









-
Shoot apical meristem (SAM), the origination of plant leaves, provides the stem cell niche to the whole of up-ground organs in higher plants, and has been divided into the central zone (CZ), renewing the meristem, as well as the peripheral zone (PZ) where the lateral organs emerge[1]. The process of leaf initiation and expansion depends on an elaborate and joint system. The regulatory networks for this procedure are constructed by various genes and transcription factors related to hormonal regulation, polarity establishment, and maintenance, as well as leaf flattening and expansion[2]. The initial cells of leaves derived from PZ grow along three asymmetric axes: proximal-distal axis, adaxial-abaxial axis, and mediolateral axis, and ultimately expand in multiple directions to a final leaf form[2−4].
A gene expression pattern distinguishing adaxial and abaxial cells are established before leaf initiation[5,6]. The formation of the adaxial domain is promoted by the genes like REVOLUTA (REV), PHAVOLUTA (PHV), and PHABULOSA (PHB) containing the HD-ZIPIII domain[7−10], as well as ASYMMETRIC LEAVES2 (AS2) with a LOB-domain[11−13]. While KANADI1 (KAN1) promotes the initiation of the abaxial domain and this prepattern shifts dynamically during leaf development[6,14].
In Arabidopsis, REVILLE (RVE) family genes negatively regulate leaf size. The rve 4 6 8 mutant has enlarged mesophyll cells, and this gene family plays a crucial role in regulating the plant's biological clock[15,16]. The Arabidopsis PIN1-type parvulin 1 (Pin1At) gene is involved in auxin polar transport and the Pin1At protein influences PINOID (PID) and Protein Phosphatase 2A (PP2A)-mediated auxin transport and polar localization related to PIN-FORMED1 (PIN1) in the stele cells[17]. Additionally, overexpression of Pin1At in Arabidopsis leads to serrated leaves and stems and accelerated flowering, indicating that the Pin1At gene regulates leaf morphogenesis and flowering time[18−20].
The WOX (WUSCHEL-related homeobox) family comprises 15 members categorized into the WUS, Intermediate, and Ancient clades[21]. These members play critical roles in various aspects of plant growth and development, including the maintenance of meristematic stem cells, embryonic development, polarization, lateral organogenesis, and organ regeneration[22]. Specifically, WOX proteins are believed to define a central domain within the growing leaf bud. This domain acts as a separator between the abaxial and adaxial domains, controlling the outgrowth of the leaf blade. This function occurs downstream of the adaxial/abaxial polarity[23,24].
In Arabidopsis, WOX1, PRS, and WOX5 redundantly regulate the expression of the auxin biosynthesis gene YUCCA (YUC), influencing regional growth along the leaf margin, thereby shaping the characteristic elliptical form of Arabidopsis leaves. Mutants lacking these genes exhibit narrower leaves[23,25]. Similar functions have been identified in various plant species, including Nicotiana, Petunia, Medicago, and garden pea[26−29]. The WOX1 mutants in tobacco and tomato show significantly more severe leaf reduction, with almost no residual leaf tissue, and WOX3 is unable to compensate for the loss of WOX1 function[28,30]. Additionally, in tomato, WOX1 not only plays a crucial role in leaf development but also regulates the initiation and growth of leaflets in compound leaves, indicating evolutionary diversity in WOX1 function during compound leaf development[30]. In Medicago truncatula, the WOX1 ortholog STENOFOLIA (STF) primarily regulates leaf expansion, while the WOX3 ortholog LOOSE FLOWER (LFL) primarily controls flower development, with minimal impact on leaf growth[31,32]. However, the expression patterns of monocots are different, like maize and rice which control leaf blade outgrowth with PRS orthologs[33−35]. WOX1 orthologs are expressed along the entire upper-lower leaf junction, while PRS orthologs have more restricted expression in the marginal meristem. These spatial differences in gene expression may explain variations in lateral leaf outgrowths between monocots and eudicots[33]. In the context of limited reporting on downstream genes associated with WOX genes, it is of paramount significance for the exploration of additional genes involved in leaf morphogenesis and the enrichment of downstream regulatory networks.
The leaf phenotypes of CsWOX1 overexpressing and mutant cucumbers have been reported to show 'butterfly-shaped' leaves and a smaller blade horizontal-vertical ratio, especially in the distal region of the leaves[36,37]. Given the limited reporting on downstream genes related to WOX genes, the exploration of additional genes involved in leaf morphogenesis and the enhancement of downstream regulatory networks is of utmost importance. In this study, a transcriptomic analysis approach was utilized to obtain a significant dataset of differentially expressed genes. By comparing the differentially expressed genes between CsWOX1-overexpression (CsWOX1-OE) lines and mutants simultaneously, candidate downstream genes associated with the CsWOX1 gene were identified.
-
Cucumber (Cucumis stativus L.) inbred CU2 was selected as the wild type for comparison and cucumber genetic transformation. CsWOX1-OE were contemporary overexpression materials obtained through transgenic technology. Spontaneous mutant mf was identified from the cucumber line 'Extra Early Majestic', and sowing the seeds of mf and wild type (AM218).
Germinated seeds and transgenic plants with regenerative roots were transferred to seedling pots and cultivated in an artificial climate chamber. The chamber's temperature was maintained at 25 °C during the day and 20 °C at night, with a light/dark cycle of 16 h/8 h. Seedlings were transplanted into a sunlight greenhouse at the 4-6 true leaf stage. Fertilization, irrigation management, and pest and disease control were conducted following standard procedures.
RNA isolation
-
RNA was extracted from 1 cm-long apical leaves at the early expansion stage, selected from 30-day-old CsWOX1-OE transgenic lines, mf mutants, and wild-type plants (CU2 and AM218). Three biological replicates were performed for each genotype sample, and the extracted RNA was used for RNA-Seq and RT-qPCR analysis. For each sample, the leaves were immersed in liquid nitrogen within a mortar and ground into a fine powder. Total RNA was extracted using kit (TIANGEN, China). The integrity and quality of the total RNA were assessed using a NanoDrop 1000 spectrophotometer and formaldehyde-agarose gel electrophoresis.
Transcriptome analysis
-
Six samples from CsWOX1-OE and CU2 were submitted to BioMarker Biomarker Technology (Beijing, China) for RNA sequencing. After verifying the purity, concentration, and integrity of the RNA samples, cDNA library construction and quality inspection were carried out. PE150 mode sequencing was performed utilizing the Illumina NovaSeq6000 sequencing platform. Clean data with high quality was obtained by filtering raw data, which removes adapter sequences and reads with low quality. Clean reads were aligned with the reference genome (Cucumis_sativus. ChineseLong_v3. genome. fa) through HISAT2 software and the reads were compared on the pair for assembly by StringTie software. After obtaining Mapped Data, library quality evaluation, variable splicing analysis, gene structure optimization, differential expression analysis, gene functional annotation, and functional enrichment were performed. Data normalization was achieved through Reads Per Kilobase of transcript, per Million mapped reads (RPKM). Differential expression genes were screened by DESeq2 software, with Fold Change (FC) ≥ 2 and False Discovery Rate (FDR) < 0.01 as the screening criteria.
RT-qPCR
-
To assess the fidelity of the RNA-seq data, six genes exhibiting opposite expression patterns between CsWOX1-OE and mf were chosen for RT-qPCR analysis. The first-strand cDNA was synthesized via reverse transcription of the total RNA extracted, employing the HiScript II 1st Strand cDNA Synthesis Kit following the manufacturer's guidelines (Vazyme, China). Subsequently, RT-qPCR was conducted using the SYBR qPCR Master Mix (Vazyme, China) on an ABI 7500 Fast Real-Time PCR System (Applied Biosystems, USA). Each gene was subjected to three biological and three technical replicates. The resulting relative expression data were normalized against the expression level of the cucumber CsActin2 gene[38]. The 2−ΔΔCᴛ method[39] was employed for calculating the relative expression levels of each gene. The primer sequences are provided in Supplementary Table S1.
