









-
There is a widespread agricultural problem worldwide, after replanting apples in the same land, abnormal tree growth and development, as well as decreased yield and fruit quality are observed, which is called apple replant disease (ARD)[1,2]. The most universal symptoms of ARD are stunted growth of sapling, damaged root systems, reduced both in yield and fruit quality[3,4], which has greatly affected the development of the apple industry. Therefore, it is particularly important to find an effective method to alleviate ARD.
ARD can be caused by many factors, among which biological factors dominated by harmful fungi in soil hold a major position[5−8]. Fusarium solani has been proven to be one of the main pathogens causing ARD in the main production areas of China. Scholars at home and abroad have put forward a variety of preventions and control measures to deal with ARD. Intercropping and crop rotation could alleviate ARD to varying degrees[9−11], crop rotation has been proven to improve the soil environment and effectively reduce the incidence of plants, but it is difficult implement in production as it is time consuming. Chemical fumigants such as methyl bromide have been shown to be highly effective against preventing and alleviating ARD[12,13], but they have been banned due to the serious effects of environmental pollution and ozone destruction[14−16]. Biological control has become an essential means of agricultural sustainable development because it can regulate soil microecology by competing with pathogenic bacteria for ecological niche[17−19]. However, biological control by antagonistic microorganisms needs continuous application because of the variety and long-term existence of pathogenic fungi in soil. Therefore, it is difficult to completely eliminate ARD through biological control[20]. Consequently, it is a new breakthrough point to explore the molecular mechanism of apple defense against F. solani.
A variety of abiotic and biological challenges occur at the same time, which puzzles the prevention and control of ARD. External unfavorable factors will trigger the inherent defense mechanism of plants[21,22]. In this process, transcription factor (TF) plays an important role. WRKY transcription factors regulated the expression of defense-related genes by combining 'W-box' sites in the promoter region of the target gene, and has been widely studied as a key regulator in the immune response from plants to various biological stresses[23−25]. AtWRKY33 can act as a positive regulator of JA- and ET-mediated defense response signals to mediate plant defense against necrotic pathogens Botrytis cinerea and Alternaria brassicicola[26]. Overexpression of GmWRKY136, 53, and 86 in soybean respectively showed increased resistance to soybean cyst nematode[27]. In tobacco, CaWRKY40 homologous gene was regulated by SA, JA, and ET signaling pathways, and coordinated the response to pepper and tobacco to R. solanacearum infection and heat stress[28]. By regulating the signal transduction mediated by SA, JA, ET, and ROS, it was found that TaWRKY49 negatively regulated the high-temperature seedling-plant resistance to Pst (HTSP) of wheat[29].
Pathogenesis-Related proteins (PRs) have been proven to participate in plant defense responses[30−32]. Studies showed that Pathogenesis-Related (PR) proteins can be divided into 17 families[33]. PRs have been identified to play a critical role in many plant species. In addition, StPR-1 has been shown to play a positive role in the infection of potato Phytophthora infestans[34]. PR2 in Arabidopsis may act as a regulator of callose and SA-dependent defense responses[35]. In apple, MdPR10-1 and MdPR10-2 were found to be related to resistance to Alternaria leaf spot[36]. Apple Pathogenesis-Related protein MdPR4 has been shown to be involved in the recognition of chitin and resistance to ARD pathogens[37]. Overexpression of MdPR10 significantly reduced the infection of Valsa mali, which is the main pathogen causing apple rot[38].
In apple, it is unknown whether the WRKY transcription factor mediates plant defense mechanisms against F. solani. In this study, the infection of F. solani increased the expression of MdWRKY20 in the root of 'M9T337', so MdWRKY20 was chosen as the research target. The purpose is to analyze the function of MdWRKY20 and its regulation mode under the infection of F. solani. Therefore, we have carried out genetic, biochemical, and physiological analysis to provide new ideas for apple to defend against F. solani and cultivate resistant rootstocks.
-
The experimental materials included tissue culture seedlings of apple (Malus domestica Borkh rootstock M9T337 with consistent growth state), and the apple callus of 'Orin' was used for pathogen infection test, cultured in subculture medium. Subculture medium: MS, 30 g/L sucrose, 1.5 mg/L 6-benzyladenine (6-BA), 0.5 mg/L 2,4-dichlorophenoxyacetic acid (2,4-D) and 7 g/L agar, pH 5.8. Selection medium: MS, 30 g/L sucrose, 1.5 mg/L 6-BA,0.5 mg/L 2,4-D, 50 μg/mL kanamycin, and 7 g/L agar, pH 5.8, and the medium was placed at 24 °C in darkness.
Seedlings of Arabidopsis Columbia (Col-0) were used for genetic transformation tests and were cultured on MS medium: MS, 30 g/L sucrose pH 5.8. Screening medium: MS, 30 g/L sucrose, 50 μg/mL kanamycin pH = 5.8. Seeds were vernalized at 4 °C for 2 d before growth at 23 °C under 16 h light/8 h dark conditions. After taking root in the culture medium, Arabidopsis was transplanted into nutrient medium (the volume ratio of vermiculite to nutrient soil was 1:1), and grew under the condition of 14 h light/10 h dark cycle, the temperature was 22 °C and the relative humidity was 60%. Nicotiana benthamiana was used in subcellular localization, and it was cultivated and grown under the conditions of 14 h light/10 h dark cycle, with a temperature of 24 °C and relative humidity of 60%.
F. solani was activated in potato dextrose agar (PDA) for 5 d at 28 °C, then the callus were infected with 0.5 cm diameter agar on which hyphae grew evenly. PDA medium without F. solani inoculation was treated as a control.
The activated F. solani was washed with sterile water to prepare spore suspension with a concentration of 105 CFU/mL, then added to PDB culture medium at a ratio of 1%, then cultured at 28 °C and 160 r/min for about 7 d. After filtering through 8-layer gauze, the spore suspension concentration was calculated using a blood cell counting plate, and the final concentration was adjusted to 105 cells/mL by adding sterile water. Fifty mL spore suspension was added to the nutrient solution, then it was poured into the 'M9T337' seedlings, and the same amount of sterile water was added to the control. The seedlings were cultured in the greenhouse, and the root samples were taken on the 0, 1, 3, 6, 9, and 12 d after inoculation, and then frozen in liquid nitrogen and stored at −80 °C.
The flower, fruit, stem, leaf, and root tissues of apple were collected under natural planting conditions and stored at −80 °C for the expression analysis of MdWRKY20.
Extraction of fermentation broth extract of F. solani and infection test of Arabidopsis
-
1% (v/v) of the target concentration of F. solani spore suspension was added to PDB medium, at 28 °C and 160 r/min for about 7 d. Spore suspension filtered with 8-layer gauze, then centrifuged at 12,000 r/min for 10 min, and the collected supernatant was mixed with ethyl acetate at 1:1 for extraction. Shaking it violently three times during this process, and then allowing it to stand until layered. The bottom liquid was collected after layering and excess water absorbed with anhydrous sodium sulfate, then concentrated in a rotary evaporator (36 °C, 80 hPa) to a powder state, and the collected powder was dissolved with methanol, and finally the concentration of fermentation broth extract was 5 mg/mL. Centrifuged at 10,000 r/min and filtered with 0.22 μm filter membrane to remove impurities, and then stored at −80 °C.
