






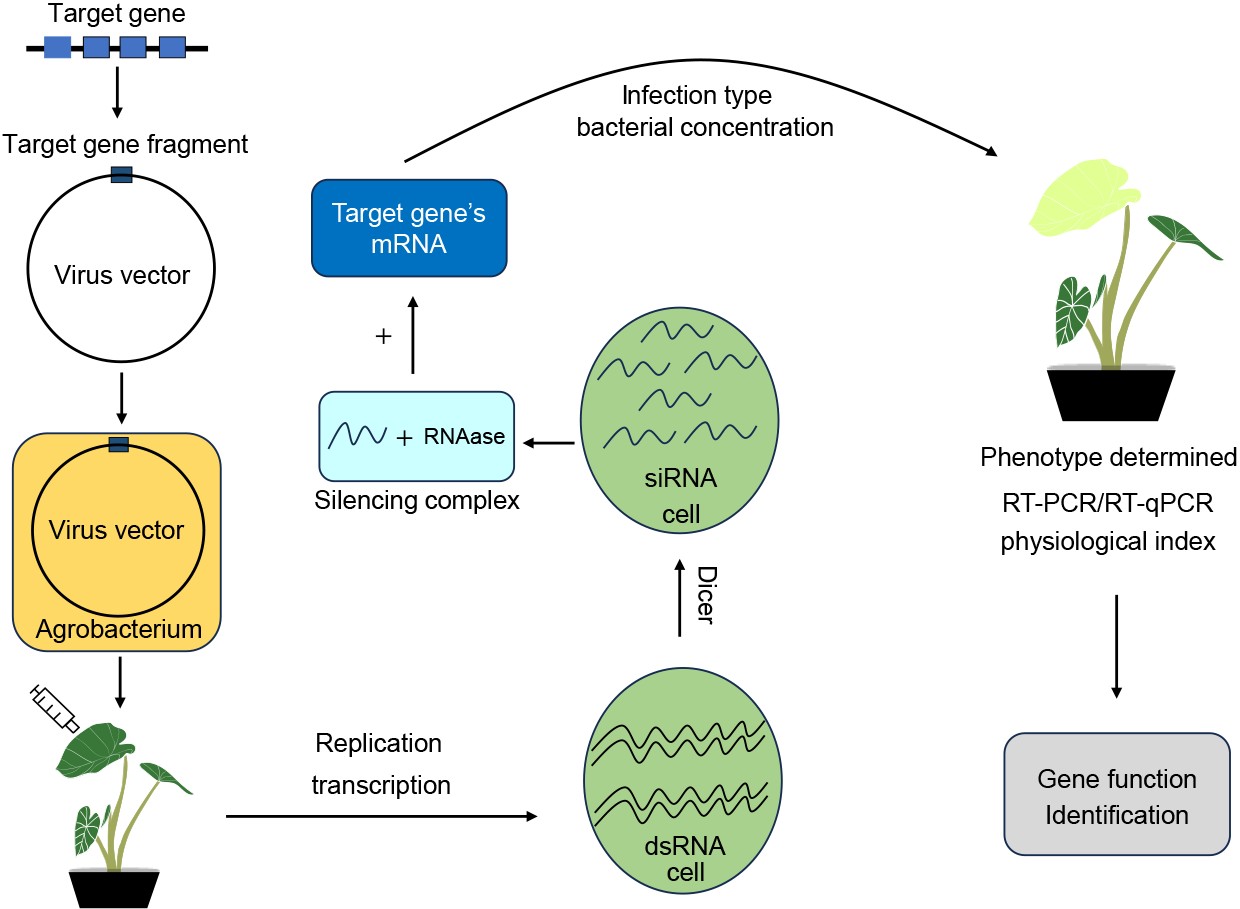



-
Taro (Colocasia esculenta (L.) Schott), from the Araceae family, is a tropical and subtropical crop, which is commonly used as food and for the industrial processing of raw material with high economic benefits. It ranks 5th among root crops and 14th among vegetable crops worldwide[1], and world annual production of taro in 2022 was estimated at 19.67 million tons (FAOSTAT 2024). Taro is typically propagated from corms with a long life cycle (harvested usually around 6 to 12 months after planting) that lead scientists to largely overlook it.
Taro is rich in mucus protein, polysaccharides, vitamins, mineral elements, and many other physiologically active substances, which have been applied to alleviate a variety of human health symptoms[2]. It can be predicted that taro will be a promising crop. In recent years, taro and its products are favored by consumers because of their unique nutritional value and healthcare functions, higher requirements are also put forward for new varieties, and clear gene function will lay the foundation for high-quality new variety breeding to further meet consumer needs.
