










-
Cultivated soybean (Glycine max) was domesticated from wild soybean (Glycine soja) approximately 5,000 years ago[1]. Soybean is one of the most economically important leguminous seed crops; it is not only a major oilseed crop, but also a source of protein for human consumption and animal feed. Currently, in China, approximately 16 million tons of soybean are used for human food annually; 45% of plant oil and 67% of animal feed are provided by soybean. Soybean is rich in nutrients such as folate, flavones, and lecithin, which are required for the human diet. To generate widely adaptable, specialized, and highly productive soybean materials, a detailed transcriptional map of seed development is needed to understand the regulation of seed production.
Seeds are the reproductive tissue of flowering plants and contain embryos that generate progeny. There are three main steps in embryo development: embryo morphogenesis, seed maturation, and seed desiccation. In dicotyledonous plants such as soybean, mature seeds are composed mainly of the seed coat and embryo, so that embryo development directly determines seed size and weight. The transcriptome is always used as a powerful tool to demonstrate complex developmental processes. In plants such as foxtail millet, wheat, maize, and barley, the transcriptome landscape of seed and embryo/endosperm development have been generated during the stages from pollination to seed maturation[2−6]. Arabidopsis has been used as a model for detailed embryo and endosperm development through transcriptome atlas at the single-cell and spatial level[7−11]. In the oilseed crop Brassica, transcriptome maps of embryo and seed coat development from the zygote stage to the mature seed stage have been generated[12]. While in soybean, the transcriptome map of seed development was generated via intact seeds, and the transcriptome datasets of embryo development included only the early stages; the fine transcription landscape for soybean embryo development from embryogenesis to seed germination is still unavailable[13−15]. Moreover, the embryo transcriptome landscape of other leguminous crops is still lacking. Therefore, a transcriptional atlas of soybean embryo development throughout the whole seed development process is needed, which could be used as a reference for other leguminous crop species.
The basic embryo development processes are similar between Arabidopsis and soybean, but several differences exist. First, embryo morphogenesis in Arabidopsis includes the globular, heart, and torpedo stages, whereas in soybean, it includes the globular, heat, and cotyledon stages[15,16]. Soybean has a more obvious cotyledon transition process. Second, during the seed maturation stage, many more secondary metabolites accumulate in soybean than in Arabidopsis, and the balance between the oil and protein contents in soybean is always an important index for high-quality soybean[17−19]. Third, soybean is more sensitive to environmental changes during seed development and yield is affected much more easily under improper temperature, light, and water conditions[20−23]. Although detailed transcription data for Arabidopsis embryos are available, a high-quality transcriptome map for soybean embryo development, especially for the embryo axis and cotyledon development is still needed to better understand and manipulate soybean production.
Soybean has experienced a long period of domestication, and the morphology and adaptability of soybean seeds have changed greatly[24]. The release of the soybean pan-genome, along with the phylogenomics of the genus Glycine, offers new opportunities to explore the details of seed development during domestication and evolution[25,26]. With the benefit of seed transcriptome data and germplasm population data, key genes and networks involved in controlling seed oil or protein content have been identified in both soybean and Brassica[27−32]. The chromatin feature changes during soybean domestication have been identified[33]. Furthermore, the transcriptomes of embryo propers and suspensors in embryos of Scarlet runner bean, common bean, soybean, and Arabidopsis revealed variations in the embryo morphology and main regulatory networks during evolution[34]. Therefore, the transcriptome landscape of soybean embryo development could largely contribute to uncovering the history of soybean domestication in terms of seed traits.
Here, a transcriptome atlas was generated for soybean embryo development from the heart embryo stage to dry seeds, including the embryo axis and cotyledon from most stages. Moreover, the samples of seed imbibition and seed germination were also included to create a comprehensive map of soybean embryo development. In this landscape, transcription factors involved in stage transition were identified, the time trajectory of the secondary metabolites as well as internal and external signal responses was determined, and RNA storage in dry seeds was detected. This transcriptional landscape revealed dynamic changes in gene expression during embryo development in soybean. In particular, several potential crucial genes in a developmental stage-dependent manner were identified. These datasets provide important resources for future functional genomic research in soybean.
-
Glycine max variety Williams82 was used in this work. The plants were grown in the field in Guangzhou, Guangdong Province (China). Pods containing embryos at the globular and heart embryo stages were collected for paraffin sectioning, and pods containing embryos from the heart embryo to later stages were collected for manual dissection. Dissection was performed using an SZ680 dissection microscope, and images were taken with a Zeiss Discovery V8. The isolated embryos, as well as the embryo axis and cotyledon, were used for RNA extraction and subsequent mRNA-seq library preparation.
Paraffin-embedded sections
-
Young ovules from early-stage pods were collected and fixed in FAA solution overnight. After dehydration with a series of graded ethanol solutions, the samples were embedded in paraffin. Sections with a thickness of 3−5 μm were prepared. The slides were stained with hematoxylin, and images were taken using a ZEISS Imager A2.
RNA-seq analysis
-
The mRNA-seq libraries were prepared using the NEBNext Ultra Il RNA Library Prep Kit and sequenced via the Illumina platform at Personalbio Company to obtain paired 150-base pair (bp) reads. The sequenced reads were subsequently mapped to Glycine max Williams82 a4 with STAR (2.7.3a). Sample correlation heatmap analysis and PCA were performed via deepTools (
https://deeptools.readthedocs.io/en/latest/content/list_of_tools.html ).Kallisto (v.0.43.0) and Sleuth (v.0.30.0) were used to obtain transcript per million (TPM) values and p values, respectively. Expressed genes are identified by TPM > 2. Differentially expressed genes (DEGs) were identified by p < 0.01 and a TPM fold-change > 2. GO annotation was performed via ShyniGO 0.80 (
http://bioinformatics.sdstate.edu/go/ ). Upset plots, volcano plots, and box plots were generated with a custom ggplot2 R script. Heatmaps were generated via online tools (www.genescloud.cn/chart/ChartOverview ,https://hiplot.com.cn/cloud-tool/drawing-tool/detail/106 ).Two genes (GLYMA.18G158900 and GLYMA.10G211600) from folate synthesis pathway were chosen for RT-qPCR confirmation, the primers used for RT-qPCR are listed in Supplementary Table S14, Actin was used as an internal reference gene for normalization.
Statistical analysis
-
In Fig. 3b and c, the genes represented for each sample were selected using the following criteria: TPM > 80 or 150 (samples with TPM > 80 are heart embryo, cotyledon embryo, early-maturation embryo axis (EA), early-maturation cotyledon (CT), germination shoot, germination root, young seedling shoot, and young seedling root; samples with TPM > 150 are middle-maturation EA, middle-maturation CT, late-maturation EA, late-maturation CT, dry-seed EA, dry-seed CT, imbibition-seed EA, imbibition-seed CT, germination CT, and young seedling CT), TPM fold-change with the comparison of each sample to young seedling shoot > 20, TPM of germination shoot < 20, and TPM of young seedling shoot < 20.
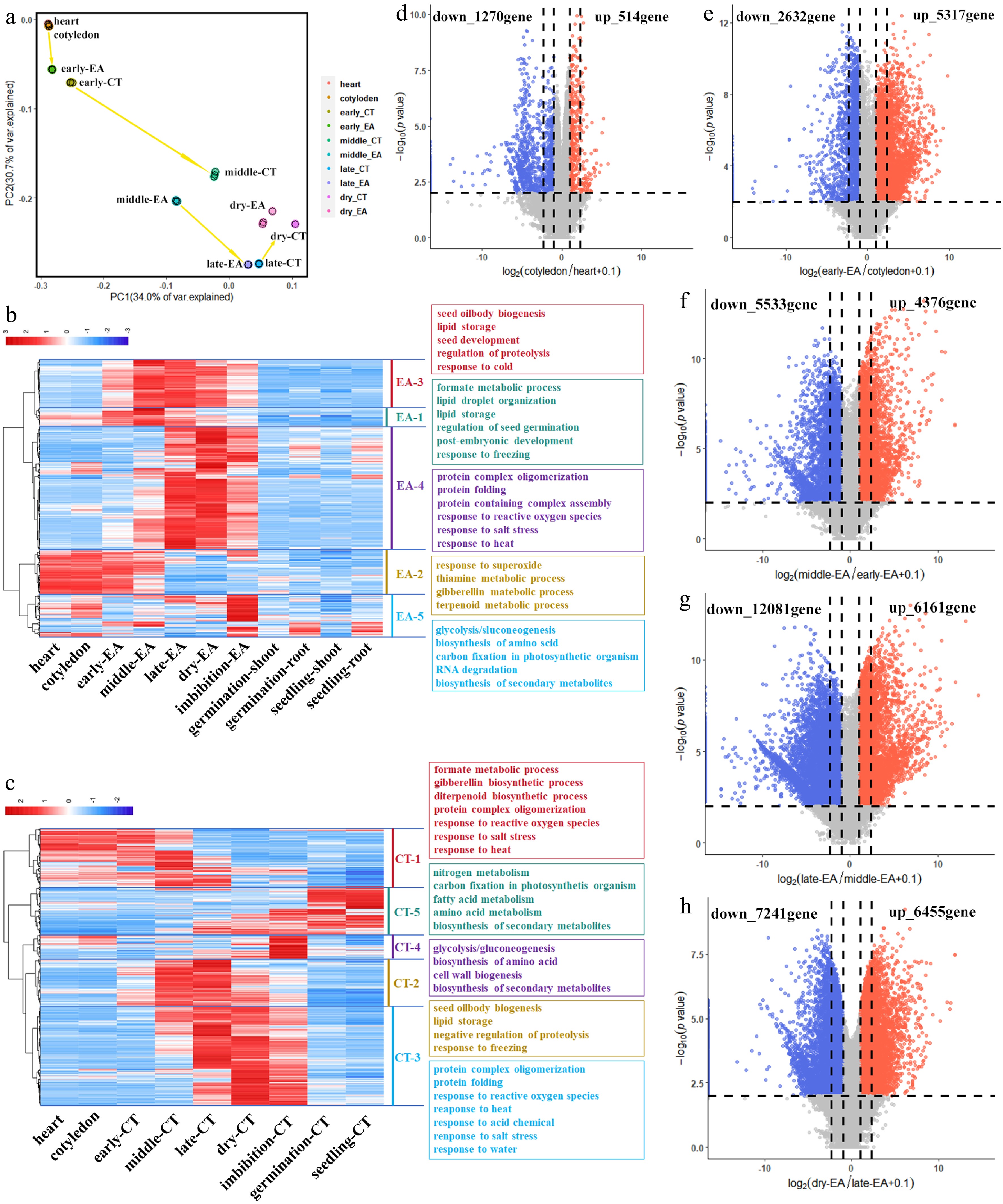
Figure 3.