Zero, six and 12 mL of fermentation broth extract was added into 300 mL of MS medium and diluted to 0, 100, and 200 mg/L respectively. After disinfection of Arabidopsis, the seeds were planted in solidified MS solid culture medium, then sealed with sealing film, Arabidopsis was cultured according to the method mentioned above.
RNA extraction and quantitative reverse transcription PCR
-
FastPure Plant Total RNA Isolation Kit (Vazyme, Nanjing, China) was employed to extract total RNA from the tissues of 'M9T337' and the callus of the apple. Then, the RNA was reverse transcripted into cDNA using HiScript III 1st Strand cDNA Synthesis Kit (Vazyme, Nanjing, China), and according to the concentration, 1 pg–1 μg total RNA was extracted for reverse transcription. The qRT-qPCR amplification reactions were performed using Taq Pro Universal SYBR qPCR Master Mix (Vazyme, Nanjin, China), and qRT-PCR was then performed on a Real-Time system (Bio-Rad, Hercules, CA, USA). Each sample was repeated three times, normalized with MdActin (CN938023) as an internal control, and the relative quantification of genes were calculated by the cycle threshold (Ct) 2−ΔΔCᴛ method[39]. Primers used for qRT-PCR analysis are listed in Supplementary Table S1.
Gene cloning and phylogenetic analysis
-
PCR-amplification (P515, Vazyme, Jiangsu, China) was used to amplify the coding sequence (CDs) of MdWRKY20 from apple leaves. Primers used for cloning CDs are listed in Supplementary Table S1. Using MEGA version 5.1 software to plot the phylogenetic tree, the WRKY20 sequence of apple and other species were derived from the NCBI database (
www.ncbi.nlm.nih.gov ). The phylogenetic tree was created by the adjacency method, and the phylogenetic tree was beautified using the online software ITOL (https://itol.embl.de/ ). DNAMAN (Lynnon Biosoft, San Ramon, CA, USA) software was used to compare protein sequences.Subcellular localization
-
The sull-length cDNAs without the stop codon of MdWRKY20 was introduced into the pRI101-GFP vector, and a 35S::MdWRKY20-GFP fusion vector was generated. The primer sequence is listed in Supplementary Table S1. The fusion vectors and empty plasmid were then introduced into Agrobacterium tumefaciens strain GV3101 and then infiltrated into tobacco leaves. After 2–3 d of infiltration, the GFP signal was observed using confocal microscopy.
Reactive oxygen species measurement diaminobenzidine (DAB) and nitroblue tetrazolium (NBT) staining of Arabidopsis leaves
-
One mg/L DAB solution and 0.5 mg/mL NBT solution were prepared with 0.01 mmol/L phosphate buffer solution (pH 7.0). After the growth of Arabidopsis for about 28 d, leaves with the same growth status were selected and placed in a centrifuge tube, which were dyed with DAB solution for 6 h and NBT solution for 2 h. Incubated at a constant temperature of 28 °C. Then, the leaves were decolored in 95% alcohol, boiled until chlorophyll was completely degraded, observed and photos taken.
Yeast one-hybrid test (Y1H) assay
-
Two vectors were used for Y1H assay, the MdWRKY20 CDs was inserted into pGADT7 vector (Clontech, TaKaRa, Japan), while the promoter fragments of MdPR1 and MdPR3 were inserted into the pHIS2 vector (BD Biosciences, NJ, China). Then the recombinant plasmids and PGADT7 empty vector were transferred into Y187 (Clontech) yeast cells. The transformed cells were cultured on screening medium (-Trp/-His) with different concentrations of 3-amino-1,2,4-triazole (3-AT) and cultured at 28 °C for about 2–3 d to select the optimal concentration. Compared with the negative control, the binding of MdWRKY20 protein to the MdPR1 promoter enables yeast strains to grow normally under conditions containing appropriate concentrations of 3-AT, while yeast containing PGADT7 empty vector cannot grow normally. Then co-transfect the recombinant plasmid containing pHIS2 and pGADT7 of the target gene. The transformed cells were cultured on deficient medium (-Trp/-His/-Leu) containing optimal 3-AT concentration, and cultured at 28 °C for about 2–3 d. Determining the binding ability of transcription factors to promoters based on yeast growth.
Electrophoretic mobility shift assays (EMSA)
-
The LightShift Chemiluminescent EMSA Kit (Thermo Scientific) was used for EMSA assay. Biotin-labeled probe primers and non-biotin labeled competitive probes were synthesized by Sangon Biotechnology (Shanghai, China) (Supplementary Table S1). Insert the MdWRKY20 CDs sequence into the pET-32a(+) (His-Tag) expression vector (Novagen, NJ, USA). Express the MdWRKY20 recombinant protein in E.coli DE3 and purify the MdWRKY20-His fusion protein using the His tagged protein purification kit (Kangwei).
The specific steps are as follows: the reaction mixture contains probe 1 μL, H2O 1 μL, LightShift 10× binding buffer 2 μL and purified protein 16 μL. The mixture was kept in the dark at 24 °C for 25 min, after adding loading buffer and mixing, nondenaturing polyacrylamide gel electrophoresis was carried out, and then the DNA-protein complex was transferred to nylon membrane. Then perform UV crosslinking, with one minute on each side. The UV Crosslinker was used for UV crosslinking, with one minute on each side. Chemiluminescence signal detection was performed using the reagents provided in the kit.
Luciferase reporter assay
-
The CDs of MdWRKY20 was inserted into pHBT AvrRpm 1 vector, and the the promoter of MdPR1 and MdPR3 were inserted into pFRK1-LUC-nos vector, respectively. After the protoplasts of 'Orin' callus tissue were extracted, co-transform two plasmids into apple callus protoplasts. The transiently transfected protoplasts were incubated at 24 °C for 6 h and suspended in 100 μL cell lysis buffer. Added 5 μL cell extract and 20 μL 1 mM 4-MUG and cultured at 37 °C for 1 h. Then added 100 μL 0.2 mol/L sodium carbonate to terminate the reaction. Luciferase reporter analysis system (Promega) was used to determine LUC activity.
Vector construction and genetic transformation
-
Insert the full-length MdWRKY20 CDs into pRI101-AN vector containing a GFP tag. Primers are listed in Supplementary Table S1. Then transfer the recombinant plasmid into Agrobacterium LBA4404 (AngYuBio, Shanghai, China) cells, and two-week-old 'Orin' callus were infected in the infection solution for about 30 min. Dry the callus with filter paper then cultured them at 24 °C for 2 d in darkness on agar-solidified MS medium without antibiotics, then transferred to selective medium containing 250 mg/L carbenicillin and 50 mg/L kanamycin. PCR amplification was used to verify overexpression of MdWRKY20.
In the transformation of Arabidopsis, the recombinant plasmid was introduced into the Arabidopsis using floral dip method[40] mediated by Agrobacterium LBA3101 (AngYuBio, Shanghai, China). After harvesting Arabidopsis seeds, the positive overexpression lines were screened with agar-solidified MS medium containing 50 mg/L kanamycin, the overexpression of MdWRKY20 was verified by PCR amplification, the T3 homozygous transgenic lines were used for subsequent phenotypic analysis.
