






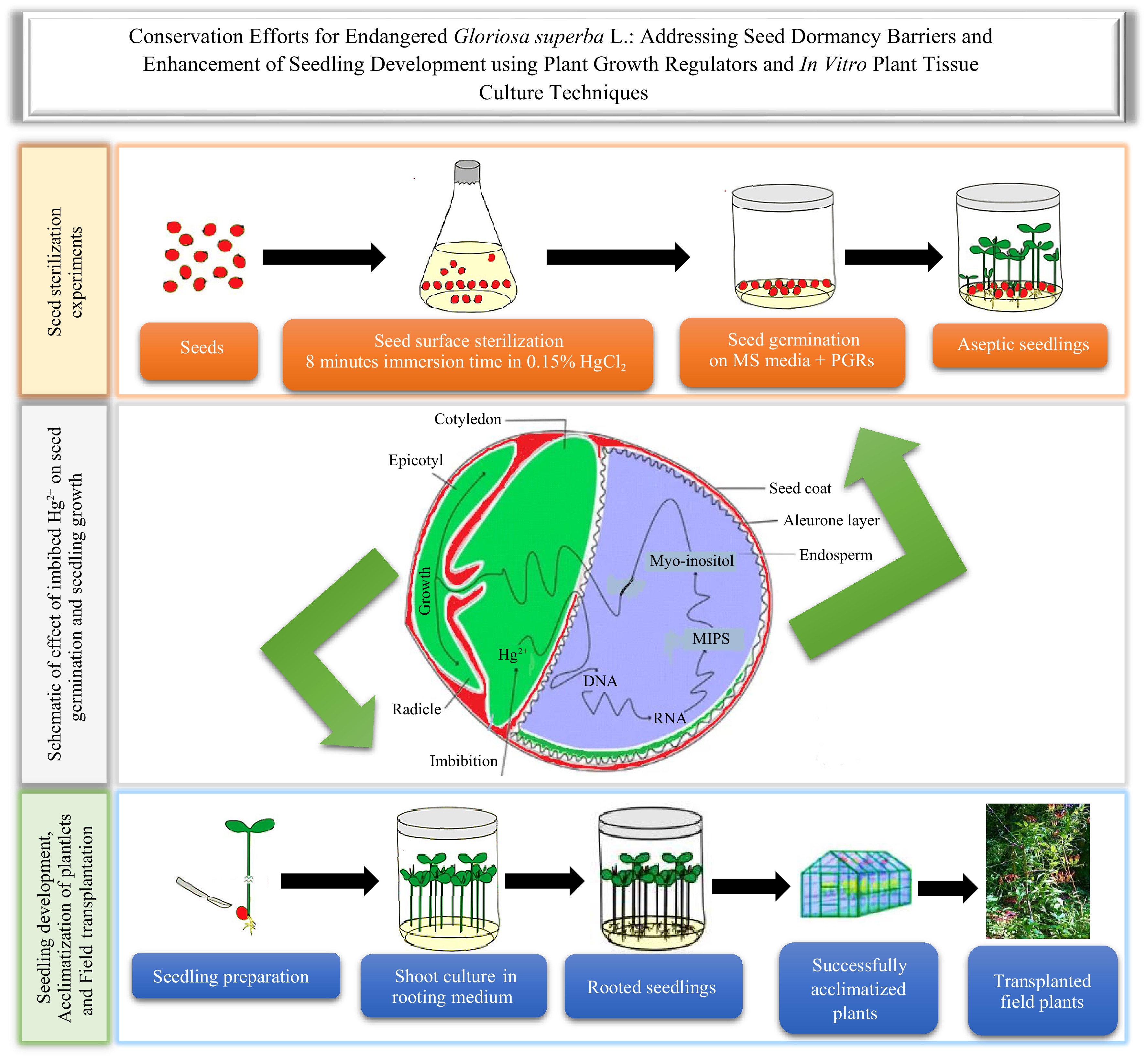


-
Efficient germination is crucial for the establishment of seedlings, the growth of plants, and the preservation of species[1]. Seed moisture, humidity, and temperature are a few factors that have a big impact on germination[2]. Ecosystems are experiencing swift changes as a consequence of environmental difficulties and global warming, leading to a significant reduction in the diversity of plant species[3]. Global conservation efforts have been carried out using both in situ and ex situ methods, with seed banking being widely adopted as an effective tool to protect genetic resources[4]. Merely gathering and retaining seeds is inadequate; for effective restoration, their eventual sprouting is crucial[5]. However, many species still lack crucial knowledge about the factors that trigger dormancy release and germination, which hinders successful propagation[6]. It is essential to have a thorough understanding of seed dormancy, which is the inhibition of germination even under ideal circumstances and can result from stimuli coming from the embryo, the seed, or the fruit coat. It is crucial to overcome these obstacles to achieve germination. There are different varieties of dormancy, each requiring certain environmental circumstances to end. This knowledge is essential for the reproduction and use of plants, and it is crucial for efforts to conserve species.
Seed germination is a complex process that begins with the absorption of water and ends with the appearance of the radicle. Plant hormones are essential for regulating seed dormancy and germination. The shift from a dormant state to an active metabolic phase is skilfully coordinated by internal gibberellins (GA), which stimulate the synthesis of hydrolytic enzymes, thus enabling the consumption of stored resources to support embryonic development[7]. Internal seed variables have an important impact on gibberellins (GA), which play a key role in seed germination[8]. Moreover, the process leading up to germination includes a substantial decrease in abscisic acid levels and a simultaneous increase in endogenous GA concentrations. The processes of imbibition and stratification facilitate this hormonal change, which is closely related to the release of dormancy[9]. Applying exogenous GA is successful in overcoming seed dormancy in certain plant species, resulting in faster seedling growth[10]. Different investigations have produced conflicting results about the presence of abscisic acid (ABA) in Gloriosa superba L. seeds. Some studies found very little or no ABA, which could mean that the seeds were physically dormant. Other studies found that chemical scarification with GA effectively overcame dormancy and sped up the germination of these seeds[10]. Nonetheless, the effectiveness of GA treatment depends on variables such as plant species, dormancy type, GA formulation, concentration, and length of treatment.
Plant growth regulators, such as cytokinins, are pivotal in seed physiology, influencing critical processes like cell division, lateral bud expansion, and germination through their modulation of ethylene release[11]. Ethylene is a hormone that plays a critical role in seed dormancy and germination. It interacts with other plant hormones and environmental factors. It counteracts abscisic acid's (ABA) effects and promotes gibberellic acid (GA) action, influencing germination. Ethylene's effectiveness depends on GA or light, and its interaction with cytokinins like kinetin can reverse dormancy. Its impact varies depending on seed dormancy type and environmental conditions, involving complex signaling pathways and interactions with other hormones[12]. Brassinosteroids (BRs) play a crucial role in regulating plant growth and development by controlling gibberellin (GA) biosynthesis and signaling pathways[13]. They are present in various plant tissues and are involved in a range of developmental processes. The BR-regulated transcription factor BES1 binds to GA promoters, regulating their expression, which is important for seedling growth. For instance, during seed germination in cereal grains, GA signaling works together with nutrient starvation signals to stimulate hydrolases, such as α-amylase, which are essential for mobilizing nutrients[14]. In contrast, auxins accumulate in embryos to suppress germination-related genes and regulate ABA metabolism, thereby maintaining dormancy until favorable conditions prevail. Auxins like indole-3-acetic acid (IAA) are integral to both germination and root formation processes[15]. As seeds imbibe water, IAA levels rise, a precursor to root elongation, particularly promoting lateral root development, illustrating their pivotal role in seedling establishment and growth[11]. Comprehending these subtleties is essential for customized and efficient approaches to seed germination[16].
A dormant seed is a seed that is unable to initiate germination despite favorable environmental conditions[17,18]. It can be classified into two main categories: physical dormancy, which occurs when palisade cells in the seed or fruit coat prevent water from entering, and morphological/physiological dormancy, which occurs when seeds have an embryo that is not fully developed[18,19]. These seeds require specific treatments to break dormancy and allow them to germinate[20]. Le Roux & Robbertse's 1997 study on the morphology and seed structures of Gloriosa superba L. revealed that seeds with an outer seed coat (sarcotesta) and sterilized with a 1% hypochlorite solution did not initiate germination. Some seed coats hindered water and oxygen absorption, causing delays[21]. However, removing the sarcotesta enhanced germination, which began 13 d after removing the sarcotesta at room temperature but with mixed results, especially at different temperatures, as germination remained unpredictable due to physiological variations among seeds. Researchers have looked into various pre-sowing techniques to enhance seed germination; however, the results remain poor[22,23].
Surface sterilization of seeds and explants is necessary to get rid of any microbes that could be harmful before in vitro regeneration techniques are used[24]. Efficiency is crucial in biotechnology-driven breeding, as it seeks to expedite the cultivation of superior plants within a limited time frame[25]. Nevertheless, the challenge of attaining efficient sterilization while maintaining plant development is a multifaceted undertaking[26]. The kind, size, and age of the explant, as well as the growing conditions, all have an impact on the effectiveness of disinfection[27]. Various disinfectants, including sodium hypochlorite (NaClO), hydrogen peroxide (H2O2), mercury II chloride (HgCl2), and others can be used depending on the degree of contamination and prevailing conditions. Extended duration and increased doses of disinfectants typically yield improved asepsis outcomes, but this may affect the viability of explants[28]. To get effective sterilization in vitro, it is important to vary the exposure time and disinfectant concentration based on the unique properties of the explants and species that were chosen.
The rising global demand for Gloriosa superba L. in export markets exacerbates its depletion and heightens the risk of extinction[29,30]. Historically, unsustainable harvesting practices have predominantly targeted tuber extraction, driven by the therapeutic benefits associated with this plant. Although countries such as India and Zimbabwe have enacted protective measures to regulate wild resource collection, illegal harvesting persists due to scarcity and escalating market demand[31]. Moreover, Gloriosa superba L. encounters challenges such as low, unpredictable, and insufficient seed germination rates, as well as susceptibility to pests in its natural habitat[32]. Meeting commercial demands requires large-scale cultivation. Although vegetative propagation is common in horticulture, it is hindered by slow growth and limited multiplication rates, which prolong the reproductive cycle. In vitro cultivation of Gloriosa superba L. seeds provides a solution to improve germination rates and accelerate growth. Seeds are preferred over vegetative parts for conservation purposes as they preserve broader genetic diversity[33].
A recent study used BET surface area analysis and 3D X-ray micro-tomography to investigate the seed coat structure of Gloriosa superba L. The study revealed that the sarcotesta and endosperm exhibit minimal porosity, rendering them impervious to water during germination[22]. Traditional techniques such as acid or mechanical scarification and water soaking are commonly used to facilitate water and oxygen absorption in hard-coated seeds, hence encouraging germination. However, despite these methods, germination rates remain low and inconsistent[22]. In vitro propagation is a versatile technique widely used in applied plant research for many purposes, such as preserving germplasm, conserving species, propagating clones on a large scale, and addressing limitations in traditional propagation methods[33,34]. It is highly beneficial for boosting seed germination by circumventing dormancy and optimizing growth conditions, resulting in reduced germination time and improved shoot and root development.
Numerous studies have researched the sterilization and germination processes of Gloriosa superba L. seeds, consistently reporting low germination rates[35]. As previously mentioned, optimizing sterilization techniques can greatly improve seed germination rates[33]. Therefore, establishing reliable protocols for sterilizing Gloriosa superba L. seeds and utilizing them for in vitro micropropagation is essential. This effort is critical for conserving and sustainably utilizing this species, particularly due to its vulnerable status in its natural habitat.
This study focuses on the crucial matter of seed dormancy in Gloriosa superba L. Although the exact dormancy classification of this species is unknown, a dual dormancy model is proposed, which includes both physical and physiological components. Through using the right seed sterilization method along with the right mix of gibberellic acid (GA3) and other plant growth regulators, this study suggests that it is possible to break dormancy and improve seed germination and seedling growth. This study seeks to develop a reliable method for sterilizing Gloriosa superba L. seeds and refine a methodology for in vitro propagation to efficiently produce Gloriosa superba L. plantlets from seeds. The findings of this research will significantly contribute to conservation efforts.
