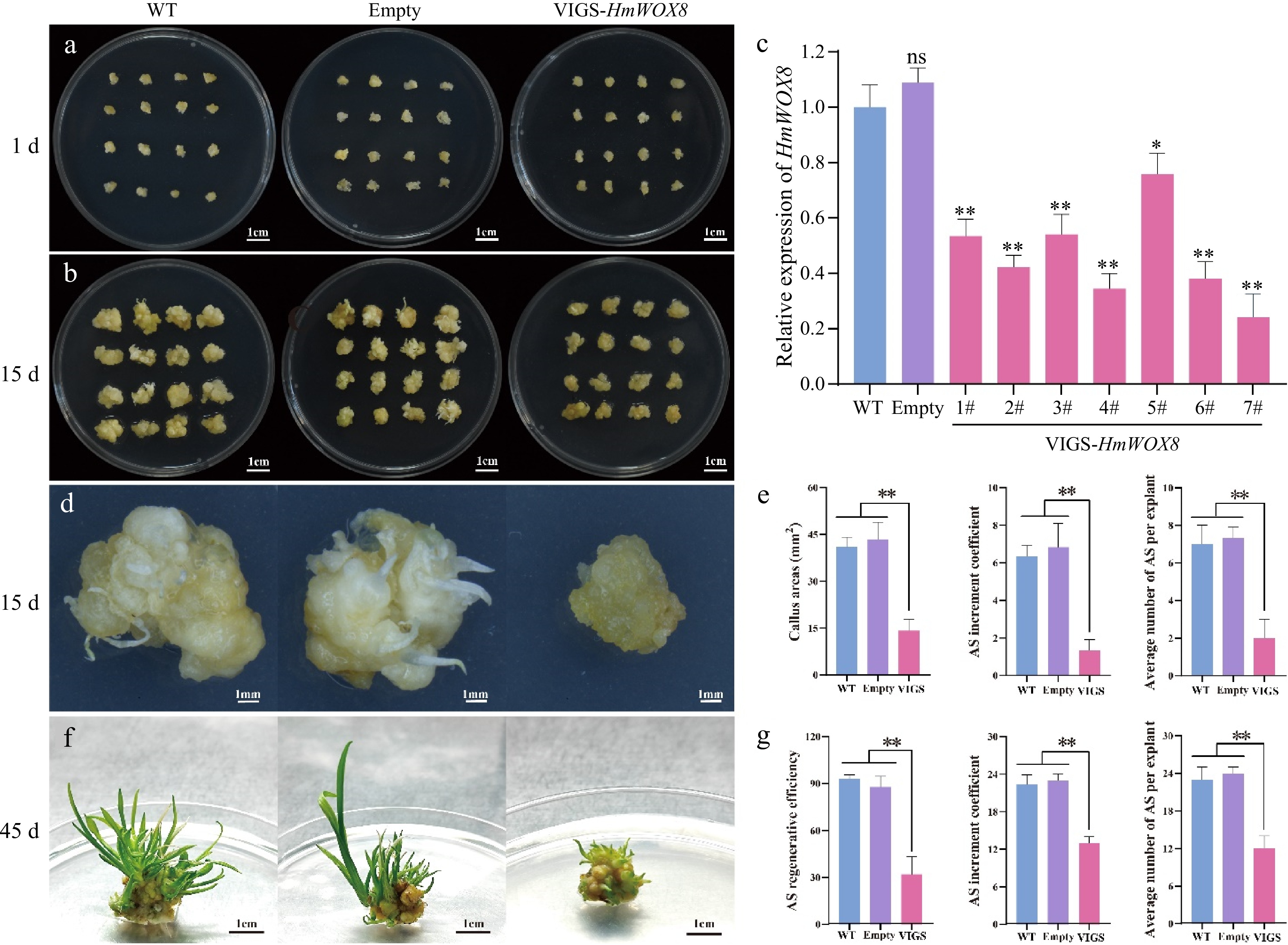
Figure 6.