Statistical analysis
-
At least three replicates were set for each experiment to ensure accuracy, and SPSS version 20.0 (IBM, Inc, Armonk, NY, USA) was used for statistical analysis. The results were compared by one-way ANOVA and Duncan test. Using GraphPad Prism version 9 (San Diego, CA, USA) for graph analysis, and p < 0.05 was statistically significant.
-
Apple rootstock 'M9T337' was infected by F. solani. Visible symptoms can be seen that the slow growth and partial browning of leaves appeared after 9 d post-infection (dpi) (Fig. 1a). On 12 dpi, the infection rate was as high as 92.5% and the mortality rate reached 37.5% (Supplementary Fig. S1). Eight WRKY family target genes were selected for transcriptional analysis according to previous research results[41]. The transcription of WRKY-TFs was induced by F. solani to different degrees (Fig. 2b–i). The infection of F. solani induced the expression of MdWRKY2/4/20/25 and inhibited the expression of MdWRKY10/33/58, but had no obvious relationship with the transcription of MdWRKY44. Among them, it was found that the transcription level of MdWRKY20 changed most significantly. Therefore, it was speculated that the transcription of MdWRKY20 may correspond to the infection of F. solani.
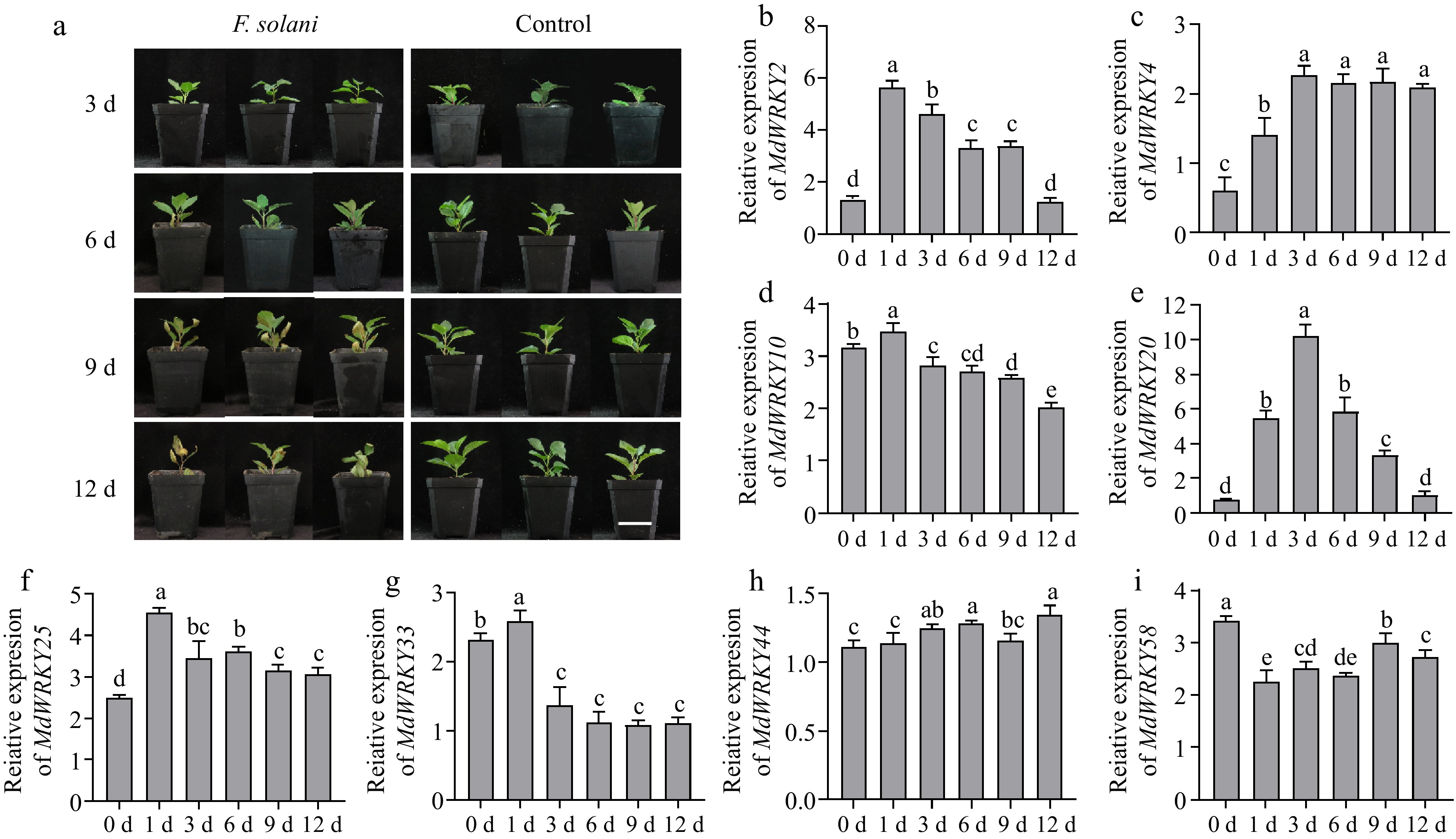
Figure 1.
Response of apple rootstock 'M9T337' to F. solani infection and expression levels of MdWRKY-TFs after F. solani infection detected by qRT-PCR. (a) Images of 'M9T337' taken on the 3, 6, 9 and 12 dpi after infection by F. solani. Scale bar = 5 cm. (b)–(i) qRT-PCR was used to detect the expression level of MdWRKY2, MdWRKY4, MdWRKY10, MdWRKY20, MdWRKY25, MdWRKY33, MdWRKY44, MdWRKY58. For (b)–(i), values were means ± s.d. of three independent biological replicates. Bars not labeled with same letters in each panel indicate values are significantly different at p < 0.05, based on one-way ANOVA and Duncan's tests.
