






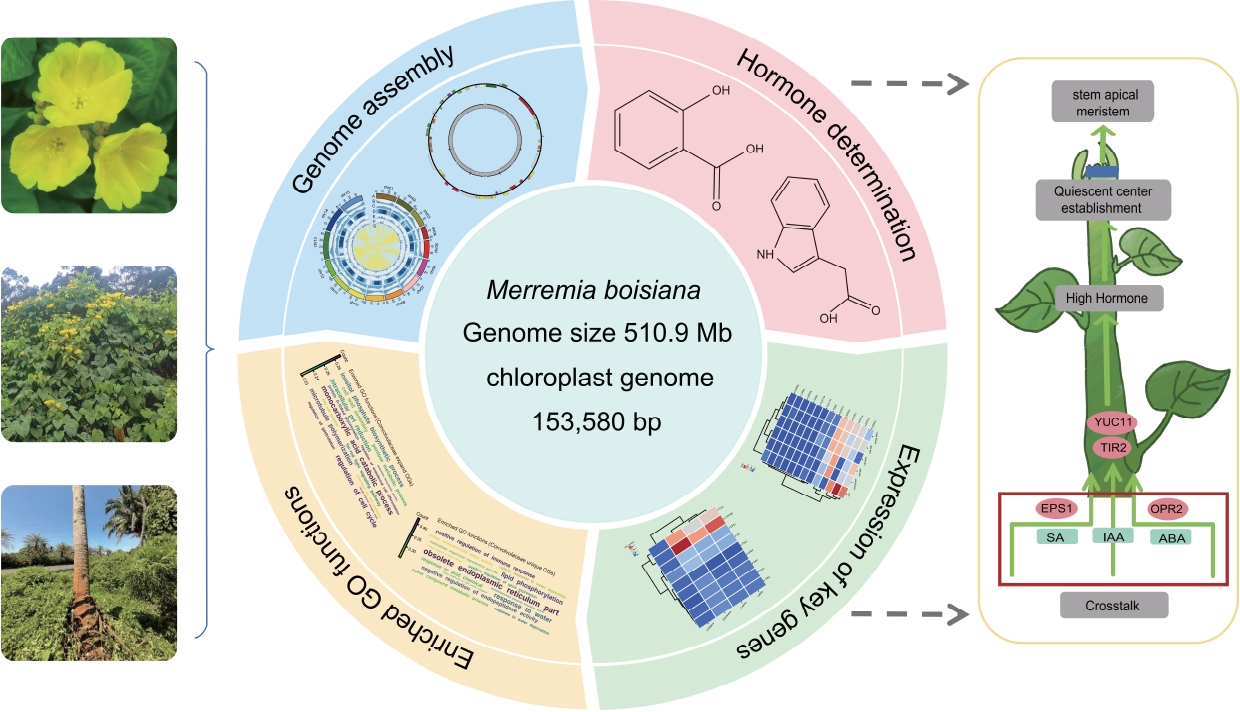



-
Merremia boisiana, previously identified as Decalobanthus boisianus, is a captivating plant species native to tropical rainforest ecosystems. It is renowned for its striking golden blossoms and remarkable growth velocity. M. boisiana occupies a broad geographical range and is predominantly distributed across the rainforests of tropical Asia and the Pacific, spanning regions such as Malaysia, Indonesia, Australia, and beyond[1,2]. Thriving in moist soils, M. boisiana exhibits a robust resilience to high humidity and temperature conditions characteristic of these tropical conditions[3]. Renowned for its swift growth, this plant boasts formidable root systems and climbing abilities, enabling it to swiftly envelop neighboring vegetation. This characteristic enables M. boisiana to compete effectively for resources such as light, water, and soil nutrients within the rainforest, swiftly adapting to and occupying new ecological niches. Within the rainforest ecosystem, M. boisiana plays a pivotal role with its dense growth patterns contributing to the formation of distinctive vegetation structures, providing habitats and food sources for a wide array of organisms. Despite its ecological importance, the current understanding of its genetics remains markedly limited. To date, only two chloroplast genomes (PRJNA832014, PRJNA883344) are available on NCBI. Notably, there are no reports on the comprehensive genome of M. boisiana, significantly hindering our comprehension of its physiological and ecological intricacies within rainforest ecosystems.
This knowledge gap underscores the urgency for further genetic research on M. boisiana, as it holds the key to unlocking the secrets of its remarkable adaptability and significant ecological contributions within tropical rainforests. M. boisiana belongs to the genus Merremia within the Convolvulaceae family, and both nuclear and plastid genetic analyses evidence its closest phylogenetic relationship with sweet potatoes (Ipomoea batatas). Although the genomes of several sweet potato varieties have been deciphered, research on their evolutionary origins and genetic improvement remains in its infancy. Due to M. boisiana's robust growth, high disease resistance, and rapid developmental rate, acquiring a complete genome for this species and exploring its superior genes hold significant value for both the study of sweet potato origins and their genetic enhancement.
Therefore, this study aims to construct a high-quality, chromosome-level reference genome for M. boisiana. By thoroughly analyzing its genetic and genomic background, we seek to identify key genes responsible for its rapid growth, high disease resistance, and strong developmental capabilities. Compared to existing research on M. boisiana, which has been largely limited to morphological descriptions and chloroplast genome sequencing, our study represents a significant advancement by providing the first comprehensive nuclear genome assembly. This enables deeper insights into the molecular mechanisms underlying its ecological adaptability and competitive growth strategies in tropical rainforests.