-
Fresh seeds of Gloriosa superba L. were carefully collected from mature and strong plants that were thriving in the Pachmarhi Biosphere Reserve (PBR) located in Madhya Pradesh, India (Fig. 1). The harvested seeds were sorted into sets of 100 and weighed accurately. These seeds were then stored in specialized containers under ambient conditions (temperature and relative humidity of 25 ± 2 °C and 50%, respectively), in preparation for further analysis. The culture medium used in this study was Murashige and Skoog (MS) medium, which was purchased from Merck (Mumbai, India). Additionally, Gibberellic acid (GA3), 6-benzylaminopurine (BAP), Indole-3-acetic acid (IAA), Indole-3-butyric acid (IBA), Kinetin (KN), 1-Naphthalene acetic acid (NAA), N6-(2-Isopentenyl) adenine (2-iP), and sucrose were sourced from Sigma-Aldrich (Bengaluru, India). Sterilizing agents such as mercuric chloride (HgCl2), sodium hypochlorite (NaClO), and hydrogen peroxide (H2O2) were obtained from Merck (Mumbai, India).

Figure 1.
Gloriosa superba L. seeds were obtained from the Pachmarhi Biosphere Reserve (Madhya Pradesh, India) and cultured using in vitro techniques. (a) Batch of pre-sterilized seeds; (b) Seed sterilization process taking place in laminar air flow (LAF); (c) Seed germination; (d) Healthy in vitro seedling germination. Scale bar = 2 cm.
Seed surface sterilization
-
The seeds of Gloriosa superba L. were surface sterilized using various concentrations (w/v) and exposure times (minutes) of three sterilants: HgCl2 (Merck), NaClO (Merck), and H2O2 (Merck) (Table 1). The seeds were first washed for 15−20 min under running tap water in conjunction with a sifting sieve to remove dirt and pulp. Subsequently, they underwent treatment with a 5% (v/v) detergent solution (Teepol), vortexed for 8 min, followed by five rinses with tap water. After that, they were immersed in a 2% (w/v) Bavistin fungicide solution, vortexed for 10 min, and then rinsed five times with tap water. Pre-sterilized seeds were transferred to a controlled environment (Laminar Air Flow (LAF)) following appropriate sterilization procedures for both the workstation and hands and washed five times with sterile distilled water to remove any traces of fungicide. The seeds were then briefly submerged in 70% (v/v) ethanol for 20 s, rinsed five times with sterile distilled water, and subjected to sterilization agents with varying concentrations (0.05%, 0.1%, 0.15%) and exposure times (2, 5, and 8 min). Afterward, the seeds were rinsed five times with sterile distilled water for 8 min to remove any residual sterilizing agents. The sterilized seeds were then ready for inoculation into the culture medium.
Table 1. Concentrations and exposure durations of mercuric chloride (HgCl2), sodium hypochlorite (NaClO), and hydrogen peroxide (H2O2) used to assess the contamination levels and survival rate of in vitro germinated Gloriosa superba L. seedlings.
Treatments Sterilant Concentration (w/v) Exposure time (min) T1 HgCl2 0.05% 2 T2 0.05% 5 T3 0.05% 8 T4 0.1% 2 T5 0.1% 5 T6 0.1% 8 T7 0.15% 2 T8 0.15% 5 T9 0.15% 8 T10 NaClO 0.5% 2 T11 0.5% 5 T12 0.5% 8 T13 1.0% 2 T14 1.0% 5 T15 1.0% 8 T16 1.5% 2 T17 1.5% 5 T18 1.5% 8 T19 H2O2 5.0% 2 T20 5.0% 5 T21 5.0% 8 T22 7.5% 2 T23 7.5% 5 T24 7.5% 8 T25 10% 2 T26 10% 5 T27 10% 8 The seeds were inspected after sterilization to ensure that they were free from unwanted after-effects of the treatment. They were then cultured in flasks containing half-strength MS medium supplemented with 1.5 mg·L−1 GA3 and 1.5 mg·L−1 BAP. The pH of the medium was set to 5.8, and it was solidified using 0.8% agar (Merck, India). Each replicate consisted of eight seeds cultivated in separate 250-mL flasks, with four replications per treatment. The cultures were maintained under a 16-h photoperiod with an 80 μmol·m−2·s−1 photosynthetic photon flux density provided by white fluorescent tubes (40 W; Philips, India). The temperature and relative humidity were set at 25 ± 2 °C and 60%, respectively. After 4 weeks, the effectiveness of different sterilization agents, concentrations, and immersion times on contamination and survival were determined (Fig. 1).
In vitro seed germination
-
The Gloriosa superba L. seeds were cleaned with the best sterilization method from the previous experiment. Then, the sterilized seeds were placed in 250-mL conical flasks containing 50 ml of full-strength MS medium supplemented with 30 g·L−1 sucrose and different concentrations of PGRs (GA3, BAP, and NAA) to evaluate their effects on in vitro seed germination (Table 2). The pH of the medium was adjusted to 5.8, and 0.8% agar (HiMedia, India) was added to solidify the medium. The flasks were sealed with non-absorbent cotton plugs and autoclaved at 121 °C for 20 min under 104 kPa pressure. Four replications were used per treatment; each replicate contained 12 seeds cultured in an individual 250-mL flask.
Table 2. Various concentrations and combinations of Gibberellic acid (GA3), 6-benzylaminopurine (BAP), Kinetin (KN), and 1-naphthalene acetic acid (NAA) used to evaluate in vitro germination of Gloriosa superba L. seeds.
Treatment PGRs (mg L−1) GA3 BAP T1 0.2 0.2 T2 0.5 0.5 T3 1.0 1.0 T4 1.5 1.5 T5 2.0 2.0 T6 2.5 2.5 GA3 KN T7 0.2 0.2 T8 0.5 0.5 T9 1.0 1.0 T10 1.5 1.5 T11 2.0 2.0 T12 2.5 2.5 GA3 NAA T13 0.2 0.2 T14 0.5 0.5 T15 1.0 1.0 T16 1.5 1.5 T17 2.0 2.0 T18 2.5 2.5 T19 (Control) 0 0 All the cultures were kept under a 10/14-h light/dark photoperiod using white fluorescent tubes (40 W; Philips, India) that provided 80 μmol·m−2·s−1 photosynthetic photon flux density. The greenhouse's relative humidity was 60%, and the temperature was maintained at 25 ± 2 °C. After 3, 5, 9, 13, 21, and 30 d, the number of germinated seeds and seedling survival were counted to assess seed germination.
In this study, seed germination was primarily defined by the emergence of a sprout, typically measuring 3 cm in length. However, regardless of the sprout's length, the appearance of specific morphological features such as the plumular leaves, cotyledonary sheath, swollen stem base, and primary root confirmed germination, accounting for variations in treatment conditions and seed health. The germination experiment was conducted for 30 d, with all seeds monitored for these key developmental markers throughout the entire duration.
Enhancement of seedling morphological traits
-
Following two weeks of seed germination on a full-strength MS medium containing 1.5 mg·L−1 GA3 and 1.5 mg·L−1 BAP, fresh seedlings were produced. The seedlings were transferred to new, full-strength MS media containing 30 g·L−1 sucrose and varying amounts of BAP, NAA, 2-iP, and IAA. The different treatment groups were used to evaluate their effects on the morphological characteristics of the seedlings (Table 3). The pH of these media was maintained at 5.8, and all the cultures were kept under a 16-h photoperiod with a photosynthetic photon flux density of 80 μmol·m−2·s−1 given by white fluorescent tubes (40 W; Philips, India). The ambient relative humidity and temperature were 60% and 25 ± 2 °C, respectively.
Table 3. Various concentrations and combinations of 6-benzylaminopurine (BAP), 1-naphthalene acetic acid (NAA), N6-(2-Isopentenyl) adenine (2-iP), and Indole-3-acetic acid (IAA) used to evaluate in vitro growth and development Gloriosa superba L. seedlings.
Treatments PGRs (mg·L−1) BAP NAA T1 0.5 0.2 T2 1.0 0.5 T3 1.5 1.0 2ip IAA T4 0.5 0.2 T5 1.0 0.5 T6 1.5 1.0 T7 (control) 0 0 All parameters, including shoot length, root length, root/shoot ratio, seedling length, and fresh weight, were measured twice. This was done before and after a 28-d seedling enhancement experiment. The only exception was the measurement of seedling biomass dry weight, which was taken once at the end of the experiment. To measure the fresh and dry weight parameters of the seedlings, an electronic balance with a precision of 0.01 mg was used. Fresh weight was measured directly (under aseptic conditions before this experiment), whereas, for dry weight measurement, seedlings were subjected to a two-step drying process. First, they were dried in an oven at 105 °C for 30 min, and then at 75 °C until a steady weight was achieved.
In vitro rooting
-
Microshoots were cut from mature seedlings and placed in culture media (half-strength MS basal media) with varying concentrations of IBA (0.5, 1.0, 1.5 mg·L−1), IAA (0.5, 1.0, 1.5 mg·L−1), and NAA (0.5, 1.0, 1.5 mg·L−1), as shown in Table 4. The pH of the half-strength MS basal media was adjusted to 5.8, and 0.8% (w/v) agar (HiMedia, India) was added to solidify the medium.
Table 4. Various concentrations of Indole-3-butyric acid (IBA), Indole-3-acetic acid (IAA), and 1-naphthalene acetic acid (NAA) used to assess in vitro root development in young shoots excised from Gloriosa superba L. seedlings.
Treatments 1/2 MS + auxins (mg·L−1) IBA T1 0.5 T2 1.0 T3 1.5 IAA T4 0.5 T5 1.0 T6 1.5 NAA T7 0.5 T8 1.0 T9 1.5 T10 (control) 0.0 The flasks with the different treatments of media were sealed with non-absorbent cotton plugs and autoclaved at 121 °C for 20 min at 104 kPa of pressure. This was done before the microshoot culture. Each treatment was replicated four times, with each replicate consisting of eight axenic microshoots cultivated in separate 250-mL flasks. Culture conditions were maintained as per the previously provided description. The cultures were checked after six weeks, and information was gathered on how many microshoots grew roots, how quickly they responded to the rooting treatment, how long it took for roots to form on each microshoot, and how long the roots were in centimeters.
Acclimatization
-
Before placing plantlets in small polyethylene bags, trays, pots, or thermocol cups (7 cm in diameter) filled with sterilized vermiculite and soil (1:1), they were rinsed with deionized water to eliminate surplus media. During the initial stage of acclimatization, the plantlets were kept under a 16-h photoperiod. They were exposed to white fluorescent tubes (40 W; Philips, India) that emitted a photosynthetic photon flux density of 50 μmol m−2 s−1. The plantlets were enclosed in polyethylene bags with small air holes to ensure high relative humidity (RH) of 90% and to prevent dehydration. The temperature in the culture room (CR) was maintained at 25 ± 2 °C. The polythene casings were regularly removed for 1 h. For two weeks, all potted plantlets were watered with 10 ml of a half-strength Murashige and Skoog (MS) basal salt solution (without sucrose and myo-inositol) every 4 d.
In the second stage, which occurred during the third week, the plantlets were transferred into medium-sized polyethylene bags, plastic cups, or thermocol cups filled with a mixture of garden soil, sand, and vermiculite in a ratio of 2:1:1 (volume to volume). The plantlets were housed in a shade net house (USNH) for 1 week, receiving regular mistings of tap water. The relative humidity (RH) was gradually decreased by 50%, and the plantlets were thereafter transplanted directly to the experimental field and home garden for a duration of 11 weeks to facilitate their continued growth and development.
When plantlets were moved, they were put in three different conditions: first, they were put in a controlled culture room for 2 weeks in a mixture of sterilized vermiculite and soil (1:1); then, they were put in a net house for 1 week in a mixture of garden soil, sand, and vermiculite (2:1:1) in the shade (USNH); finally, they were put out in the field for 11 weeks in direct sunlight (DSL). Information was gathered for a maximum duration of 14 weeks after the initiation of microplantlet acclimation. After transplanting the microplantlets onto the stated potting mixtures, weekly observations were conducted. The overall count of microplantlets recorded per treatment was 56 (with four replicates, each containing 14 microplantlets). The survival percentage of the regenerated plantlets was calculated using the formula:
$ \begin{split} & \rm{S}urvival\; rate\; (\text{%})= \\ &\rm{\dfrac{No.\; of\; surviving\; regenerated\; plants}{Total\; no.\; of\; transplanted\; regenerated\; plants}}\times 100\text{%} \end{split} $ The data represents the mean and standard error (SE).