As research continues, the elucidation of gene function is urgent, but the genetic transformation system is still unstable in taro, which hinders the gene functional verification and greatly limits the utilization of excellent taro germplasm resources. An effective gene function verification system is urgently required. Due to virus-induced gene silencing (VIGS) advantages of stability, high efficiency, simplicity, low cost, high throughput and short cycle, it has been widely applied in the gene function research of vegetable crops[3]. The robust VIGS system establishment in taro can not only verify gene function, but also greatly promote the utilization of germplasm resources, which has an important role in promoting the improvement and breeding of taro.
At present, VIGS technology has been widely applied in the study of gene function related to growth and development, stress, substance synthesis, and metabolic regulation of vegetable crops[3−5]. However, the silencing efficiency induced by viral vectors is host-depended. Based on previous studies, several viral vectors have been successfully applied in VIGS, including tobacco mosaic virus (TMV), potato virus X (PVX), tobacco rattle virus (TRV), tomato golden mosaic virus (TGMV), and cabbage leaf curl virus (CbLCV)[6]. The TRV virus vector has been widely used in gene silencing experiments because of its high silencing efficiency, long silencing duration, and mild virus symptoms[7,8]. During the VIGS system establishment process, phytoene desaturase (PDS) is often selected as the target gene to indicate the success of gene silencing.
TCP TFs are named after the first known members (TB1, CYC, and PCFs) that share a highly conserved TCP domain, which harbors a non-canonical basic-helix-loop-helix (bHLH) structural motif[9,10]. They are classified into two divergent groups; TCP class I and TCP class II, the latter is further divided into two clades, CIN and CYC/TB1[9,11]. As a plant-specific transcription factor, they play a crucial role in plant growth and development, including regulating flower organ development, leaf morphology and lateral branch growth[11]. Currently available evidence has shown that most TCP class I genes are activators of plant development, whereas TCP class II members often function as a repressor of various growth and development pathways[12−14]. The function of TCP TFs in taro have however not yet been reported.
Although VIGS technology has been widely used to verify plant gene function, there are no reports of VIGS technology in taro. The establishment of a taro VIGS technology system would lay the foundation for rapid verification of gene function, and clarify the gene function that controls important agronomic traits in taro, which would promote germplasm resource utilization and taro breeding improvement.
-
Ganyu No. 1 and Ganyu No. 2 were used for VIGS system construction, corms with 1 cm diameter were used as test materials. pTRV1 and pTRV2 were selected as VIGS vectors and stored in the laboratory (Fig. 1a & b). Escherichia coli DH5α and Agrobacterium GV3101 receptor cells were purchased from Huayueyang Biotechnology Co., Ltd (Beijing, China).
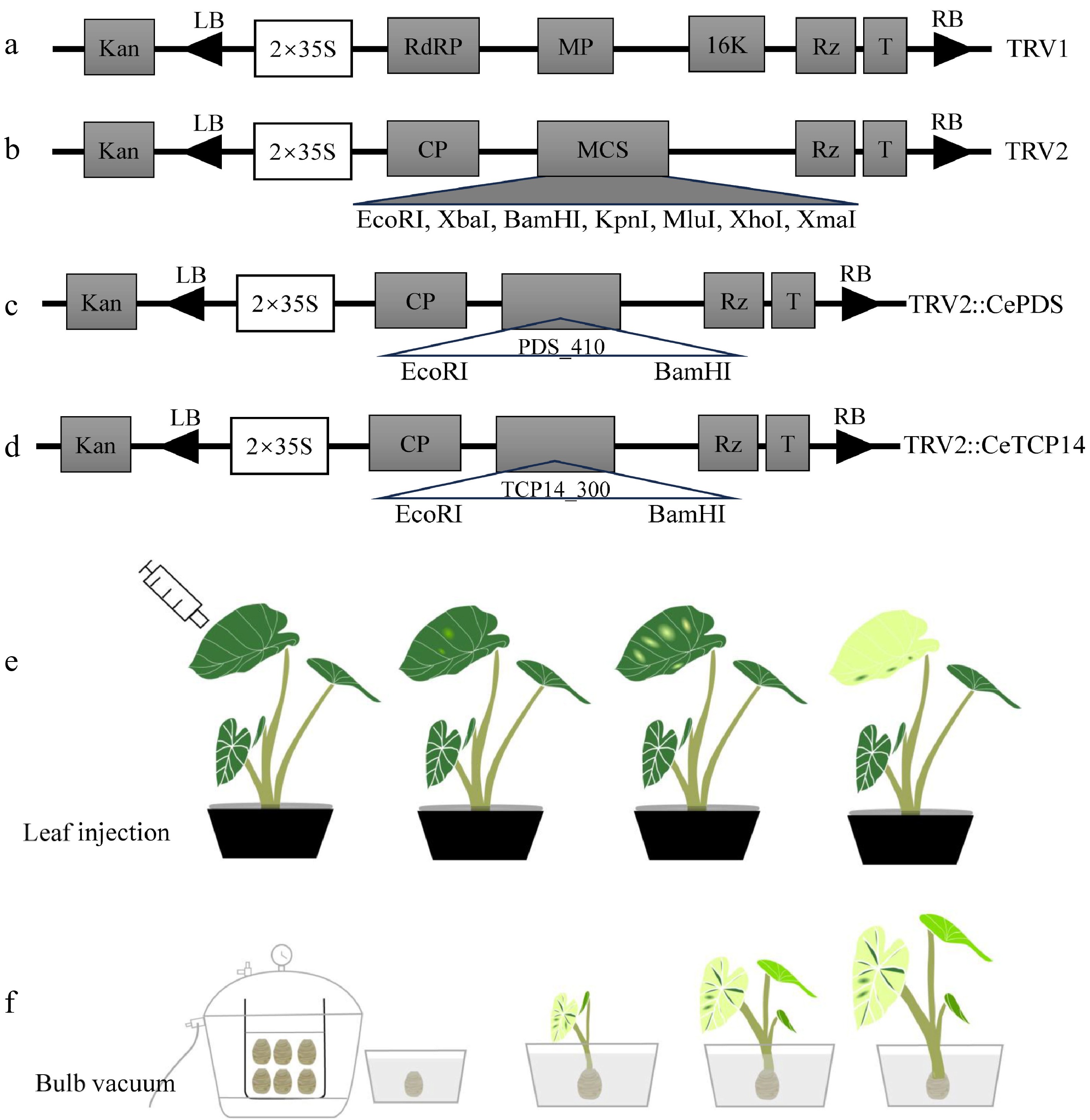
Figure 1.