Moreover, our findings have direct implications for sweet potato research. Given the close phylogenetic relationship between M. boisiana and sweet potatoes, our analysis of key growth and resistance genes can serve as a valuable resource for comparative genomics. Specifically, the identification of genes associated with robust root development, rapid biomass accumulation, and enhanced stress tolerance in M. boisiana may offer novel genetic targets for improving sweet potato breeding programs. By leveraging these insights, our study not only enhances our understanding of this enigmatic rainforest species but also provides crucial scientific evidence for sweet potato genetic improvement and stress resilience research.
-
Sequencing materials for M. boisiana were collected in the proximity to Guanlan Lake, located in Haikou City, Hainan Province, China. For sequencing purposes, DNA was extracted from young leaves, while transcriptome materials were sourced from various tissues including roots, stems, and functional leaves of M. boisiana. All transcriptome libraries were sequenced using the Illumina platform[4], generating paired-end reads. To enrich the transcriptome sequencing data, we further collected stems from different individuals, which were then segmented, and sequenced. For Nanopore sequencing[5], collected DNA samples underwent agarose gel electrophoresis along with other necessary preparations. Subsequently, the SQK-LSK109 kit[6] was employed for DNA extraction, resulting in approximately 59.5 Gb of sequencing data. Regarding Hi-C sequencing, we utilized the high-throughput sequencing platform DNBSEQ to generate the corresponding data.
To obtain RNA sequencing (RNA-Seq) data, RNA was extracted from coarse roots, root tips, stems, and leaves of M. boisiana using the RNeasy Plant Mini Kit (Qiagen). This extraction process yielded 15 sets of relevant RNA-Seq data, thereby providing a comprehensive overview of the transcriptome across different tissues of this plant.
Genome assembly and annotation
-
In this study, we focused on M. boisiana and employed Fastp v0.23.4[7] software to screen and clean 59.5 Gb of high-quality Oxford Nanopore Technologies (ONT) data, followed by de novo genome assembly using Nextdenovo v2.5.2[8,9]. After initial assembly, we refined and polished the 68 Gb of high-quality Illumina data using Polish v4.3.1[10] software to further enhance the accuracy and quality of the genome assembly. Subsequently, BWA v0.7.18[11] software was utilized to align the reads to the assembled genome. For the 141 Gb of Hi-C paired-end sequences, we carried out a series of processing steps including trimming, clustering, filtering, and optimization using 3DDNA[12], and Juicebox v2.20.00[13] software (Supplementary Fig. S1), ultimately leading to the construction of a complete, chromosome-level genome assembly arranged in the correct order and orientation. To assess the quality of the assembly, we comprehensively evaluated the genome using Busco v5.1 tool[14] with the embryophyta_odb10 database, examining the presence and completeness of orthologs. As a result, we obtained a high-quality, well-assembled genome sequence for M. boisiana, laying a solid foundation for subsequent functional studies.
For annotation, we initially identified and built repeat sequence models from the whole-genome sequence using RepeatModeler v2.0.5[15] through a de novo prediction approach. These models were then utilized in conjunction with RepeatMasker v2.0.5[16] to efficiently and accurately identify repetitive sequences by comparing the M. boisiana DNA sequence against established databases. We compared the repeat sequence content of M. boisiana with that of other Convolvulaceae species, including Cuscuta australis, Ipomoea aquatica, and Ipomoea cairica. This comparative analysis allowed us to better understand the structural variations and gene expression patterns of M. boisiana, facilitating the identification of key genes associated with its important traits. Additionally, gene prediction was performed ab initio using Augustus v3.5.0[17], and GeneScan v1.0[18], with gene model parameters specifically trained for this species. To further refine the annotation, we leveraged annotated information from homologous species such as Cuscuta australis, Cuscuta campestris, Ipomoea cairica, and Ipomoea triloba, to accurately determine exon boundaries and splice sites through sequence alignment[19]. Finally, Hisat v2.2.1[20] software was employed to annotate the genome based on transcriptome data. The results from these three annotation methodologies were integrated using EvidenceModeler (EVM) v1.0.0[21] to produce a comprehensive gene structure annotation.
Assembly and annotation of the chloroplast genome
-
Using the software GetOrganelle v1.7.7.1[22], we assembled the Illumina paired-end sequencing data by specifying the option -k with values of 21, 45, 65, 85, and 105; respectively. The resulting two FASTA files were then utilized as input files for annotation on the OGDRAW platform[23] (
https://chlorobox.mpimp-golm.mpg.de/OGDraw.html ). Following the platform's guidelines, we annotated the organellar genome and opted for the output in GenBank (GB) format. To visualize the annotated genome, we accessed the 'upload' section of the OGDRAW online tool and submitted the GB file containing the genomic sequence of M. boisiana. After selecting the appropriate options, we submitted the file for processing. The OGDRAW then generated and displayed the annotated organellar genome in a graphical format, facilitating a clear representation of the organellar genetic architecture.Expression level calculation
-
For the calculation of transcriptome expression levels in M. boisiana, we first subjected the obtained Illumina reads to a rigorous filtering process. We employed HiSat2[20], to construct an index using the '-build' option, which incorporates critical information regarding exons, splice sites, and transcripts to facilitate an accurate alignment. This constructed index was then employed during the alignment process to ensure the accuracy of the results. After alignment, we converted the results into suitable format using SamTools[24]. We then performed differential expression analyses with the Rsubread software[25], allowing us to identify genes with significant changes in expression levels.
Phylogenetic analysis and divergence time estimation
-
We prepared protein data from 62 species encompassing the Rosidae, Asteridae, and Magnoliidae subclasses, as well as various monocots and gymnosperms. Utilizing OrthoFinder v2.5.5[26], a tree-based methodology, we identified high-quality orthologous groups (Orthogroups) across these species. The OrthoFinder outputs were refined with TRIMAL, applying the -gt 0.6 option to remove short sequences within each Orthogroup and using a -cons 60 threshold to retain only the most conserved sites. Subsequently, we performed a maximum likelihood (ML) analysis on the concatenated protein sequences from these species using the parallelized version of RAxML v8.2.13[27], namely raxmlHPC-PTHREADS. This analysis aimed to infer the phylogenetic relationships within the context of M. boisiana, providing a clearer understanding of its evolutionary positioning relative to other plant species.