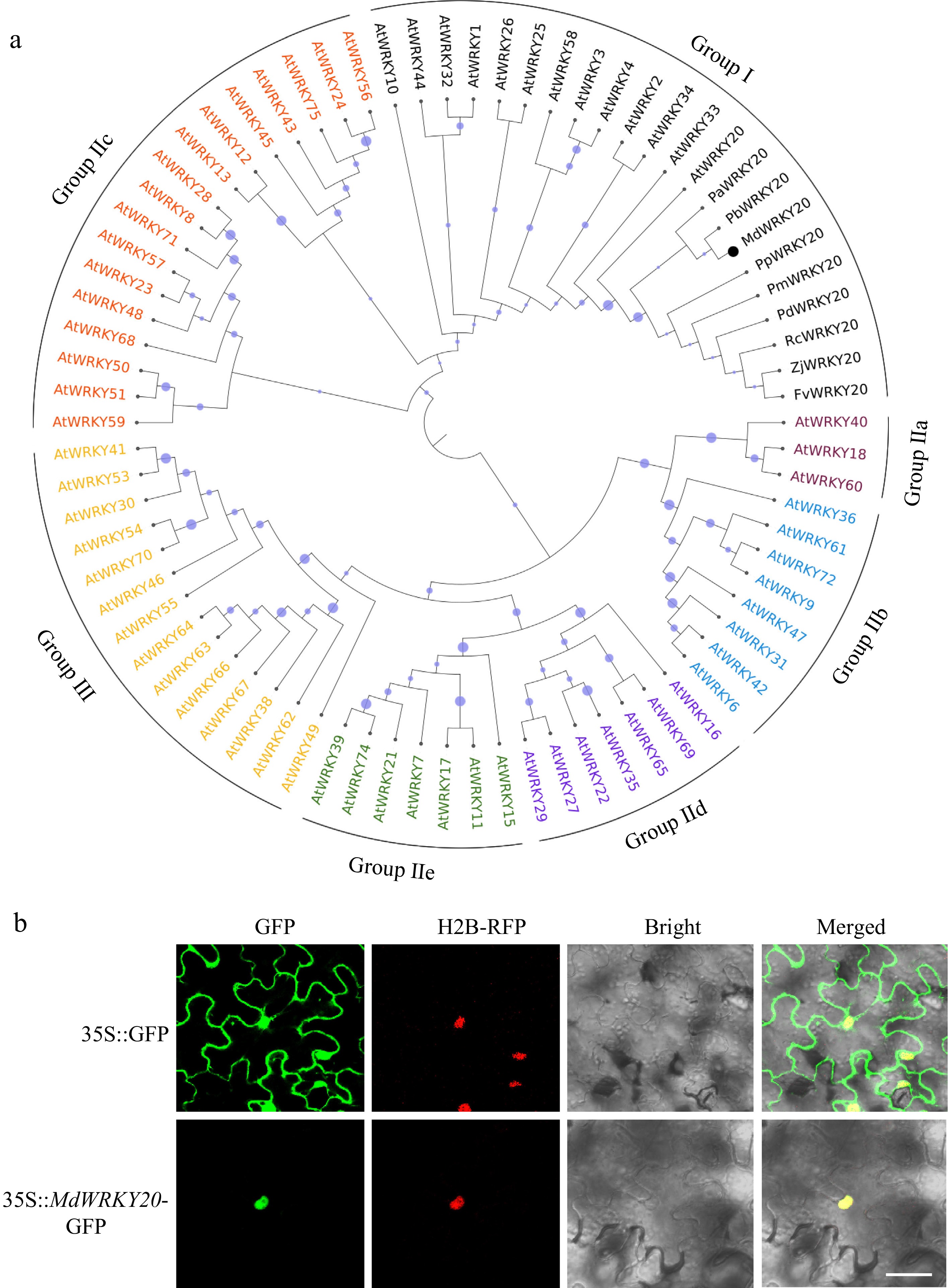
Figure 2.
Phylogenetic analysis and subcellular localization analysis of MdWRKY20. (a) Phylogenetic relationship and subgroup classification of MdWRKY20, AtWRKY, and other Group I WRKYs proteins. The WRKY20 sequence of apple and other species was derived from NCBI database (
www.ncbi.nlm.nih.gov ), and the WRKY protein sequence of Arabidopsis was derived from TAIR database (www.arabidopsis.org ). The phylogenetic tree was created using MEGA software (version 5.1) by the adjacency method. MdWRKY20 is highlighted with black circles in the figure. (b) 35S::MdWRKY20-GFP vectors were transformed into the leaves of Nicotiana benthamiana. The nucleus were labeled with red fluorescent protein H2B. Three biological repeats were performed. Scale bar = 50 μm.Phylogenetic analysis and subcellular localization analysis of MdWRKY20
-
MdWRKY20 was expressed in every tissue of apple, with the highest expression in root and fruit (Supplementary Fig. S2). To examine the evolutionary relationship of MdWRKY20, the phylogenetic tree was generated by using the full-length amino acid sequences of Arabidopsis WRKYs and various species of Group I WRKYs (Fig. 2a). MdWRKY20 belongs to group 1, along with several other Group I WRKY20 proteins. The subcellular localization results showed that green fluorescence could be observed throughout the whole cell in the empty GFP, whereas the green fluorescence of MdWRKY20-GFP was confined to the nucleus (Fig. 2b).
Overexpression of MdWRKY20 improves the resistance of 'Orin' callus to F. solani
-
Recombinant pRI101-AN vector carrying MdWRKY20 was transformed into 'Orin' apple callus, and the callus tissue overexpressing MdWRKY20 obtained (Fig. 3a). Compared with the wild type (WT), it was observed that fungal elongation of the strain in MdWRKY20-OE callus were significantly reduced four days after inoculation with F. solani (Fig. 3b), and the diameter of the plaque extension decreased by 56.32%. The expression of MdWRKY20 in transgenic callus were significantly higher than WT (Fig. 3c). This result indicates that MdWRKY20 has a positive effect in the response of apple callus to F. solani infection.
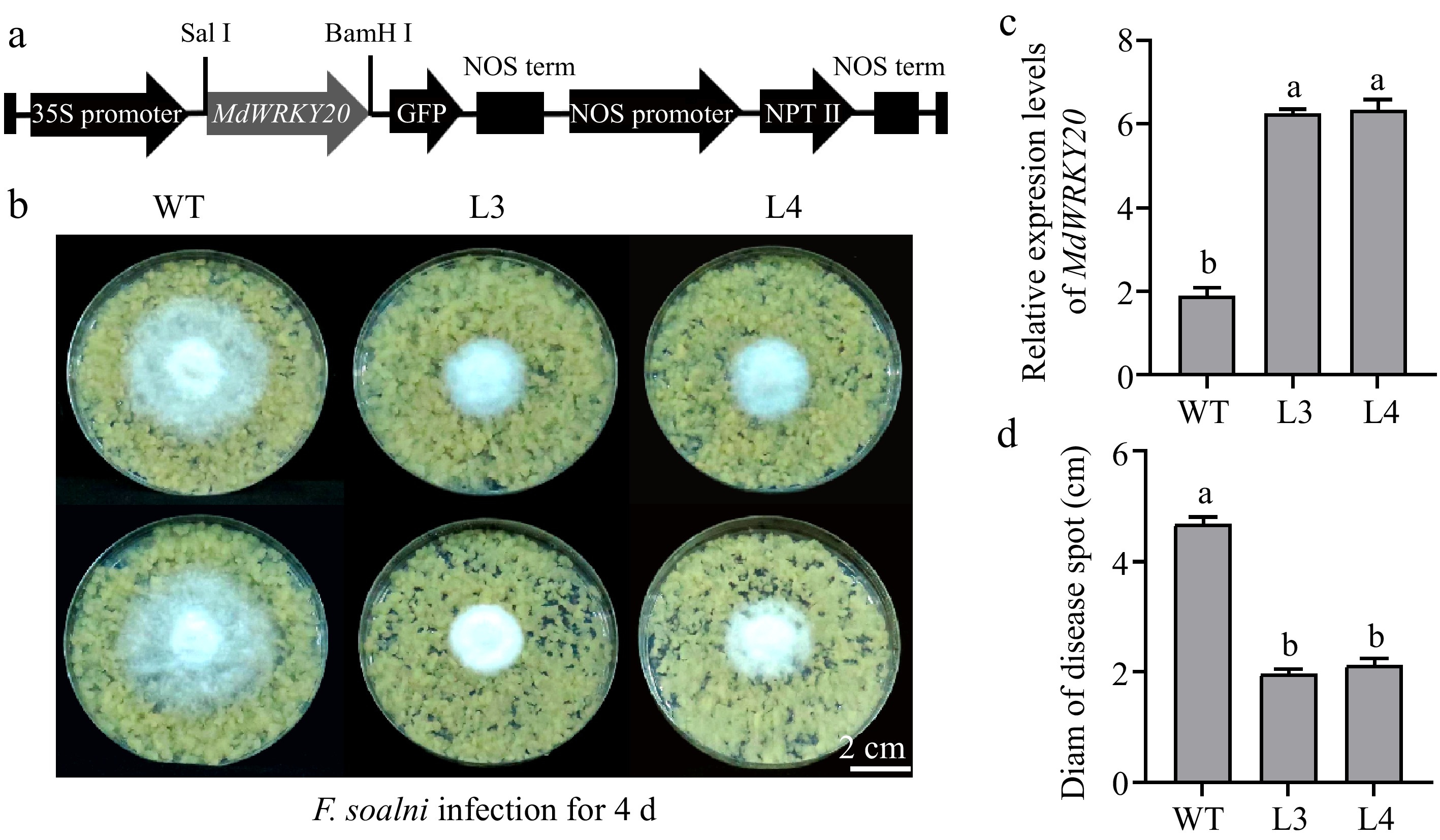
Figure 3.