Transcription dynamics during soybean embryo development. (a) PCA analysis shows correlation of samples from heart embryo to dry seed. (b) Heatmap of significant genes during EA development, and five gene clusters were clarified to show the main cell biological process in EA. (c) Heatmap of significant genes during CT development, and five gene clusters were clarified to show the main cell biological process in CT. Volcano plot and DEG numbers for the comprison of (d) cotyledon embryo vs heart embryo, (e) early-maturation EA vs cotyledon embryo, (f) middle-maturation EA vs early-maturation EA, (g) late-maturation EA vs middle-maturation EA, and (h) dry-seed EA vs late-maturation EA, respectively.
In Fig. 4b, the EA-enriched transcription factors (TFs) were selected based on the following criteria: TPM of early-maturation EA > 10, TPM of early-maturation CT < 5, TPM of middle-maturation CT < 5, TPM of dry-seed CT < 5, TPM fold-change of early-maturation EA / early-maturation CT > 4, and TPM fold-change of middle-maturation EA / middle-maturation CT > 4.
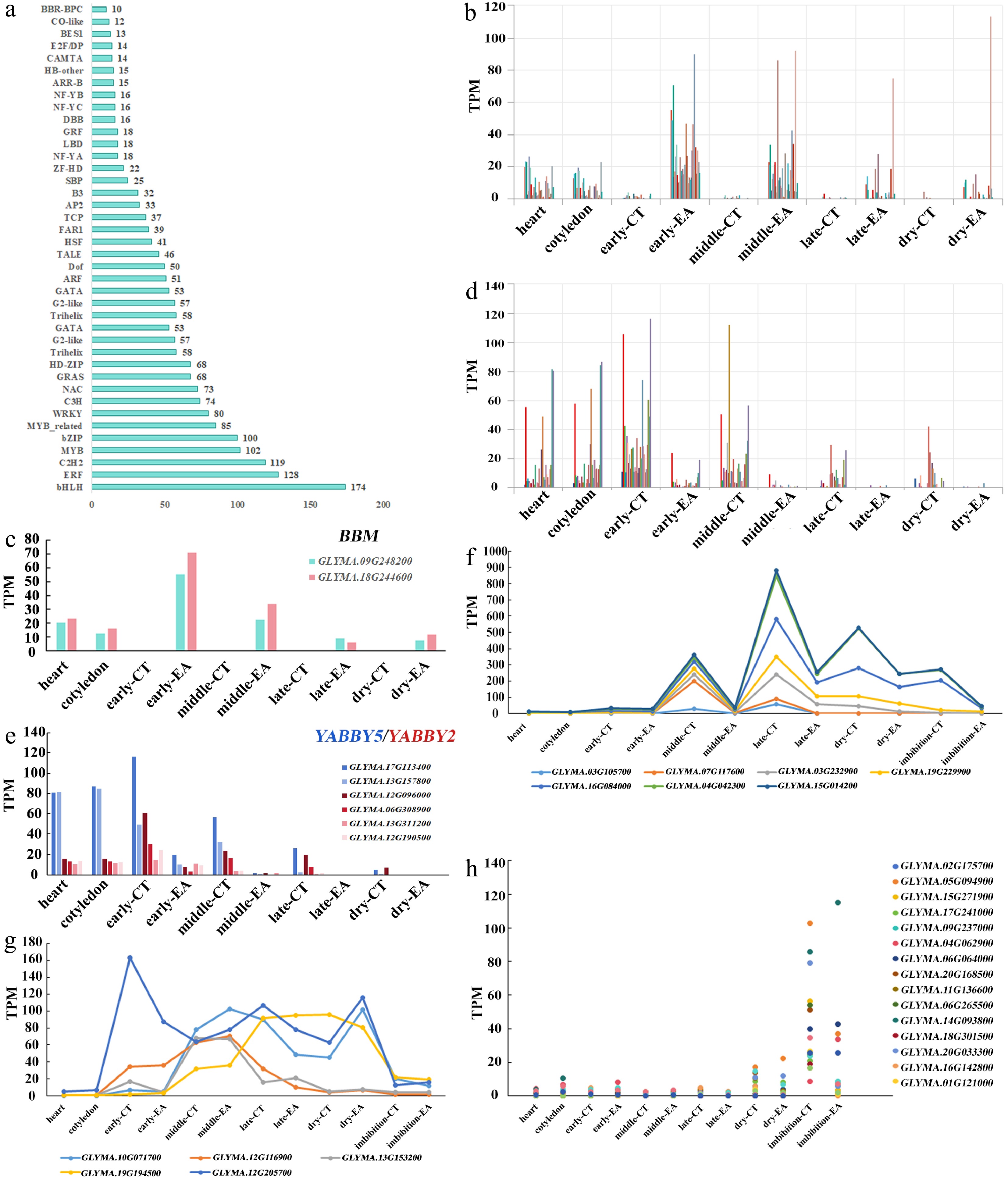
Figure 4.
TFs involved in embryo morphogenesis and development. (a) Family and numbers of TFs expressed during soybean embryo development. (b) Expression pattern of EA-enriched TFs. (c) Expression of two BBM genes. (d) Expression pattern of CT-enriched TFs. (e) Expression of six YABBY genes. (f) Expression pattern of maturation-enriched TFs with high level in CT. (g) Expression pattern of maturation-enriched TFs with high level in a certain stage. (h) Expression pattern of TFs enriched in imbibition seed.
In Fig. 4d, the CT-enriched TFs were selected on the basis of the following criteria: TPM of early-maturation CT > 10, TPM fold-change of early-maturation CT / early-maturation EA > 4, and TPM fold-change of middle-maturation CT / middle-maturation EA > 4.
In Fig. 4f and g, the maturation-enriched TFs were selected based on the following criteria: TPM of middle-maturation CT > 15, TPM of late-maturation CT > 15, TPM fold-change of middle-maturation CT / cotyledon > 4, TPM fold-change of late-maturation CT / cotyledon > 4, TPM of young seedling bud < 10, and TPM of young seedling root < 10.
In Fig. 4h, imbibition-enriched TFs were selected according to the following criteria: TPM of late-maturation CT < 5, TPM of late-maturation EA < 5, TPM of middle-maturation CT < 5, TPM of middle-maturation EA < 5, TPM of young seedling bud < 5, TPM of young seedling root < 5, TPM of imbibition-seed EA > 15, and TPM of imbibition-seed CT > 15.
In Supplementary Fig. S3d, early embryo-enriched TFs were selected based on the following criteria: TPM of heart embryo > 10, TPM of cotyledon embryo > 10, TPM fold-change of heart / early-maturation EA > 2, and TPM fold-change of heart / early-maturation CT > 2.
-
Plant embryos are initiated from fused egg-sperm cells. In soybean, this process takes place inside the papilionaceous flower before floral opening. Zygotes then undergo cell division to generate embryo proper and suspensor[34]. The embryos are located in the embryo sac near the micropyle, with easily recognized structures from a globular shape at the beginning to a heart shape (Fig. 1a). The subsequent seed maturation stage is divided into three parts: the 'early maturation' stage, with distinct structures of the embryo axis (EA) and the cotyledon (CT), accompanied by a large increase in cell number; the 'middle maturation' stage, with a gradual accumulation of nutrients and dehydration substrates resulting in increased seed size; and the 'late maturation' stage, with seeds turning yellow and dry, ending with the highest level of metabolite accumulation (Fig. 1b). The 'imbibition' stage occurs when the dry seeds are fully rehydrated (Fig. 1c). The 'germination' seeds have hook hypocotyls and closed cotyledons, whereas young seedlings have straight hypocotyls and open cotyledons (Fig. 1c). Whole embryos from heart and cotyledon embryos, as well as EA and CT from maturation-, dry-, and imbibition-seeds, were isolated to generate transcription data (Fig. 1d). The samples of CT, shoot, and root from germinated seeds were also included (Fig. 1d). Thus, a total of 18 samples (each with three replicates) were used to generate the transcriptome landscape for soybean embryo development.