To estimate divergence times among closely related species, including C. australis, C. campestris, C. epithymum, I. triloba, I. trifida, I. aquatica, I. cairica, I. triloba, and M. boisiana, we utilized single-copy gene protein sequences from the Solanaceae, Asteraceae, and Rubiaceae families. We employed the mcmctree v4.9 tool[28] from the PAML software package for the divergence time estimation. Following this analysis, the estimated divergence times were integrated into a tree reconstructed using IQTREE v2.3.5[29], facilitating a comprehensive understanding of the evolutionary relationships among species.
Gene family expansion and contraction
-
Based on the results obtained from OrthoFinder, we constructed a species tree encompassing 18 selected species. To estimate the birth and death rate of genes, we employed the maximum likelihood (ML) method implemented in CAFE5 v1.1 software[30]. Subsequently, we applied the Viterbi algorithm to calculate the expansion probabilities of each orthologous group across various branches, setting a significance threshold of 0.01 to identify orthologous groups exhibiting accelerated expansion rates. Furthermore, we conducted a lineage-specific analysis of gene family expansion and contraction among the orthologous groups of the 18 species. The proteins within the 18 genomes were annotated by referencing the Pfam database to identify their functional domains. We documented the occurrences (presence/absence) and abundances of these domains within each genome. By comparing the data between the two groups, we aimed to deduce the patterns of domain gain, loss, and combination, thus enriching our understanding of the evolutionary dynamics at play within these species.
Synteny and whole-genome duplication analysis
-
We selected protein sequence data from I. cairica, I. trifida, I. aquatica, and M. boisiana as the foundation for our synteny analysis. To ensure the reliability of our results, we performed self-alignments and cross-alignments within and between genomes using BLASTP v2.9.0-2 tool[31], setting an E-value threshold of 1e-5 to ensure the reliability of the results. Following this, we used MCScanX v1.0.0[32] software to identify high-confidence syntenic blocks. To visualize the synteny information, we employed Circos v0.52[33], which effectively illustrated the collinear regions among chromosomes. For the analysis of whole-genome duplication (WGD) events, we applied WGD v1.1.054[34] to calculate WGD occurrences and used Datacolor[35] software to plot the distribution of Ks values, thereby estimating the timing and frequency of whole-genome duplication events.
Polyploidy and karyotype analysis
-
We preprocessed the protein data from Ipomoea aquatica, I. cairica, I. trifida, and M. boisiana using BLAST v2.9.0-2[36], selecting the output file format '-outfmt 6' and '-num_alignment 2'. Subsequently, we employed WGDI v0.6.5[37] with the '-km' option to construct the Ancestral Convolvulaceae Karyotype (ACK). Based on the protein sequence information, we inferred the modern karyotypes of the species under investigation. To quantify the fission and fusion events that occurred during the evolutionary history of these species, we enumerated all syntenic blocks and calculated the corresponding fission and fusion counts.
-
Based on a flow cytometry estimate of 523 Mb for the M. boisiana (Fig. 1a) genome (Fig. 1b), we initiated a comprehensive genome sequencing project. A total of 68 Gb of paired-end sequencing data was generated, resulting in a genome coverage of 130%. Through Genomescope v2.0[38], we confirmed that M. boisiana is a diploid species with a heterozygosity rate of 1.02% and a repeat sequence content of 60.9%. Additionally, Oxford Nanopore Technologies sequencing yielded 59.5 Gb of data, representing a 113× coverage depth of the genome. The genome assembly resulted in 141 contigs with a total length of 514.66 Mb, with a contig N50 size of 21.24 Mb, and an average contig length of 50,755 bp. To achieve a high-quality, chromosome-level reference genome, we leveraged 141 Gb of Hi-C paired-end sequencing data to scaffold the contigs, successfully organized into 15 chromosomes (Fig. 1c). The final assembled genome size was determined to be 510 Mb, closely aligning with the initial flow cytometry estimate of 523 Mb.