Vectors used and infection flow chart in this study. The plasmid profile of (a) pTRV1, (b) pTRV2, (c) pTRV2::PDS410, (d) pTRV2::CeTCP14. Color change of leaves by (e) leaf injection and (f) bulb vacuum injection methods.
Vector construction for VIGS
-
First, the protein sequence of SlPDS (Solyc03g123760) from tomato and AtPDS (AT4G14210) from Arabidopsis were used to blast in the taro reference genome Taro_JAAS_v1.0 (BioProject: PRJNA587719)[15]. Then, the obtained sequences were validated in the transcriptome of Ganyu No. 1, which has been published previously[16]. Finally, the CePDS (EVM0008568.1) sequence with 1,743 bp was obtained and verified, CeTCP14 (EVM0000825.2) was screened out based on RNA-seq analysis, the gene sequences are shown in the Supplemental File 1. The mRNA from young leaves was reverse transcribed into cDNA, and then used as a template, the primers used in this study are listed in Table 1, pTRV2-CePDS was constructed by restriction enzymes (EcoRI and BamHI), pTRV2-CeTCP14 was constructed by homologous recombination, and the vector construction was verified by sanger sequencing (Fig. 1c & d).
Table 1. Primers used in this study
Primer name Primer sequence (5'−3') Length (bp) Purpose CePDS-EcoRI-F GGAATTCATGGGCTTTACCAGTTCTCTTTCGG 410 Used for fragments amplified of CePDS and CeTCP14 inserted in TRV2 vector CePDS-BamHI-R CGGGATCCTCCAGCAATATAGGCTTATGACCTG TRV2-TCP14_F GTGAGTAAGGTTACCGAATTCATGGGGGAGAGCCACCAG 300 TRV2-TCP14_R CGTGAGCTCGGTACCGGATCCATCGACGGCCTTGCTGGG qCePDS-F GGTCGTTGGGGAGGAAGC 140 Used for the qRT-PCR of CePDS qCePDS-R TCTAGTCGGGCGTGGTGA qCeTCP14_F CCACACCGCCATCCAGTT 110 Used for the qRT-PCR of CeTCP14 qP_TCP14_R CGAGCTCGTCTATGGCGG CeActinF CTAGTGGTCGCACAACAGGT 191 Used for the qRT-PCR of reference genes CeActinR TTCACGCTCAGCAGTGGTAG TRV1F1 CGTGTTGCATTTCGATGAA 525 Used for the detection of RdRp in TRV1 TRV1R1 GACAACGCCACGATTAAGT TRV2F1 GTTGAAGAAGTTACACAGCA 407 Used for the detection of coat protein in TRV2 TRV2R1 TCTTCAACTCCATGTTCTCT pTRV2_F TGTCAACAAAGATGGACATTGTTAC 198 / 480 Used for the detection of TRV2 and TRV2-CeTCP14 expression pTRV2_R ACACGGATCTACTTAAAGAA TRV2_F TGTTACTCAAGGAAGCACGATGAGCT − Used for vector construction sequencing and colony PCR detection TRV2_R GTACAGACGGGCGTAATAACGCTTA Note: underline indicates cleavage sites, bold indicates protective bases. Vector transformation
-
Plasmids pTRV1, pTRV2, TRV2-CePDS and pTRV2-CeTCP14 were transferred into agrobacteria GV3101 using the freeze-thaw method[17]. First, 5 ml YEB solution containing rifampicin (25 mg/L) and kanamycin (50 mg/L) was used to culture bacteria by shaking, and colony PCR detection was performed with specific primers to obtain positive monoclones (Table 1). Then 500 μL of bacteria solution was transferred to 50 mL of YEB solution containing MES (10 mM), acetosyringone (20 μM) and corresponding antibiotics for overnight culture. The bacteria were centrifuged at 3000× when the OD reached 1.2, and collected with MS containing MES (10 mM), acetyl syringone (200 μM) and MgCl2 (10 mM) at pH = 5.6 and then stood at room temperature for 1−2 h. Afterwards, pTRV1 was mixed with pTRV2, TRV2-CePDS and PTRV2-CeTCP14 resuspension in equal volume respectively, the mixed solution was used for infection in subsequent research.