TRV-mediated VIGS of the HmWOX8 gene in H. middendorffii. (a) Callus morphology of VIGS-HmWOX8 lines cultured on induction medium for 1 d compared with WT (uninfected callus) and empty (infection with pTRV1+pTRV2 agrobacterium). Scale bar = 1 cm. (b) Callus morphology of VIGS-HmWOX8 lines cultured on induction medium for 15 d compared with WT and empty. Scale bar = 1 cm. (c) HmWOX8 expression levels in WT, empty and VIGS-HmWOX8 lines. (d) Morphology of callus and AS regeneration in WT, empty, and VIGS-HmWOX8 in 15 d. Scale bar = 1 cm. (e) Callus areas, AS increment coefficient and the average number of per explant of WT, empty, and VIGS-HmWOX8 lines in 15 d. (f) Morphology of AS regenerated from callus in WT, empty, and VIGS-HmWOX8 for 45 d. Scale bar = 1 cm. (g) AS regenerative efficiency, AS increment coefficient and average number of AS per explant of WT, empty, and VIGS-HmWOX8 lines in 45 d. Error bars = standard deviation. *p <0.05, **p <0.01 (Student's t-test).
Interaction between HmWOX8 and HmCUC2
-
Through the String database, the interaction between OsWOX8 and OsCUCs was predicted in rice. At the same time, it was found that the CUC3 gene was upregulated 7.85-fold in HmWOX8-OE plants based on rice transcriptome analysis, and its expression pattern is similar to HmWOX8. To test this possibility, the homologous gene CUC2 from H. middendorffii was cloned. Then, a yeast two-hybrid (Y2H) assay was conducted using full-length HmCUC2 as bait, and HmWOX8 as prey. The yeast color test with X-α-gal showed that the combinations of yeast co-transformed with AD and BK plasmids were blue (Fig. 7a), confirming the strength of interactions between the HmWOX8 protein and HmCUC2 protein. To confirm the interaction between HmWOX8 and HmCUC2 in living plant cells, the BiFC assays were conducted using the split GFP system. The results indicated that HmWOX8 and HmCUC2 interacted in the nucleus as evidenced by green fluorescence, while negative controls showed no signals in plant cells (Fig. 7b).
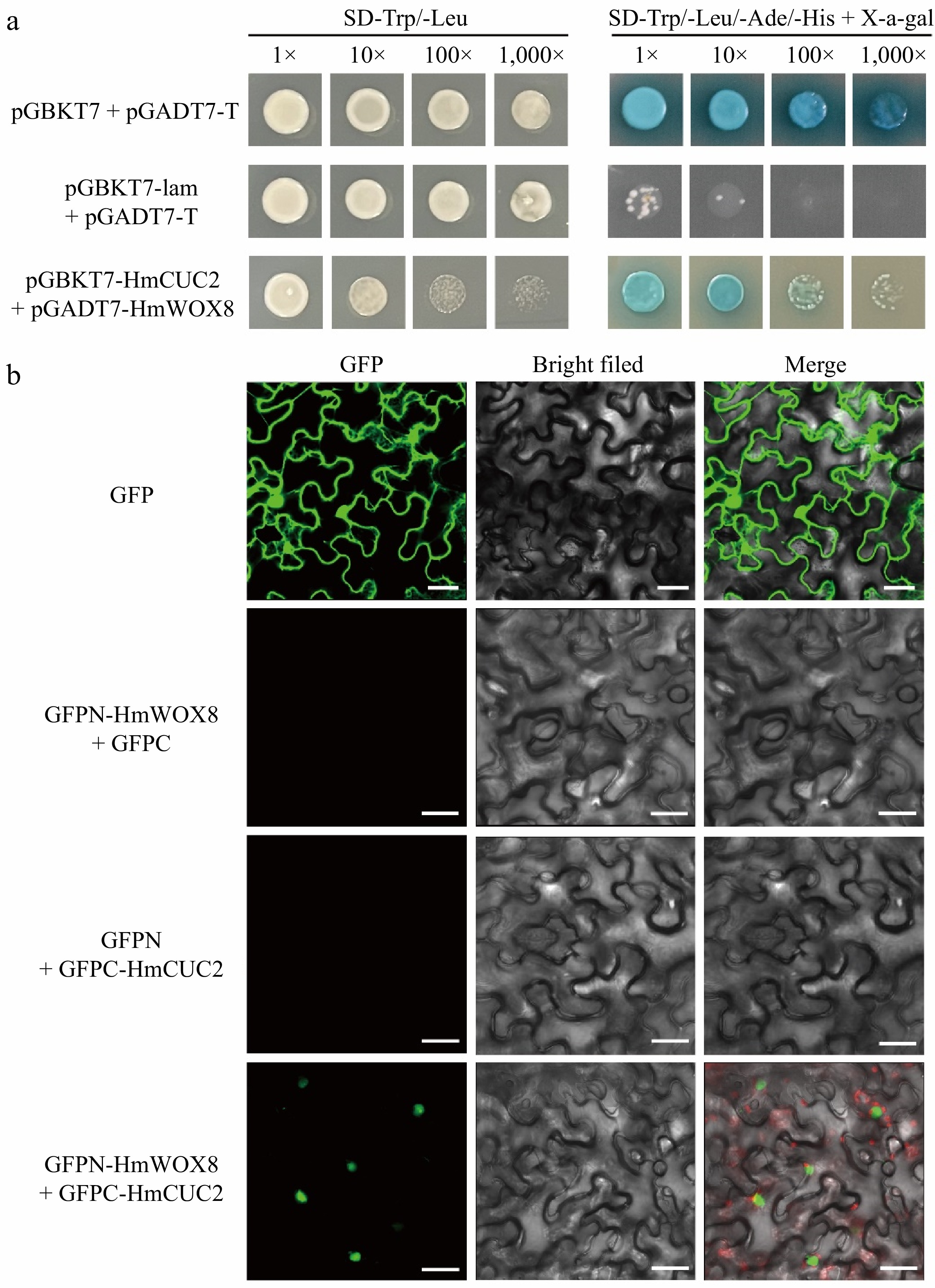
Figure 7.