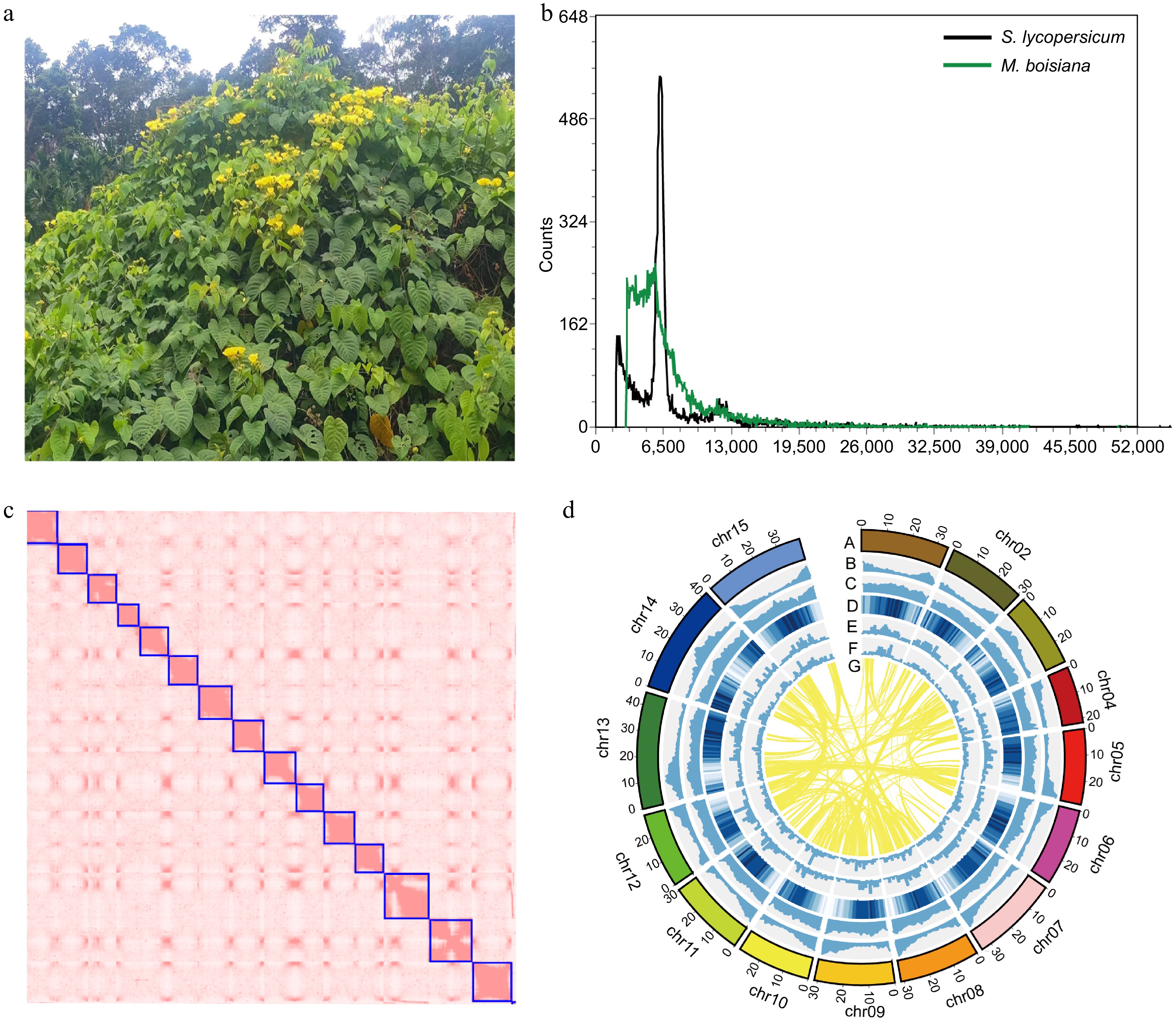
Figure 1.
Morphological traits and genome characterizations of M. boisiana. (a) The flower and leaf of M. boisiana. (b) Flow cytometry analysis results. (c) The Hi-C heatmap of the genome assembly. (d) Genome features of M. boisiana. (Chromosome karyotypes; Gene density; GC content per Mb; Repeat content per Mb; Gypsy content per Mb; Copia content per Mb; The syntenic regions between different chromosomes were identified)
The assembly quality was evaluated using BUSCO revealing a completeness of 98.7%, including 92.6% single-copy genes and 6.1% duplicated genes. Only 0.5% of the genome was fragmented, and 0.8% of genes were missing. Further assessments, including the LAI score[39] (11.27), and the Merqury v1.3 (33.2) quality index[40], confirmed that this assembly meets the standard required for a reference genome (Table 1). In summary, these results demonstrate the high quality of our M. boisiana genome assembly, rendering it suitable for evolutionary studies and gene mining.
Table 1. The genome features of M. boisiana.
Genomic feature Merremia boisiana Estimated genome size (Mb) 523.64 Genome size (Mb) 510.9 Heterozygous (%) 1.02 Contig N50 (Mb) 21.24 Length of N50 (bp) 50,755 Number of contigs 141 Number of chromosomes 15 Repeat sequence content (%) 60.93 GC content (%) 36.23 Number of genes 37,389 Genome completeness (BUSCO) 98.7 Gene completeness (BUSCO) 99.2 Genomic LAI 11.27 (reference level) Genomic Merqury 33.2 (reference level) Repeat sequence characterization and genome annotation
-
After employing both de novo and homology-based approaches for identifying repetitive sequence annotations, we discovered that M. boisiana harbors a substantial repetitive sequence content, comprising 60.93% of the genome. This includes 20.58% retrotransposons and 2.66% DNA transposons, with Long Terminal Repeat (LTR) sequences alone accounting for 18.78% of the genome. The whole-genomic GC content reached 36.23% (Fig. 1d). Through a comprehensive integration of de novo prediction, homology-based alignment, and transcriptome annotation, we identified a total of 37,389 protein-coding genes, achieving a BUSCO v5.1[14] completeness of 99.2% for the genome (Table 1). Remarkably, the gene count in M. boisiana surpasses that of the diploid sweet potato (Ipomoea. trifida, 28,456 genes)[41] and Cuscuta australis (19,671 genes)[42], suggesting that M. boisiana likely encodes an expanded repertoire of stress tolerance and developmental genes. This provides a valuable genetic resource for sweet potato breeding studies, potentially enabling the introduction of novel genes for enhanced resistance and growth traits.
Gene family contraction and expansion in evolutionary context
-
We constructed a phylogenetic tree based on the protein sequences of 62 different species, including M. boisiana, and identified that the sister group of M. boisiana is Ipomoea. Through this phylogenetic framework, we estimated the stem age of M. boisiana to be approximately 93 million years ago (Mya), the crown age to be around 59 Mya, and the divergence time between M. boisiana and its closest Ipomoea relative to be 20 Mya (Fig. 2a, b). This indicates that M. boisiana has retained some ancestral traits while also acquiring unique adaptive features. Its close phylogenetic relationship with Ipomoea aligns with their shared morphological and physiological characteristics, such as rapid growth, climbing ability, and strong adaptation to tropical environments.