Agrobacterium infection
-
Leaf injection[18] and bulb vacuum infiltration[8] were applied in agrobacterium infection. Infection solution containing these four combinations: pTRV1 + pTRV2, pTRV1 + pTRV2-PDS, pTRV1 + pTRV2-CeTCP14, negative control (suspensions). In the leaf injection method, 1 mL syringe without needle was used, and plant material with 2−3 fully unfolded leaves were selected for infection material, about 1/3−1/2 of the leaf area were injected, and then cultured in a seedling room with 16 h of light and 8 h of darkness at 22−24 °C (Fig. 1e). For the bulb vacuum method, the cleaned corms were punctured by needles that facilitate infection liquid entry, the pressure was 750 mm Hg for 30 min, then the surface of the taro was washed after infection. The plants were planted in pots (10 cm × 10 cm) and moved to the seedling room (Fig. 1f). The temperature and light conditions were the same as those for the leaf injection method. Leaf color change was continuously observed.
Determination of gene expression, chlorophyll content, and starch content
-
After 20 d of infection, the photobleaching phenotype appeared, the leaves/corms sample were collected, the RNA was extracted using the polysaccharide polyphenol plant RNA extraction kit (0416-50), Huayueyang Biotechnology Co. Ltd (Beijing, China), and the expression levels of CePDS and CeTCP14 were detected by RT-qPCR, actin with stable expression level was selected as the internal reference, and the relative expression level of genes was normalized by the 2−ΔΔCᴛ method with three biological repeats[19]. The primers are listed in Table 1.
After the photobleaching phenotype occurred in the VIGS-CePDS group, leaves without coarse leaf veins were collected. The samples were cut into small strips, and about 0.2 g of fresh samples were placed into 50 mL centrifuge tubes with three biological repeats, 25 mL of 95% ethanol was added, and left to stand for 36 h until the leaves become white under dark conditions. The well-mixed supernatant was taken for spectrophotometer determination at wavelengths of 649 and 665 nm respectively, OD values, A649 and A665 were recorded. chlorophyll a and chlorophyll b were calculated as follows: Ca = 13.95 × A665 − 6.88 × A649, Cb = 24.96 × A649−7.32 × A665, total chlorophyll content = Ca + Cb[19].
After 20 d of VIGS-CeTCP14 suspension infection, 0.5 g fresh corms flesh samples at the same sites were collected to determine starch content with three biological repeats. The methods were as those reported by Gao[20].
-
To confirm whether the TRV-based vector could induce gene silencing in taro, Ganyu No. 1 was selected as material for preliminary testing with CePDS as the indicator by leaf injection at OD600 = 0.6. After 20 d, the leaves began to show photobleaching phenomenon (Fig. 2b), about 10 d later, almost the entire leaf turned white (Fig. 2c), there was no colour change in the negative control group and the empty-vector group (Fig. 2a, d). Then the total number of plants showing the photobleaching phenomenon were counted, and the average value was 3.67 (30 plants each time with three biological replicates). Therefore, the TRV-based vector could induce gene silencing in taro, but the proportion with photobleaching phenotype was low (12.23%).