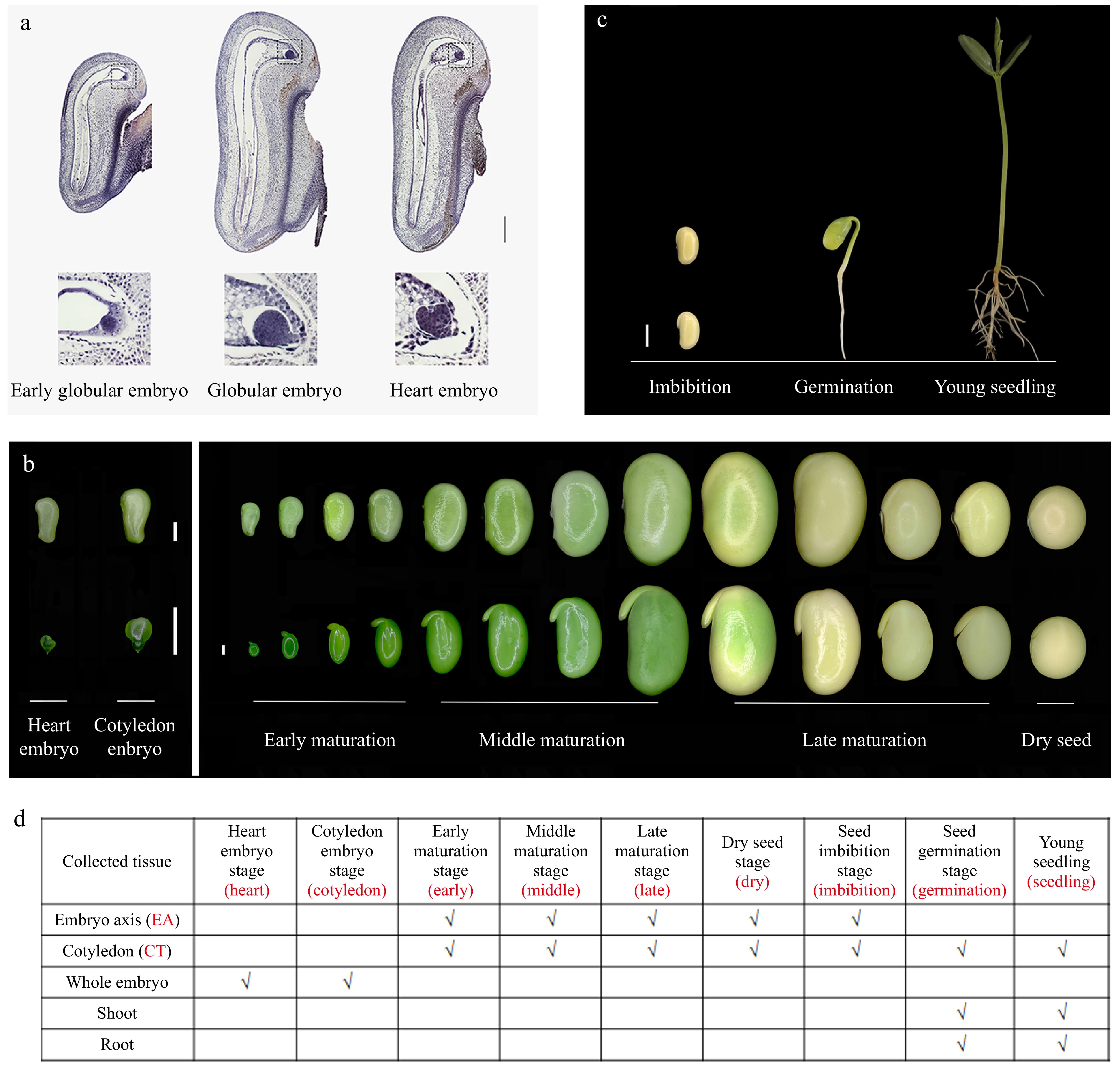
Figure 1.
Morphological view of soybean seed development and germination process. (a) Paraffin section of globular and heart embryo stage seeds. The upper panel shows the longitudinal section of the ovule with embryos inside the embryo sac, the lower panel shows the enlarged view of embryos. Scale bar = 100 μm. (b) Dissected embryos from the corresponding seeds during soybean seed development. The upper panel shows the whole seeds, the lower panel shows the isolated embryos. Scale bar = 1 mm. (c) Morphological view of imbibition seed, germinated seed, and young seedling. Scale bar = 1 cm. (d) The samples used for RNA-seq library preparation. '√' represents the selected samples. The words in red represent the short name for the stages and samples in later analysis.
A transcriptome atlas of the embryo axis and cotyledon development in soybean seeds
-
Each RNA-seq library had a mean value of 22.5 million 150 bp paired reads, with an average unique mapping rate of 94.48% (Supplementary Table S1). The three replicates within each sample were highly correlated (Supplementary Fig. S1a, 1b). The total number of expressed genes in each sample (with TPM > 2) was between 16,808 and 26,941. The dry seeds presented obvious valleys, as the number of expressed genes was 18,800 in EA and 16,808 in CT (Fig. 2a & b). The number of detected expression genes are nearly the same in heart (23,063) and cotyledon (23,056) embryos. The expressed genes reached the highest level in early-maturation embryos, with 25,473 in EA and 24,229 in CT, and then gradually decreased to the lowest level in dry seeds. After rehydration, the number of expressed genes increased again in the imbibition and germination embryos (Figs. 2a & b). In each sample, nearly 2/3 of the genes presented a transcription level between TPM of 2 and 100, whereas 1/3 of the genes were not expressed (TPM < 2) (Fig. 2c). EA had more genes with TPM values of 2−100 than CT in each stage, whereas CT had more highly expressed genes (TPM > 100) than EA (Fig. 2c). The expression of genes with different transcription levels generally decreased to the lowest level in dry seeds (Fig. 2c). These results revealed dynamic transcription features during seed development and germination.
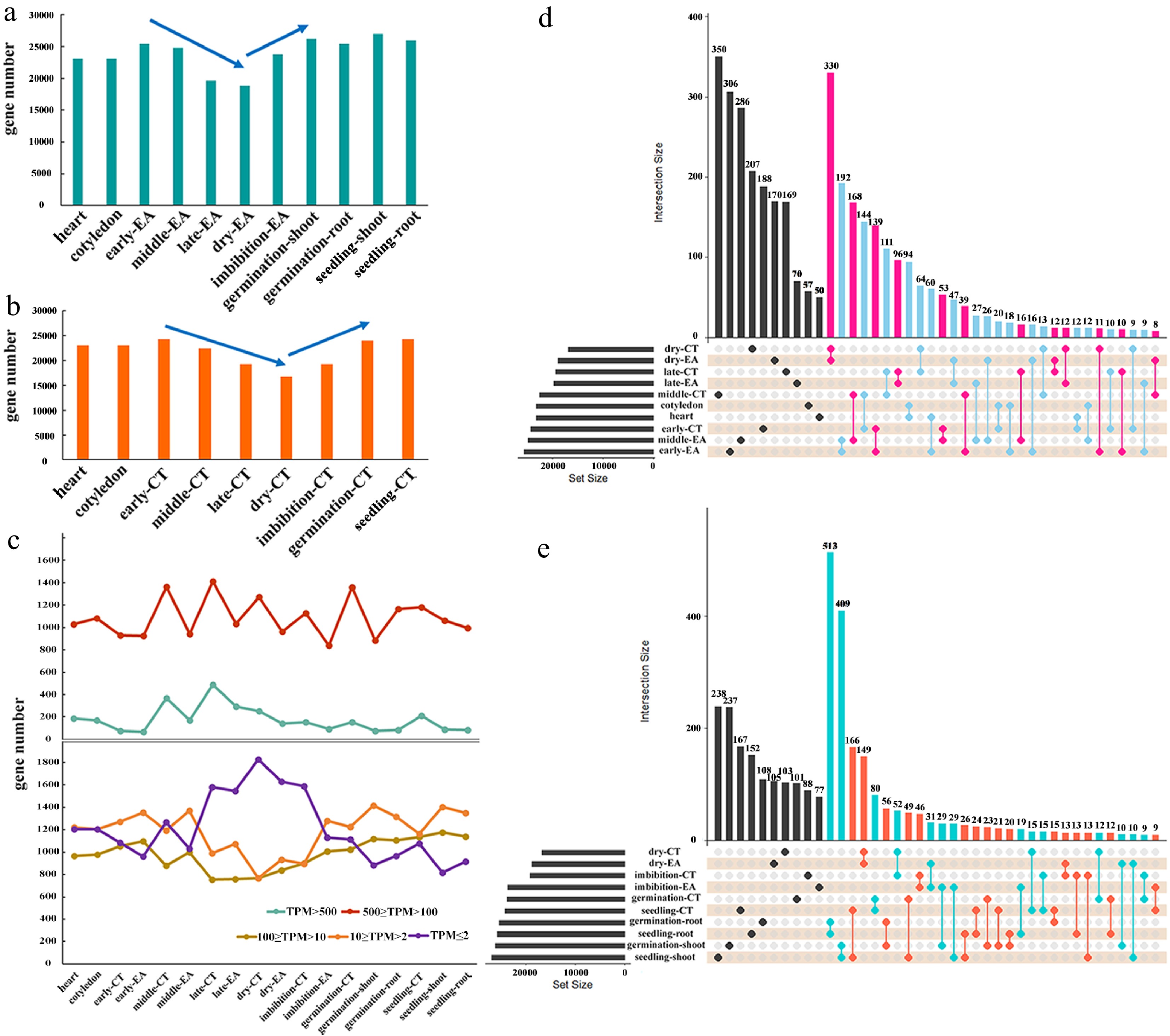
Figure 2.