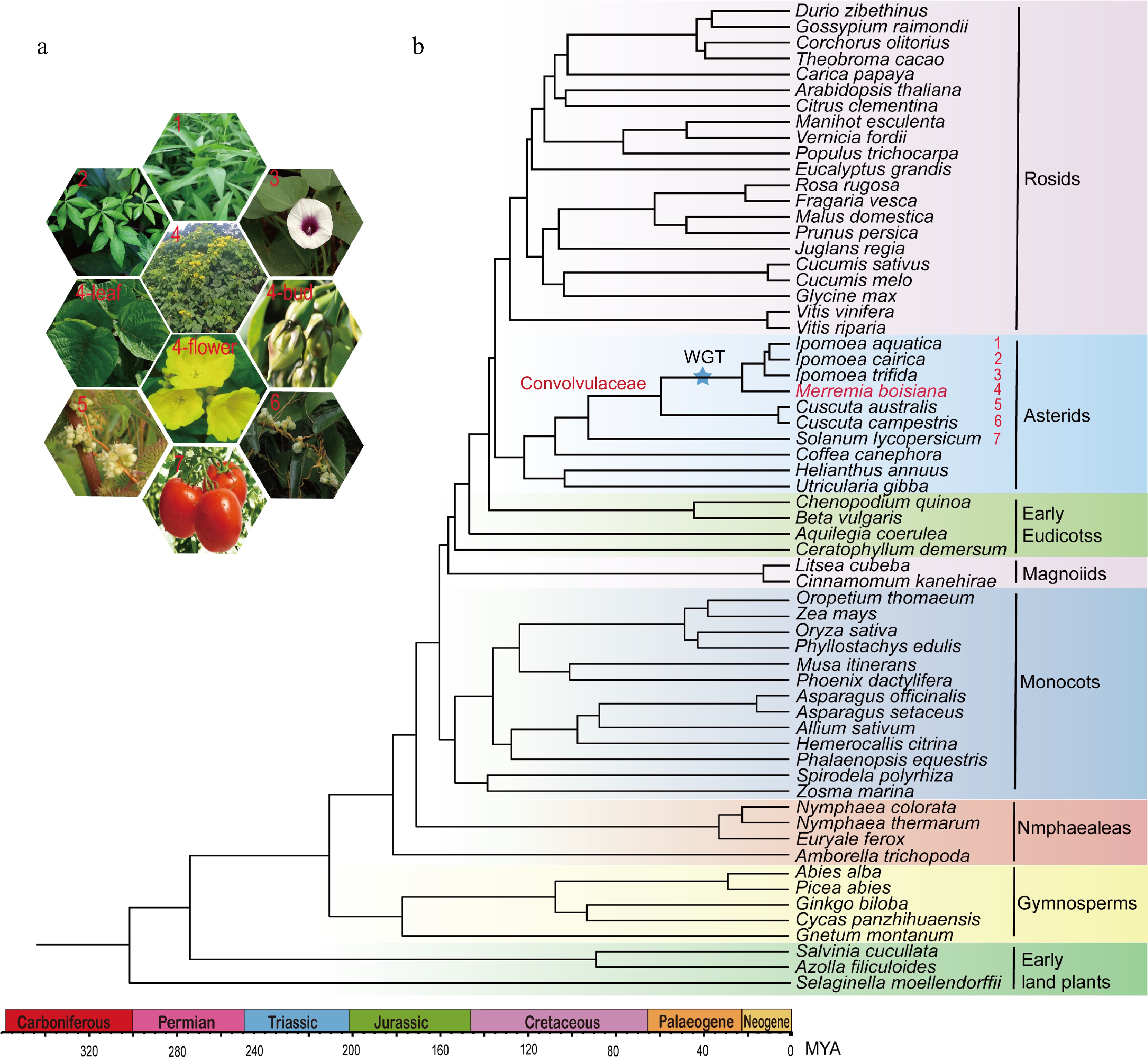
Figure 2.
Phylogenomic relationships of M. boisiana. (a) Pictures of six plants in the Convolvulaceae family. (b) Phylogeny and divergence time estimation of 60 plant species.
By conducting a detailed phylogenetic analysis of species closely related to M. boisiana, we examined 18 taxa from families such as Convolvulaceae, Solanaceae, Rubiaceae, and Asteraceae. Among Convolvulaceae species, we observed that M. boisiana has undergone a notable gene expansion, with an additional 1,377 genes, contrasting sharply with other members such as I. trifida (67 genes), I. triloba (77 genes), I. aquatica (981 genes), and I. cairica (636 genes). The expansion of gene families in M. boisiana may be closely linked to its invasive characteristics. The substantial increase in 1,377 genes, compared to its close relatives in Ipomoea, suggests that gene expansion could have played a crucial role in its aggressive growth, climbing ability, and adaptability to tropical environments. Notably, I. batatas, a hexaploid species, exhibited even more extensive gene expansion (12,937 genes), highlighting the role of genome duplication and expansion in evolutionary adaptation (Fig. 3a).