Yeast one-hybrid assay
-
CsWOX1 coding sequences were cloned into the pB42AD plasmid. Subsequently, the promoter fragment of approximately 2.0-kb from CsRVE6, CsRVE8, CsABCB, and CsPin1At were cloned into the pLacZi2u plasmid. The primer sequences are detailed in Supplementary Table S1. Following this, the pB42AD-CsWOX1 and pLacZi2u-pCsRVE6/pLacZi2u-pCsRVE8/pLacZi2u-pCsABCB/pLacZi2u-pCsPin1At constructs were co-transformed into the yeast EGY48 strain and cultured at 30 °C for 2–4 d on SD/-Trp/-Ura plates. Monoclonal yeast transformants were selected and cultured on SD/-Trp/-Ura/X-gal plates for 2–4 d to ascertain protein-protein interactions. The observation of both normal colony growth and the development of a blue coloration indicates the plausible occurrence of mutual interactions between the two proteins.
Dual-luciferase reporter assay
-
A 952 bp sequence located upstream of the translation initiation start site of CsRVE6 was integrated into the transient expression vector, pGreenII 0800-Luc, thereby generating a ProCsRVE6: LUC reporter construct. Subsequently, the CDS sequence of CsWOX1 were inserted into pGreenII 62-SK, resulting in the creation of 35S:CsWOX1 effectors. The reporter and effectors were independently transformed into GV3101-pSoup. For each effector, a transient co-infiltration was conducted into N. benthamiana leaves alongside the pGreenII 62-SK vector and ProCsRVE6:LUC reporter serving as a negative control, maintaining an effector : reporter ratio of 9:1[40]. Each experimental condition encompassed three biological replicates and three technical replicates, followed by the execution of the dual-luciferase reporter assay[36]. The activities of firefly luciferase (LUC) and Renilla luciferase (REN) were quantified using a Dual-Luciferase reporter gene detection kit (Yeasen Biotech, Shanghai, China), adhering to the manufacturer's instructions. The primer sequences utilized are provided in Supplementary Table S1.
-
To elucidate the downstream regulatory network of CsWOX1 gene and identify potential candidate genes related to leaf morphogenesis, transcriptome analysis was performed on CsWOX1-OE transgenic lines and wild-type CU2 utilizing RNA-Seq. Six samples, divided into two groups with three biological replicates each, were processed for transcriptome sequencing, generating 47.09 Gb of clean data. At least 6.80 Gb clean data were generated for each sample with a minimum 93.67% of clean data achieving a quality score of Q30 (Supplementary Table S2). Clean reads of each sample were mapped to the specified reference genome. The mapping ratio ranged from 95.38% to 97.56% (Supplementary Table S3).
Principal Component Analysis (PCA) based on the transcriptome data from CsWOX1-OE and the wild type (CU2) revealed clear separation between the two groups, highlighting significant differences. The proximity of biological replicates within each group indicated similar sample compositions with minimal variations (Fig. 1a). Between CsWOX1-OE and the wild type, a total of 3,757 differentially expressed genes (DEGs) were identified, comprising 2,517 upregulated and 1,240 downregulated genes (Fig. 1b). All the DEGs were functionally annotated in eight databases: COG, GO, KEGG, KOG, NR, Pfam, Swiss-Prot, and eggNOG. The number and percentage of DEGs annotated in each database are shown in Fig. 1c. Prediction of alternative splicing, gene structure optimization analysis and novel gene discovery was processed on top of mapping results, during which 955 were discovered and 422 novel genes were annotated with a putative function.
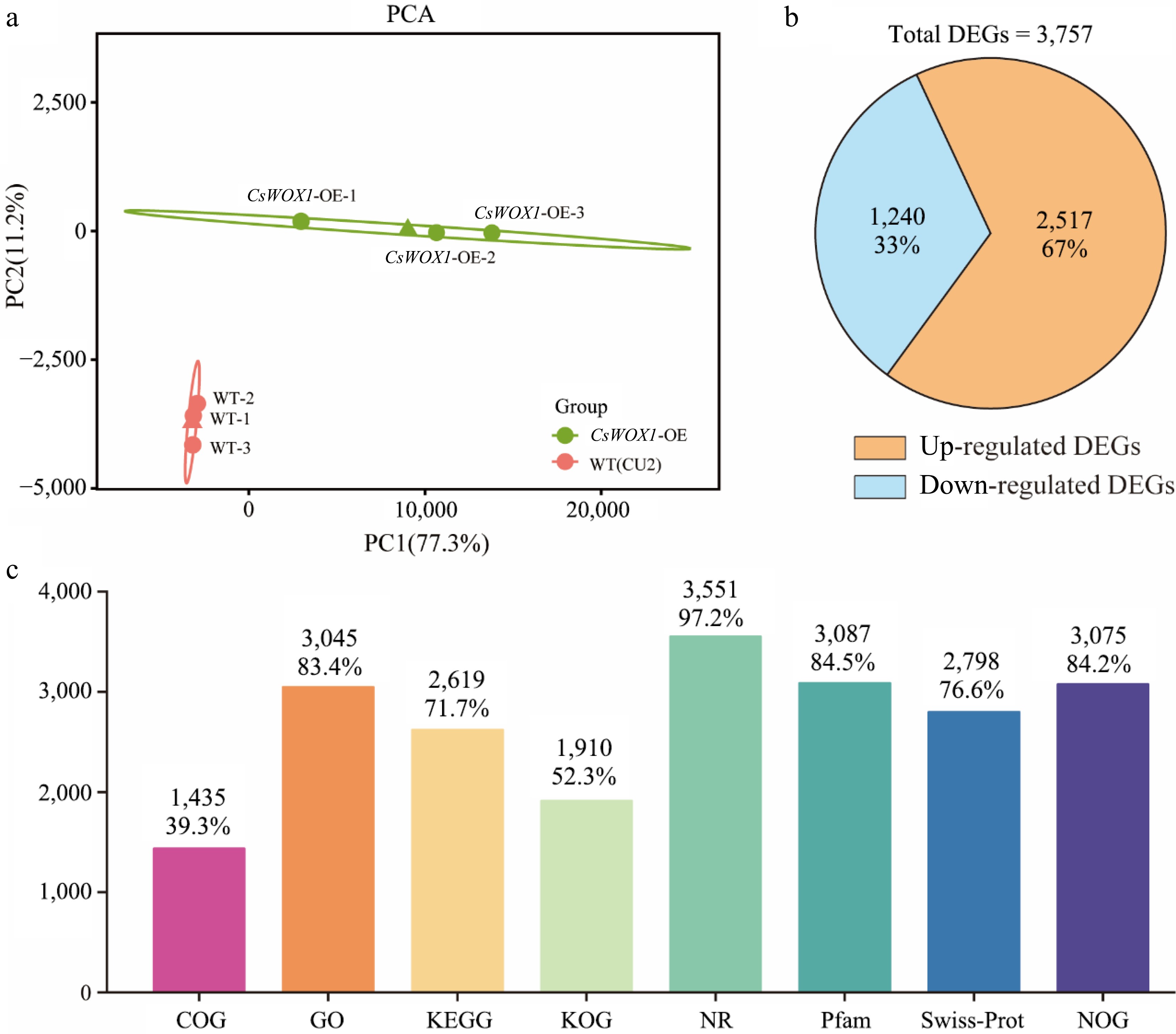
Figure 1.