The transcriptome atlas for soybean embryo development. (a) The number of expressed genes in EA of each stage, and in root/shoot of the germinated plants. (b) The number of expressed genes in CT of each stage. (c) The number of genes from five different expression levels in all the samples. Shared genes and specific genes in samples from (d) heart embryo to dry seed, and from (e) dry seed to young seedling.
The expressed genes were initially compared between different samples and it was found that the number of genes shared between EA and CT from the same stage was always greater than that between EAs or CTs at different stages (Fig. 2d & e). For example, 330 genes were shared between dry-seed CT and dry-seed EA, but 64 genes were shared between dry-seed CT and late-maturation CT, 13 between dry-seed CT and middle-maturation CT, 47 between dry-seed EA and late-maturation EA, and 26 between dry-seed EA and middle-maturation EA (Fig. 2d). These data suggested that, without consideration of gene expression levels, gene expression patterns in EA and CT from the same embryo are generally more similar; thus, the variations in EA/CT between different stages were the main focus in the subsequent analysis.
Transcription dynamics during soybean embryo development
-
The sample correlation clearly revealed a pattern of embryo development from heart embryo to dry seed (Fig. 3a). Heart and cotyledon embryos are closely correlated, whereas early-maturation embryo is more closely correlated with earlier stages than with later stages (Fig. 3a). EA and CT from the same stage were clustered together, which is consistent with the results in Fig. 2d. Imbibition seed and dry seed were clustered together, and germination seed and young seedling were clustered together, which is consistent with the developmental process (Supplementary Fig. S2a). Featured genes of EA from each stage were identified, and these genes were combined to show the general process of EA development (Fig. 3b). Five core cell biological processes were detected on the basis of gene expression patterns (from 'EA-1' to 'EA-5') (Fig. 3b). According to the annotation, the postembryonic development process (in 'EA-1') occurs together with the gibberellin response (in 'EA-2') in the heart and cotyledon embryos, the metabolic substrate biogenesis and environment signal response starts from the early embryo until the dry seed (from 'EA-1' to 'EA-4'), and lipid biogenesis takes place in the early embryo ('EA-1'), while protein production is enriched in the middle maturation stage ('EA-4') (Fig. 3b). The biological processes in CT are similar to those in EA (Fig. 3c).
When differentially expressed genes (DEGs) were compared between adjacent stages, notably, from the heart to cotyledon embryo, only 514 genes were upregulated, whereas 2,170 genes were downregulated (Fig. 3d). There was a dramatically distinct trend of DEG changes between EA and CT in the early-maturation embryo; in EA, there were 5,317 upregulated genes and 2,632 downregulated genes, whereas in CT, there were 4,672 and 4,232 up- and downregulated genes, respectively (Fig. 3e; Supplementary Fig. S2b). In the later stages, the DEG variations were similar between EA and CT. In the middle maturation stage and dry seed, approximately 1,000 more DEGs were downregulated than upregulated; in the late maturation stage, nearly 5,000 more genes were downregulated than upregulated, which was associated with significant gene depression during seed desiccation (Fig. 3e−h; Supplementary Fig. S2c−S2e). The typical biological processes in EA and CT are consistent, and the KEGG terms 'circadian rhythm' and 'biosynthesis of secondary metabolites' exist extensively in early-, middle-, and late-maturation embryos. 'Flavonoid biosynthesis' is specified in early- and middle-maturation EA, and 'ubiquitin mediated proteolysis' and 'plant hormone signal transduction' are specified in late-maturation CT (Supplementary Fig. S2f−S2k). These results revealed key cell biological processes during soybean embryo development, which are coincident with the seed morphogenesis process.
Transcription factors involved in soybean embryo development
-
Transcription factors (TFs) play key roles in cell fate determination and developmental stage transition. In total, 3,747 TFs were identified in soybean according to family assignment rules and classified into 57 families (
https://planttfdb.gao-lab.org/index.php?sp=Gma ). A total of 2,190 TFs existed in our transcriptome landscape, among which 1,922 TFs were expressed (TPM > 2). These 1,922 TFs are arranged on the basis of the families and gene numbers in each family (Fig. 4a). The top five TFs are the basic helix-loop-helix (bHLH), ethylene responsive factor (ERF), C2H2 zinc finger, myeloblastosis (MYB), and basic leucine zipper (bZIP) families, followed by the WRKY, C3H zinc finger, NAM, ATAF1/2, and CUC2 (NAC), GAI-RGA-, and -SCR (GRAS) families (Fig. 4a). There are 174 bHLH family TFs expressed during embryo development, accounting for 31.75% (174/548) of the total bHLH family. For some TFs, more than half of the family of genes are expressed during embryo development; for example, 49.33% (74/150) of the genes in the C3H zinc finger family, 55.77% (58/104) of the genes in the Trihelix family, 57.61% (53/92) of the genes in the GATA family, and 50.61% (41/81) of the genes in the heat shock transcription factor (HSF) family are active in soybean embryos.TFs involved in soybean and Brassica seed development have been reported previously[35,36], but detailed information on TFs enriched in each embryo stage is lacking. TFs involved in embryo morphogenesis and stage transition were detected in the datasets. Thirty two EA-enriched TFs were identified that are relatively highly expressed in EA during embryo development (Fig. 4b; Supplementary Table S2). Two BABY BOOM (BBM) genes (GLYMA.09G248200 and GLYMA.18G244600) and four SHOOT MERISTEMLESS (STM) genes (GLYMA.14G047000, GLYMA.09G007500, GLYMA.15G111900, and GLYMA.07G263600) were in this group, and these genes function in embryogenesis[37−39] (Fig. 4c; Supplementary Fig. S3a). Two WUSCHEL RELATED HOMEOBOX 11 (WOX11) genes (GLYMA.03g007600 and GLYMA.19g118400) are expressed in late EA and function in organ regeneration and seed dormancy[40,41] (Supplementary Fig. S3b).
Twenty eight CT-enriched TFs were identified that were highly expressed in CT (Fig. 4d; Supplementary Table S3). The YABBY family is involved in abaxial cell fate specification in lateral organs[42]. Two YABBY5 genes (GLYMA.17G113400 and GLYMA.13G157800) and four YABBY2 genes (GLYMA.12G096000, GLYMA.06G308900, Glyma.13G311200, and Glyma.12G190500) were identified in this group (Fig. 4e). The Teosinte branched1/Cincinnata/proliferating cell factor (TCP) family of proteins are reported to be involved in heterochronic regulation of leaf differentiation[43,44]. It was found that two TCP5 genes (GLYMA.17G079900 and GLYMA.05G019900), four TCP2 genes (GLYMA.13G219900, GLYMA.15G092500, Glyma.05g142000, and Glyma.08G097900), and four TCP3 genes (GLYMA.06G232300, Glyma.12G158900, Glyma.13G271700, and Glyma.12G228300) are highly expressed in early-maturation CT (Supplementary Fig. S3c).
Nineteen early embryo-enriched TFs were identified with high expression in heart and cotyledon embryos but decreased expression in the later stages (Supplementary Fig. S3d; Supplementary Table S4). The AINTEGUMENTA (ANT) family regulates growth and cell numbers during organogenesis, six ANT genes (GLYMA.14G089200, GLYMA.06G049200, Glyma.04G047900, Glyma.17G232290, Glyma.05G108600, and Glyma.17G158300) were found that are enriched in this group[45,46] (Supplementary Fig. S3e).
Soybean has experienced a long period of polyploidization and domestication, and duplicated genes are the main products[24−26,33,47]. Thus, the transcriptional divergence of the duplicated genes were checked for these TFs mentioned above. There are three copies of BBM genes in soybean; two copies are highly expressed during embryogenesis (Fig. 4c), whereas the third copy (Glyma.10G171400) was undetectable in the whole landscape. Among the six STM genes in soybean, four of which are enriched in EA at early embryo stages (Supplementary Fig. S3a), one (Glyma.17G010200) has relatively low transcription (Supplementary Fig. S3a), and the expression of the last one (Glyma.02G269900) is undetectable. The WOX11 genes have two copies in soybean (Supplementary Fig. S3b). The YABBY5 and YABBY2 genes have two and four copies in soybean, respectively, all of them are highly expressed in CT (Fig. 4e). Each of the TCP5, TCP2, and TCP3 gene has four paralogs, except two TCP5 genes (Glyma.06G204300 and Glyma.04G161400), the remaining genes are highly expressed in CT (Supplementary Fig. S3c). There are six copies of ANT genes in soybean, all of which have similar expression patterns (Supplementary Fig. S3e). According to these results, the genes in soybean could have as many as six paralogs, and both divergence and redundancy exist in these duplicated genes. The data revealed that soybean embryo development involves a more complicated regulatory network than Arabidopsis and rice.