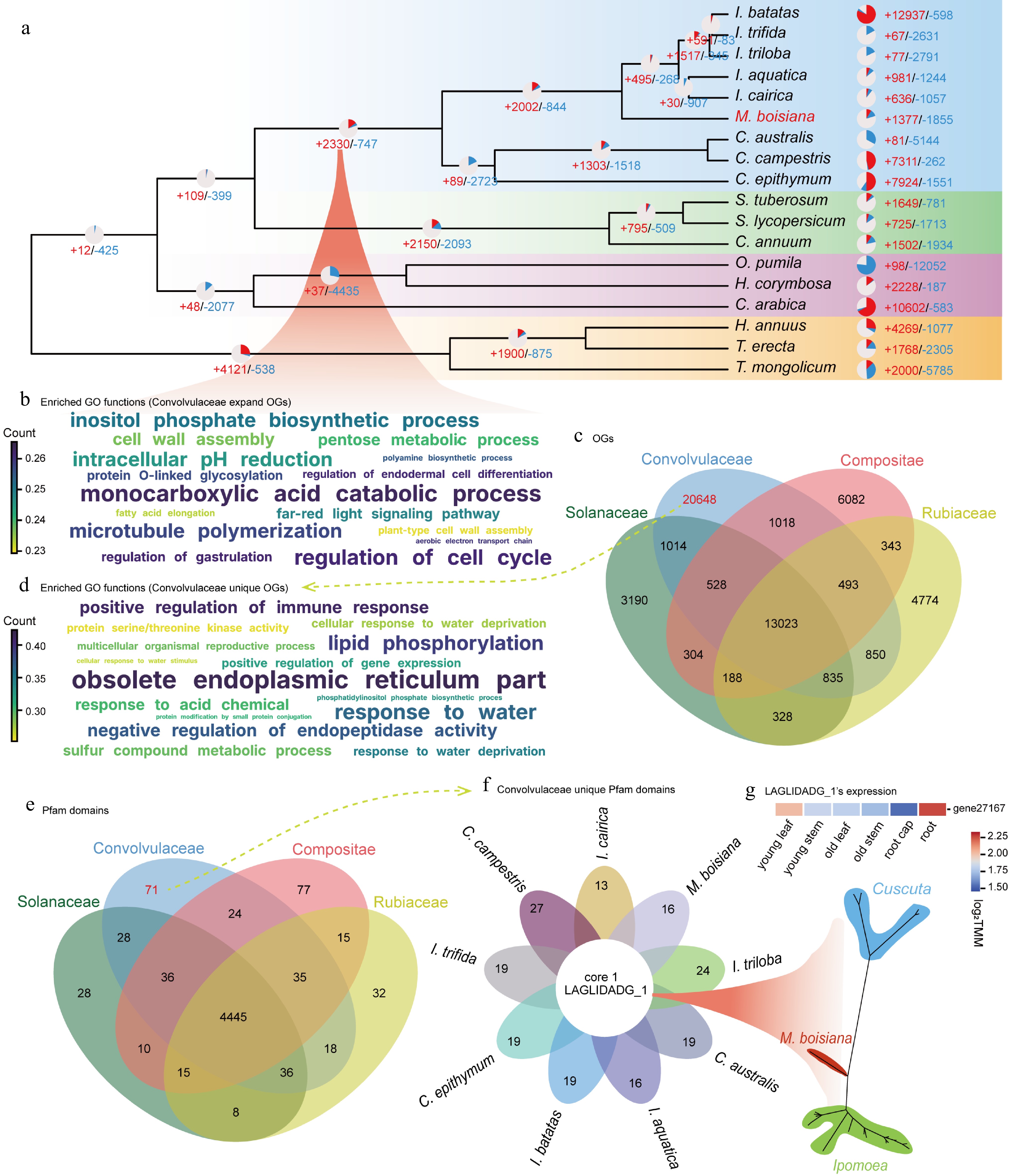
Figure 3.
Enriched orthogroups and domains in the Convolvulaceae family (a) Gene family expansion and contraction of M. boisiana. (b) Functional (GO) enrichment for the Convolvulaceae node. (c) Functional (GO) enrichment for the Convolvulaceae unique. (d) OGS overlap among Convolvulaceae, Solanaceae, Rubiaceae, and Asteraceae. (e) LAGLIDADG_1's expression of M. boisiana. (f) Convolvulaceae unique Pfam domains. (g) Pfam domains overlap among Convolvulaceae, Solanaceae, Rubiaceae, and Asteraceae.
For the last common ancestor (LCA) of Convolvulaceae, we inferred orthogroups (OGs) and analyzed Pfam domains, revealing that Convolvulaceae LCA had enriched Gene Ontology (GO) terms related to the regulation of cell cycle, polyamine biosynthetic processes, and far-red light signaling pathways (Fig. 3b). Among the 20,648 orthologous (OGs) unique to the Convolvulaceae (Fig. 3c), we identified significant enrichments in categories linked to immune response, gene expression, and responses to water availability (Fig. 3d). Compared to other non-invasive close relatives, we performed GO annotation specifically on the expanded genes of M. boisiana and found that its invasiveness may partially stem from its unique plant hormone crosstalk mechanism (Fig. 5a). By regulating hormone pathways to enhance root development, M. boisiana accelerates resource acquisition and suppresses the growth of native plants, ultimately gaining an ecological competitive advantage. After analyzing the 71 Pfam domains exclusively found within the Convolvulaceae family (Fig. 3e), we discovered a uniquely shared Pfam domain, LAGLIDADG_1 (Fig. 3f). Notably, LAGLIDADG_1 exhibited high specificity of expression in the roots of M. boisiana (Fig. 3g), suggesting its potential involvement in root development within the Convolvulaceae family, although the underlying mechanisms remain unclear. These finding provides a basis for our further research into the rapid growth and invasive mechanisms of M. boisiana.