Transcriptome information and analysis of CsWOX1-OE. (a) The principal component analysis (PCA) of differently expressed transcripts from two groups. (b) Statistics on the number of up-regulated and down-regulated DEGs. (c) The number and proportion of DEGs annotated in different databases.
Combined analysis of CsWOX1-OE and mf transcriptome sequencing results
-
Based on the published RNA-Seq data of mf[37], a combined analysis of the transcriptome data from the CsWOX1-OE and the mutant was conducted. Compared with the 3,757 DEGs found in CsWOX1-OE, only 200 DEGs were found in the RNA-Seq results of mf, with 59 upregulated genes and 141 downregulated genes[37].
The DEGs of CsWOX1-OE and mf were analyzed by Gene Ontology (GO), and the GO classification was divided into three main categories: biological process (BP), cellular component (CC), and molecular function (MF). The results, as shown in Fig. 2, showed that in CsWOX1-OE and mf, cellular and cellular fractions accounted for the largest proportion of cellular components, with 1,643 and 95 DEGs annotated, respectively. Binding and catalytic activities were the two most dominant molecular functions of DEGs. Cell process, metabolic process, and single-organism process were the biological processes with the highest number of defined DEGs. In addition, KEGG pathway enrichment analysis was performed on the DEGs of CsWOX1-OE and mf. This analysis revealed the impact of the CsWOX1 gene on various biological processes and metabolic pathways in cucumber (Fig. 3). The pathways annotated with DEGs were classified into five categories: Cellular Processes, Environmental Information Processing, Genetic Information Processing, Metabolism, and Organismal Systems, among which the metabolic pathways annotated the largest number of DEGs, both in CsWOX1-OE and mf. In mf, the pathway with the most DEGs was the starch and sucrose metabolic pathway, which accounted for 13.64% of all DEGs. In CsWOX1-OE, the plant-pathogen interaction pathway and the plant hormone signaling pathway annotated the first and second place with 9.88% of 140 DEGs and 8.82% of 125 DEGs, respectively. This indicates that the elevated expression level of CsWOX1 expression significantly affects plant disease resistance and hormone signal mechanisms.
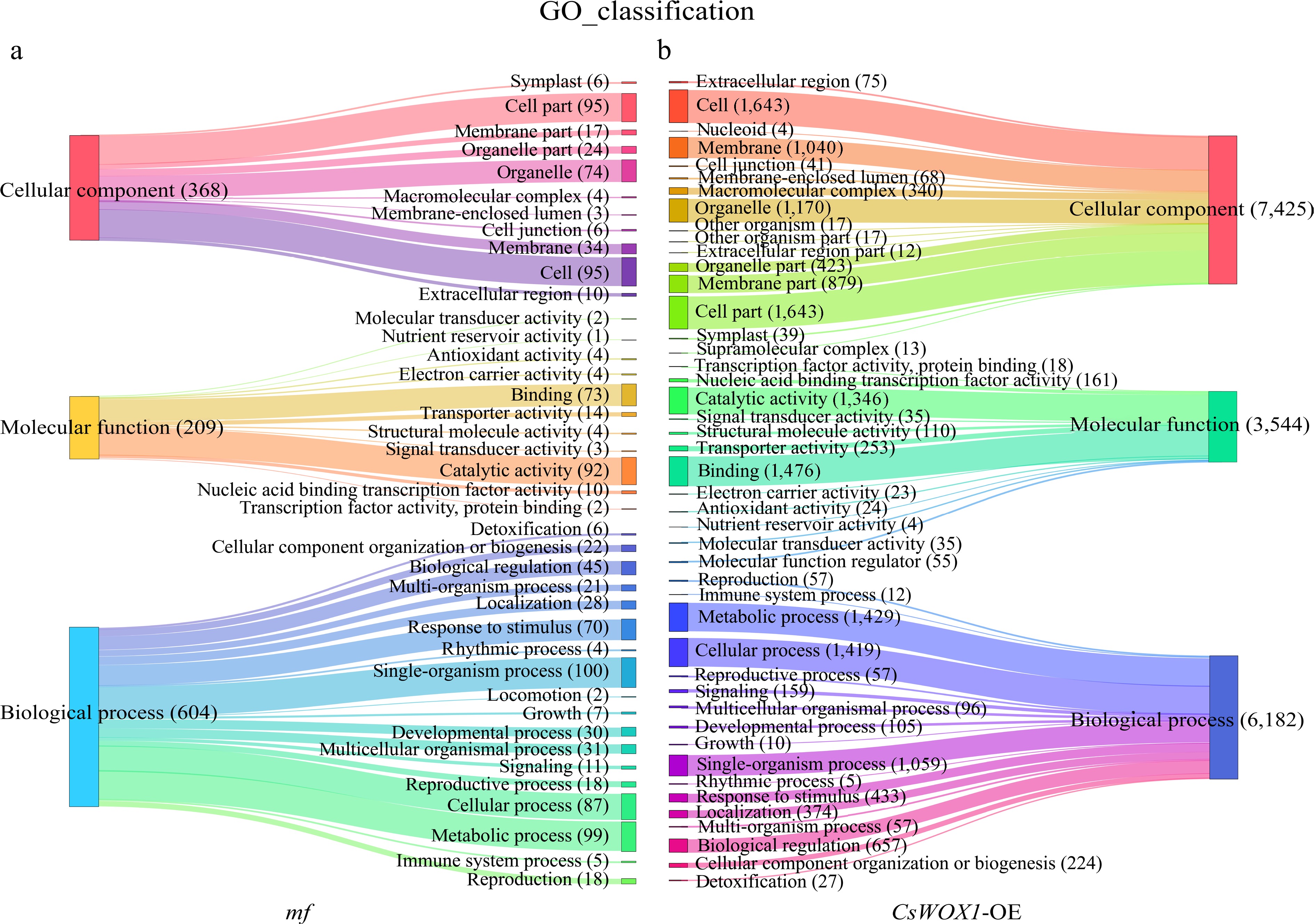
Figure 2.
The GO classification analysis results show the differences in the number and categories of annotated DEGs between the mf and CsWOX1-OE, with three main GO categories: Cellular component, Molecular function, and Biological process. (a) GO classification of DEGs in the mf sample. (b) GO classification of DEGs in the CsWOX1-OE sample. Each band represents the annotation status and trends of different GO terms and the numbers in parentheses indicate the number of genes in each category.