The TFs involved in seed maturation can be classified into two groups on the basis of the transcription pattern: the first group has a high expression peak in CT and a low expression peak in EA, and the second group has expression peaks covering both CT and EA within a specific stage (Fig. 4f, g). For example, HSF family members are known heat shock factors that respond to heat, and four HSF genes (GLYMA.17G053700, GLYMA.13G105700, GLYMA.20G156800, and GLYMA.09G143200) are highly expressed in both the EA and CT of late-maturation and dry seeds (Supplementary Fig. S3f). Three ABA response TFs (GLYMA.07G083500, GLYMA.08G056700, and GLYMA.06G010200) were highly expressed either in middle- and late-maturation CT or in the middle/late stages, covering CT and EA (Supplementary Fig. S3g−S3i). Finally, 17 germination seed-specific TFs were identified, among which four bHLH family genes (GLYMA.02g175700, GLYMA.05g094900, GLYMA.15g271900, and GLYMA.17g241000) were highly expressed (Fig. 4h; Supplementary Fig. S3j). The well-known TFs were present in the predicted tissue and stage. Moreover, soybean genes with little functional annotation were detected in our transcriptome atlas, which is important for future soybean functional genomics research (Fig. 4f−h; Supplementary Tables S2−S4).
Metabolite synthesis during soybean embryo development
-
Soybean is an important crop because of its abundant metabolite production. Modern breeding aims to create specialized soybeans with the help of biosynthesis technology, so metabolite synthesis details during embryo development are needed. Soybean has been domesticated to have a relatively high oil content, and there is a competence between oil content and protein content. It was found that the expression patterns of oil and protein synthesis genes during embryo development are quite similar, the core synthesis periods are focused in the maturation stage and dry seed stage, and genes generally have higher expression levels in CT than in EA (Fig. 5a & b). Soybean has a high content of flavonoids, and the genes from the flavonoid synthesis pathway do not exhibit biased expression in EA or CT (Fig. 5c). Instead, they present two clear waves: one wave has high expression before the late maturation stage, and the other wave has high expression from the late maturation stage until seed imbibition (Fig. 5c). Soybean oil utilization is determined by fatty acid composition, and fatty acid synthesis genes have expression patterns similar to those of flavonoids (Supplementary Fig. S4a). In soybean, the plants with higher steroid levels had increased yield under drought stress conditions[48]. The steroid-related genes in soybean are expressed during whole embryo development even in imbibed seeds, and their expression peaks are before the late embryo stage (Supplementary Fig. S4b).
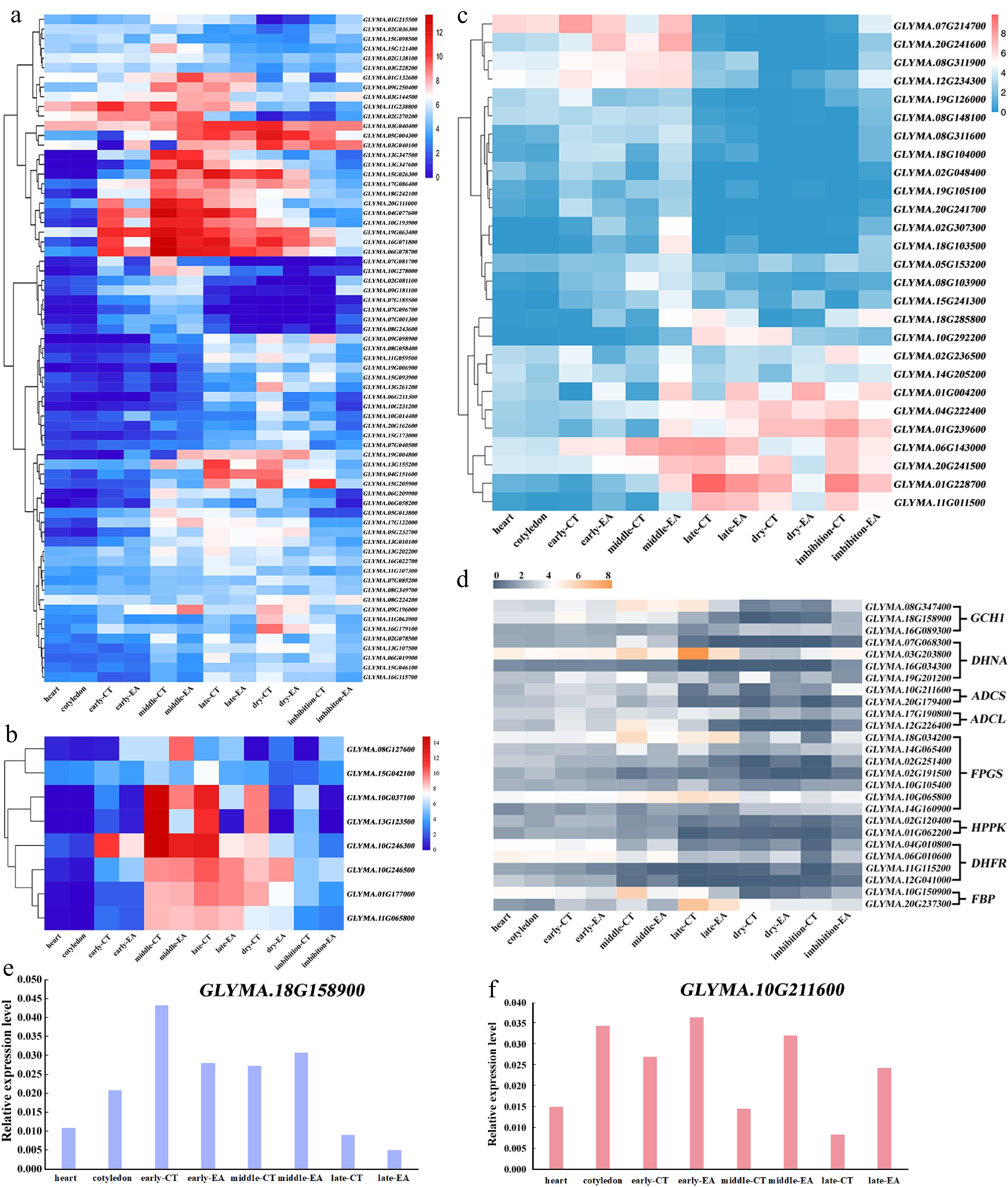
Figure 5.
Metabolites synthesis during soybean embryo development. Heatmap of gene expression patterns from (a) lipid, (b) conglycinin and glycinin, (c) flavonoid, (d) folate synthesis network during embryo development, respectively. RT-qPCR results for two genes, (e) GLYMA.18G158900, and (f) GLYMA.10G211600, which are involved in folate synthesis.
Soybean is also a good source of folate. The main folate synthesis pathway included GTP CYCLOHYDROLASE I (GCH1), DIHYDRONEOPTERIN ALDOLASE (DHNA), AMINODEOXYCHORISMATE SYNTHASE (ADCS), 4-AMINO-4-DEOXYCHORISMATE LYASE (ADCL), FOLYLPOLYGLUTAMATE SYNTHETASEs (FPGSs), HYDROXYMETHYLDIHYDROPTERIN PYROPHOSPHOKINASE (HPPK), DIHYDROFOLATE REDUCTASE THYMIDYLATE SYNTHASEs (DHFR-TSs), and FOLATE BINDING PROTEIN (FBP) (Supplementary Table S5). According to the transcription map, it is obvious that most of the folate synthesis genes have a core expression stage before dry seeds (Fig. 5d). The expression pattern was checked for one copy of GCH1 gene (GLYMA.18G158900) and one copy of ADCS gene (GLYMA.10G211600) by RT-qPCR, the results showed consistent expression patterns with the transcriptome data (Fig. 5e & f). Furthermore, it was found that gene divergence and redundancy also occurs in folate synthesis-related genes. Each of these genes have two to five paralogous copies, and the copies have variant transcription patterns; for example, two of the ADCL gene, Glyma.06G082700 and Glyma.13G274402, do not have detectable transcription in soybean embryos (Supplementary Table S5).