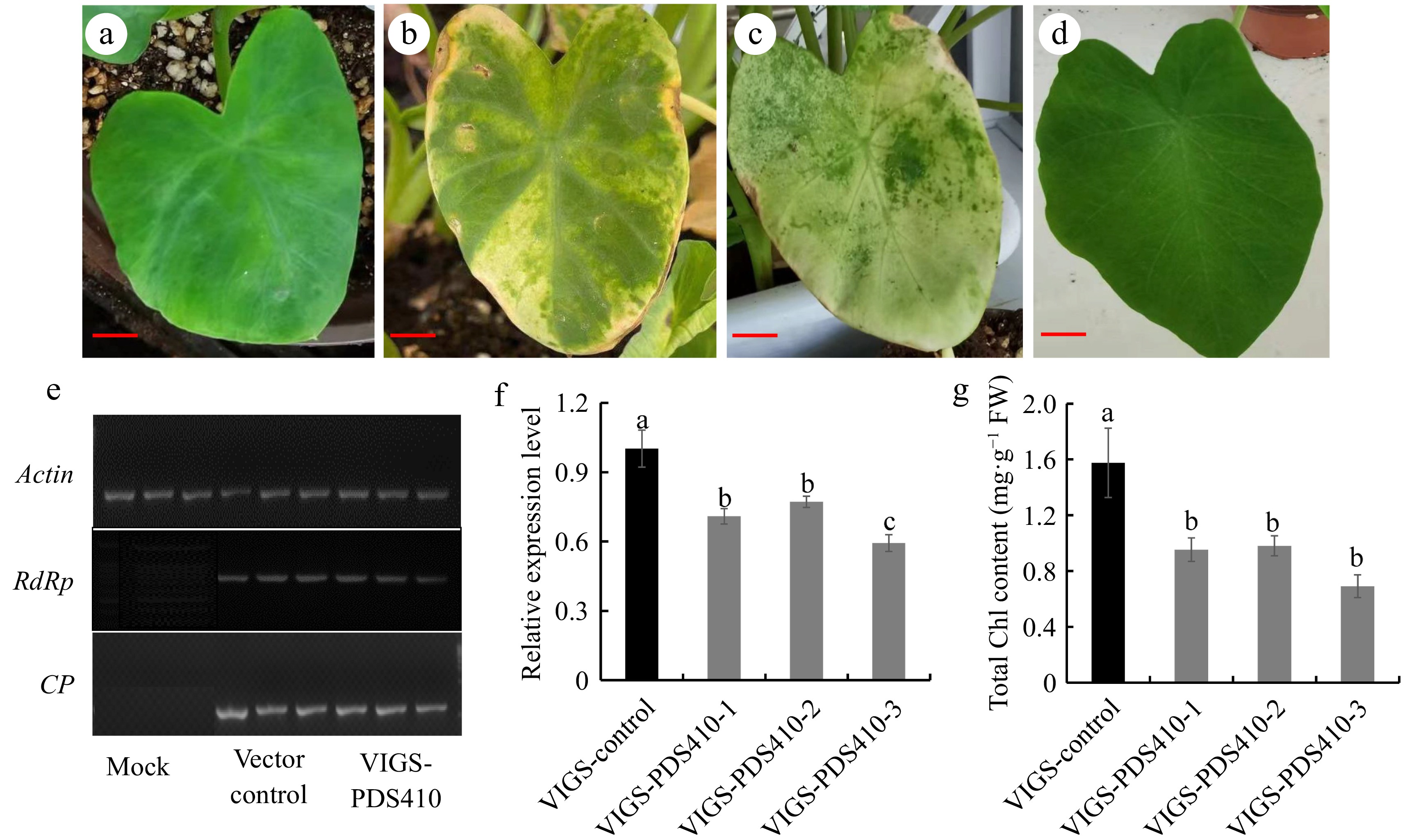
Figure 2.
VIGS system constructing in taro. (a) Empty vector phenotype at 20 d of leaf infection with OD600 = 0.6, (b) VIGS-PDS410 phenotype at 20d of leaf infection OD600 = 0.6, (c) VIGS-PDS410 phenotype at 30 d of leaf infection with OD600 = 0.6, (d) mock phenotype after 20 d infection, (e) RdRp gene expression in TRV1 and CP gene expression in TRV2 were detected by RT-PCR with actin as the internal reference, (f) detection of CePDS expression level for photobleaching phenotype plants, (g) chlorophyll content detection for photobleaching phenotype plants. Scale bars = 3 cm.
Detection of CePDS silent seedlings in taro
-
To verify that the phenotypic changes were caused by endogenous CePDS silencing through RNA expression derived from pTRV1 and pTRV2-PDS, RT-PCR was applied for detection. Transcriptional analysis of pTRV1 coat protein (CP) and RNA-dependent RNA polymerase (RdRp) from pTRV2 confirmed the successful expression of pTRV1 and pTRV2 vectors in taro leaves (Fig 2e). The expression level of CePDS were about 59.34%−77.18% in VIGS-CePDS plants compared to the control through further detection (Fig. 2f), and the chlorophyll content decreased by 37.80%−56.11% in VIGS-CePDS plants (Fig. 2g). Phenotypic and molecular results of VIGS-CePDS seedlings clearly showed that CePDS expression in taro was targeted and silenced after VIGS, but the number of silenced lines was low and needed to be further improved.