Functional characteristics of 'Orin' callus overexpressing MdWRKY20. (a) MdWRKY20 CDs was inserted into PRI101-AN vector with CaMV 35S promoter and green fluorescent protein (GFP) sequence. (b) Phenotype of wild type (WT) and MdWRKY20-OE callus 4 d after the infection of F. solani. (c) RT-PCR based validation of MdWRKY20-OE in 'Orin' callus. (d) The diameter of the plaque extension of different callus after infection with F. solani. (c), (d), Values are means ± s.d. of three independent biological replicates. Bars not labeled with the same letters in each panel indicate values are significantly different at p < 0.05, based on one-way ANOVA and Duncan's tests.
Overexpression of apple MdWRKY20 promotes the expression of PR protein gene
-
To further investigate the mechanism by which MdWRKY20 and PRs enhance the tolerance of apple callus to F. solani, the expression levels of PR genes in apple callus overexpressing MdWRKY20 and WT were measured after 7 d of inoculation with F. solani. It was found that there was no significant change in the expression of MdPR2, MdPR3, and MdPR4 in the MdWRKY20-OE lines, but the expression of MdPR1 was significantly higher than that in WT under stress conditions (Fig. 4a–d). Therefore, the results indicated that MdWRKY20 may play a role by positively activating the expression level of MdPR1.
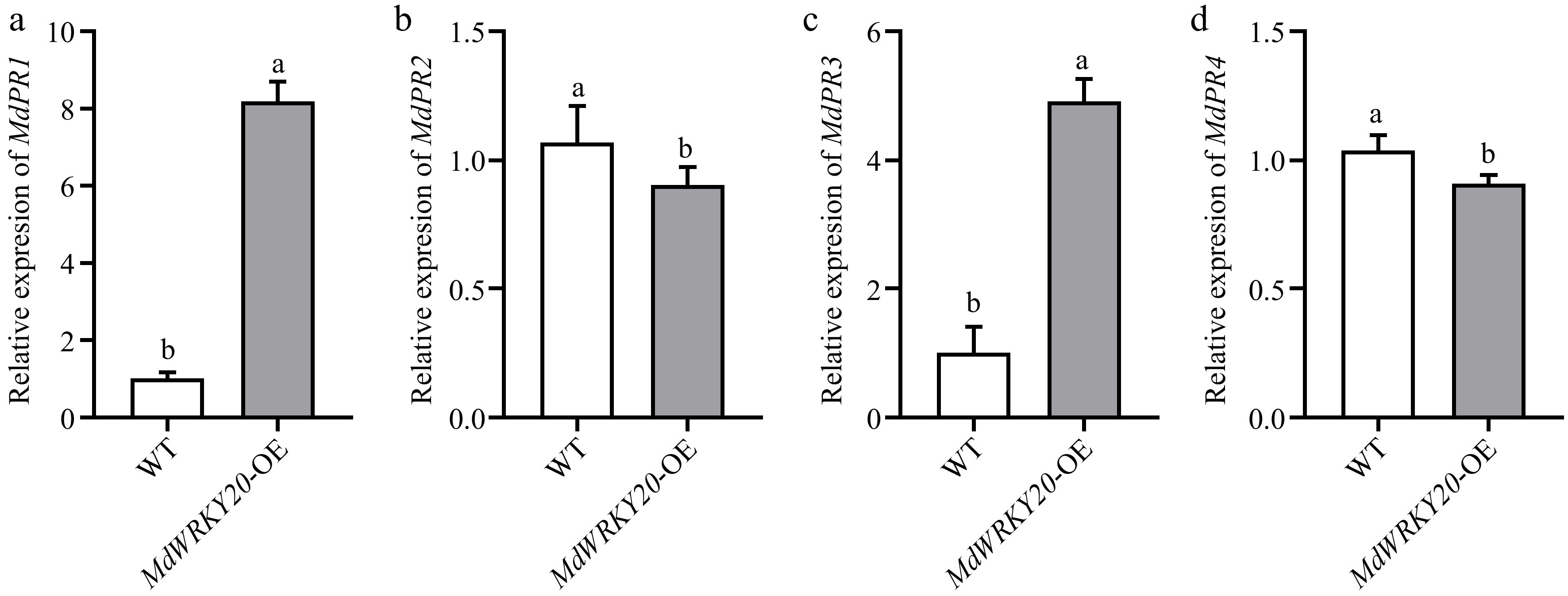
Figure 4.
qRT–PCR analysis of MdPR1, MdPR2, MdPR3, and MdPR4 expressions in MdWRKY20-OE and wild type apple callus with F. solani infection. Values are means ± s.d. of three independent biological replicates. Bars not labeled with the same letters in each panel indicate values are significantly different at p < 0.05, based on one-way ANOVA and Duncan's tests.
MdWRKY20 binds to the promoters of MdPR1
-
To verify the specificity of MdWRKY20 binding to the promoters of MdPR1 after F. solani infection, Y1H assay was carried out. It was found that MdWRKY20 interacted with the MdPR1 promoter, which was screened at a suitable 3-AT concentration of 100 mM (Fig. 5a). By analyzing the cis-acting elements in the promoter sequences, it was found that there are two speculated W-box motifs in the MdPR1 promoter (Fig. 5c). Therefore, EMSA demonstrated that MdWRKY20 could bind to the W-box II motif in the promoter of MdPR1 (Fig. 5d). With the increase of cold probe concentration, the binding between MdWRKY20 to the MdPR1 promoter was weakened (Fig. 5d). To further explore the effect of MdWRKY20 on the activity of the MdPR1 promoter, LUC activity assay was performed, and the results showed that MdWRKY20 has transactivation activity towards the MdPR1 promoter (Fig. 5e).