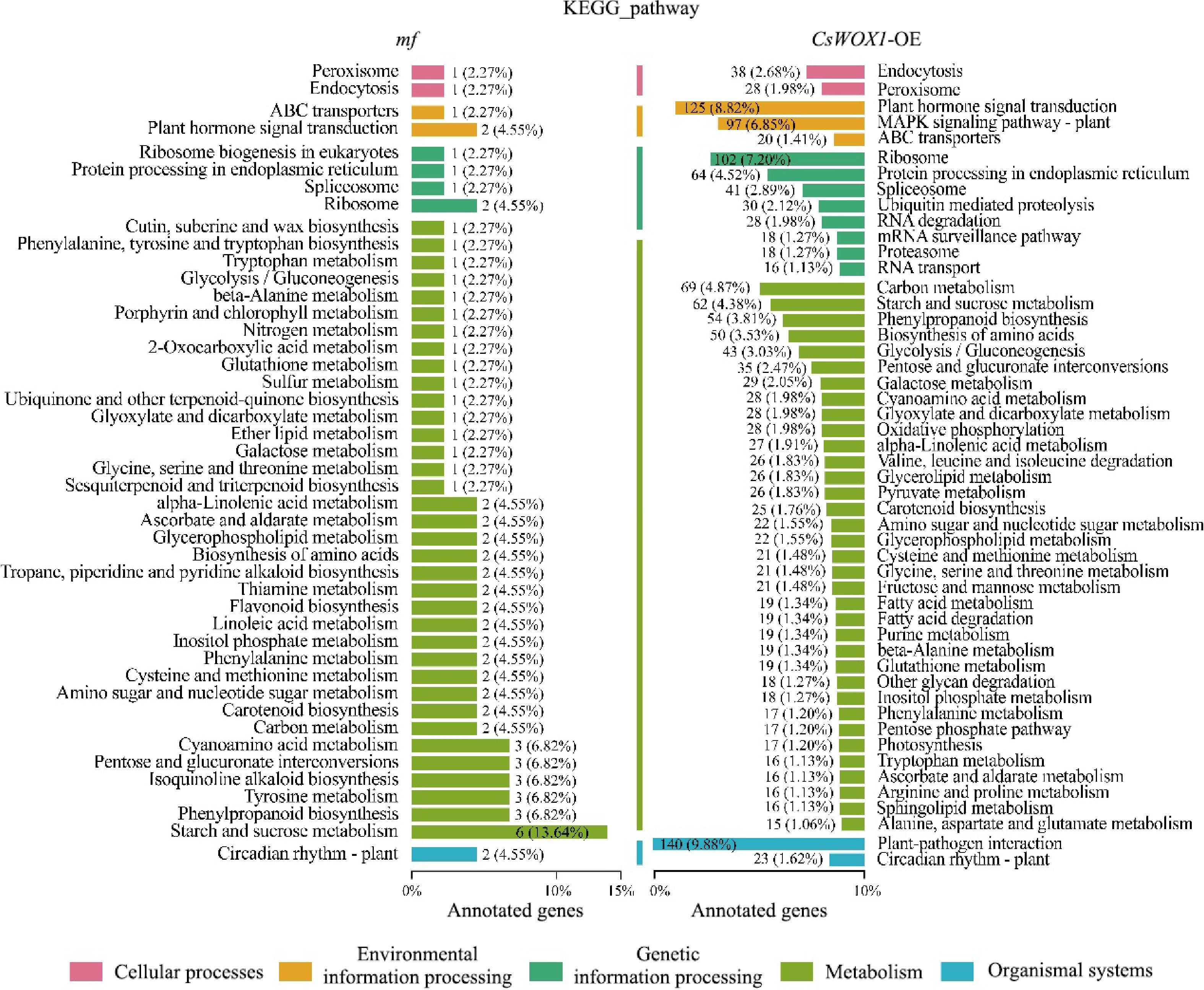
Figure 3.
KEGG pathway analysis of the DEGs from mf and CsWOX1-OE shows the number and proportion of DEGs annotated in different metabolic pathways. The KEGG pathways are categorized into five groups: cellular processes, environmental information processing, genetic information processing, metabolism, and organismal systems.
Through joint comparative analysis, we found that there were 80 identical genes in the DEGs of CsWOX1-OE and mf, called common DEGs (Fig. 4a). By analyzing the changes in the expression trends of these 80 common DEGs, it was found that 20 of these genes showed the same change trends in CsWOX1-OE and mf, either increasing or decreasing together, while the other 60 genes showed opposite expression changes. The expression levels of these 20 DEGs with the same trend and 60 DEGs with opposite trends in CsWOX1-OE and mf were shown in Fig. 4b and c, respectively, and the heatmaps were plotted by the Log2 FC values of the DEGs. Since CsWOX1 expression was opposite in CsWOX1-OE and mf, this study focusses on the 60 common DEGs with opposite changes in expression trends. The KEGG pathway analysis revealed that these 60 common DEGs were annotated into 26 pathways, of which three pathways, namely, circadian-plant (ko04712), MAPK signaling pathway-plant (ko04016), and plant-pathogen interaction (ko04626), all three pathways were enriched with more than two common DEGs (Table 1).
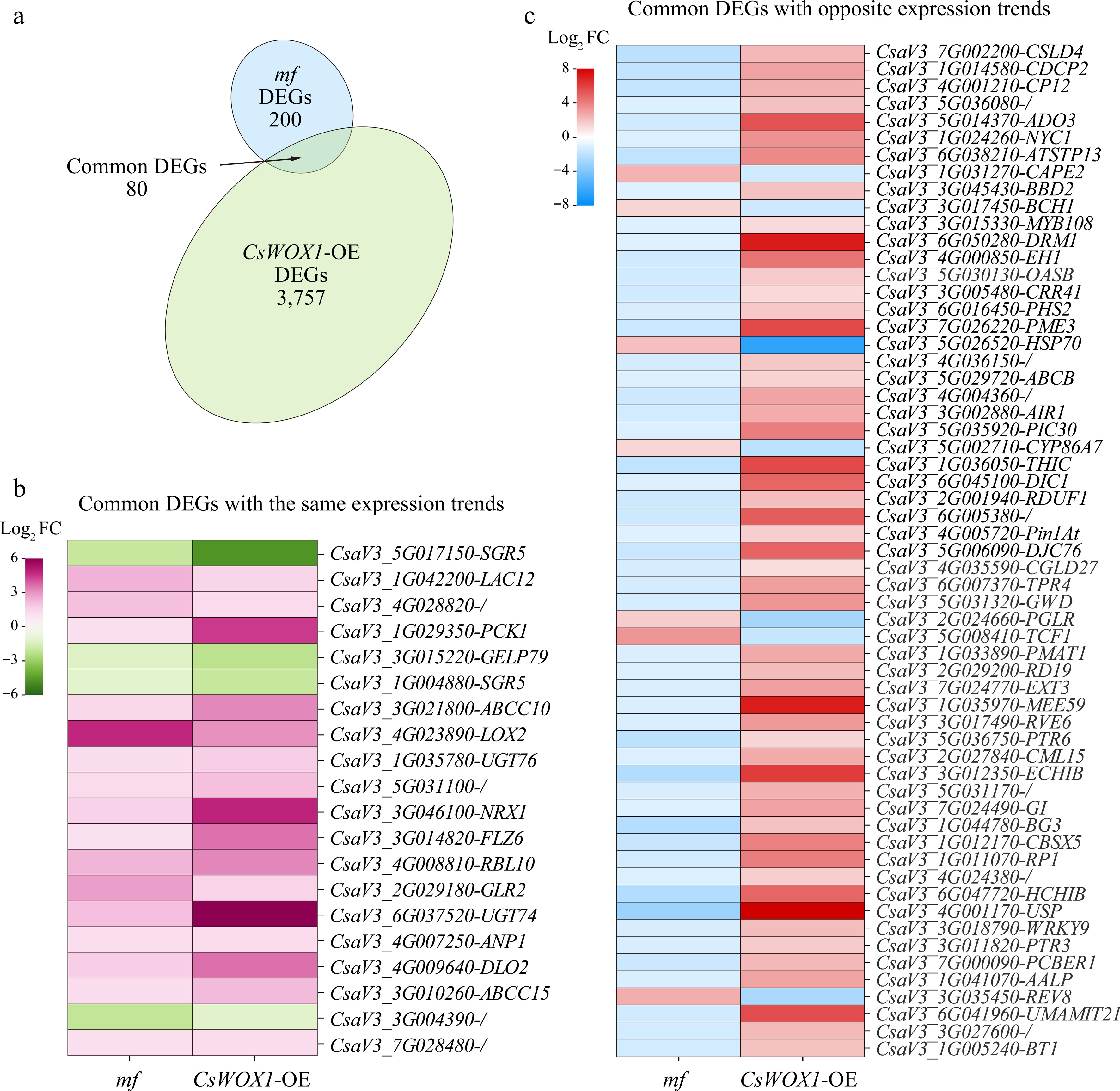
Figure 4.