VIGS system optimization in taro
-
Studies have shown that factors such as infecting solution concentration and infecting method would influence the silencing effect[8,18,21]. The VIGS system was therefore optimized based on these two factors. Based on photobleaching phenotype, the results showed that when OD600 = 1.0, the number of silent lines was the largest compared to OD600 = 0.6 and OD600 = 0.8, the proportion reached 27.77%, but there was no significant difference compared with D600 = 1.2 at 30 d after infection (Table 2). Subsequently, the bulb vacuum infection and leaf injection infection were compared, the number of photobleached lines showed no significant difference (Table 3). The above results indicate that the concentration of infective bacterial solution was a key factor affecting gene silencing in taro.
Table 2. Bacterial concentrations optimization for the VIGS system.
OD600 Total number of inoculation plants Number of photobleaching phenotype The percentage of
photobleaching phenotype0 10 0 d 0 0.6 30 3.00 ± 1.00 c 10% 0.8 30 5.33 ± 0.58 b 17.77% 1.0 30 8.33 ± 0.58 a 27.77% 1.2 30 7.67 ± 0.58 a 25.57% Table 3. Comparison of the VIGS system based on infection type.
Infection type Total number of inoculation plants Number of photobleaching phenotype The percentage of
photobleaching phenotypeLeaf injection 30 8.33 ± 0.58 a 27.77% Bulb evacuation 30 8.00 ± 0.58 a 26.67% Verification of the function of CeTCP14 based on the established VIGS system
-
In our previous work, it was found that CeTCP14 presented obvious upregulated trend during the early stage of corm expansion, this gene was further selected to verify the robustness of the constructed VIGS system. CeTCP14 was silenced with OD600 = 1.0 and the bulb vacuum infiltration method to identify its function. RT-PCR results confirmed the successful expression of pTRV1 and pTRV2 vectors in taro corms (Fig. 3a), the expression level of CeTCP14 in VIGS-CeTCP14 plants decreased by 36.66%~56.06% (Fig. 3b), this resulted in significant starch content decrease, only 70.88%~80.61% of the control (Fig. 3c). In maize, ZmTCP7 regulated the starch content through binding to the ZmBt2 promoter[22], CeTCP14 and ZmTCP7 belong to the same TCP class II, the factors interacting with CeTCP14 in taro to regulate starch content need further study.
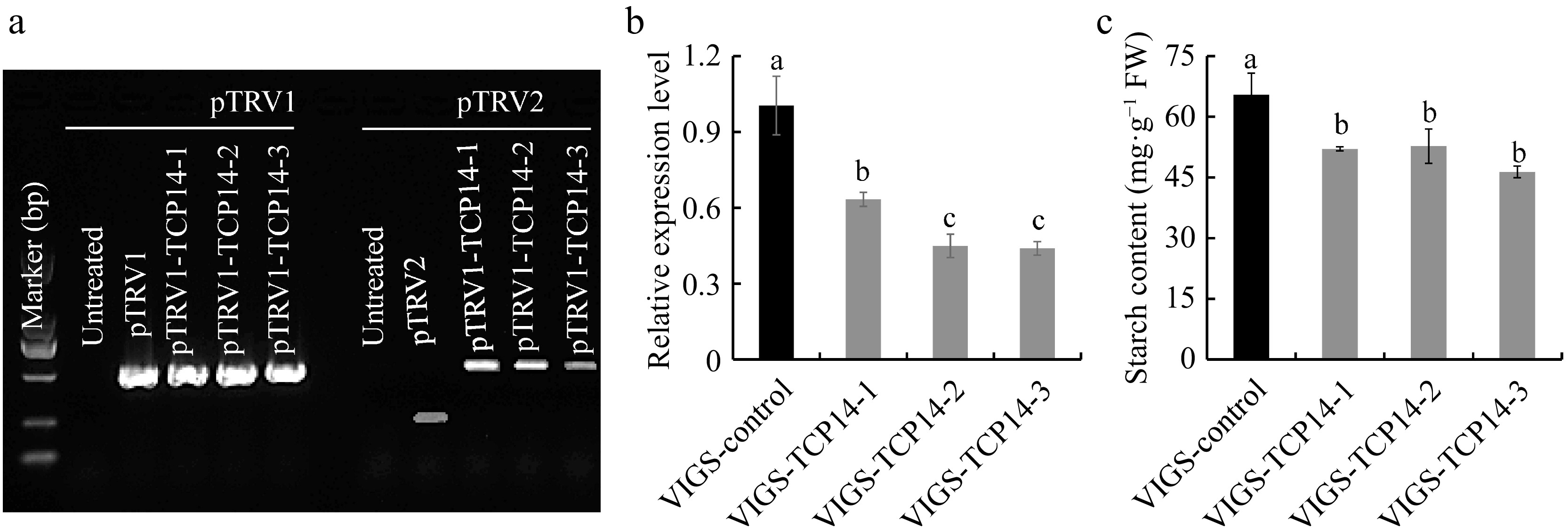
Figure 3.