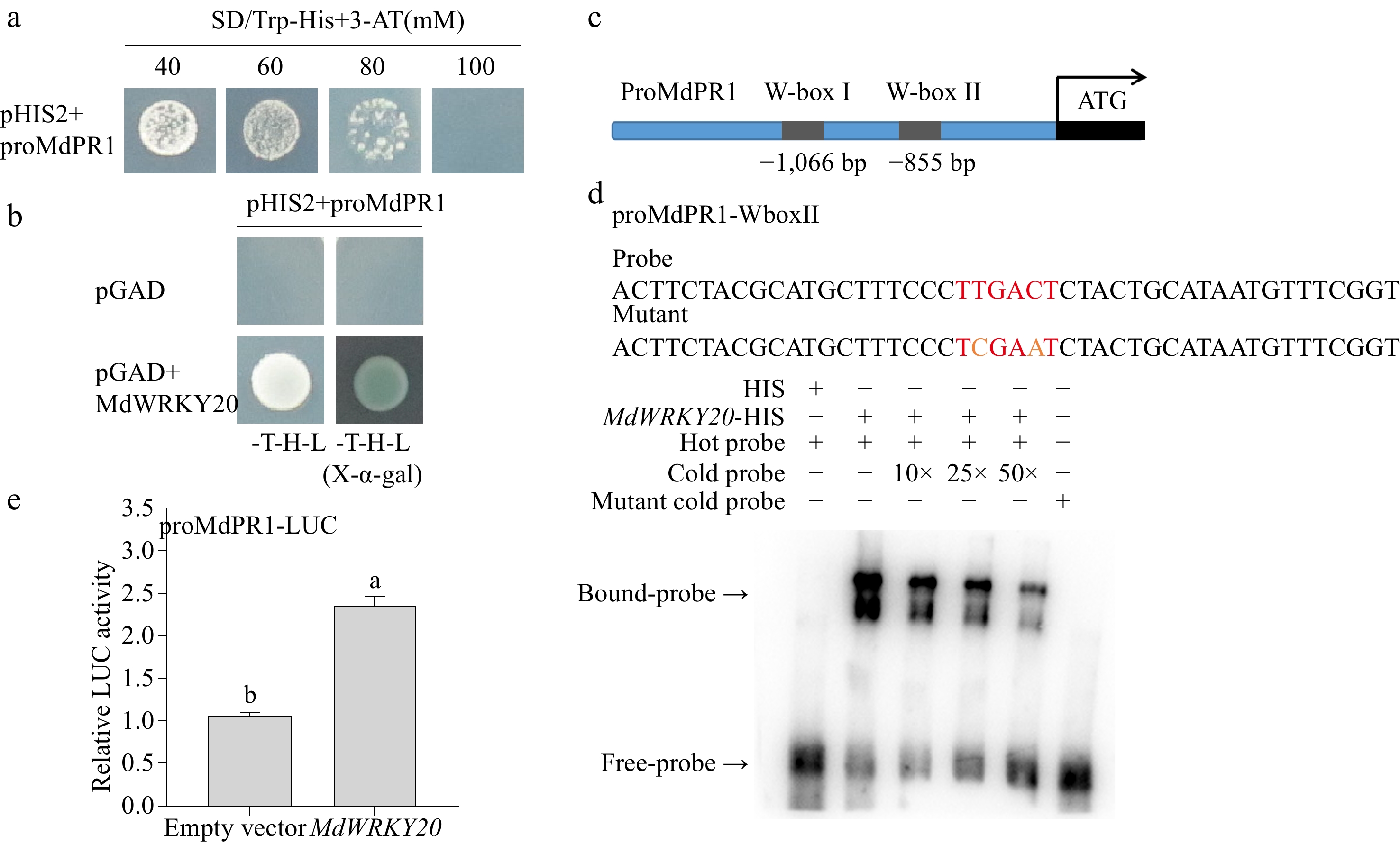
Figure 5.
MdWRKY20 binds to the MdPR1 promoter. (a) The optimal 3-AT concentration was determined by cloning proMdPR1 into the pHIS2 vector. (b) MdWRKY20 interacted with MdPR1 promoter fragments as per the Y1H assay. (c) The W-box I and W-box II elements were used in the EMSA. (d) EMSA analysis revealed that MdWRKY20 binds to the W-box II of the MdPR1 promoter (e) Luciferase reporter (LUC) assays showed the MdWRKY20-mediated activation of proMdPR1.
Overexpression of MdWRKY20 improves the germination rate and F. solani resistance of Arabidopsis
-
To further explore the function of MdWRKY20, the expression vector was constructed to transform Arabidopsis and obtained MdWRKY20-OE Arabidopsis (Supplementary Fig. S3). Inoculate Arabidopsis on MS medium, which contains extracts of fermentation broth extract of F. solani with concentrations of 0, 100, and 200 mg/L, and observe the phenotype of Arabidopsis after 7 d. It could be seen that the growth of Arabidopsis were uniform and flourishing under control conditions. Under the treatment of F. solani, the growth of Arabidopsis were inhibited to varying degrees. When the concentration of extracts of fermentation broth extract of F. solani is 100 mg/L, the growth of Col were significantly inhibited, while plants overexpressing MdWRKY20 were less inhibited (Fig. 6d). When the concentration of extracts of fermentation broth extract of F. solani reached 200 mg/L, the germination rate of Col seed decreased and the growth rate slowed down and the leaves turned purple (Supplementary Fig. S4), while the transgenic lines showed better tolerance, mainly reflected in more lush leaves and healthier growth state (Fig. 6f). By contrast, the MdWRKY20-OE lines showed better resistance to F. solani infection.
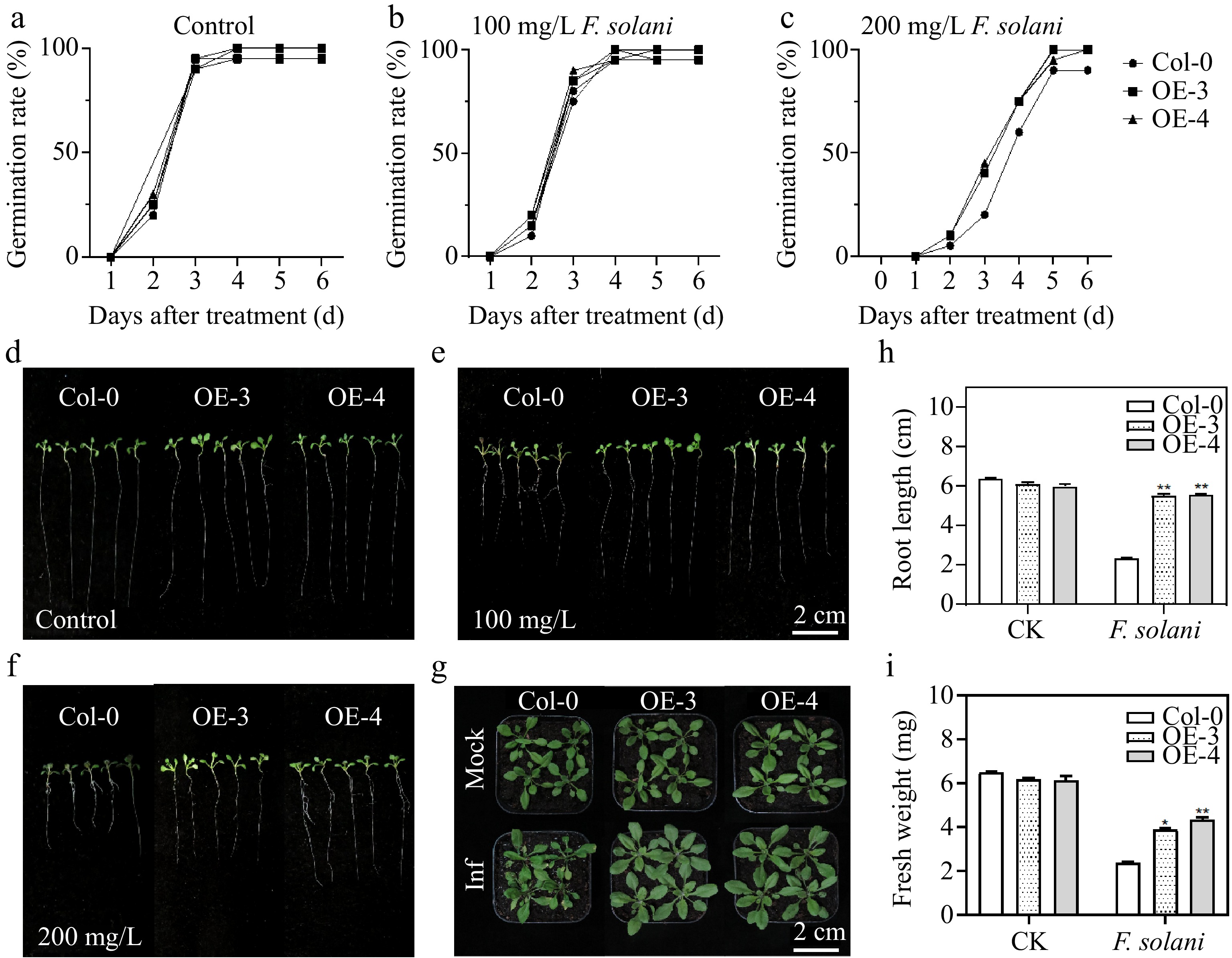
Figure 6.