Combined analysis of mf and CsWOX1-OE DEGs. (a) Identification of eight common DEGs between mf and CsWOX1-OE. (b) Heatmap of gene expression levels of 20 common DEGs with the same trend of change. (c) Heatmap of gene expression levels of 60 common DEGs with opposite trends. The colored legend positioned to the left of each map indicates fold changes (Log2 value).
Table 1. Joint KEGG pathway analysis of common DEGs in CsWOX1-OE and mf.
Pathway ID Pathway name Common DEGs ko00906 Carotenoid biosynthesis CsaV3_1G014580, CsaV3_3G017450 ko04712 Circadian rhythm - plant CsaV3_5G014370, CsaV3_3G017490,
CsaV3_7G024490, CsaV3_3G035450ko00860 Porphyrin and chlorophyll
metabolismCsaV3_1G024260 ko04016 MAPK signaling pathway - plant CsaV3_1G031270, CsaV3_6G047720,
CsaV3_3G018790ko04075 Plant hormone signal transduction CsaV3_1G031270 ko04626 Plant-pathogen interaction CsaV3_1G031270, CsaV3_2G029200,
CsaV3_2G027840, CsaV3_3G018790ko00590 Arachidonic acid metabolism CsaV3_4G000850 ko04146 Peroxisome CsaV3_4G000850 ko00270 Cysteine and methionine metabolism CsaV3_5G030130 ko00920 Sulfur metabolism CsaV3_5G030130 ko01200 Carbon metabolism CsaV3_5G030130 ko01230 Biosynthesis of amino acids CsaV3_5G030130 ko00500 Starch and sucrose metabolism CsaV3_6G016450, CsaV3_1G044780 ko00040 Pentose and glucuronate interconversions CsaV3_7G026220, CsaV3_2G024660 ko03040 Spliceosome CsaV3_5G026520 ko04141 Protein processing in endoplasmic
reticulumCsaV3_5G026520 ko04144 Endocytosis CsaV3_5G026520, CsaV3_7G024770 ko02010 ABC transporters CsaV3_5G029720 ko00073 Cutin, suberine and wax biosynthesis CsaV3_5G002710 ko00730 Thiamine metabolism CsaV3_1G036050 ko04120 Ubiquitin mediated proteolysis CsaV3_5G008410 ko00943 Isoflavonoid biosynthesis CsaV3_1G033890 ko00944 Flavone and flavonol biosynthesis CsaV3_1G033890 ko00071 Fatty acid degradation CsaV3_3G012350 ko03008 Ribosome biogenesis in eukaryotes CsaV3_5G031170 ko00520 Amino sugar and nucleotide sugar metabolism CsaV3_6G047720 Validation of differentially expressed genes
-
To verify the accuracy of Illumina sequencing data, six common DEGs with opposite expression trends in CsWOX1-OE and mf were selected for RT-qPCR to detect their expression changes. Four genes involved in the plant circadian pathway were selected, GIGANTEA (CsGI, CsaV3_7G024490), FLAVIN-BINDING KELCH REPEAT F-BOX 1 (CsFKF1, CsaV3_5G014370), REVILLE6 (CsRVE6, CsaV3_3G017490), and REVILLE8 (CsRVE8, CsaV3_3G035450), as well as two genes involved in growth hormone transport, ATP BINDING CASSETTE subfamily B (CsABCB, CsaV3_5G029720), and Arabidopsis PIN1-type parvulin 1 (CsPin1At, CsaV3_4G005720).
The relative expression levels of these six genes in the wild type, CsWOX1-OE, and mf are shown in Fig. 5. The relative expression levels of the genes were calculated by the 2−ΔΔCᴛ method and analyzed for significant differences, which showed that the expression level of the CsRVE8 gene was significantly decreased in CsWOX1-OE and significantly increased in mf. In contrast, the expression levels of the other five genes were significantly increased in CsWOX1-OE and significantly decreased in mf. Although the fold change of genes differed between the transcriptome data and the RT-qPCR results due to the sensitivity of the different techniques, the trends in the expression levels of the six genes detected by RT-qPCR were consistent with the RNA-Seq results, which proved that the data from RNA-Seq were accurate and reliable.

Figure 5.
RT-qPCR analyses of (a) CsRVE6, (b) CsRVE8, (c) CsFKF1, (d) CsPin1At, (e) CsABCB, and (f) CsGI in CsWOX1-OE and mf. Significance analyses compared to WT were performed with the two-tailed Student's t-test. Different letters (a, b, c) indicate significant differences between groups (p < 0.05). The values represent the mean ± SD (n = 3).
CsWOX1 regulates auxin polar transport-related genes
-
Previous studies have shown significant changes in leaf size and shape in CsWOX1-OE and mf that are associated with the polarity transport of auxin and the response to auxin polarity signaling[36,37,41]. By analyzing the KEGG pathway in mf and CsWOX1-OE, it was found that many DEGs have been annotated to the phytohormone signaling pathway (ko04075) and most of the DEGs involved in the auxin transduction showed up-regulation. AUXIN RESISTANT1 (AUX1, K13946), auxin influx carrier, expressed increasingly both in CsWOX1-OE and mf, which were regulated by CsaV3_3G034000 and CsaV3_2G015870, respectively. The upregulated gene in mf (CsaV3_2G013720) affected the regulation of the auxin-responsive protein IAA: AUX/IAA (K14484), which was jointly controlled by four upregulated genes (CsaV3_2G004120, CsaV3_2G013230, CsaV3_3G023550, and CsaV3_3G048770) and one downregulated gene (CsaV3_3G012650) in CsWOX1-OE. As for the AUXIN RESPONSE FACTOR (ARF, K14486), it was only upregulated in CsWOX1-OE, regulated by CsaV3_1G023020 and CsaV3_6G036480.
Joint analysis of the DEGs involved in the polarity transport of auxin were conducted, identifying two common DEGs: the homologs genes of the ABCB and Pin1At. The ABCB encodes phosphor glycol proteins (PGP), and they have been shown to participate in plant auxin efflux by stabilizing PIN proteins in the plasma membrane[42,43]. In CsWOX1-OE transgenic lines, the expression of the CsABCB gene increased by 2.71 times and its expression level was reduced by 2.07 times in mutants. Pin1At, peptidyl-prolyl cis/trans isomerase gene[20], affects PIN1-related auxin transport and polarity localization in mesocolumnar cells mediated by PID and PP2A as well as catalyzes the conformational change of the phosphorylated Ser/Thr-Pro motif in PIN1[17]. CsPin1At exhibited up-regulation in CsWOX1-OE transgenic lines and down-regulation in mf mutants.
Both of these genes exhibited a consistent expression pattern, with an upregulation in the CsWOX1-OE and a downregulation in the mf. This result suggests a correlation between the CsWOX1 gene and auxin transport and distribution, which could potentially influence leaf development.
CsWOX1 regulates the genes controlling plant rhythms and leaf expansion
-
In CsWOX1-OE, plenty of core circadian clock-related genes annotated in rhythm pathways expressed differently from the wild type, such as the upregulated genes: Early Flowering 3 (ELF3), Phytochrome-Interacting Factor 3 (PIF3), PSEUDO-RESPONSE REGULATOR 5 (PRR5), and PSEUDO-RESPONSE REGULATOR 7 (PRR7). As for the LATE ELONGATED HYPOCOTYL (LHY) transcription factor with an MYB-like domain, it's homologous gene RVE6 was upregulated while RVE8 was downregulated. Furthermore, the expression of CRYPTOCHROME (CRY), which receives blue light signals, was increased, and the expression of CONSTITUTIVE PHOTOMORPHOGENIC 1 (COP1), which is inhibited by CRY, decreased. The expression of GI, FKF1, CYCLING DOF FACTOR 1 (CDF1), and CONSTANS (CO) all increased, and they collectively regulate plant flowering.