Detection of taro VIGS system for CeTCP14 in Ganyu No. 2. (a) RdRp gene expression in pTRV1 and fragement expression in pTRV2 were detected by RT-PCR. (b) Detection of CeTCP14 expression at 20 d after bulb vacuum infiltration. (c) Starch content detection for VIGS-CeTCP14 plants.
-
The genome of taro has been previously reported[23], which facilitates the gene mining that controls important agronomic traits. CePDS was cloned as an indicator gene from taro based on homologous alignment and a VIGS system was constructed, for color changes are the most intuitive way after gene silencing or gene editing[19,24−26].
Virus silence-inducing vector is another important factor affecting the success of VIGS system construction. Although a variety of viral vectors have been used in VIGS, including RNA viruses, DNA viruses, and satellite viruses, the silencing efficiency induced by each viral vector is host-dependent. For example, the potato virus X (PVX) is an RNA virus, studies in tomatoes have found that in vitro transcription is required before RNA infection[27]. DNA viral vectors are simple to construct, easy to operate, and do not require in vitro transcription, such as beet curl top virus (BCTV) and tomato curl leaf virus (ToLCV) can be effectively used for gene silencing[28,29]. Satellite viruses have a small genome, replicate quickly in the host and are easy to inherit. Chinese tomato yellow leaf curl virus (TYLCV) and tobacco stalk curl virus (TbCSV) of these virus vectors have been successfully used to verify gene function[30,31]. The virus-silencing induction vector selected in this study is TRV, which belongs to the RNA virus and is widely used because of its long silencing time, mild virus symptoms and high silencing efficiency[7,8]. In this study, the TRV-based vector could produce silencing effects in Ganyu No. 1 and Ganyu No. 2 which further verified that the TRV vector is suitable for gene silencing in taro, but the percentage of photobleaching phenotype is relatively low compared to banana. In banana VIGS research, another RNA virus, cucumber mosaic virus (CMV) performed better gene silencing effect with a 95% infection rate and reduced target gene transcripts to 10%−18% of the control, the success of developing a banana VIGS system could be partially credited to the choice of utilizing a CMV isolate naturally infecting bananas during its construction[32], while in taro, TRV is not a serious taro-hosting virus, and greatly affected the infection rate, one way that can be used to improve the silencing effect is to increase the concentration of bacterial solution. It is also reported that CMV could infect taro[16], both banana and taro are monocots and exhibit similar growth habits, it might be worth testing CMV in taro as well as in the subsequent optimization experiments. The infecting bacterial solution concentration and infection method were also important factors affecting the silencing effect in VIGS. In the present study, the best infection effect was achieved with OD600 = 1.0, and this concentration was also applied in other plants used in VIGS system construction[18,21]. It may be related to bacterial vitality and infection ability at this concentration. But recent research of engineered TRV vectors on tobacco resulted in a OD600 value of bacterial suspensions of 1 × 10−5 still achieving good silencing effect through a one-strain/two-vector approach[33], these JoinTRV vectors that used pLX as back-bones have been tested in a range of agrobacterium strains, are notably more compact than any other TRV vector system currently documented, except for TRV are tobacco-hosting viruses, perhaps compact structure is another key factor to high efficiency at low concentration. The main VIGS inoculation methods are vacuum penetration[34,35], friction inoculation[36], root-filling method[37], and leaf injection[38]. In corm crops, it usually turns out that vacuum penetration is superior to leaf injection[8,38]. However, the results in taro bulbs showed no significant difference between these two methods (Table 3). The possible reason is that taro bulbs are firm and full of starch with multiple buds. Buds with less infection would display obvious growth advantages, this may be an important factor resulting in no significant differences in the photobleaching phenomenon.
Currently available evidence has shown that most TCP class I genes are activators of plant development, whereas TCP class II members often function as a repressor of various growth and development pathways[12−14]. In maize, ZmTCP7 affects the accumulation of starch by targeting the promoter of ZmBt2[22], besides, ZmTCP7 lacks transactivation activity in yeast systems[10,12], this means that ZmTCP7 may dimerize with other regulatory protein(s) or TF(s) to regulate ZmBt2 to control the accumulation of starch. In this research, it was found that after CeTCP14 expression level was downregulated by VIGS, the starch content decreased above 20% (Fig. 3), this means CeTCP14 also contributes to the accumulation of starch. In VIGS-CeTCP14 plants, the expression level of CeTCP13, CeTCP10, and starch biosynthesis gene (ADP-glucose pyrophosphrylase (AGPL), granule-bound starch synthase (GBSS), soluble starch synthase (SSS), starch branching enzyme (SBE), isoamylase (ISA1) were also detected in taro corms, they all presented a significant down-regulated trend (data not shown), these results indicate that CeTCP14 is an upstream regulatory gene in the starch biosynthesis pathway, but whether it interacts with CeTCP13 and CeTCP10 to regulate the starch pathway remains to be further investigated.