Germination percentage and phenotypic analysis of Arabidopsis under control and F. solani treatment. The germination rates of Arabidopsis seeds treated with (a) 0 mg/L , (b) 100 mg/L, and (c) 200 mg/L fermentation extract of F. solani. The phenotype of Arabidopsis was recorded by taking photos on the (d)−(f) 7 dpi, and (g) 21 dpi treated with (d) 0 mg/L, (e) 100 mg/L, and (f) 200 mg/L fermentation extract of F. solani. For (d)–(f) bars = 2 cm. (h) Root length of Arabidopsis after 7 d of treatment with control and F. solani. (i) Fresh weight of Arabidopsis after 7 d of treatment with control and F. solani. * 0.01 ≤ p < 0.05, ** 0.001 < p ≤ 0.01.
ROS level, antioxidant enzyme activity, and MDA content of Arabidopsis
-
Further, the ROS level, antioxidant enzyme activity and MDA content of Arabidopsis leaves under control and F. solani treatment were detected (Fig. 7). The results showed that under control conditions, there were no significant differences in MDA content, ROS levels, and antioxidant enzyme activity among transgenic Arabidopsis and Col. However, after treatment with F. solani, the ROS level and MDA content of MdWRKY20-OE Arabidopsis were significantly lower than Col, and the antioxidant enzyme activities were significantly higher than that of Col (Fig. 7a–f). The results of histochemical staining showed that, unlike Col, the accumulation of hydrogen peroxide (H2O2) and superoxide radical (O2−) in MdWRKY20-OE lines were lower, which showed that transgenic plants had better antioxidant capacity (Fig. 7g, h).
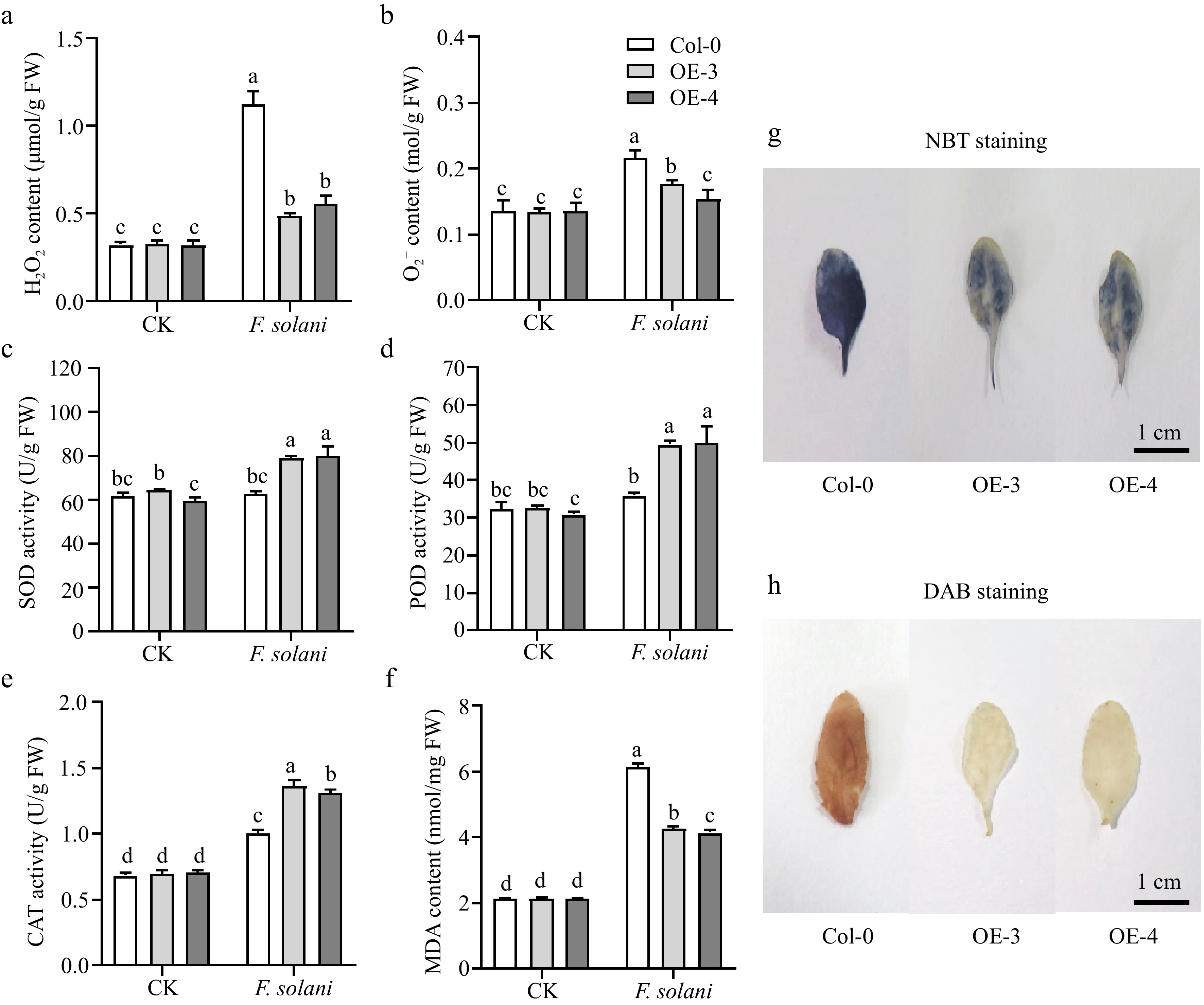
Figure 7.
Effects of F. solani treatment on several parameters of Arabidopsis leaves. The influence of F. solani treatment on (a), (b) active oxygen levels, (c)−(e) the activity of antioxidant enzymes, and (f) the content of malondialdehyde in Arabidopsis leaves. The (g) NBT staining, and (h) DAB staining of Arabidopsis leaves. For (a)–(f), values were means ± s.d. of three independent biological replicates. Bars not labeled with the same letters in each panel indicate values are significantly different at p < 0.05, based on one-way ANOVA and Duncan's tests. For (g) and (h), the deeper the color, the more content of O2− or H2O2 in plant tissues. Bars = 1 cm.