Statistical analysis
-
A fully randomized experimental design was utilized for all trials, wherein seeds and seedlings were randomly chosen and organized into groups for each treatment. The seed sterilization, seed germination, seedling development, and rooting studies were conducted using four replicates for each treatment level. Each replication consisted of eight seeds, 12 seeds, 12 2-week-old seedlings, and eight microshoots, respectively. The experiments were performed twice.
After 4 weeks, data was collected for all parameters in each experiment. The percentage response to treatment was determined by dividing the total number of explants or microshoots that exhibited a response by the total number of replicates and then multiplying the result by 100. The seed germination percentage was determined by dividing the number of healthy seedlings by the total number of replicates and then multiplying the result by 100. The seed contamination rate was determined by dividing the total number of contaminated seeds by the total number of replicates and then multiplying the result by 100. The seedling survival rate was determined by dividing the total number of surviving seedlings by the total number of replicates and then multiplying the result by 100.
The determination of normality was conducted using the Shapiro-Wilk test. If the normality test had a p-value greater than or equal to 0.05, a parametric test, specifically a one-way ANOVA at a significance level of 0.05, was employed to compare the means. Conversely, if the p-value was less than or equal to 0.05, a non-parametric test, specifically a Kruskal-Wallis test at a significance level of 0.05, was used to compare the means. The data were gathered and subjected to one-way analysis of variance (ANOVA) and/or the Kruskal-Wallis test using R software (version 4.4.0). The mean separation was performed using Tukey's honestly significant difference (HSD) test with a significance level (α) of 0.05. The data were shown as mean values with a standard error. Different letters in the figures indicated significant differences at a significance level of p < 0.05.
-
The highest contamination frequency (68.75%) and lowest contamination frequency (0.00%) were found at 5% H2O2 for 2 min and 10% H2O2 for 5 min, and 8 min, respectively, which were significantly different (p < 0.001) (Fig. 2a, b). Furthermore, there were significant differences in explant viability between treatments (p < 0.001) (Fig. 2c, d). Explant viability was highest (96.88%) in 7.5% H2O2 for 8 min of immersion.
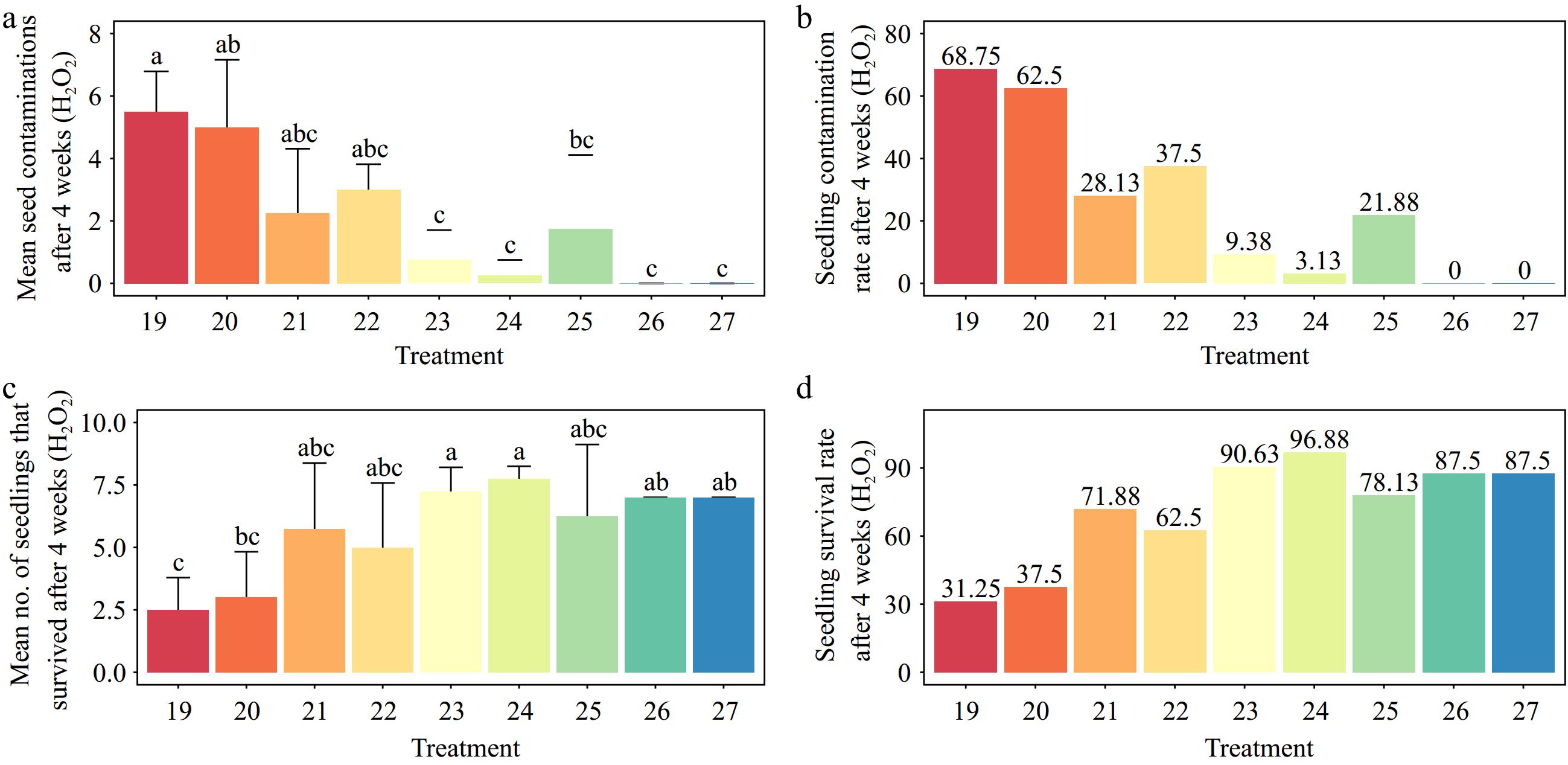
Figure 2.
Effects of various concentrations of hydrogen peroxide (H2O2) and immersion times on Gloriosa superba L. seed sterilization after 4 weeks of culture. (a) Mean seed contamination, (b) seedling contamination rate, (c) mean number of seedlings that survived, and (d) seedling survival rate are shown. Treatments are T19: 5.0% H2O2 for 2 min; T20: 5.0% H2O2 for 5 min; T21: 5.0% H2O2 for 8 min; T22: 7.5% H2O2 for 2 min; T23: 7.5% H2O2 for 5 min; T24: 7.5% H2O2 for 8 min; T25: 10% H2O2 for 2 min; T26: 10% H2O2 for 5 min; T27: 10% H2O2 for 8 min. Bars indicate mean ± SE. Different letters indicate significant difference by Tukey's test at p ≤ 0.05.
Effects of various concentrations of HgCl2 and immersion times on Gloriosa superba L. seed sterilization
-
The different treatments demonstrated a significant difference in contamination and explant viability (p < 0.001). The highest contamination was recorded for 0.05% HgCl2 for 2 min of immersion, while no contamination was recorded for 0.15% HgCl2 for 8 min of immersion (Fig. 3a, b). The treatment with 0.15% HgCl2 for 8 min of immersion had the highest explant viability (100.00%). However, the lowest explant viability (21.87%) was observed after exposure to 0.05% HgCl2 for 2 min (p < 0.001) (Fig. 3c, d).
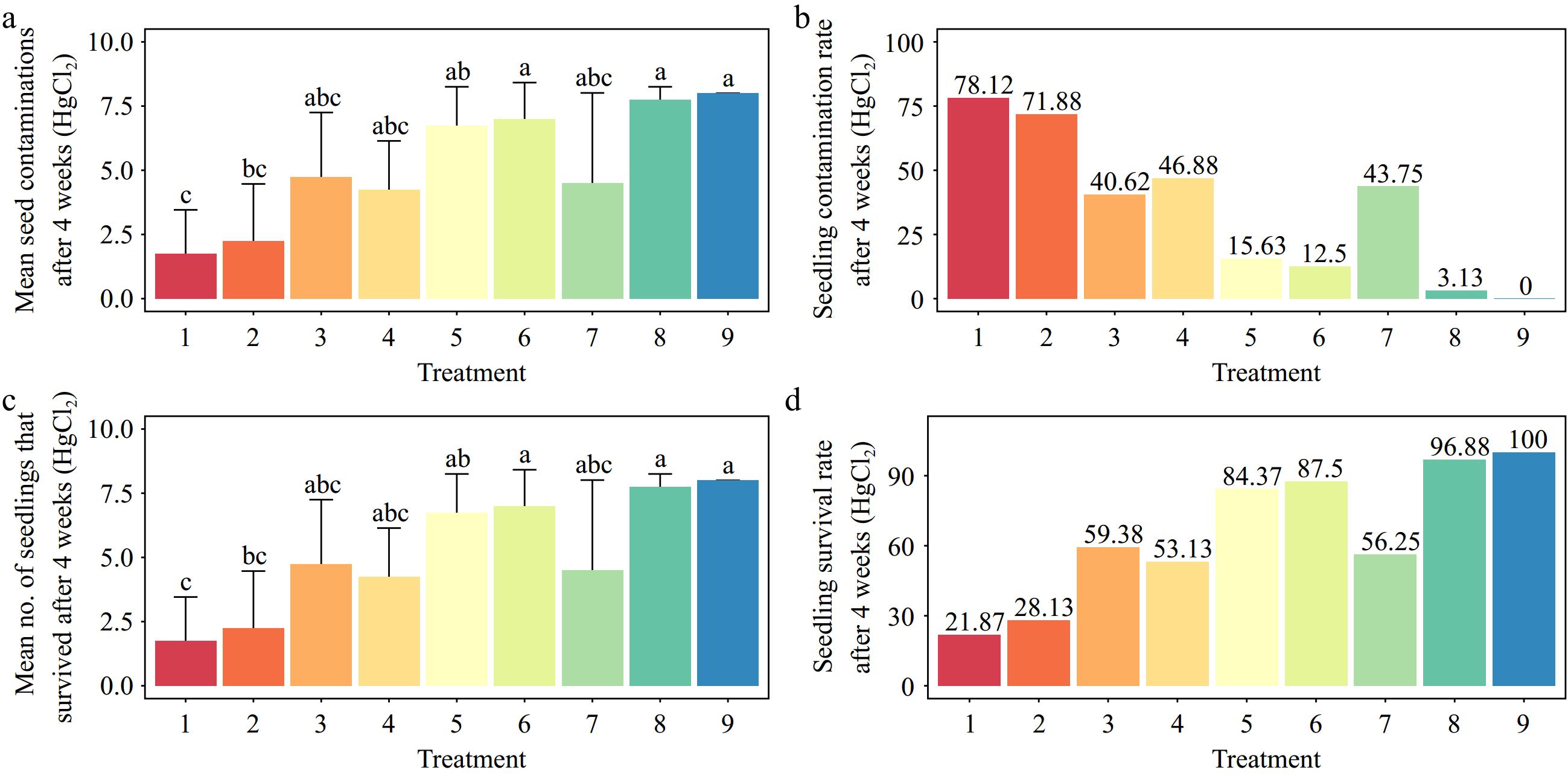
Figure 3.
Effects of various concentrations of mercuric chloride (HgCl2) and immersion times on Gloriosa superba L. seed sterilization after 4 weeks of culture. (a) Mean seed contamination, (b) seedling contamination rate, (c) mean number of seedlings that survived, and (d) seedling survival rate are shown. Treatments are T1: 0.05% HgCl2 for 2 min; T2: 0.05% HgCl2 for 5 min; T3: 0.05% HgCl2 for 8 min; T4: 0.1% HgCl2 for 2 min; T5: 0.1% HgCl2 for 5 min; T6: 0.1% HgCl2 for 8 min; T7: 0.15% HgCl2 for 2 min; T8: 0.15% HgCl2 for 5 min; T9: 0.15% HgCl2 for 8 min. Bars indicate mean ± SE. Different letters indicate significant difference by Tukey's test at p ≤ 0.05.