Internal and external signal response during soybean embryo development
-
The phytohormones gibberellin (GA) and abscisic acid (ABA) have antagonistic effects on seed development[49]. GA promotes cell division and growth in the early embryogenesis stage, although ABA has a short high-level window in this process[18]. In the seed maturation stage, ABA mainly acts to promote cell enlargement and seed filling; later, ABA induces dormancy and inhibits germination in mature seeds by upregulating its own levels and downregulating GA synthesis[18]. The GA/ABA-related genes were divided into GA/ABA biosynthesis pathway genes and signal transduction pathway genes. The GA biosynthesis related genes mainly included ENT-COPALYL DIPHOSPHATE SYNTHETASE (CPS), ENT-KAURENE SYNTHASE (ent-KS), ENT-KAURENE OXIDASE (ent-KO), ENT-KAURENOIC ACID HYDROXYLASE (ent-KAO), GIBBERELLIN 20/3/2 OXIDASE (GA20ox/GA3ox/GA2ox), and ELONGATED UPPERMOST INTERNODE 1 (EUI1) (Supplementary Table S6). The GA signal transduction related genes mainly included GA-INSENSITIVE (GAI), REPRESSOR of GA1-3 (RGA), GA-INSENSITIVE DWARF1 (GID1) and SLEEPY1 (SLY1) (Supplementary Table S6). The transcription patterns showed that most of these genes involved in GA biosynthesis and signal transduction had intensive and relatively high expression before the late embryo maturation stage (Fig. 6a). Moreover, the ABA biosynthesis pathway included a group of genes such as ZEAXANTHIN EPOXIDASE (ZEP), 9-CIS-EPOXYCAROTENOID DIOXYGENASE (NCED), ALDEHYDE OXIDASE 3 (AAO3), and ABA 8'-hydroxylase (CYP707A1 and CYP707A2) (Supplementary Table S7). The ABA signal transduction pathway genes included PYRABACTIN RESISTANCE 1 (PYR1), PYR1-LIKE (PYL), PROTEIN PHOSPHATASE 2C (PP2C), SNF1-RELATED PROTEIN KINASE 2 (SnRK2s), ABA INSENSITIVE (ABIs), FUSCA 3 (FUS3), LEAFY COTYLEDON 1 (LEC1), and other factors (Supplementary Table S7). It was found that several genes (ABAs, CYP707As, AHGs, and SnRK2s) had increased expression levels from late embryo maturation stage until imbibed seeds, however, the variation level was weaker than GA-related genes (Fig. 6b). These results revealed that the expression of the GA and ABA-related genes are temporally separated during embryo development, with the trend in GA-associated genes were more significant.
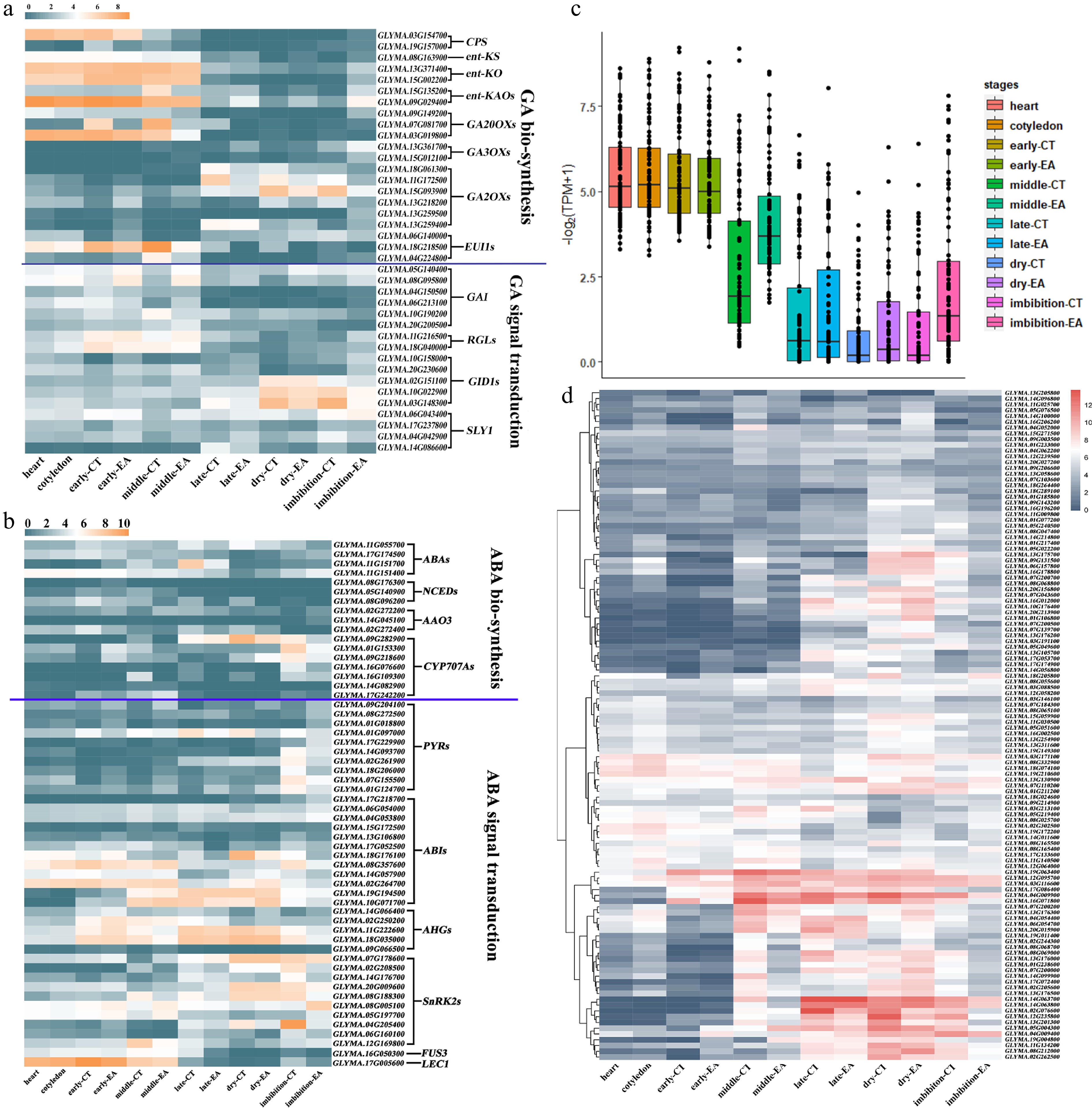
Figure 6.
Internal and external signal response in soybean embryo development. Heatmap of gene expression patterns from (a) GA, and (b) ABA synthesis and signal transduction network during embryo development. (c) Expression pattern of cell cycle associated genes. (d) Heatmap of gene expression patterns from water and heat response network expression during embryo development.
In early embryo development, which includes heart embryo, cotyledon embryo, and early maturation embryo, cell cycle genes as well as DNA replication genes are highly expressed compared with those in later stages, which shows that the cell number in seeds is determined at the early embryo stage (Fig. 6c; Supplementary Fig. S5a; Supplementary Tables S8 & S9). Accordingly, GmCYP87 was reported to regulate seed size in soybean, and three copies of GmCYP87 genes were highly expressed in the early embryo development stages[45] (Supplementary Fig. S5b).
The seed desiccation process takes place during seed maturation, and this process reaches the highest level in dry seeds. The gene expression patterns of the water and heat responsive networks revealed that the main reactions occurred from the late maturation stage to dry seeds, and the responses continued to imbibed seed (Fig. 6d). Within the 117 water and heat responsive genes, 50 of them were from the GO term 'protein processing in endoplasmic reticulum', four of them were from the GO term 'cellular iron ion homeostasis', and 17 were from 'regulation of transcription-DNA-templated'. The superoxide response pathway had a similar expression pattern (Supplementary Fig. S5c). These results revealed that the seeds at the maturation stage were sensitive to environmental changes.
RNA storage in embryos of dry seeds
-
Although the expressed gene number and gene expression level were the lowest in dry seeds, 6,455 and 8,075 genes were still upregulated in the EA and CT of dry seeds, respectively, compared with those in the late maturation stage (Fig. 3h; Supplementary Fig. S2e). Among these upregulated genes, the KEGG term 'spliceosome' appeared in both CT and EA, which was unexpected (Fig. 7a & b). These 'spliceosome' genes from dry seeds were subsequently selected to determine their expression in imbibed seeds. In the CT of the imbibed seeds, the transcription levels of most of these genes remained unchanged (100 genes unchanged from 144 genes in total); in the EA of the imbibed seeds, the transcription levels of half of these genes (49 genes) remained unchanged, whereas those of the remaining 39 genes were downregulated (88 genes in total) (Fig. 7c & d). When all the genes associated with the KEGG term 'spliceosome' are presented, a group of genes with high expression levels in both dry seeds and imbibition-seed CT are present, and the expression was quickly decreased in the following germination stage (Fig. 7e). For this group of genes, the expression level in imbibition-seed EA was much lower than imbibition-seed CT (Fig. 7e). This phenomenon was also observed for the KEGG term 'ribosome assembly' (Supplementary Fig. S6). Since RNA storage in dry seeds has been reported in different plants, these results revealed that these 'spliceosome' and 'ribosome assembly' transcripts might be stored in dry soybean seeds.