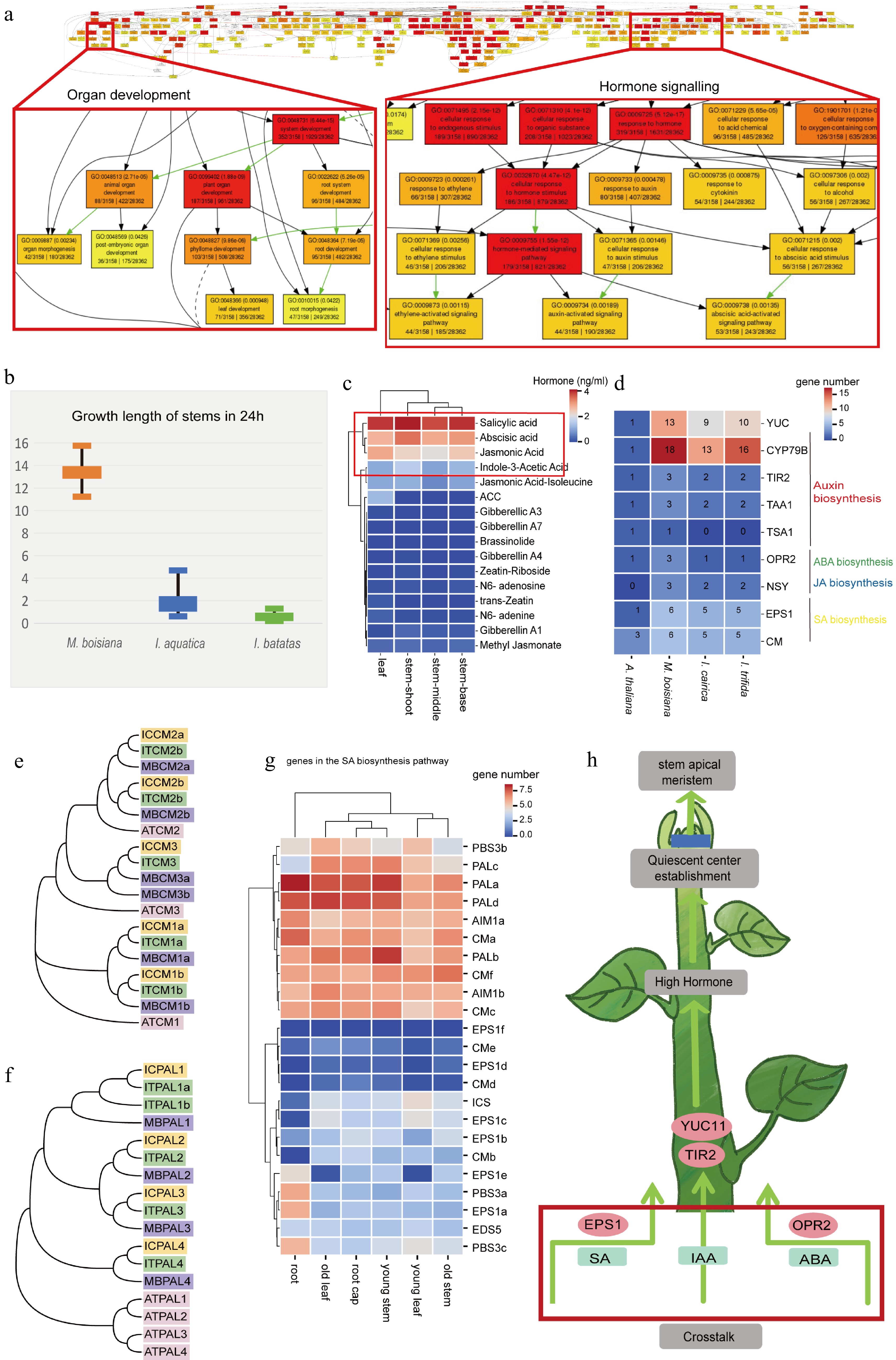
Figure 5.
The phytohormones regulate the rapid growth of roots in M. boisiana. (a) Go annotation of M. boisiana, a focus on organ development and hormone signalling in red boxes. (b) The rapid growth of M. boisiana, I. aquatica, and I. batatas. (c) The hormone content in different organs, among which four kinds of hormones (SA, ABA, JA, and IAA) have higher content across all organs. (d) Compared to plants such as I. aquatica, I. cairica, and A. thaliana, there is an expansion of key genes related to hormone synthesis in M. boisiana. These key genes promote root development. (e) Phylogenetic tree of M. boisiana, and A. thaliana CM. (f) Phylogenetic tree of M. boisiana, and A. thaliana PAL. (g) Highly expressed genes involved in SA hormone synthesis. (h) These key genes promote stem development.
Paleo whole-genome triplication and the chromosomal evolution
-
To identify potential paleo whole genome duplication (WGD) and whole genome triplication (WGT) events, our analysis reported no recent duplication events in M. boisiana or Ipomoea. Instead, it appears that their most recent common ancestor underwent an ancient hexaploidization event (Fig. 4a). The ancestral Convolvulaceae karyotype (ACK) was composed of 18 chromosomes. Following a WGT event, a series of chromosomal fusions and breaks occurred, resulted in the current configuration of 15 chromosomes, which represent the basic ploidy (x) across all species within the Convolvulaceae family. In detail, the ACK originally comprised 18 chromosomes, which after the WGT event, resulting in an expanded set of 54 chromosomes. Subsequently, these 54 chromosomes were subjected to a series of chromosomal rearrangements, including one reciprocal translocation (RTA) (chromosomal rearrangement event, not further specified), one nested chromosome fusion (NCF) (a non-specific chromosomal fusion or fission event, here assumed to be a fusion for context consistency), two end-to-end joining (EEJ) events (chromosomal end-to-end joins, effectively fusions), one chromosomal loss, and two fission events. These rearrangements ultimately led to the formation of the current karyotype of Ipomoea, characterized by 15 chromosomes. These large-scale genome rearrangements may have facilitated the retention and neofunctionalization of key genes, as well as the expansion of essential gene families, particularly those involved in hormone regulation, stress resistance, and growth development. We found a significant expansion of genes related to phytohormone biosynthesis in M. boisiana. This gene expansion may confer enhanced root growth capacity, strengthening its ability to compete for resources and ultimately increasing its invasiveness. The enrichment of genes related to immune responses, environmental stress adaptation, and water utilization in the genome of M. boisiana suggests that WGT and subsequent chromosomal rearrangements may have contributed to its improved disease resistance and abiotic stress tolerance, allowing it to gain a competitive advantage and become a dominant invasive species. Moreover, I. aquatica, a sister species to Ipomoea, evolved further from this karyotype through a series of additional changes, including three NCFs and EEJs, two chromosomal losses, and five fission events (Fig. 4b).