Effects of different NaClO concentrations and immersion times on Gloriosa superba L. seed sterilization
-
The results showed that 0.5% sodium hypochlorite for 2 min of immersion and 1.5% sodium hypochlorite for 8 min of immersion resulted in the highest (84.38%) and lowest (6.25%) contaminations, respectively (p < 0.001) (Fig. 4a, b). Also, the explant viability was highest (93.75%) when it was immersed in 1.5% sodium hypochlorite for 8 min. This was statistically significantly different from the lowest explant viability (15.63%) (p < 0.001) (Fig. 4c, d).
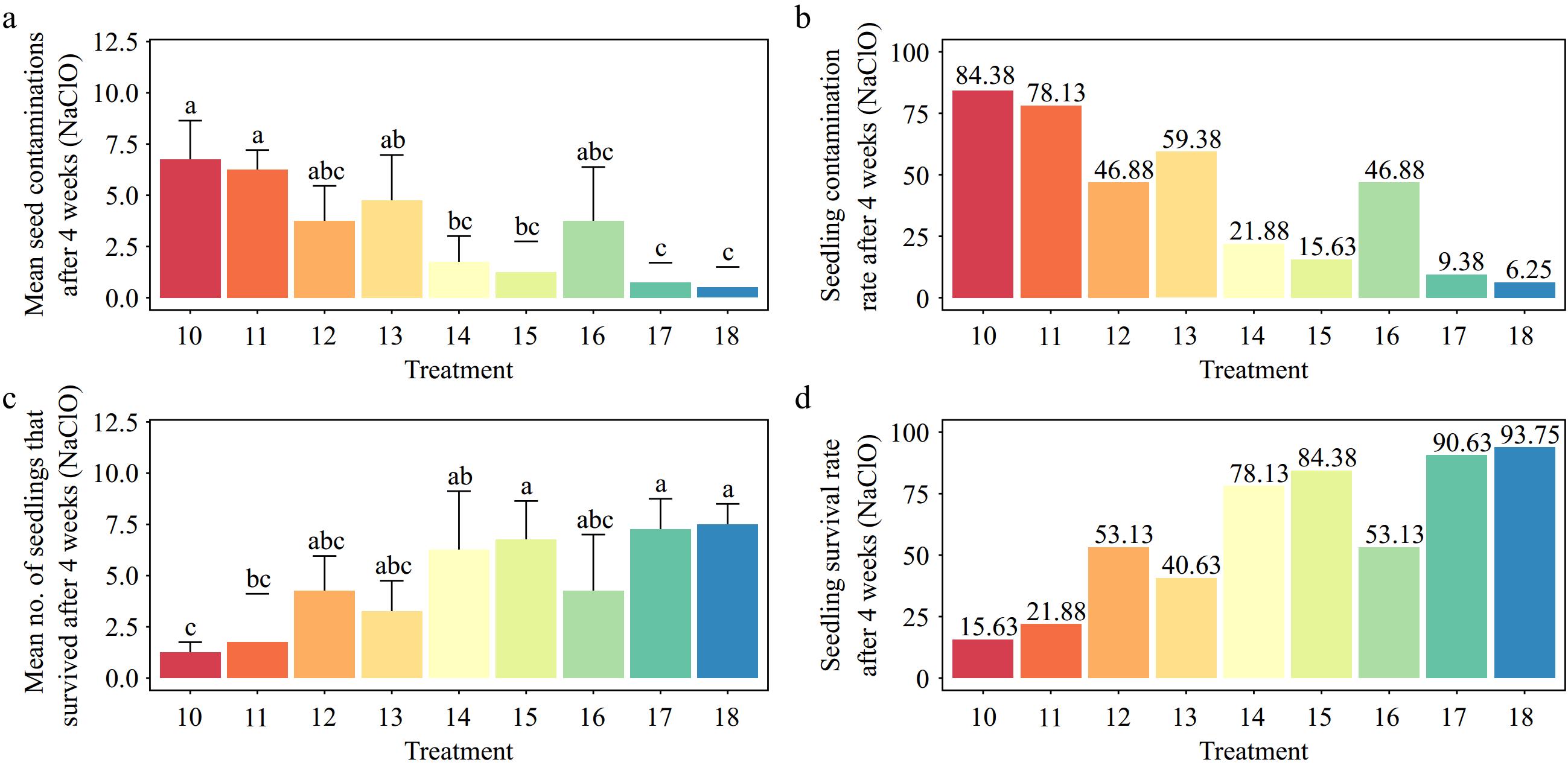
Figure 4.
Effects of different sodium hypochlorite (NaClO) concentrations and immersion times on Gloriosa superba L. seed sterilization after 4 weeks of culture. (a) Mean seed contamination, (b) seedling contamination rate, (c) mean number of seedlings that survived, and (d) seedling survival rate. Treatments are T10: 0.5% NaClO for 2 min; T11: 0.5% NaClO for 5 min; T12: 0.5% NaClO for 8 min; T13: 1.0% NaClO for 2 min; T14: 1.0% NaClO for 5 min; T15: 1.0% NaClO for 8 min; T16: 1.5% NaClO for 2 min; T17: 1.5% NaClO for 5 min; T18: 1.5% NaClO for 8 min. Bars indicate mean ± SE. Different letters indicate significant difference by Tukey's test at p ≤ 0.05.
Effect of plant growth regulators on Gloriosa superba L. in vitro seed germination
-
For 30 d, Gloriosa superba L. seeds showed increased germinability in response to the various treatments. Except for the 3rd day (p < 0.4557), the different treatments showed a significant difference only after the 5th day of germination (p < 0.001) (Fig. 5a–g). On the 3rd, 5th, 9th, 13th, 21st, and 30th days, treatment with 1.5 mg·L−1 GA3 and 1.5 mg·L−1 BAP resulted in the highest average seed germination of 0.25, 3.00, 5.75, 7.25, 9.25, and 9.25, respectively (Figs 6, 7). Treatment with 1.5 mg·L−1 GA3 and 1.5 mg·L−1 BAP showed the highest seedling survival rate (77.08%), while treatment with 0.2 mg·L−1 GA3 and 0.2 mg·L−1 NAA showed the lowest (50.00%).
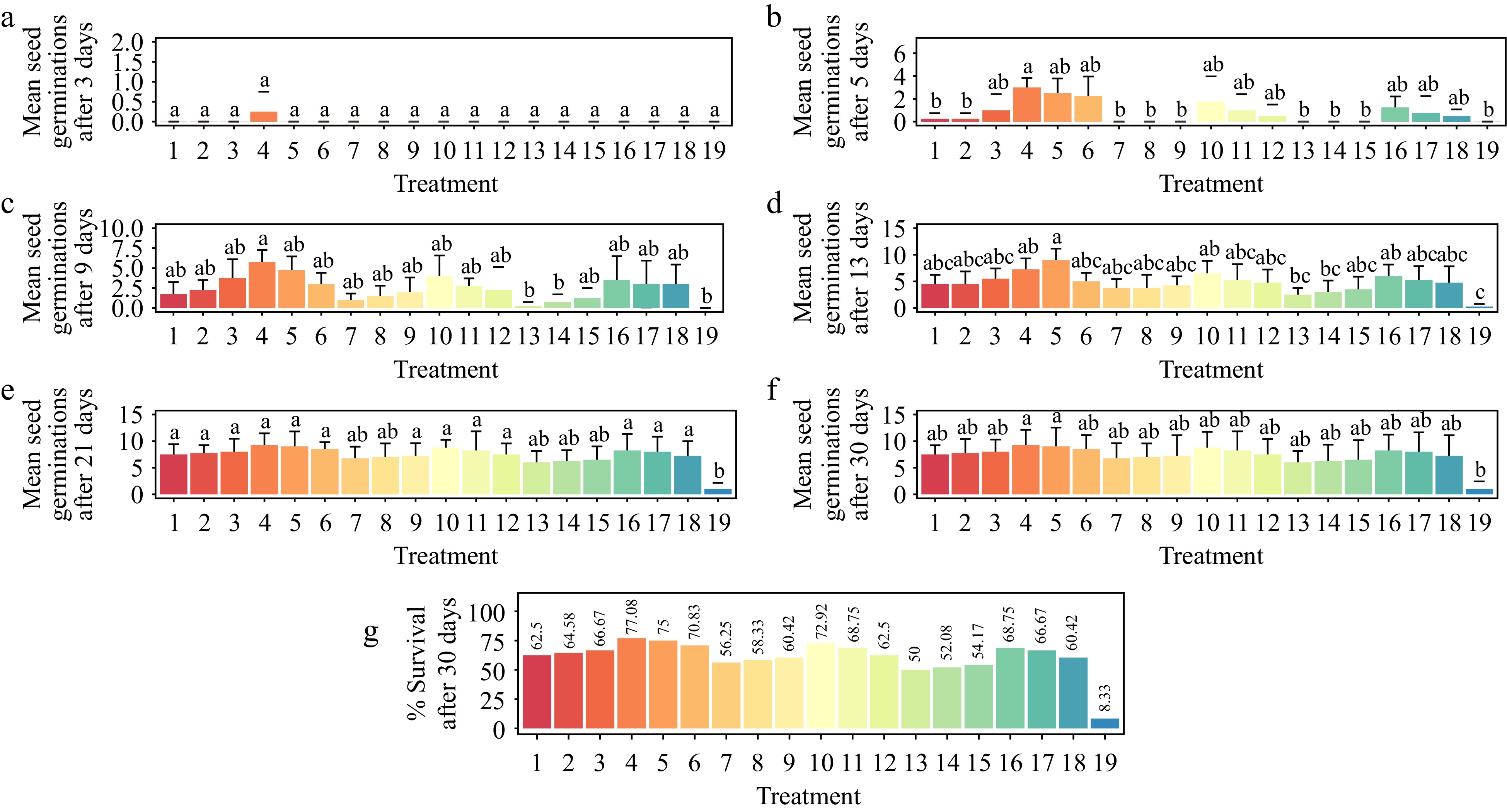
Figure 5.
Effect of plant growth regulators (PGRs) on Gloriosa superba L. seed germination after 30 d of culture. (a) Mean seed germination after 3 d, (b) mean seed germination after 5 d, (c) mean seed germination after 9 d, (d) mean seed germination after 13 d, (e) mean seed germination after 21 d, (f) mean seed germination after 30 d, and (g) seedling survival rate after 30 d are shown. Treatments are T1: 0.2 mg·L−1 GA3 (Gibberellic acid), 0.2 mg·L−1 BAP (6-benzylaminopurine); T2: 0.5 mg·L−1 GA3, 0.5 mg·L−1 BAP; T3: 1.0 mg·L−1 GA3, 1.0 mg·L−1 BAP; T4: 1.5 mg·L−1 GA3, 1.5 mg·L−1 BAP; T5: 2.0 mg·L−1 GA3, 2.0 mg·L−1 BAP; T6: 2.5 mg·L−1 GA3, 2.5 mg·L−1 BAP; T7: 0.2 mg·L−1 GA3, 0.2 mg·L−1 KN (Kinetin); T8: 0.5 mg·L−1 GA3, 0.5 mg·L−1 KN; T9: 1.0 mg·L−1 GA3, 1.0 mg·L−1 KN; T10: 1.5 mg·L−1 GA3, 1.5 mg·L−1 KN; T11: 2.0 mg·L−1 GA3, 2.0 mg·L−1 KN; T12: 2.5 mg·L−1 GA3, 2.5 mg·L−1 KN; T13: 0.2 mg·L−1 GA3, 0.2 mg·L−1 NAA (1-Naphthalene acetic acid); T14: 0.5 mg·L−1 GA3, 0.5 mg·L−1 NAA; T15: 1.0 mg·L−1 GA3, 1.0 mg·L−1 NAA; T16: 1.5 mg·L−1 GA3, 1.5 mg·L−1 NAA; T17: 2.0 mg·L−1 GA3, 2.0 mg·L−1 NAA; T18: 2.5 mg·L− 1 GA3, 2.5 mg·L−1 NAA; T19: Control (media without PGRs). Bars indicate mean ± SE. Different letters indicate significant difference by Tukey's test at p ≤ 0.05.

Figure 6.
Gloriosa superba L. seed germination. (a) & (b) Imbibition, seed swelling, and protrusion of the radicle. (c) & (d) Seed and seedling germination. Scale bar = 2 cm.