Direct interaction between HmWOX8 and HmCUC2. (a) Yeast two-hybrid assay. pGBKT7 + pGADT7-T was used as a positive control, and pGBKT7-lam + pGADT7-T was used as a negative control. (b) Bimolecular fluorescence complementation assay. GFP was used as a positive control, GFPN-HmWOX8 + GFPC and GFPN + GFPC-HmCUC2 were used as a negative control. Scale bar = 30 μm.
-
Understanding the fate, dynamics and regulatory mechanisms of cells during callus proliferation and shoot regeneration is essential for plant developmental biology[47]. An efficient shoot regeneration system is the main factor leading to successful genetic transformation[21,48]. Little work has been done on improving the shoot regeneration ability in H. middendorffii. WOX proteins are key regulators implicated in stem cell proliferation and maintenance in different types of meristems, thereby serving as promising developmental regulators for plant regeneration[49]. In this study, a crucial role for HmWOX8 in the regulation of callus proliferation and shoot regeneration was uncovered.
WOX gene family is highly conserved in plants. According to the phylogenetic analysis, HmWOX8 gene is similar to other WOX8 genes from different species. Multiple sequence alignment and conserved motif analyses further validated that HmWOX8 proteins contained a highly conserved DNA-binding HOX domain, and WOX8 genes were conserved in plants. The subcellular localization of HmWOX8 was determined in rice protoplasts, indicating that the protein is located in the nucleus. Previous studies have shown that the WOX gene family members have crucial functions in many growth and development processes, such as stem cell maintenance, somatic embryogenesis, and organ formation in plants[50−53]. Studies on WOX8 have focused on its function in the regulation of early embryonic development[38,41], and little is known of its role in callus proliferation and shoot regeneration. To explore the developmental function of HmWOX8, the HmWOX8 overexpression vector was transformed into Arabidopsis and rice. The results revealed that HmWOX8 promotes callus proliferation in vitro by increasing callus areas. In addition, callus areas and shoot regenerative efficiency of VIGS-HmWOX8 lines were lower than that in WT and empty in H. middendorffii. In summary, HmWOX8 was positively correlated with callus proliferation and shoot regeneration.