In the plant circadian rhythm regulation pathway, only GI, FKF1, RVE6, and RVE8 are co-DEGs, and their trends of expression change showed exactly opposite directions in CsWOX1-OE and mf. GI and FKF1 participate in the blue light signal transduction pathway and the regulation of photoperiod flowering[44,45]. RVE 6 and RVE 8 have been determined to promote clock pace in a partially redundant manner with their homologs REVILLE 4, as well as control leaf surface area and cell size, resulting in a 30% increase in the average area of mesophyll cells in the rve4 rve6 rve8 mutant[15]. The relative expression changes of the homologous genes CsRVE6 and CsRVE8 in CsWOX1-OE and mf not only exhibit opposite trends but also have a complementary relationship. Specifically, when CsRVE6 expression increased in CsWOX1-OE, CsRVE8 expression decreased; in mf, when CsRVE6 was downregulated CsRVE8 was then upregulated.
A large number of DEGs were identified in the plant circadian regulatory pathway and there were four co-DEGs, of which CsRVE6 and CsRVE8 were associated with the formation of leaf surface area. This provides a basis for the involvement of the CsWOX1 gene in plant circadian regulation and establishes a new link between rhythm-related genes and leaf development.
CsWOX1 binds and transcriptionally activates the promoter of CsRVE6 and CsPin1At
-
Through comparative analysis of the transcriptomic data between CsWOX1 overexpression and mf, significant changes in the expression levels of genes involved in auxin polar transport and plant circadian rhythm regulation were found. To test whether CsWOX1 directly binds to the promoters of six common DEGs (CsRVE6, CsRVE8, CsGI, CsFKF1, CsPin1At, and CsABCB), yeast one-hybrid experiments were conducted with the promoter sequences (about 2,000 bp) of these genes and the CsWOX1 transcription factor. The results indicated that the CsWOX1 transcription factor binds only to the promoters of CsRVE6 and CsPin1At, causing the corresponding yeast colonies to turn blue in SD/-Ura/-Trp/X-gal medium (Fig. 6). Regarding the remaining four genes, they didn't exhibit transcriptional regulation by CsWOX1 transcription factor.
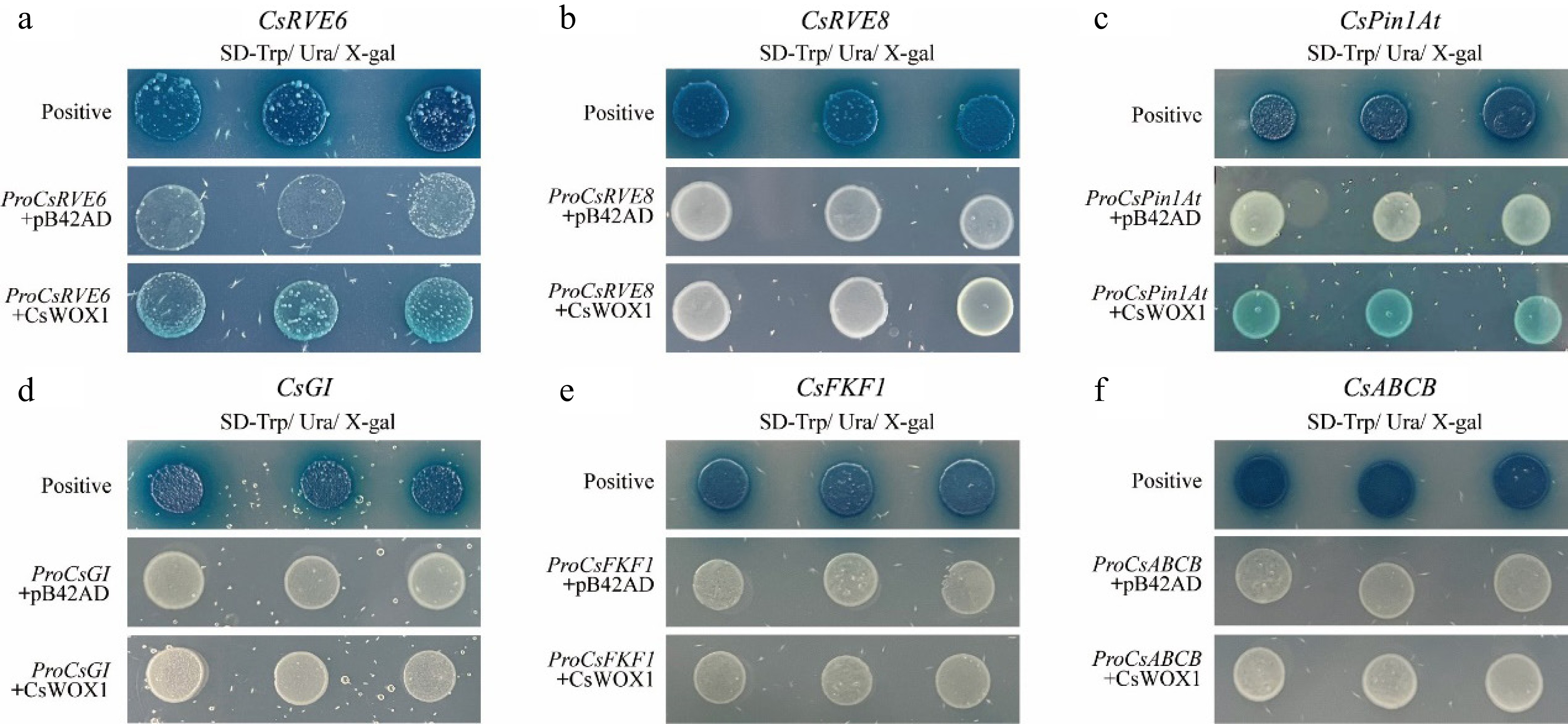
Figure 6.
Yeast-one hybrid validation between CsWOX1 transcription factors and candidate genes, which showed that CsWOX1 bound to the promoters of CsRVE6 and CsPin1At.
A dual-LUC reporter assay was employed to examine the impact of CsWOX1 on the activity of the firefly luciferase gene (LUC) driven by the promoters of CsPin1At or CsRVE6 in N. benthamiana leaves. The co-expression of 35S:CsWOX1 with proCsPin1At:LUC or proCsRVE6:LUC vectors resulted in a significant increase in LUC/REN signals of approximately 15-fold and 5-fold, respectively, compared to expression of proCsPin1At: LUC or proCsRVE6:LUC vectors alone (Fig. 7). This observation suggests that the CsWOX1 transcription factor activates the expression of CsPin1At and CsRVE6.