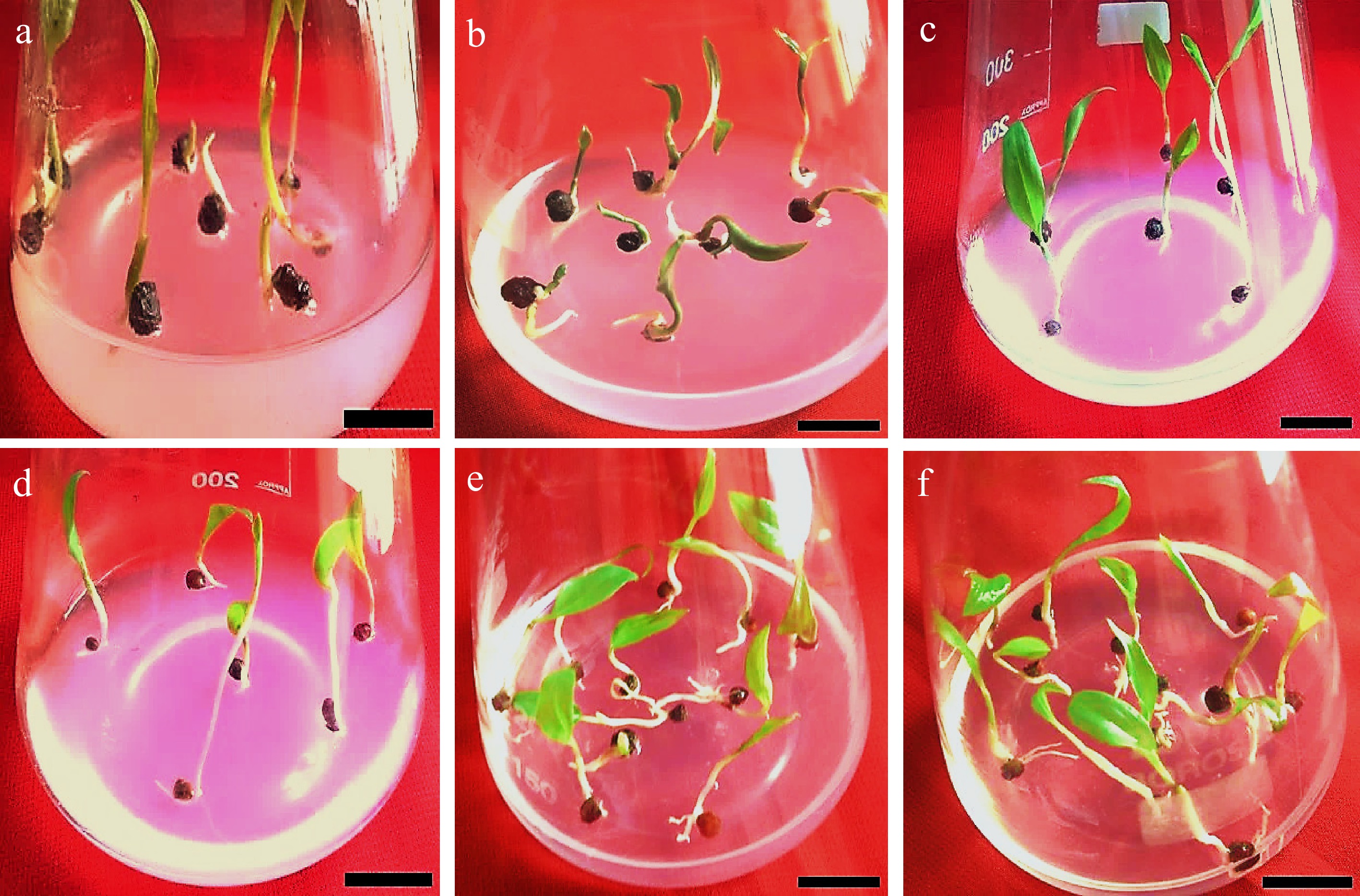
Figure 7.
Effects of various concentrations of PGRs (GA3, BAP, and NAA) on Gloriosa superba L. seed germination in full-strength MS medium supplemented with 30 g·L−1 sucrose after 30 d of culture. (a) & (b) Initial seedling developmental stage on 1.5 mg·L−1 GA3 (Gibberellic acid) + 1.5 mg·L−1 NAA (1-Naphthalene acetic acid) fortified medium. (c) & (d) Initial seedling developmental stage on 1.5 mg·L−1 GA3 + 1.5 mg·L−1 KN (Kinetin) fortified medium. (e) & (f) Initial seedling developmental stage on 1.5 mg·L−1 GA3 + 1.5 mg·L−1 BAP fortified medium. Scale bar = 2 cm.
Effect of plant growth regulators on the development of Gloriosa superba L. in vitro seedling morphological traits
-
Except for the shoot-to-root ratio (p < 0.515), the different PGR concentrations resulted in significant differences in seedling shoot length (p < 0.0155), seedling length (p < 0.0034), seedling root length (p < 0.00228), fresh weight (p < 0.001), and dry weight (p < 0.001) (Fig. 8a–f). The treatment of seedlings with 1.5 mg·L−1 BAP and 1.0 mg·L−1 NAA showed the highest average seedling shoot length (4.2 cm), seedling length (5.83 cm), seedling root length (4.08 cm), fresh weight (334 mg), and dry weight (39.1 mg) except for the shoot-to-root ratio, which showed no significant difference (p < 0.515) (Fig. 9).
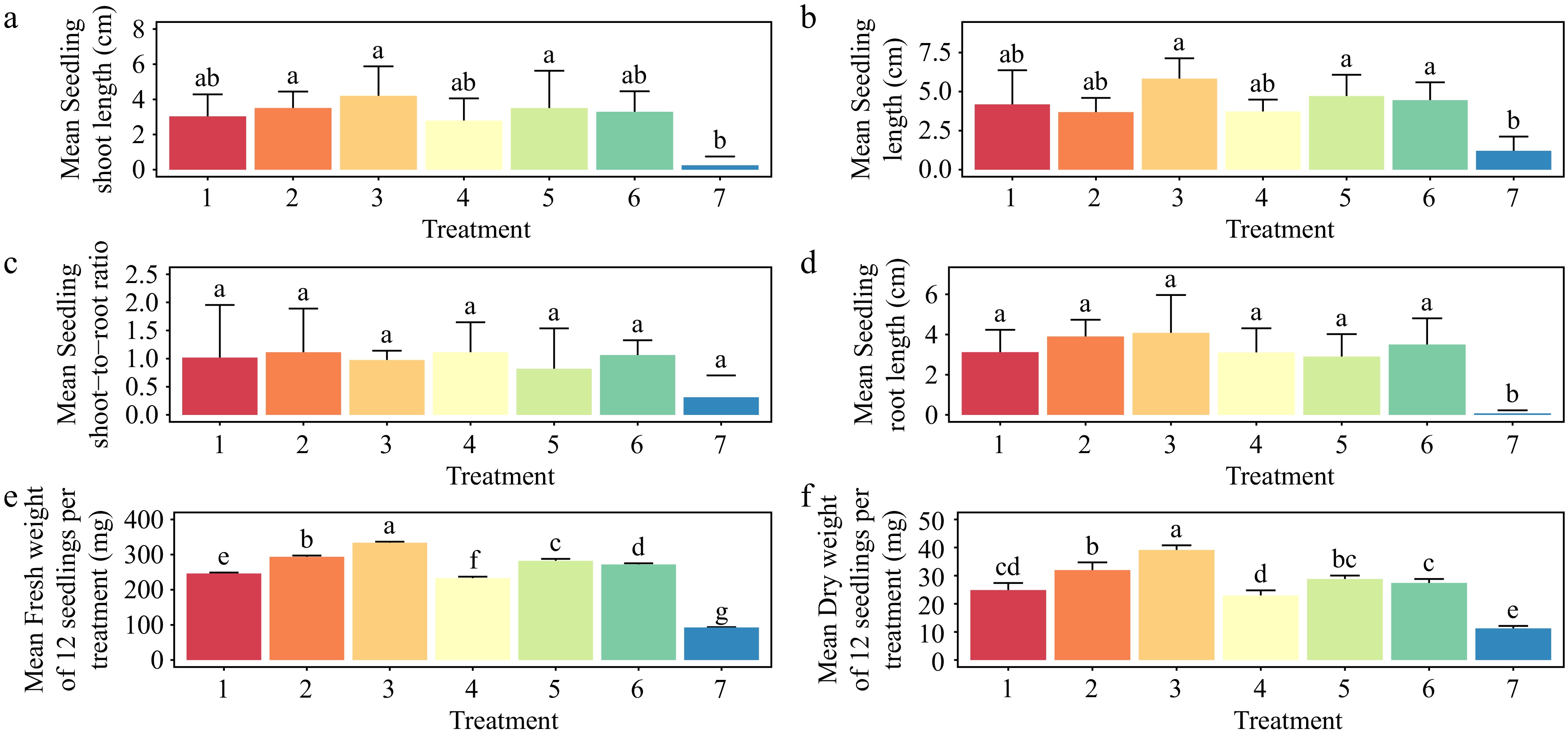
Figure 8.
Effect of plant growth regulators (PGRs) on Gloriosa superba L. seedling morphological traits after four weeks of culture. (a) Mean seedling shoot length, (b) mean seedling length, (c) mean seedling shoot-to-root ratio, (d) mean seedling root length, (e) mean seedling fresh weight, and (f) mean seedling dry weight are shown. Treatments are T1: 0.5 mg·L−1 BAP (6-benzylaminopurine), 0.2 mg·L−1 NAA (1-Naphthalene acetic acid); T2: 1.0 mg·L−1 BAP, 0.5 mg·L−1 NAA; T3: 1.5 mg·L−1 BAP, 1.0 mg·L−1 NAA; T4: 0.5 mg·L−1 2iP (N6-(2-isopentenyl) adenine), 0.2 mg·L−1 IAA (Indole 3-acetic acid); T5: 1.0 mg·L−1 2iP, 0.5 mg·L−1 IAA; T6: 1.5 mg·L−1 2iP, 1.0 mg·L−1 IAA; T7: control (media without PGRs). Bars indicate mean ± SE. Different letters indicate significant difference by Tukey's test at p ≤ 0.05.

Figure 9.
Seedlings of Gloriosa superba L. cultured in shooting and seedling enhancement media to test the effects of different plant growth regulator combinations on the morphological traits and biomass of seedlings. (a) Germinated seeds before transfer into seedling enhancement media. (b) Initiation of seedling development on 1.5 mg·L−1 BAP (6-benzylaminopurine) + 1.0 mg·L−1 NAA (1-Naphthalene acetic acid). (c)−(d) Seedling growth on 1.5 mg·L−1 BAP + 1.0 mg·L−1 NAA after 4 weeks. Scale bar = 2 cm.
Treatment with 1.0 mg·L−1 2iP and 0.5 mg L−1 IAA produced the second-best results for increasing average seedling length (4.71 cm) (Fig. 8b), while treatment with 1.0 mg·L−1 BAP and 0.5 mg·L−1 NAA produced the second-best results for increasing average seedling shoot length (3.51 cm), seedling root length (3.90 cm), fresh weight (294 mg), and dry weight (32 mg). Both treatments (1.0 mg·L−1 BAP, 0.5 mg·L−1 NAA, and 0.5 mg·L−1 2iP, 0.2 mg·L−1 IAA) produced the highest average shoot-to-root ratio (1.11) (Fig. 8c).
Effect of plant growth regulators on in vitro rooting of Gloriosa superba L. microshoots
-
Young seedlings of Gloriosa superba L. transferred from the seedling enhancement treatments were cultured in half-strength MS media supplemented with various concentrations of IBA (0.5–1.5 mg·L−1), IAA (0.5–1.5 mg·L−1) and NAA (0.5–1.5 mg·L−1) for rooting (Fig. 10). The responses to root formation changed depending on the type and amount of auxin added to the medium. These changes were significant (p < 0.001), but the average root length stayed the same. The best treatment for rooting was 1.0 mg·L−1 IBA, with a rooting rate of 84.37% (Fig. 10b). 1.0 mg·L−1 IBA induced healthy adventitious roots; the rooting speed was the fastest (within 7 d) (Fig. 10c), and no calluses appeared at the bases of the adventitious roots.