It is well known that shoot regeneration is dependent on cell division and cell response to plant hormones, especially auxin and cytokinin responses. A high ratio of CK to auxin promotes shoot regeneration, whereas a high ratio of auxin to CK promotes root regeneration[11]. Transcriptome analysis uncovered that HmWOX8 overexpression changed the expression of hormone signaling pathway genes (Fig. 5a). ARF is an important regulatory factor in the auxin signaling pathway, which connects auxin with the target gene of downstream response. It is worth noting that the four ARF genes were upregulated in the HmWOX8-OE lines (Fig. 5b), suggesting there may be downstream genes of HmWOX8 that promote AS regeneration. Evidence has shown that MdARF9 is the downstream target gene of MdAIL5 in apple, and positively affects AS regeneration[13]. CK response is critical for de novo stem cell initiation and shoot meristem establishment. Type-B ARRs are transcriptional activators of cytokinin signaling, which maintains the signaling homeostasis by directly regulating type-A ARRs[19]. Correspondingly, type-A ARRs negatively regulate cytokinin signaling[54]. Previous research revealed that overexpression of ARR12 (B-ARRs) increases shoot regeneration, ARR7 and ARR15 (A-ARRs) overexpression results in the suppression of shoot regeneration[20,55]. In the current study, the expression level of B-ARRs was clearly increased, and multiple A-ARRs expression levels were decreased in the HmWOX8-OE lines (Fig. 5b), indicating that HmWOX8 can enhance the regenerative ability of explants by regulating the negative feedback loop in CK signaling pathway. Overall, the increased auxin and CK biosynthesis and sensitivity may play an indispensable influence in promoting AS regeneration of HmWOX8-OE lines. Previous studies found that ethylene exerts both positive and negative impacts on shoot regeneration, depending on the sensitivity of explants to ethylene signaling[21]. The JA-signaling pathway is involved in de novo shoot regeneration[56]. However, the molecular mechanism underlying the role of ABA, BR and SA signaling pathways in shoot regeneration is still largely unclear.
Shoot and root regeneration depend on the activity of stem cells at the stem cell niche to establish apical meristem primordium, a process which is stimulated and regulated by a number of specific regulators[57]. Studies have shown that the regulators of root meristem formation such as WOX5/7/11, LBD16, as well as SCARECROW (SCR) are involved in the acquisition of competency for shoot regeneration[36,58]. In the current study, the expression levels of WOX5/7 and SCR14/30 in the HmWOX8-OE lines were significantly increased (Fig. 5c), indicating that HmWOX8 could enhance shoot regeneration by upregulating these pluripotent factors. BBM is a member of the APETALA2/ETHYLENE RESPONSE FACTOR (AP2/ERF) family and its expression has been shown to improve plant transformation and regeneration[22]. One upregulated BBM gene was detected in transcriptome data (Fig. 5c). AILs, PLT1, and PLT2 promote shoot regeneration through the activation of CUC genes[30]. The CUC proteins are indispensable for the establishment of shoot promeristem, the CUC3 gene were clearly upregulated in the HmWOX8-OE lines in this study (Fig. 5). The CUC2 activity is required once shoot progenitors are regenerated and it is essential to initiate the regeneration of lateral organs at the periphery of shoot progenitors[31]. The Y2H and BiFC assays showed that HmWOX8 can interact with HmCUC2 protein (Fig. 7).
Based on these results, it is speculated that HmWOX8 promotes callus proliferation and shoot regeneration through two pathways (Fig. 8). (I) HmWOX8 activates the expression of ARF, B-ARR, SnRK2, ETR1/2, BKI1, and inhibits GH3, A-ARR, CTR1, EIN2, BRI1, JAR1, MYC2, TGA expression to regulate crossing among different hormone signaling pathways, thus ensuring signal integration for efficient callus proliferation and AS regeneration. (II) HmWOX8 can upregulate the expression of some shoot development-related genes, including, WOX5/7, BBM, AIL5/7, PLT1, PIN6, CUC3 and SCR14/30, to regulate AS formation. In addition, studies have found that HmWOX8 can directly interact with HmCUC2. Therefore, it is suspected that HmCUC2 may be a key regulator of AS regeneration in plants, and this needs to be proven in subsequent studies. This study provides a theoretical foundation for clarifying the mechanism of HmWOX8-mediated promotion of callus proliferation and AS regeneration.