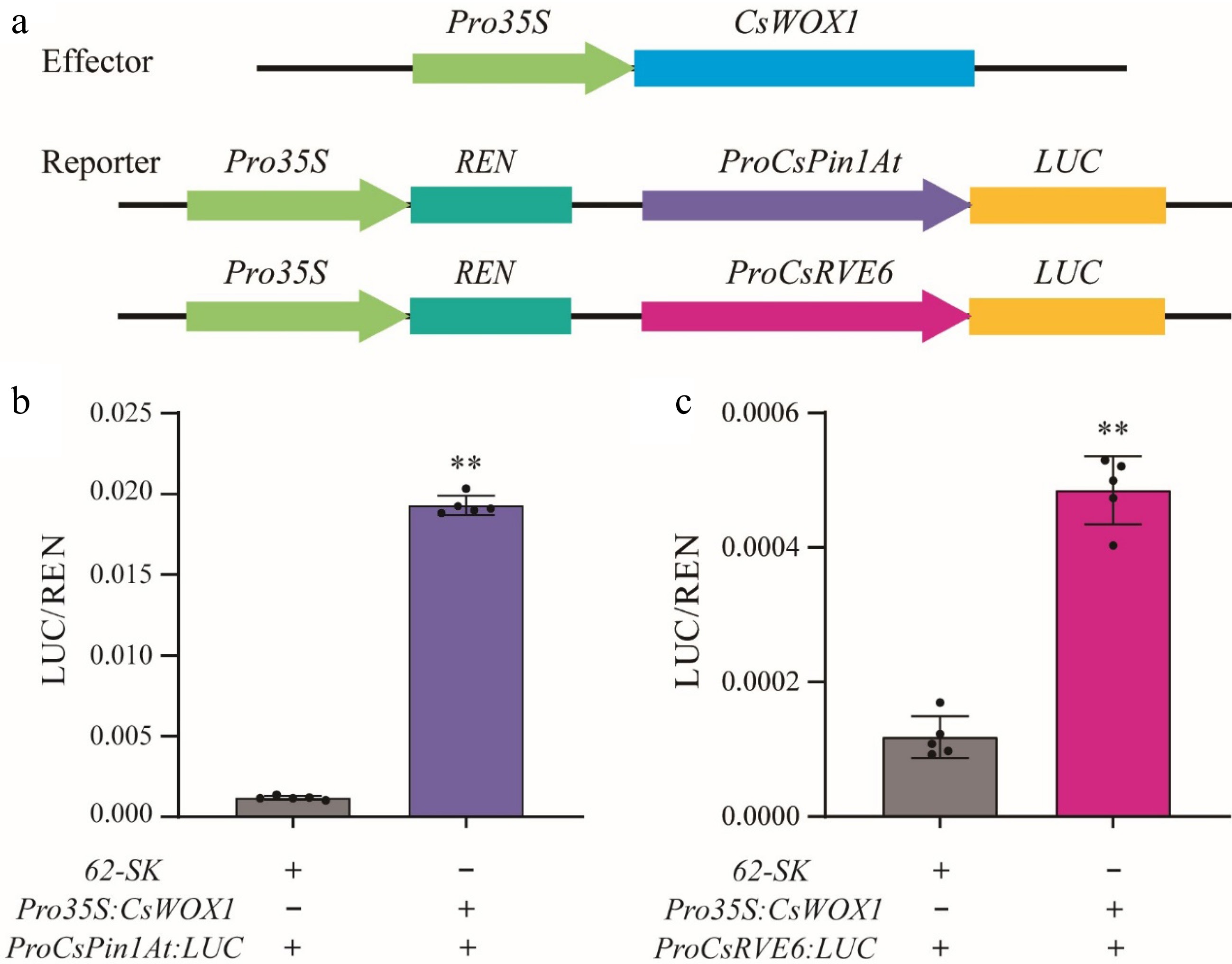
Figure 7.
CsWOX1 actives the expression of CsRVE6 and CsPin1At. (a) Structural diagram of the reporter and effector constructs used for LUC/REN assays. LUC activity measurement after transient expression of 35S:CsWOX1 with (b) ProCsPin1At:LUC and (c) ProCsRVE6:LUC in tobacco leaves. The LUC/REN ratio from the empty vector (62-SK) combined with ProCsPin1At:LUC and ProCsRVE6:LUC were used as the calibration. Two-tailed Student's t-test was performed and statistically significant differences were indicated by ** p < 0.01. Values are mean ± SD (n = 5).
To elucidate the expression patterns of CsRVE6 and CsPin1At, an analysis of their gene expression in various cucumber organs was conducted, including the root, stem, leaf, petal, sepal, tendril, and apical bud, using RT-qPCR (Supplementary Fig. S1). The results revealed that CsRVE6 is predominantly expressed in the petal, sepal, tendril, apical bud, and leaf, whereas CsPin1At exhibits high expression in the petal, sepal, tendril, and root. These differential expression patterns hint at the potential functions of CsRVE6 and CsPin1At in diverse cucumber organs, offering valuable insights for future research into their specific contributions to plant development.
-
In the present investigation, RNA-seq technology was employed to identify downstream DEGs of CsWOX1-OE and joint analysis performed with transcriptome sequencing results of the mutant mf. The RNA-Seq dataset substantially augmented our comprehension of the regulatory network involving CsWOX1 by furnishing a comprehensive collection of differentially expressed genes. This enriched our insight into the involvement of WOX1 in diverse growth and developmental pathways in cucumber. Given the substantial number of DEGs and the objective of pinpointing downstream genes intricately linked to CsWOX1 expression variations, a cross-analysis of the 3,657 DEGs in CsWOX1-OE and the 200 DEGs in mf was conducted, identifying 80 common DEGs. Subsequently, an in-depth analysis was performed to unravel the profound implications of the WOX1 gene in organ development, hormonal regulation, and various facets of plant growth and development pathways. Utilizing a comprehensive analytical approach encompassing GO, KEGG pathway analysis, gene expression trends, and RT-qPCR, six differentially expressed genes were identified as potential candidate genes.
Initially, it is noteworthy that both CsWOX1-OE and mutant cucumbers demonstrated significant alterations in leaf morphology. The mf displayed a marked reduction in the leaf tip area and an acicular constriction on either side of the leaf veins, whereas the overexpressing plants displayed a butterfly-like leaf structure. Consequently, investigating the expression changes of the ABCB and Pin1At genes became the focus of the study.
Through homology analysis, it was identified that CsaV3_5G029720 shares the highest homology with AtABCB21 in Cluster II of the ABCB subfamily in Arabidopsis[46]. It has been established that ABCB21 plays a pivotal role in controlling auxin concentration in plant cells, serving as a conditional input/output transporter. Specifically, when the cytoplasmic IAA concentration is low, ABCB21 mediates the inward transport of auxin, whereas in cases of high cytoplasmic IAA concentration, ABCB21 facilitates outward auxin transport[46]. Notably, during the developmental stage of 7−10 d in Arabidopsis seedlings, ABCB21 exhibits specific expression on the abaxial side of cotyledons and in the root region[46,47], concomitant with the expression of WOX1 in these same areas[41]. Furthermore, ABCB21 has been implicated in the lateral distribution of auxin in Arabidopsis leaf blades. When abcb21-1 (a weak allele of ABCB21) is further mutated on the background of abcb1 abcb19 mutants, the resulting phenotype shows reduced leaf length and smaller epidermal cells[47−49], consistent with the reduced leaf cell size observed in mf mutant plants[23,41]. These observations lead us to speculate that in cucumber, although the WOX1 gene does not directly interact with ABCB, it may still affect auxin transport and regulate plant leaf cell growth and expansion.
The Pin1At gene has been shown to control the floral transition in plants by accelerating the cis/trans isomerization of phosphorylated Ser/Thr-Pro motifs in two MADS-box transcription factors[18,19], SUPPRESSOR OF OVEREXPRESSION OF CO 1 (SOC1), and AGAMOUS-LIKE 24 (AGL24)[17,20]. Overexpression of Pin1At in Arabidopsis leads to an expedited flowering process, accompanied by the emergence of serrated leaves and cauline leaves[20], indicating that Pin1At influences flowering time and leaf morphology in plants. Additionally, CsPin1At exhibits higher expression levels in petals, sepals, lateral branches, and roots, which may be related to the development of floral organs, lateral meristems, and root apical meristems.