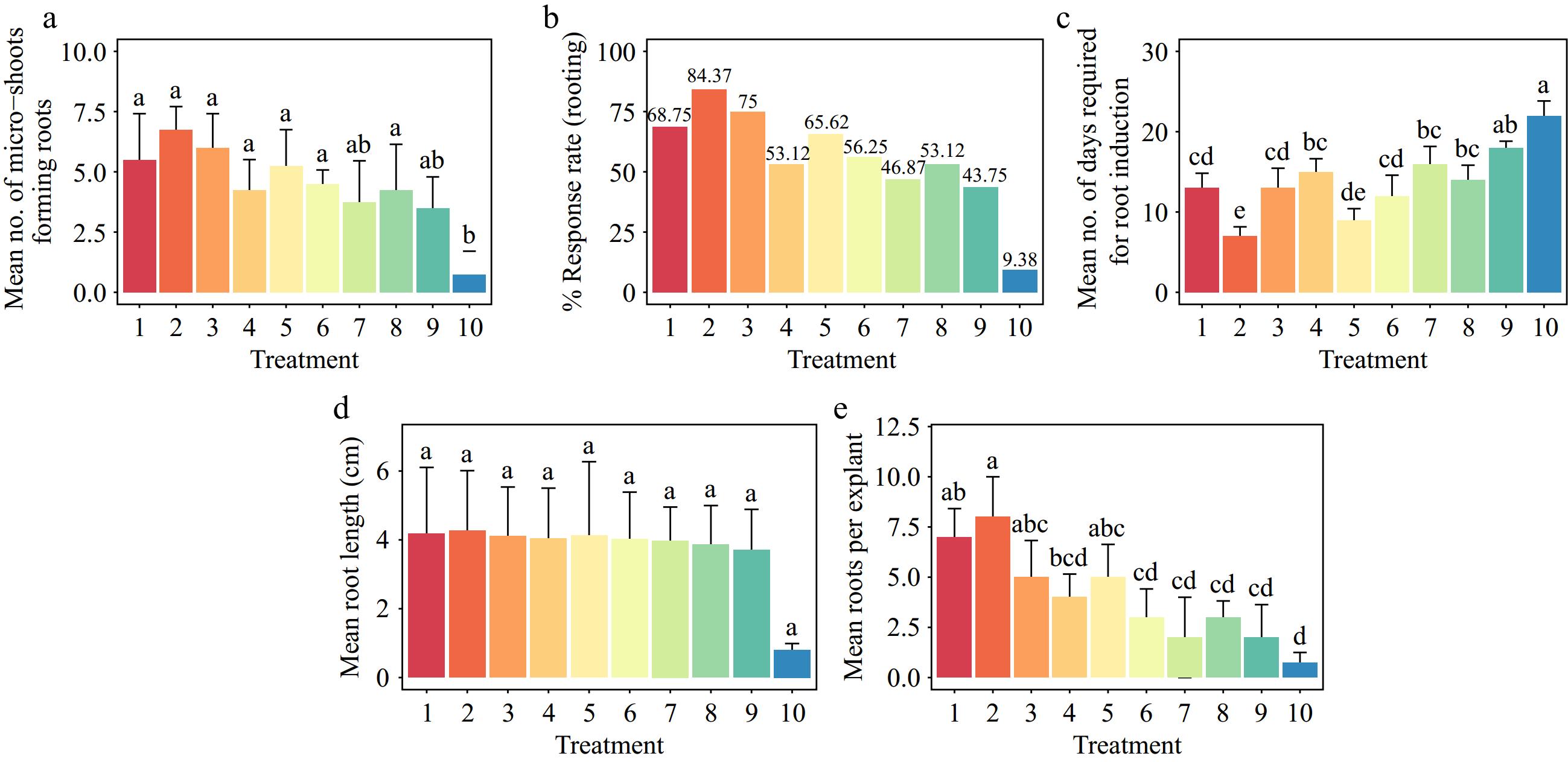
Figure 10.
Effect of PGRs on Gloriosa superba L in vitro morphogenetic response to root induction from seedling derived microshoot explants. (a) Mean microshoots forming roots, (b) response rate to rooting treatment, (c) mean days required for root induction, (d) mean root length, and (e) mean root per explant. Treatments are T1: 0.5 mg·L−1 IBA (Indole-3-butyric acid); T2: 1.0 mg·L−1 IBA; T3: 1.5 mg·L−1 IBA; T4: 0.5 mg·L−1 IAA (Indole 3-acetic acid); T5: 1.0 mg·L−1 IAA; T6: 1.5 mg·L−1 IAA; T7: 0.5 mg·L−1 NAA (1-Naphthalene acetic acid); T8: 1.0 mg·L−1 NAA; T9: 1.5 mg·L−1 NAA; T10: Control (media without PGRs). Bars indicate mean ± SE. Different letters indicate significant difference by Tukey's test at p ≤ 0.05.
Ex vitro acclimatization and plant growth
-
Plantlets transferred ex vitro on a substrate of sterilized vermiculite-soil 1:1 (v/v) and grown in a culture room for 14 d, followed by 7 d under shade in a net house on a substrate of garden soil-sand-vermiculite 2:1:1 (v/v), were readily acclimatized at a rate of 100% (Fig. 11a, 12a–e). During the next 11 weeks of growth in the field under direct sunlight, the survival rate dropped to 60% (Fig. 11a, 12f, g). All of the surviving plants flowered, produced microtubers, grew to normal plant height, and had multiple healthy leaves (p < 0.001) (Figs 11b–e; 12h–j).
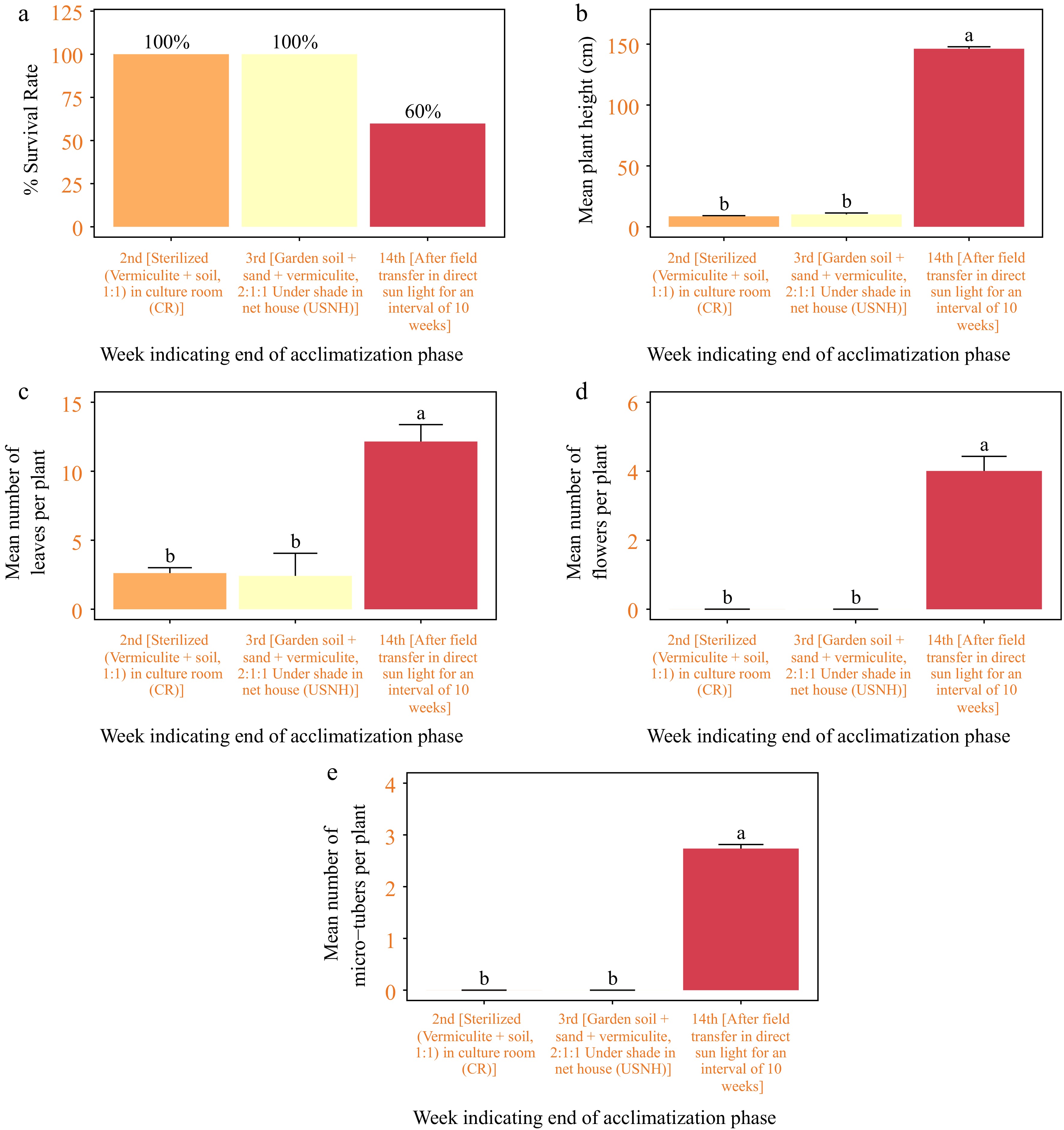
Figure 11.
Acclimatization of in vitro regenerated seedlings of Gloriosa superba L. (a) Plant survival, (b) plant height in cm, (c) number of leaves per plant, (d) number of flowers per plant, and (e) number of microtubers per plant, measured after 2 weeks of transplant in sterilised (vermiculite + soil, 1:1) grown in culture room (CR); 1 week of transplant in garden soil + sand + vermiculite, 2:1:1 under shade in net house (USNH), and finally, 11 weeks of transplant in the field under direct sun light (DSL).

Figure 12.
Acclimatization of in vitro regenerated seedlings of Gloriosa superba L. (a) Microshoots transplanted into sterilized substrate (vermiculite + soil, 1:1) grown in culture room (CR). (b) Plantlets in chill trays containing a mixture of garden soil + sand + vermiculite, 2:1:1 under shade in net house (USNH). (c) Direct transplantation in the field under direct sun light (DSL). (d) Fully grown plant with developed flowers at the later stage of acclimatization in the field under direct sun light (DSL). (e) Some microtubers harvested at the later stage of acclimatization. Scale bar = 2 cm.
-
This work focused on enhancing the in vitro germination of Gloriosa superba L. seeds, aiming to resolve the problem of seed dormancy and sluggish and inconsistent seed germination commonly encountered in natural and traditional cultivation approaches. The use of an effective seed sterilization method, combined with the strategic application of GA3 and other plant growth regulators, resolved seed dormancy and resulted in significant improvements in both seed germination and seedling growth, thus demonstrating the superiority of in vitro plant tissue culture techniques in micropropagation via seed explants and conservation.
Effects of various concentrations of sterilant and immersion times on Gloriosa superba L. seed sterilization
-
The presence of microorganisms, including bacteria, yeast, and fungi, is the primary source of losses in plant in vitro cultivation due to contamination. These microorganisms engage in a competition for resources with plant tissues, which often leads to a higher rate of mortality in the culture. Nevertheless, their existence can also lead to inconsistent growth, tissue death, decreased shoot multiplication, and diminished root development. Surface sterilization of explants can be challenging, and even small errors during the sterilization procedure can lead to a waste of time, effort, and resources. This can have significant financial repercussions if not addressed effectively.
Furthermore, living materials must retain their biological activity while undergoing sterilization, with only contaminants being destroyed. Therefore, explants are subjected to surface sterilization using sterilants at suitable concentrations for a given duration. Sen et al. discovered that the negative effects on germination (%) and plantlet development became more noticeable after an 8-min treatment. This suggests that growth is significantly slowed after this time. Therefore, a standardized immersion period limit of 8 min was implemented for all seed sterilization trials[36].
Sodium hypochlorite, mercuric chloride, and hydrogen peroxide are frequently employed as sterilants to achieve surface sterilization of plant and seed material from different species[37]. Nevertheless, these compounds sometimes prove inadequate for successfully eliminating contaminants, particularly when it comes to seeds that have been harvested from an open field and stored under unsterile conditions. The current study aimed to determine the optimal sterilization protocol for Gloriosa superba L. seeds by testing various concentrations of mercuric chloride (0.05%, 0.1%, and 0.15%), sodium hypochlorite (0.5%, 1.0%, and 1.5%), and hydrogen peroxide (5.0%, 7.5%, and 10%) at different immersion times (2, 5, and 8 min).