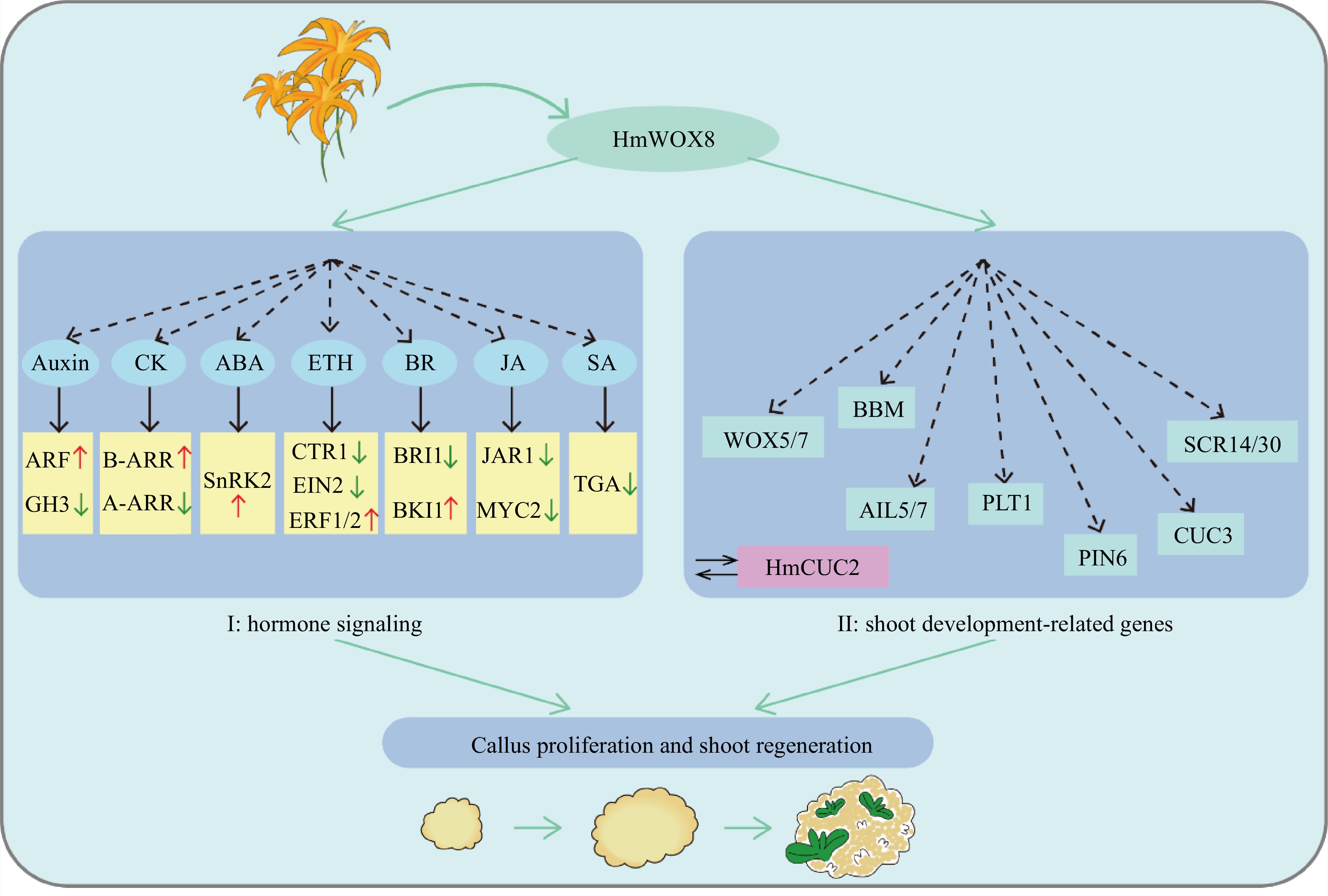
Figure 8.
Hypothetical model of HmWOX8 regulating callus proliferation and shoot regeneration.