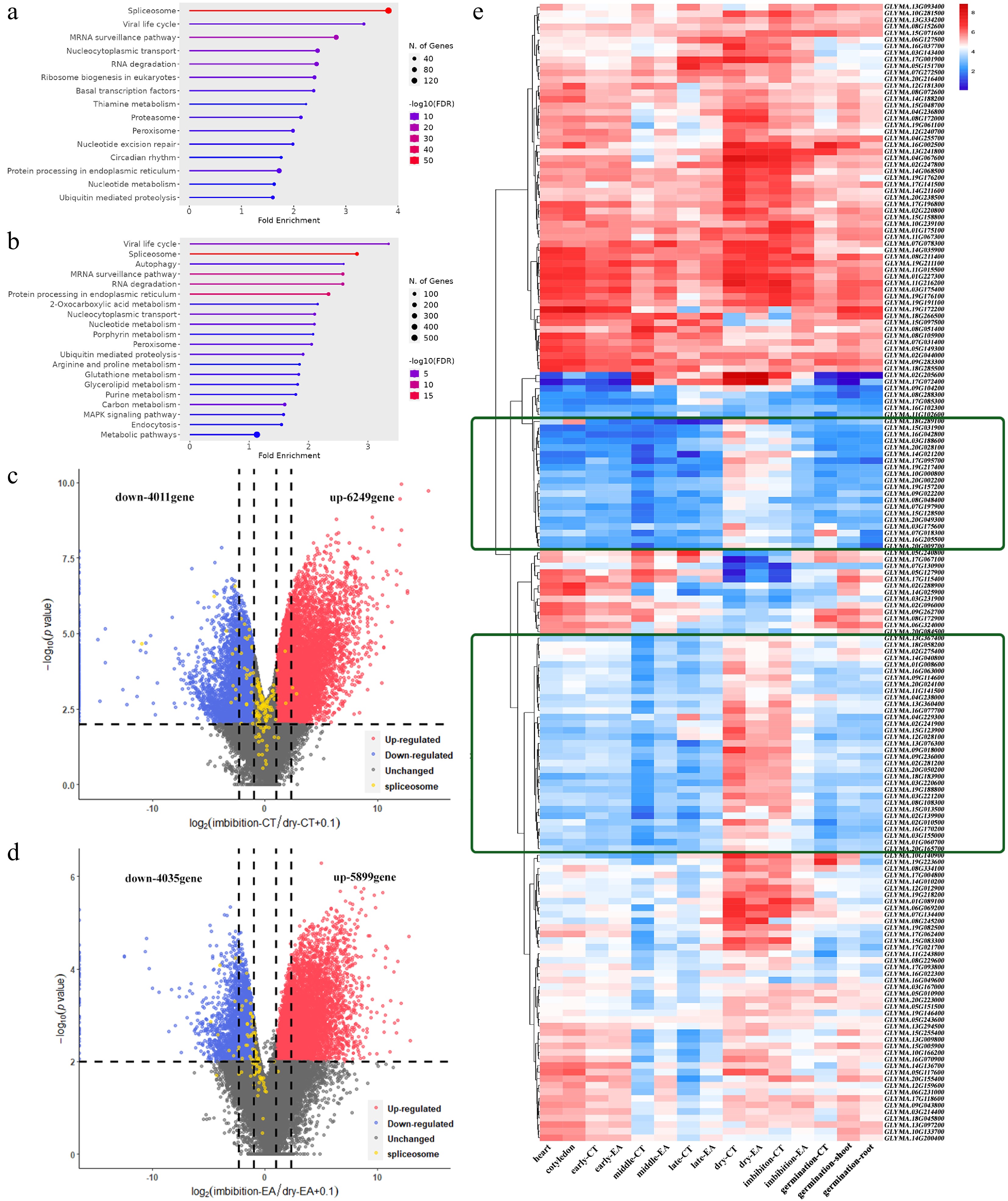
Figure 7.
RNA storage in embryo of dry seeds. KEGG items of upregulated genes in comparison of (a) dry-seed CT vs late-maturation CT, and (b) dry-seed EA vs late-maturation EA. Volcano plot and DEG numbers for comparison of (c) imbibition-seed CT vs dry-seed CT, and (d) imbibition-seed EA vs dry-seed EA. (e) Heatmap of genes expression patterns from KEGG item 'spliceosome' during embryo development. The green box shows the group of genes with significantly high expression in dry seeds.
Domestication selection of dehydration genes in soybean seed maturation
-
As mentioned above, the seed dehydration process is accompanied by a water/heat response and the accumulation of desiccation proteins (Fig. 6d). Drought stress can lead to a reduction in seed number and size[20,21,50,51]. The global temperature has continued to increase since the Industrial Revolution, and we are wondering whether desiccation genes are positively selected during soybean domestication in response to high temperatures. Four groups of genes associated with desiccation were selected: dehydrin, LATE EMBRYOGENESIS ABUNDANT (LEA), HEAT SHOCK PROTEIN (HSP), and oleosin[52,53] (Supplementary Tables S10−S13). The transcription levels of the dehydrin, LEA, and HSP genes increased gradually from early maturation to dry seeds reached the highest level in dry seeds, and then decreased in imbibed seeds (Fig. 8a−c). The oleosin genes presented the highest expression level in the middle maturation stage (Fig. 8d). The haplotypes of these genes were analyzed (
https://yanglab.hzau.edu.cn/SoyMD/#/ ). More than 70% of the dehydrin and oleosin genes and more than 86% of the HSP and LEA genes presented dominant haplotypes during soybean domestication, which revealed that these dehydration genes were positively selected (Supplementary Fig. S7a). There are two main types of haplotype selection: one type has a dominant haplotype that is selected positively and gradually from wild soybean to landrace and cultivar (Supplementary Fig. S7b & S7c), and the other type has a dominant haplotype with 100% in landrace and cultivar (Supplementary Fig. S7d & S7e). Furthermore, the expression of these four groups of genes was checked in developing seeds from soybean germplasm resources, including three wild soybean, nine landrace, and 14 cultivar[25] (https://yanglab.hzau.edu.cn/SoyMD/#/ ). The LEA groups presented more expressed genes and higher expression levels from wild soybean to cultivar soybean, and the results were similar to those of the dehydrin and HSP groups (Fig. 8e; Supplementary Fig. S7f & S7g). These findings revealed that dehydration proteins, which are important for soybean seed maturation and desiccation were under positive selection during domestication.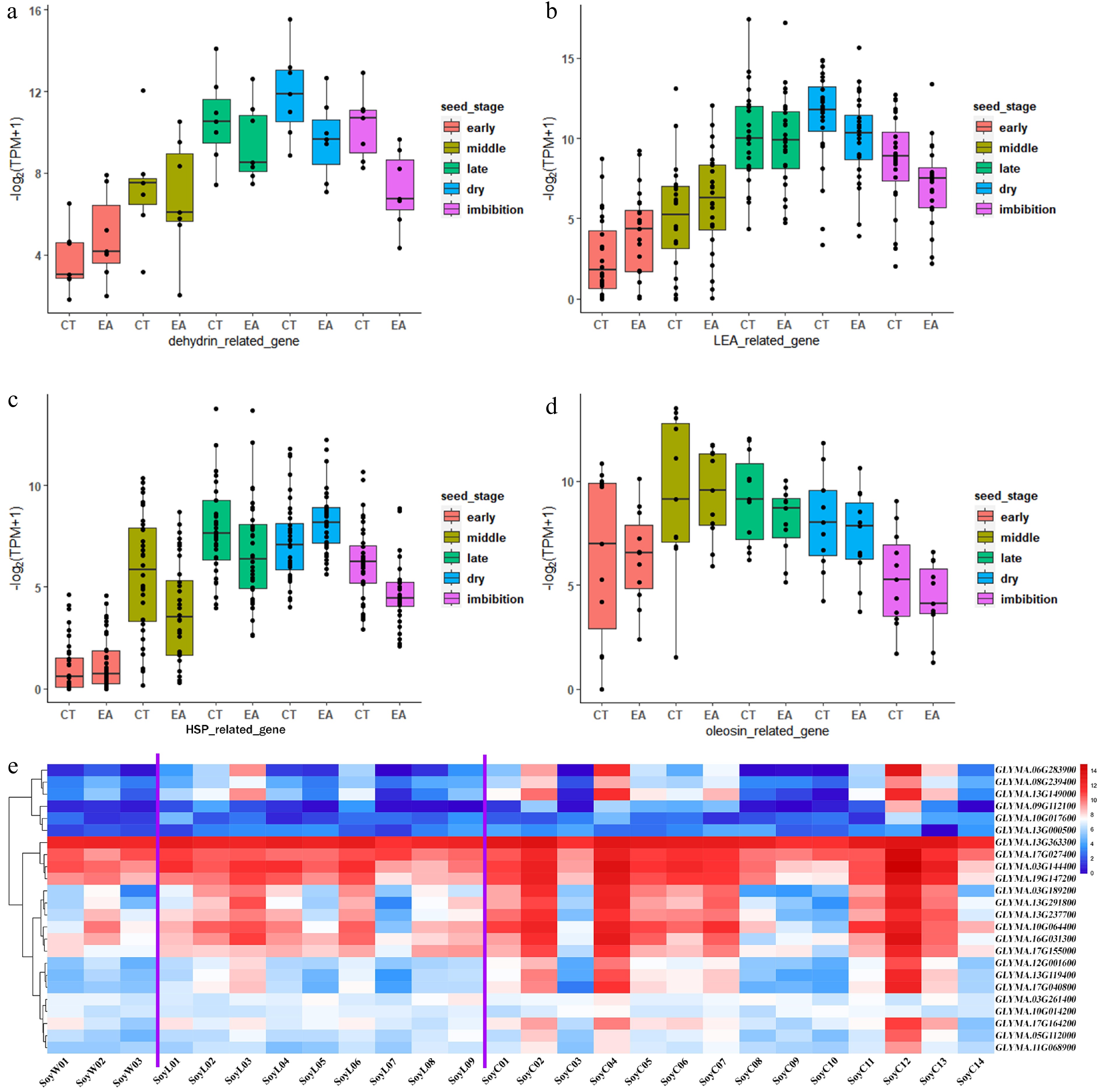
Figure 8.