Mercuric chloride exhibits potent antibacterial properties, effectively eliminating both fungi and bacteria. However, it can also be detrimental to seeds and plant materials, perhaps causing their demise. It is potentially the most efficient agent for sterilizing soil-borne and epiphytic fungi in seeds. It is favored over NaClO and H2O2 due to its higher level of activity. Mercuric chloride is extremely poisonous, but sterilants like hypochlorite are consistently safer, even when used in high quantities[38]. Unlike earlier studies that used a 0.1% concentration of HgCl2, this one showed that the seeds were not sterilized even when they were exposed to this concentration and for as long as they were submerged[39]. Instead, the best benefits came from utilizing a slightly higher concentration (0.15%) and the longest possible immersion time (8 min). This confirmed that the tissue samples were fully functional and free from any contamination. This highlights the possibility of enhancing the effectiveness of sterilization by raising both the concentration and duration of immersion. Gloriosa superba L. seeds had a strong sarcotesta that protected them from the possible harmful effects of high HgCl2 concentrations and immersion durations.
The use of hypochlorite salts for disinfection can be traced back to the mid-18th century[40]. Most disinfection products primarily consist of chlorine-based industrial solutions[41]. Of all the NaClO concentrations that were tested, the best results were seen when the highest concentration (1.5%) was combined with the longest immersion time (8 min). This led to the highest percentage of seedling survival (93.75%) and the lowest percentage of contamination (6.25%). In contrast to HgCl2, where phytotoxicity had a direct effect on the viability of explants, the success of sterilization had a bigger effect on the survival of seedlings in NaClO. Significantly, seedling survival was negatively impacted when the immersion time with NaClO was less than 5 min, irrespective of concentration. This emphasizes the crucial importance of the duration of immersion. This discovery is consistent with prior studies on Ficus religiosa seeds, which showed that higher concentrations of NaClO and longer periods of immersion had a substantial positive effect on sterilization[42]. Similarly, a study conducted on Achyranthes aspera seeds found that higher doses of NaClO were effective in sterilizing the seeds[36].
Hydrogen peroxide is a widely recognized compound that exhibits strong oxidizing properties[43]. According to the study's findings, placing seeds in a 7.5% H2O2 solution for 8 min led to the highest rate of seedling survival (96.88%) and a contamination rate of 3.13%. On the other hand, exposing the seeds to a greater concentration of H2O2 (10%) for a minimum of 5 min resulted in complete sterilization, but it led to decreased seedling survival. This suggests that the concentration of H2O2 affects the effectiveness of sterilization, with higher amounts having a detrimental effect on the viability of the explant. The results align with earlier studies that have shown that greater concentrations of H2O2 can decrease contamination but may also harm the viability of the explants[44]. While several studies indicate that sodium hypochlorite is more efficacious in contamination control, alternative viewpoints contend that hydrogen peroxide may be preferable, leading to reduced contamination levels and increased germination rates[37]. Data showing that H2O2 is a potent sterilizing agent and also aids seed germination by suppressing germination inhibitors support this viewpoint[43]. This phenomenon could perhaps elucidate the reason behind the unusually high survival of the explants, even when exposed to higher concentrations (up to 10%) and longer immersion durations (up to 8 min), without any evidence of contamination.
Seed development in Gloriosa superba L.
-
Gloriosa superba L. seeds are pale orange, sarcotestal, and desiccated, with a dormancy phase lasting approximately four months. Under optimal conditions, mature seeds initiate germination approximately 2 weeks following water absorption[45]. The linear embryo is 2.0–2.5 mm in length and is enclosed within the mostly proteinaceous endosperm. During germination, the haustorial cotyledon undergoes elongation, while the embryo axis is expelled from the seed[46] (Fig. 6). Hypogeal germination is characterized by the elongation of the stem axis, keeping the cotyledon below the ground. Root primordia originate internally within the stem tissue, resulting in the development of a widespread adventitious root system. The cotyledon gives rise to a well-developed vascular system that extends into the primary root and the initial leaf primordia responsible for photosynthesis (Fig. 6). Once the seedling has grown a minimum of two photosynthetic leaves, the lower part of the stem begins to enlarge. The seedling undergoes a rosette stage, characterized by the presence of two or more leaves, prior to the elongation of the internode below the apical bud. The swelling stem area predominantly comprises two buds, occasionally three or four, depending on the number of leaves in the rosette stage. The tuberous hypopodium initially extends horizontally, then responds to gravity and transforms into a bloated, cylindrical subterranean tuberous structure with a vertical orientation. An 'anatropous bud' is formed when the basal part of the bud is pushed towards the terminal position due to hyponastic development[45]. Intercalary growth makes the leaf bases longer to match the hypopodium's growth, creating a leaf-like sheath with veins around the tuber. In the subsequent growing season, the apical meristem of the dormant bud becomes active, giving rise to an aboveground shoot and a root system that develops from an unusual location. The parent tuberous section of the previous season eventually depletes its reserves and shrinks, likely providing mainly starch reserves to subsequent plants[45].
Effect of plant growth regulators on Gloriosa superba L. in vitro seed germination
-
A previous study by Le Roux & Robbertse[21] revealed that Gloriosa superba L. seeds with the sarcotesta intact never germinated and were heavily contaminated by fungi. After the sarcotesta was removed, seeds germinated at an ambient temperature of 23 ± 2 °C, achieving a germination percentage of 21.5%. However, germination was significantly lower at 30 °C and nonexistent at 35 °C. Germination remained irregular across all temperature ranges, with the highest germination rate observed at 23 ± 2 °C after 31 d[21]. These findings suggest that Gloriosa superba L. seeds germinate best at ambient temperatures, and the removal of the seed coat significantly alleviates physical dormancy. However, this intervention does not guarantee the alleviation of morphological seed dormancy, as overall germination rates remain poor regardless of temperature.
This study examined how different plant growth regulators (GA3, BAP, NAA, and KN) affected the germination of Gloriosa superba L. seeds. The most effective treatments for breaking seed dormancy and commencing germination in Gloriosa superba L. seeds were the application of 1.5 mg·L−1 GA3 and 1.5 mg·L−1 BAP. Germination started as early as the 3rd day after the seeds were treated with these substances. The treatment exhibited the highest mean seed germination throughout all recorded time intervals (days 3, 5, 9, 13, 21, and 30), resulting in an overall survival percentage of 77.08% by the 30th day. In addition, the application of different concentrations of GA3 in combination with KN and NAA led to survival rates of 72.92% and 68.75%, respectively. The results of this study are the first to show that using the right amounts of GA3 and cytokinin, specifically BAP, together is the best way to break the dormancy of Gloriosa superba L. seeds and help them germinate. This treatment outperforms the effects of GA3 when combined with KN or NAA. Numerous earlier research projects have shown that cytokinins and gibberellins work together to make tomatoes grow faster[47]. Giving Nannochloropsis oceanica cytokinin and GA3 together led to more growth, more lipid production, and more polyunsaturated fatty acids and eicosapentaenoic acid being made. This highlights the potential of this combined effect in various plant species[48]. It has been shown that the interaction between cytokinins and gibberellins has a big impact on cell division and cytodifferentiation. This shows how important this interaction is in many areas of plant biology[49]. Surprisingly, the seedling survival rate obtained during the germination experiment was unexpectedly lower compared to the remarkable survival rate attained during the seed sterilization experiment. However, it should be noted that the seed survival rates in the germination experiment were calculated based on a 30-d duration. This unexpected observation suggests potential factors, such as abiotic stresses, that could have impacted seedling growth beyond the first 30 d. Additionally, the meticulous separation, measurement, and use of different seed batches in this study could explain the variations in seedling survival rates due to disparities in seed viability.
Seed soaking is a well-established technique for enhancing germination rates by softening the seed coat and removing germination-inhibiting substances[22]. Additionally, pre-treatment with 0.15% HgCl2 for 8 min during sterilization can chemically scarify seeds and induce molecular changes due to the entry of Hg2+ during water imbibition. This chemical scarification helps break seed dormancy and promotes more efficient germination. As noted previously, the use of sterilants not only sterilizes seeds but could also potentially aid germination by neutralizing germination inhibitors[33,43]. The results of this study suggest that an efficient method of seed sterilization can weaken the seed coat and indirectly promote chemical scarification. This is a necessary step for speeding up the process of breaking seed dormancy when combined with a specific formulation of GA3 and other plant growth regulators, thus improving the effectiveness of seed germination (Fig. 6). In a prior study comparing the efficacy of potassium nitrate and gibberellic acid in breaking seed dormancy in both coated and uncoated seeds of Agrimonia eupatoria L., researchers found that the treatments were more effective in promoting seed germination in uncoated seeds compared to coated ones. Furthermore, the study indicated that combining seed coat removal with potassium nitrate application not only increased seed germination rates but also enhanced seedling length[50]. The authors hypothesized that potassium ions from potassium nitrate enhanced cell wall permeability, leading to heightened enzyme activity and cellular metabolism. Their research also suggested that potassium nitrate might help seeds germinate by changing the balance of hormones inside the seed, which could lower inhibitors like abscisic acid and end physiological dormancy[50]. Another prior study highlighted contrasting responses in Vigna radiata (mungbean) when exposed to mercury stress, with seedling growth and root elongation proving more sensitive compared to seed germination[51]. Mercury treatments across all concentrations significantly reduced seed germination, shoot and root length, and seedling dry weight relative to the control, indicating a detrimental impact on mungbean germination and growth[51].
Recent studies underscore the critical role of aquaporins in seed imbibition and subsequent germination[52]. It has been demonstrated that abiotic stresses, including water deficit, salinity, and heavy metals, modulate the expression of aquaporins across different plant organs[53]. These membrane proteins are particularly abundant in regions of cell division and enlargement, making them critical for facilitating water transport between adjacent cells during seed germination. Seed aquaporin activity closely links to water uptake and the initiation of metabolic processes within the seed, thereby promoting enhanced germination rates[52]. Furthermore, heavy metals such as cadmium (Cd), copper (Cu), and mercury (Hg) can significantly impact the gene expression and function of D-myo-inositol-3-phosphate synthase (MIPS) in plants[53]. MIPS is essential for de novo inositol synthesis and is highly expressed in developing seeds. It plays a critical role in stress responses and regulates the synthesis of phytate, the major phosphorus storage compound in seeds. Moreover, MIPS is integral to complex plant stress response mechanisms and participates in various biochemical and physiological processes, including intracellular signal transduction, membrane construction, protein anchoring, cell wall construction, and auxin storage and trafficking within plant seeds[53]. Arabidopsis possesses three MIPS genes, with MIPS1 being the most extensively characterized. Researchers have shown that a loss-of-function mutant in mips1 causes deformed cotyledon development[54]. Among the three genes, MIPS1 exhibits the highest expression during seed development. Double mutants, mips1 mips2+/− and mips1 mips3, display severe embryogenesis defects, leading to altered cotyledon numbers and deformed shapes. Homozygous triple mutants are embryonically lethal, underscoring the critical role of de novo synthesis of myo-inositol in proper development[54].
These findings highlight the differential impact of heavy metal ions on pathways associated with seed germination and seedling growth. While germinating seeds are highly sensitive and their germination can be inhibited by heavy metals like mercury, which block aquaporins, these metals also can modulate gene expression and the function of myo-inositol phosphate synthase (MIPS) in various plant species. The potential blocking effect of aquaporins in seeds due to prolonged sterilization in mercuric chloride could have been mitigated or negated by the in vitro growth conditions. The gel-like consistency of the media plays a crucial role in regulating water absorption by seeds by retaining water, maintaining surface moisture, aiding nutrient transport, and ensuring a stable pH. This controlled environment is essential for seed germination and early seedling growth. Furthermore, the density of the gel in plant tissue culture media significantly influences seed germination rates by managing water absorption and moisture levels. In this study, the gel density was optimal, as excessively dense gels can limit oxygen availability and gas exchange, while moderate gel loading enhances germination speed and synchrony. Achieving the optimal gel density was critical for balancing moisture and aeration, thereby ensuring optimal seed growth and germination[55]. Furthermore, depending on the plant species and genotype, this modulation can potentially bypass the aquaporin-blocking effect, resulting in either positive or negative impacts on seed germination and seedling growth[52]. For instance, in Pisum sativum, the reduction of root hydraulic conductivity (Lpr) due to HgCl2 treatment was accompanied by an increase in the expression of plasma membrane intrinsic protein (PIP), suggesting a compensatory mechanism for the blocked aquaporins. Conversely, in Populus deltoides roots subjected to copper stress, genes encoding plasmalemma (PIP) and tonoplast (TIP) aquaporins were downregulated under Cu application[52]. Although the specific effect of HgCl2 treatment on MIPS in Gloriosa superba L. seeds was not evaluated through molecular studies, it is plausible that Hg ions played a critical role in upregulating MIPS pathway-related genes in these seeds. This upregulation could have significantly influenced seed germination, thereby enhancing subsequent seedling growth and development.