-
In the current study, the HmWOX8 gene was isolated from H. middendorffii, and its function characterization explored using transient overexpression and VIGS analysis. The results indicated that HmWOX8 positively regulates callus proliferation and shoot regeneration in Arabidopsis and rice. Additionally, silencing the HmWOX8 gene in H. middendorffii exhibits lower callus proliferation efficiency and shoot regeneration ability. The transcriptome analyses in HmWOX8-OE and WT rice were conducted to elucidate the potential mechanisms involved in plant regeneration. The results revealed that HmWOX8 enhances the efficiency of callus proliferation and shoot regeneration through two different regulation paths, including hormone signaling pathways and shoot development-related genes. This study provides key insights into the functional diversification of the WOX gene family during plant regeneration. However, more work would be needed to further excavate the HmWOX8 gene of upstream regulatory factors and downstream target genes in shoot regeneration process.
-
The authors confirm contribution to the paper as follows: study conception and design: Liu Y, Chen Y; data collection: Gao Z, Hou F; analysis and interpretation of results: Chen A; helpful discussion provided: Zhao X; manuscript review and editing: Zhao X. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
This research was funded by the National Natural Science Foundation of China (No. 32102420).
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Xueying Zhao, Along Chen
- Supplemental Table S1 qRT-PCR primer sequences.
- Supplemental Table S2 qRT-PCR primer sequences.
- Supplemental Fig. S1 PCR identification of Arabidopsis transformed with HmWOX8 gene.
- Supplemental Fig. S2 GUS staining verification, and HmWOX8 gene relative expression in WT and OE of rice.
- Supplemental Fig. S3 GO enrichment analysis of differentially expressed genes (DEGs) between HmWOX8-OE and WT
- Supplemental Fig. S4 qRT-PCR validation of DEGs in WT and HmWOX8-OE. The bars represented standard deviation.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhao X, Chen A, Gao Z, Hou F, Chen Y, et al. 2024. Overexpression of HmWOX8 promotes callus proliferation and shoot regeneration by regulating hormone signaling and shoot development-related genes. Ornamental Plant Research 4: e026 doi: 10.48130/opr-0024-0024 

Overexpression of HmWOX8 promotes callus proliferation and shoot regeneration by regulating hormone signaling and shoot development-related genes
- Received: 03 May 2024
- Revised: 07 July 2024
- Accepted: 29 July 2024
- Published online: 04 September 2024
Abstract: Shoot regeneration capacity is essential for prosperous genetic transformation. Previous studies have shown that WUSCHEL-related homeobox (WOX) transcription factor plays a crucial role in callus growth, shoot regeneration, and root development. However, the mechanisms and functions of shoot regeneration related to WOX8 remain unclear. In the current research, the HmWOX8 gene was isolated from Hemerocallis middendorffii by RACE (Rapid-amplification of cDNA ends) technology. Overexpression of HmWOX8 improved callus proliferation and shoot regeneration ability of Arabidopsis and rice, whereas silencing HmWOX8 in H. middendorffii resulted in the inverse correlation. Transcriptome analysis revealed that HmWOX8 enhances the efficiency of callus proliferation and shoot regeneration through two different ways of regulation, including hormone signaling pathways and shoot development-related genes. (I) HmWOX8 regulates crosstalk among different hormone signaling pathways by activating and inhibiting the expression of different genes in these pathways, thus ensuring signal integration for efficient callus proliferation and shoot regeneration. (II) HmWOX8 can upregulate the expression level of shoot developmental genes, including WOX5/7, BBM, AIL5/7, PLT1, PIN6, CUC3, and SCR14/30, to regulate shoot emergence and outgrowth. In addition, Yeast two-hybrid assays and Bimolecular fluorescence complementation assay suggested that HmWOX8 directly interacts with HmCUC2, thereby promoting shoot regeneration. The present research improves the understanding of molecular mechanisms for HmWOX8-mediated regeneration and provides valuable gene resources for breeding programs to promote plant regeneration.
-
Key words:
- WOX8 gene /
- Overexpression /
- Callus proliferation /
- Shoot regeneration