Additionally, yeast one-hybrid assay indicated that the LHY homologous gene, CsRVE6, was transcriptionally activated by CsWOX1, whereas CsRVE8 was not. RVE6 and RVE8 transcription factors are homologous to CIRCADIAN CLOCK ASSOCIATED1 (CCA1) and LHY[50,51]. CCA1 and LHY are homologous transcription factors with MYB-like domains and play pivotal roles in the plant circadian clock[52,53]. CCA1 and LHY suppress the expression of TIMING OF CAB EXPRESSION1 (TOC1), forming a negative feedback regulation loop[54,55]. CCA1 and LHY inhibit TOC1 expression, and TOC1, in turn, inhibits the expression of CCA1, LHY, and other genes from the PRR family[56−58]. RVE8, on the other hand forms an additional feedback loop within the circadian clock. RVE8 positively regulates PRR5, and PRR5 protein, in turn, suppresses the expression of RVE8[16,50,51].
In Arabidopsis, the RVEs partially redundantly promote the circadian rhythms[16] and negatively regulate leaf cell growth rate and expansion area[15,16]. The leaves of adult triple rve mutants (rve 4 6 8) and quintuple rve mutants (rve 3 4 5 6 8) are approximately 30% larger than those of wild-type Arabidopsis, and this change is attributed to a 30% increase in cell area in leaves of the same age. Further research suggests that the RVE gene family negatively regulates plant responses to sucrose at concentrations ranging from 0.5% to 6%[15,59]. This, to some extent, explains the phenomenon of increased leaf area in rve multiple mutants. In cucumber, CsRVE6 is highly expressed in petal and sepal, tendril, apical bud, and leaf, suggesting CsRVE6 may play a critical role in the growth and development of these tissues, particularly in flower development and the growth of lateral branches and shoot apices.
Moreover, the rve multiple mutants exhibit a delay in flowering time[15]. Notably, the transition from vegetative to reproductive growth in plants is intricately linked, with photoperiodic rhythms governed by the circadian clock playing a crucial role in this process[60]. Under long-day conditions, the precise regulation of CO protein expression is pivotal for inducing flowering[61,62]. Previous studies have elucidated that FKF1 contributes to the stabilization of CO protein expression through two distinct pathways: one involving FKF1 itself, and the other involving CDF1, which cooperates with GI to alleviate the inhibition on CO and FT transcription[63,64].
Noteworthy is the observation that within the entire flowering regulation pathway, spanning from the light-sensitive CRY, through ELF3, GI, FKF1, CDF1, and CO genes, all of them, except for COP1, exhibited upregulated expression in CsWOX1-OE plants. As CsWOX1 expression increased, the genes that inhibit CDF1 also exhibited an upregulation in expression, resulting in an upregulation of CDF1 expression. This may be a regulatory mechanism through which the plant seeks to maintain equilibrium in flowering regulation. Conversely, when the CsWOX1 gene was mutated, both GI and FKF1 genes down-regulated in this pathway. However, the expression levels of other genes in this pathway did not show significant differences, possibly due to the upregulation of the LHY homolog gene, RVE8, in the mutant. RVE8 establishes a feedback loop with the PRRs family genes, thereby activating the expression of PRR7 and PRR9, which in turn inhibit CDF1. This intricate network ensures that the expression of CO and FT in the mutant remains largely unaltered.
However, research on the association between WOX family genes and plant circadian rhythms is limited. When the homologous gene of Loquat, EjWUSa, is overexpressed in Arabidopsis, it exhibits early bolting by 10 d and precocious flowering by 9 d compared to the wild type, suggesting that EjWUSa can promote flowering in Arabidopsis[65]. Although the specific mechanisms by which the CsWOX1 gene interacts with genes involved in plant circadian regulation and how it influences cucumber circadian regulation remained unknown, the present transcriptomic data and analysis has identified a significant number of differentially expressed genes related to plant rhythms. These findings lead us to propose that CsWOX1 likely exerts some degree of influence on the plant's circadian clock, thereby laying the groundwork for further investigations into this area.
This research was funded by the National Key Research and Development Program of China (2022 YFD1602000), the National Natural Science Foundation of China (32202519, U22A20498), the Science and Technology Innovation Team of Shaanxi (2021TD-32), and the Key Research and Development Program of Shaanxi (S2023-YF-YBNY-0530). The authors would like to thank Dr. Hu Wang for providing valuable transcriptomic data. Thank you to all those who made this research possible.
-
The authors confirm contribution to the paper as follows: study conception and design: Li Z, Shen J, Zhang A, Jiang Y; data collection: Zhang A, Li X, Zhang H; analysis and interpretation of results: Zhang A, Li X, Zhang H, Jiang Y, Li Q; draft manuscript preparation: Zhang A, Zhang H, Li X, Jiang Y, Li Q. All authors reviewed the results and approved the final version of the manuscript.
-
The data that support the findings of this study are available in the NCBI repository. These data were derived from the following resources available in the public domain: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1138173.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 The primers used in this study.
- Supplementary Table S2 Quality statistics of sequencing data.
- Supplementary Table S3 Mapping statistics of RNA-seq reads to the reference genome.
- Supplementary Fig. S1 RT-qPCR analysis of CsRVE6 and CsPin1At expression in various cucumber organs (root, stem, leaf, petal, sepal, tendril, lateral branch, apical bud). (a) CsRVE6 relative expression level. (b) CsPin1At relative expression level. Different letters (a−e) indicate significant differences between groups (p < 0.05). The values represent the mean ± SD (n = 3).
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang A, Li X, Zhang H, Jiang Y, Li Q, et al. 2025. Transcriptome analysis of CsWOX1 mutant and overexpressing lines reveals downstream genes regulating cucumber leaf morphogenesis. Vegetable Research 5: e004 doi: 10.48130/vegres-0024-0038 

Transcriptome analysis of CsWOX1 mutant and overexpressing lines reveals downstream genes regulating cucumber leaf morphogenesis
- Received: 28 August 2024
- Revised: 19 October 2024
- Accepted: 28 October 2024
- Published online: 11 February 2025
Abstract: WUSCHEL-related homeobox 1 (WOX1), categorized into the WUS clade, has been determined as the essential gene for leaf expansion as well as promoting leaf outgrowth along the mediolateral axis. The phenotypic characteristics of transgenic cucumber plants with CsWOX1 overexpression (CsWOX1-OE) and mutants (designated as mango fruit, mf) have been reported. Specifically, CsWOX1-OE cucumber plants exhibit leaves with a distinctive 'butterfly-shaped' appearance, while mf mutants show a rapid reduction in the area of their leaf tips. In this study, RNA-Seq was employed to sequence the transcriptome of CsWOX1-OE to reveal its differentially expressed genes (DEGs). Combined with the transcriptome sequencing data of the mf mutant, GO classification, and KEGG pathway enrichment analyses were performed, revealing new links between the WOX1 protein and many core plant regulatory pathways. The further joint analysis identified 80 common DEGs between CsWOX1-OE and mf, with a significant proportion of these common DEGs being annotated to the circadian pathway and the auxin polarity transport pathway. Six genes (CsGI, CsFKF1, CsRVE6, CsRVE8, CsABCB, and CsPin1At) were selected for Y1H and Dual-LUC interaction validation experiments. These experiments demonstrated that CsWOX1 directly targets the promoters of CsRVE6, which regulates leaf expansion, and CsPin1At, which is involved in auxin polarity transport, thereby promoting their transcription. In conclusion, these findings provide a foundation for exploring the potential regulatory mechanisms associated with CsWOX1, expecting to contribute to the construction of a more comprehensive gene network for leaf morphogenesis.
-
Key words:
- Cucumber /
- CsWOX1 /
- RNA-Seq /
- Leaf morphogenesis