Effect of plant growth regulators on the development of Gloriosa superba L. in vitro seedling morphological traits
-
The present study aimed to evaluate the impact of different combinations of various types and concentrations of plant growth regulator (PGR) treatments on seedling growth parameters. Under carefully controlled in vitro conditions, seedlings grew much faster after being treated with plant growth regulators (PGRs) as compared with the group that did not receive PGR treatment. It was observed that adding 1.5 mg·L−1 BAP and 1.0 mg·L−1 NAA to MS media with 30 g·L−1 sucrose and a 16-h photoperiod enhanced seedling growth after 4 weeks of culture. This treatment significantly improved seedling length, seedling root length, and seedling biomass. This result shows that, when used at the right concentrations, the combined impact of BAP and NAA on the growth of Gloriosa superba L. seedlings is greater than that of 2iP and IAA (Fig. 8a−f). A previous study on Cymbidium aloifolium L. found that adding BAP and NAA to the MS medium accelerated seedling growth compared to when the medium lacked plant growth regulators[56]. For Scutellaria bornmuelleri, the optimal plant growth regulator combination for growth and development was TDZ and BAP[57]. Conversely, 2iP and IAA were the most effective in achieving direct organogenesis. This highlights the varied impacts that different types and combinations of plant growth regulators can have on the growth and developmental processes of different plant species[57].
Effect of plant growth regulators on in vitro rooting of Gloriosa superba L. microshoots
-
The experiment on seedling growth showed that the best combination of plant growth regulators (1.5 mg·L−1 BAP and 1.0 mg·L−1 NAA in MS media with 30 g·L−1 sucrose and a 16-h photoperiod) made many seedling traits much better. The only trait that wasn't significantly better was the shoot-to-root ratio, which was the same as the control (Fig. 8c). The seedling roots showed a non-adventitious characteristic, with an average length of 4.08 cm, and had limited growth. This observation fits with how the species usually germinates in the hypogeum, which is to focus on the first growth of the tuberous hypopodium while the roots of the seedling grow. These tubers serve as crucial nutrient stores for the plant, housing inactive buds that subsequently become active, resulting in the emergence of new shoots and root systems. Therefore, following the results of the seedling growth experiment, it was determined in this study to excise the shoots of the fully grown young seedlings. Subsequently, these shoots underwent a rooting treatment to promote further root growth before proceeding with the hardening procedure.
Several studies have looked at how auxin-group hormones (IAA, IBA, and NAA) affect how roots form and how plants grow in general[58]. This study examined the effect of auxin hormones on root induction and development in microshoots derived from in vitro-grown Gloriosa superba L. seedlings. This study emphasized the efficacy of 1.0 mg·L−1 IBA for rooting. This concentration exhibited the fastest rate of root formation and the shortest time for root initiation, and it performed exceptionally well in terms of average root length and the number of roots per explant. This observation aligns with the findings documented in prior investigations[59,60]. The process of shoot excision and rooting demonstrated exceptional efficacy. Although the average root length yielded comparable outcomes to the seedling development experiment, this method resulted in the growth of robust adventitious roots. Significantly, the quantity of roots produced per shoot was considerably greater in comparison to the initial seedling development experiment (Fig. 10e).
Ex vitro acclimatization and plant growth
-
Hardening is a crucial stage in the multiplication of tissue culture plants. It entails gradually acclimating plantlets from laboratory settings to soil, which signifies the ultimate achievement of the procedure. The plantlets, which had developed strong shoots and roots, were withdrawn and thoroughly washed to remove any agar residue. They were then transplanted into vessels filled with a sterile mixture of vermiculite and soil in a 1:1 volume ratio. The containers were initially placed in a controlled culture environment for 2 weeks, during which they adapted to their new substrate. Afterward, they spent an extra week in a net house with shade, where they were planted in a mixture of garden soil, sand, and vermiculite at a ratio of 2 parts soil to 1 part sand to 1 part vermiculite (volume/volume). Surprisingly, all plantlets survived without exception during this time of acclimation. However, after being transplanted onto the field around 11 weeks later, the survival rate decreased to 60%. According to Mosoh et al.[29], the main reason for the drop in survival rates is that the plantlets are exposed to a lot more photosynthetically active radiation (PAR) when they move from vessels with garden soil, sand, and vermiculite in the shaded net house to the open field. Moreover, it is possible that the three-week time adjustment was insufficient to provide the required resilience for immediate transplanting into the field. This situation corresponds to the more general occurrence observed when plants produced in a controlled environment are transferred to soil and exposed to conditions outside of the controlled environment. This can lead to reductions in plant survival rates due to changes in the environment that have a negative impact[29]. Notably, there were no noticeable variations in the physical or developmental traits of the surviving plantlets.
-
In this study, sterilizing with 0.15% mercuric chloride (HgCl2) for 8 min removed all contaminants and had an amazing 100% survival rate for the seedlings. This led to the production of seeds that were free of pests and had strong seedling growth. The best treatment used Murashige and Skoog (MS) medium with 1.5 mg·L−1 GA3 and 1.5 mg·L−1 BAP, along with 4% sucrose and a 16-h photoperiod. This resulted in great seed germination (9.25 out of 12 seed explants) and a great overall seedling survival rate of 77.08% after a month. Also, seedlings that were 2 weeks old were grown on MS medium with 1.5 mg·L−1 BAP and 1.0 mg·L−1 NAA, 30 g·L−1 sucrose, and a 16-h photoperiod. After 4 weeks, the seedlings were the longest (5.83 cm), and the roots were the longest (4.08 cm). Moving cut seedling shoots to half-strength MS medium with 1.0 mg·L−1 IBA increased the rate of root formation by 84.37% and helped roots grow even more. Subsequently, these in vitro-grown plantlets were successfully acclimatized and transplanted under field conditions, achieving a commendable 60% survival rate after 11 weeks.
Effective sterilization of seeds is crucial for removing contaminants and facilitating important processes such as seed coat softening and chemical scarification. Integration with plant growth regulators accelerates the breaking of seed dormancy, thereby enhancing germination rates. Precise seed treatment protocols are essential for advancing seed propagation and conservation practices in plant science. This study provides a robust methodology for the mass in vitro propagation of Gloriosa superba L., alleviating strain on the species' wild populations. Additionally, evaluating supply chains, market dynamics, and prospects is likely to benefit both small-scale stakeholders and the phytopharmaceutical industry.
In the future, it is crucial to prioritize research that thoroughly investigates the complex mechanistic elements underlying the effectiveness of sterilization in breaking seed dormancy. This research should prioritize elucidating the intricate molecular mechanisms responsible for seed dormancy release and the enhancement of germination facilitated by heavy metal ions, such as Hg2+, and potentially other metals. Understanding these mechanisms is pivotal for advancing our comprehension of how environmental factors impact seed physiology and germination processes, with implications spanning agriculture, conservation, and ecosystem management. By delving into seed biology at a molecular level, such studies will lay the groundwork for developing precise treatment protocols. This, in turn, promises to elevate germination success rates and refine propagation strategies, thereby bolstering efforts in sustainable agriculture and biodiversity conservation.
-
The authors confirm their contribution to the paper as follows: study conceptualization and design: Khandel AK; data collection and curation: Mosoh DA, Khandel AK; analysis and interpretation of results: Mosoh DA; funding Acquisition: Mosoh DA, Khandel AK, Vendrame WA; investigation: Mosoh DA, Khandel AK, Verma SK, Vendrame WA; methodology: Mosoh DA, Khandel AK, Verma SK; project management: Mosoh DA, Khandel AK; resources: Mosoh DA, Khandel AK; software: Mosoh DA; supervision: Mosoh DA, Khandel AK, Verma SK, Vendrame WA; validation: Mosoh DA, Khandel AK, Vendrame WA; visualization: Mosoh DA; original draft manuscript preparation: Mosoh DA; revised – manuscript: Mosoh DA, Vendrame WA. All authors reviewed the results and approved the final version of the manuscript.
-
The data that support the findings of this study are available on request from the corresponding author.
The USDA National Institute of Food and Agriculture has provided support for this study, specifically under Hatch project 7001563. In the early stages of the project, Dr. Rohit Sharma from the Centre for Biodiversity Exploration and Conservation (CBEC) provided crucial support, for which we are grateful. His contributions have played a crucial role in determining the course of our research. We express our profound gratitude to Mr. Tetu Acha Samuel (MD, USA) and his colleague, Mr. Adamou Musa (TX, USA), for their prompt assistance with appropriation.
-
The authors declare that they have no conflict of interest.
-
Received 29 May 2024; Accepted 29 July 2024; Published online 12 September 2024
-
Gloriosa superba L. is a critically endangered tropical plant with significant medicinal, cultural, and ornamental value. However, seed dormancy represents a significant barrier to natural regeneration in wild plant populations.
This study found that sterilizing seeds with 0.15% mercuric chloride (HgCl2) for 8 minutes effectively eliminated contamination, ensuring 100% seedling survival.
Pre-treatment with HgCl2 weakened the seed coat and induced chemical scarification, enhancing in vitro seed germination. Moreover, the imbibed Hg2+ ions likely induced the upregulation of MIPS genes in the seeds, which subsequently complemented the action of plant growth regulators (PGRs) in enhancing seed germination, seedling growth, and development.
The combination of GA3 and BAP was most effective for in vitro seed germination. The combination of BAP and NAA was most effective for seedling development and elongation, while IBA was the best for rooting microshoots derived from seedlings.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Mosoh DA, Khandel AK, Verma SK, Vendrame WA. 2024. Overcoming dual seed dormancy and enhancing in vitro seedling development of Gloriosa superba (L.) with a targeted sterilization approach and plant growth regulator synergy. Tropical Plants 3: e031 doi: 10.48130/tp-0024-0033 

Overcoming dual seed dormancy and enhancing in vitro seedling development of Gloriosa superba (L.) with a targeted sterilization approach and plant growth regulator synergy
- Received: 29 May 2024
- Revised: 13 July 2024
- Accepted: 29 July 2024
- Published online: 12 September 2024
Abstract: The status of Gloriosa superba L. in the wild has been declining due to over-collection and habitat destruction. Intrinsic severe seed dormancy and low germination rates hinder the cultivation process. To conserve this plant, in vitro culture protocols have been developed to enhance seed germination and seedling growth. An effective sterilization method involving 0.15% mercuric chloride (HgCl2) for 8 min was found to eliminate contamination and yield a 100% survival rate, resulting in disinfested seeds and robust seedling growth. The most successful treatment consisted of Murashige and Skoog (MS) medium with 1.5 mg·L−1 GA3 and 1.5 mg·L−1 BAP, along with 4% sucrose under a 16-h photoperiod, which achieved the highest average in vitro seed germination (9.25 out of 12 seed explants) and an impressive overall seedling survival rate of 77.08% after 30 d. Subsequent growth of two-week-old seedlings on MS medium with 1.5 mg·L−1 BAP and 1.0 mg·L−1 NAA supplemented with 30 g·L−1 sucrose and a 16-h photoperiod resulted in the greatest average seedling length (5.83 cm) and seedling root length (4.08 cm) after four weeks. Transferring the excised shoots of in vitro-grown seedlings to half-strength MS medium with 1.0 mg·L−1 IBA led to maximum root induction (84.37%) and further enhanced root development. These in vitro-grown plantlets were successfully acclimatized and transplanted under field conditions, with a 60% survival rate after 11 weeks.