-
F. solani is a common plant pathogenic fungi, which has been proved to be one of the main pathogens causing ARD[42]. Replant disease is widespread in orchards worldwide. It is a disease based on the soil microbial community and harmful to plant physiology and morphological response, especially in Rosaceae plants. This disease frequently occurs in warm and humid environments, which causes great losses. Therefore, we are committed to finding a new method to effectively alleviate ARD.
Various fluctuating abiotic environmental factors accompany the growth process of plants, such as drought, salinity, threshold temperatures, nutrient starvation, and so on[43−46]. These factors may occur in many stages of plant growth and may limit the growth and development of plants, which has attracted wide attention because of the negative impact on agricultural production. Plants have their own strategies to deal with various stresses. They have evolved complex mechanisms at different levels to cope with the changing external environment.
At the molecular level, transcription factors play a vital role in this process. WRKY transcription factors have been widely reported in immunity triggered by various microbial-related molecular patterns. For example, in Brassica napus, BnWRKY33 upregulates the expression of genes regulated by salicylic acid (SA) and jasmonic acid (JA), positively regulating resistance to Sclerotinia sclerotiorum pathogens[47]. In addition, SpWRKY6 reduced cell membrane damage by regulating ROS level and expression level of PR genes[48]. In this study, it was found that the expression of MdWRKY20 was upregulated after infecting apple rootstocks with F. solani, indicating that MdWRKY20 may be related to apple rootstock's defense against F. solani infection, but the regulation mechanism of apple under F. solani infection needs to be further explored and supplemented.
The DNA binding domain of WRKY protein can activate or inhibit its expression by binding to the W-box element of the target gene promoter[49]. In Arabidopsis, AtWRKY33 mediated resistance targets NCED3 and NCED5 directly[40]. In addition, the W-box element in the promoter of WRKY protein can also be targeted by other WRKY proteins. For example, the synergistic effect of WRKY70 and WRKY54 has a negative impact on the response of Arabidopsis to Pectobacterium carotovorum and Botrytis cinerea[50]. After the callus were inoculated with F. solani, it was observed that the diameter of plaque extension of the callus with overexpression of MdWRKY20 was obviously smaller than that of the wild type. Transgenic Arabidopsis also showed increased resistance. MdWRKY20 may exert its function by binding to the W-box of downstream target genes and activating its expression.
Studies have shown that the PR gene was one of the most promising candidate genes for the cultivation of crop varieties resistant to multiple stresses[51−53]. The overexpression of PR genes in plants alone or in combination greatly improved the level of plant defense response to various pathogens[14,54,55]. Under stress conditions, the expression of MdPR1 was significantly upregulated after overexpression of MdWRKY20 in apple callus compared with wild type. Based on the analysis of Y1H, EMSA, and LUC, we have confirmed that MdWRKY20 participate in the defense response to F. solani infection by promoting the expression of MdPR1.
Active resistance refers to the host defense response caused by pathogen infection, and reactive oxygen species (ROS) burst is the earliest defense response produced by plants when pathogens invade[56]. Pathogen infection would unbalance ROS metabolism in plants and increase ROS production, and lead to the destruction of cell membrane structure[57]. In this study, after Arabidopsis were infected with F. solani, the H2O2 and O2− production rates in the leaves of Col were significantly increased, which were significantly higher than that in the MdWRKY20-OE lines. Appropriate increase of ROS plays an active role in plants' response to pathogen infection, but when ROS is excessive, it will lead to membrane lipid peroxidation, decreased enzyme activity, and cell metabolism disorders, which may eventually lead to the death of host cells[58,59]. Defense-related enzymes such as SOD, POD, and CAT could remove excessive ROS[60,61] from plant leaves. The present results showed that the contents of SOD, POD, and CAT in MdWRKY20-OE Arabidopsis leaves were significantly increased after F. solani infection, while the increase range of the Col were not significant. This may be due to the cell membrane damage of Col being severe, while transgenic lines could respond to the infection of F. solani, so their cell membrane damage was relatively light. The MDA content in leaves of Arabidopsis increased after infection with F. solani, but the MDA content in MdWRKY20-OE lines were significantly less than the Col, which also proved that the overexpression of MdWRKY20 had strong tolerance to F. solani infection.
-
In this study, MdWRKY20 was isolated from the apple rootstock 'M9T337' and its role in resisting F. solani infection reported. The plants with high expression of MdWRKY20 showed enhanced resistance to F. solani infection. MdWRKY20 promoted its expression by combining with the promoters of disease course related proteins MdPR1 to improve the resistance of plants, which was the positive regulatory factor for resistance of apple to F. solani. This work has provided a scientific basis for the prevention and control of ARD and can be used to introduce durable resistance to replant disease in apples.
This research was funded by the National Natural Science Foundation of China (Grant No. 32072510), China Agriculture Research System of MOF and MARA (Grant No. CARS-27), Taishan Scholar Funded Project (No. ts20190923, No. tsqn202408119), Key RandD program of Shandong Province (Grant No. 2022TZXD0037), and the National Key Research and Development Program (Grant No. 2023YFD2301003).
-
The authors confirm contribution to the paper as follows: experiment design: Mao Z, Yin C; research performed: Zhao L, Liu Y, Wang M and Xiang L; data analysis: Wang H; manuscript preparation: Jiang H, Chen X. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Lei Zhao, Yusong Liu
- Supplemental Table S1 Primer Sequence used in the experiment.
- Supplemental Fig. S1 Infection rate and mortality rate of apple rootstock 'M9T337' infected with F. solani after 12 d.
- Supplemental Fig. S2 Relative expression of MdWRKY20 in different tissues of apple.
- Supplemental Fig. S3 Gene expression analysis of transgenic Arabidopsis lines.
- Supplemental Fig. S4 Germination of Arabidopsis under control and F. solani treatment.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhao L, Liu Y, Wang M, Xiang L, Wang H, et al. 2025. MdWRKY20-MdPR1 module mediates resistance of apple to Fusarium solani. Fruit Research 5: e001 doi: 10.48130/frures-0024-0033 

MdWRKY20-MdPR1 module mediates resistance of apple to Fusarium solani
- Received: 09 September 2024
- Revised: 30 September 2024
- Accepted: 12 October 2024
- Published online: 06 January 2025
Abstract: Apple replant disease (ARD) caused by the pathogen Fusarium solani is a destructive disease in apple planting areas worldwide, which leads to the decline of apple quality and yield. WRKY transcription factors are involved in the process of plants responding to various environmental stresses, but the function of WRKY TFs in ARD is unclear. In this study, the expression of MdWRKY20 was significantly increased after infection of apple rootstock 'M9T337' with F. solani. Transgenic analysis showed that the resistance of apple callus and Arabidopsis to F. solani increased after overexpression of MdWRKY20. Ectopic expression of MdWRKY20 also significantly enhanced antioxidant capacity in Arabidopsis under treatment with F. solani. Then, MdWRKY20 was found to bind directly to the W-box II of the MdPR1 promoter and significantly promoted its expression. In summary, MdWRKY20 plays a positive role in regulating the resistance of apples to F. solani.
-
Key words:
- Apple replant disease /
- Fusarium solani /
- MdWRKY20 /
- MdPR1 /
- Disease resistance