Domestication selection of dehydration genes in soybean seed maturation. Box plot shows expression pattern of (a) dehydrin family, (b) LEA family, (c) HSP family, and (d) oleosin family during seed development. (e) Heatmap shows expression of LEA family genes in developing seed from wild soybeans, landraces and cultivars. The purple lines separate wild soybean, landrace and cultivar into three groups. 'SoyW' represents wild soybean, 'SoyL' represent landrace, 'SoyC' represents cultivar soybean.
-
Plant embryo development is strongly associated with seed number, seed size, and seed quality, which are directly related to yield and nutrient supply. The knowledge of crop embryo development could facilitate functional genomics research and modern intelligent breeding. However, the embryo development data of oilseed crops and leguminous crops are scarce compared with those of other crops, such as maize, rice, and wheat[2−6]. The available soybean embryo transcription data were obtained only at early development stages before seed maturation[13−15]. The transcriptional landscape of embryo development throughout the whole seed development process in soybean is still unknown. In this work, a soybean embryo transcriptome atlas was constructed covering embryo morphogenesis, seed maturation, seed dominance, and seed germination. Moreover, data on the embryo axis and cotyledon were included, which provides a reference for other leguminous crops.
Soybean seed weight is a major factor for yield and is mainly controlled by the number of embryo cells and substrate accumulation in each cell. On the one hand, gene networks related to the cell cycle, phytohormone reactions, and morphogenesis directly determine the embryo cell number. On the other hand, nutrient accumulation largely affects seed weight and quality. The dynamic transcription patterns for each of these networks are listed in the present work, which provides more targets for future seed design. As a key TF in initiating embryogenesis, BBM is a well-known target for maternal haploid induction[37]. In the present datasets, several TFs involved in embryogenesis and cell fate determination were discovered, which provide potential targets suitable for soybean haploid induction. Since the seed maturation process is quite sensitive to environmental stress, we also generated transcription maps of stress response networks. Among these networks, not only well-known genes but also genes with predicted roles were identified, which provided resources for the study of soybean functional genomics.
Interestingly, it was found that 'spliceosome' and 'ribosome assembly' transcripts exhibit high expression in dry soybean seeds and keep maintenance in the imbibition stage. In the work by Jia et al., the authors profiled full-length RNA with poly(A) tail and identified transcripts with long poly(A) tail in the tissue of pollen and seed[54]. As more poly(A) binding proteins could bind to the long poly(A) tail, the authors predicted that these transcripts in seeds and pollen are more stable, which are stored for seed and pollen germination[54]. Another group also identified full-length long-lived mRNAs in the aleurone and embryo cells from dry seeds of common wheat, their results showed that long-lived mRNAs may be related to cell survival and seed longevity[55]. Here, the 'spliceosome' and 'ribosome assembly' transcripts were enriched in dry seeds and imbibed seeds then decreased dramatically in the germinated seeds, suggesting that these transcripts might be stored for quick transcript splicing and fast protein synthesis during the rehydration process. Moreover, we found, in the imbibed seeds, these two groups of genes decreased faster in EA compared to CT, suggesting different rehydration rates in these two tissues.
Soybean has a long history of polyploidization and domestication, and duplicated genes always have divergent or redundant functions[33,47]. The transcription patterns of the duplicated genes of genes involved in embryo morphogenesis and folate synthesis were clearly examined. It was found that there could be as many as six paralogous genes in soybean and that the transcription patterns of these genes are not always similar. These data revealed that the regulation of embryo development in soybean is far more complicated than that in Arabidopsis and rice, which provides a new model for gene function studies under genome duplication.
Transcriptome data and germplasm genotyping data are always used together to trace genes controlling important traits[56,57]. It was found that gene networks regulating seed desiccation were positively selected under domestication, and the expression of these genes continued to increase from wild soybean to cultivars. Therefore, the embryo landscape generated in this work will facilitate a better understanding of key seed trait formation and selection under domestication.
-
In this work, a full-scale soybean embryo transcriptome atlas was generated from the heart embryo stage to germinated seeds, including the embryo axis and cotyledon from most stages. This landscape shows transcription dynamics during embryo development from the aspects of TFs, phytohormones, the stress response, metabolite accumulation, and RNA storage, which will help to better understand leguminous embryo development and design of future soybean seeds.
This work was supported by the Guangdong Laboratory for Lingnan Modern Agriculture (NG2022002), the Guangdong Ninth Pearl River Talent Program 'Team of plant meiosis recombination and germplasm innovation' (2021ZT09N333) and National Natural Science Foundation of China (32470344). We thank Chenjiang You and Yuan Fang (South China Agricultural University) for help with data analysis and Changkui Guo (South China Agricultural University) for help with soybean field organization.
-
The authors confirm the contributions to the paper as follows: study conception and design: Wang Y, Liu Y, Peng H; data collection: Chen Z, Hou J, Zhuang B, Huang J, Han J; analysis and interpretation of results: Chen Z, Wei Y, Hou J, Zhu X; draft manuscript preparation: Wang Y, Liu Y. All the authors reviewed the results and approved the final version of the manuscript.
-
The data supporting this study's findings are deposited in BioProject ID PRJNA1155808. All the data generated or analyzed during this study are included in this published article and its supplementary information files.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Zhengkun Chen, Yanni Wei, Jiamin Hou, Jing Huang
- Supplementary Table S1 All the RNA-seq libraries used in this work and the mapping rate of each library.
- Supplementary Table S2 List of EA-enriched TFs.
- Supplementary Table S3 List of CT-enriched TFs.
- Supplementary Table S4 List of early embryo-enriched TFs.
- Supplementary Table S5 List of folate biosynthesis pathway related genes.
- Supplementary Table S6 List of genes associated to GA synthesis and signal transduction.
- S2k
- Supplementary Table S7 List of genes associated to ABA synthesis and signal transduction.
- Supplementary Table S8 List of cell cycle associated genes.
- Supplementary Table S9 List of DNA replication associated genes.
- Supplementary Table S10 List of dehydrin family genes.
- Supplementary Table S11 List of LEA family genes.
- Supplementary Table S12 List of HSP family genes.
- Supplementary Table S13 List of oleosin family genes.
- Supplementary Table S14 List of primers used for RT-qPCR.
- Supplementary Fig. S1 Correlation of the RNA-seq data.
- Supplementary Fig. S2 Transcription dynamics during soybean embryo development.
- Supplementary Fig. S3 TFs involved in embryo morphogenesis and development.
- Supplementary Fig. S4 Metabolite synthesis during soybean embryo development.
- Supplementary Fig. S5 Internal and external signal response in soybean embryo development.
- Supplementary Fig. S6 Heatmap of gene expression patterns from the KEGG term "ribosome assembly" during embryo development. The red box represents the group of genes whose RNA was stored in dry seeds.
- Supplementary Fig. S7 Domestication selection of dehydration genes in soybean seed maturation.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Hainan Yazhou Bay Seed Laboratory. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Chen Z, Wei Y, Hou J, Huang J, Zhu X, et al. 2024. Transcriptional atlas for embryo development in soybean. Seed Biology 3: e022 doi: 10.48130/seedbio-0024-0021 

Transcriptional atlas for embryo development in soybean
- Received: 03 October 2024
- Revised: 16 November 2024
- Accepted: 29 November 2024
- Published online: 13 December 2024
Abstract: Soybean is an economically important leguminous seed crop and provides plant oil and protein for human food and animal feed. Seeds are derived from embryos, whose development directly determines the sizes and weights of seeds. In recent decades, the molecular mechanisms underlying embryo development have been extensively studied in Arabidopsis, maize, and rice. To date, there is no available transcriptome landscape for the whole embryo development in leguminous crops. Here, a comprehensive transcriptional atlas was generated for soybean embryo development from the heart embryo to germinated seed, which exhibits a dynamic transcription during embryo development. Then, transcription factors involved in the specification of embryo axis and cotyledon, as well as in the maintenance and transition of specific stages were identified. Furthermore, those differential expression of genes related to synthetic stages and tissues for different secondary metabolites during embryo development, including flavonoids, folate, etc were detected. It was found that the genes associated with gibberellin synthesis and signal transduction were highly expressed before the middle maturation stage, which supports the known effect of gibberellin on early seed development. Potential transcript storages in dry seeds were also exhibited. Interestingly, it was found that embryo dehydration genes are positively selected during soybean domestication. Taken together, these transcriptome datasets not only provide global insight into gene networks during soybean embryo development but also generate resources for the study of soybean functional genomics.
-
Key words:
- Soybean /
- Transcriptome /
- Embryo development