VIGS is a powerful tool used in plant molecular biology to study gene function. VIGS allows for high-throughput functional analysis of plant genes. By silencing individual genes and observing the resulting phenotypic changes, researchers can elucidate the roles of specific genes in various biological processes such as development, stress response, and disease resistance.
-
In this study, we constructed a VIGS system initially in taro with TRV as vector and CePDS as indicator gene, combined with phenotype change, gene expression, and chlorophyll content determination. Through the optimization of bacterial solution concentration and infection mode, it was found that the silencing effect was the best at OD600 = 1.0, and the silencing plants rate reached 27.77%. There was no significant difference in the silencing plant numbers between leaf injection method and bulb vacuum infiltration method. After silencing the CeTCP14 based on the established system, the starch content in the bulb is reduced by more than 20%, preliminarily verified CeTCP14 could promote starch accumulation in taro, which laid a foundation for rapid gene function verification in taro.
-
The authors confirm contribution to the paper as follows: study conception and design, writing - review & editing: Zhou Q, Xiao Y; experiment design: Cui J, Pan R, Zhu Q, Huang Y, Zhou Q (directing), Xiao Y (directing); experiment execution, writing - draft manuscript: Gui Y, Li B, He Y, Zhang Y, Zhou Q, Xiao Y; data analysis: Gui Y, Li B, He Y, Zhang Y, Zhou Q, Xiao Y; analysis and interpretation of results: Cui J, Pan R. All authors have read and approved the final manuscript.
-
The CePDS and CeTCP14 sequence used in this study are available in the taro reference genome Taro_JAAS_v1.0 (BioProject: PRJNA587719), and the gene numbers are EVM0008568.1 and EVM0000825.2, respectively.
This research was supported by Jiangxi Provincial Key Research and Development Project of China (Grant No. 20212BBF61001), the National Natural Science Foundation of China (32060683), the Natural Science Youth Foundation of Jiangxi Province (20224BAB215025),and the Youth Foundation of Jiangxi Provincial Department of Education (GJJ210442).
-
The authors declare that they have no conflict of interest.
-
accompanies this paper at (https://www.maxapress.com/article/doi/10.48130/tp-0024-0025)
-
Received 25 March 2024; Accepted 7 June 2024; Published online 11 September 2024
-
CePDS was used as an indicator gene to construct a VIGS system in taro.
The silencing plant rate of VIGS system increased significantly by optimized bacterium concentration.
CeTCP14 was selected to further verify the robustness of the constructed system.
-
# Authors contributed equally: Yanling Gui, Bicong Li
- Supplemental File 1 Gene IDs and sequences for CePDS and CeTCP14.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Gui Y, Li B, Zhu Q, He Y, Zhang Y, et al. 2024. Construction and application of a virus-induced gene silencing system in taro. Tropical Plants 3: e030 doi: 10.48130/tp-0024-0025 

Construction and application of a virus-induced gene silencing system in taro
- Received: 25 March 2024
- Revised: 01 June 2024
- Accepted: 07 June 2024
- Published online: 11 September 2024
Abstract: Virus-induced gene silencing (VIGS) technique is an important means for rapid identification of plant gene function, and a stable VIGS system in taro can provide technical support for rapid and efficient gene function verification. A VIGS system was constructed with a tobacco rattle virus (TRV)-based vector, with phytoene desaturase (CePDS) as the indicator gene, then CeTCP14 was silenced to further verify the robustness of this system. Ganyu No.1 was used as the material to construct the taro VIGS system using the leaf injection method with CePDS at OD600 = 0.6, and the silencing plant rate was 12.23%, the expression level of CePDS was about 59.34%−77.18% compared to the control, and the chlorophyll content decreased 37.80%−56.11% in CePDS silencing plants. The silencing plant rate increased significantly by leaf injection at OD600 = 1.0, and reached 27.77%, but there was no significant difference in silencing plant rate between the leaf injection method and bulb vacuum treatment. CeTCP14 was further silenced with bacteria solution OD600 = 1.0 and bulb vacuum treatment in Ganyu No.2, the silencing plant rate was 20%, and the expression level of CeTCP14 was 43.94%−63.34% of the control. Meanwhile, the starch content in the corms decreased significantly to 70.88%−80.61% compared to the control. In conclusion, the results indicated that a TRV-based VIGS system is effective in taro. The concentration of bacterial solution is a key factor affecting the VIGS system, CeTCP14 can affect starch accumulation in taro bulbs. A robust VIGS system establishment in taro could lay a good foundation for the subsequent rapid gene function verification.