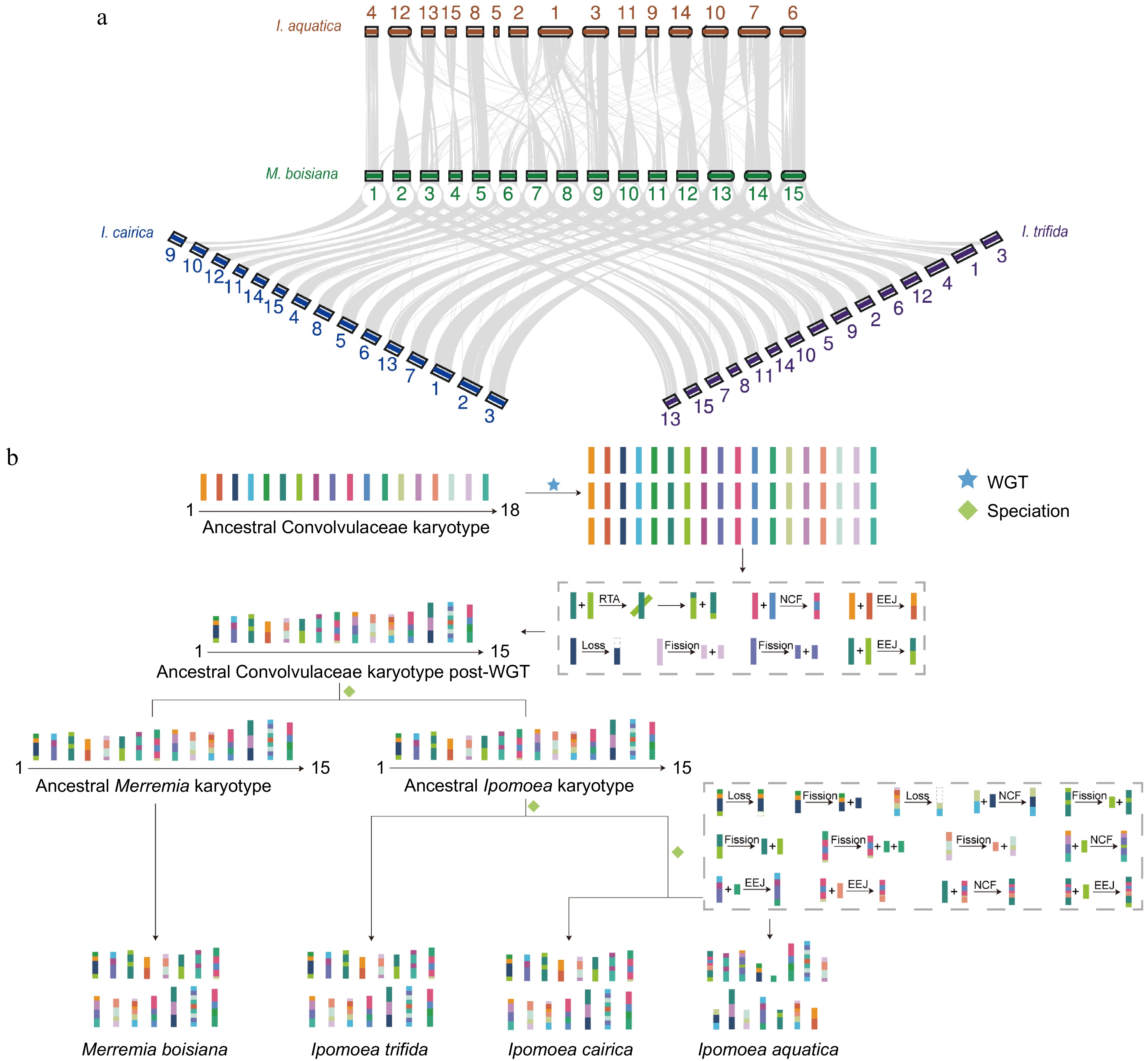
Figure 4.
Collinearity and karyotype analysis of M. boisiana. (a) Collinearity between M. boisiana and I. aquatica, I. cairica, and I. trifida. (b) WGD and chromosome-level genomic evolution of M. boisiana.
Chloroplast assembly
-
The chloroplast genome of M. boisiana consists of a double-stranded circular DNA structure comprising a large single-copy region (LSC), a small single-copy region (SSC), and two inverted repeat regions (IRA and IRB)[43]. The total length of this genome is 153,580 base pairs (bp) and it encodes a total of 96 genes, with the highest number of genes belonging to the RPS gene family, accounting for 15 genes. In comparison, the chloroplast genomes of related species such as Ipomoea alba[44] exhibit a greater gene account, comprising 130 coding genes over a length of 161,353 bp. Other species, including Ipomoea nil (161,747 bp), Ipomoea purpurea (161,629 bp), Ipomoea hederacea (161,354 bp), Ipomoea lacunosa (161,492 bp), and Ipomoea triloba (161,750 bp), each encoding 112 genes[45] (Supplementary Fig. S2).
Four key hormones are involved in M. boisiana's rapid growth
-
M. boisiana has emerged as a rainforest killer in Southeast Asia and southern China, posing a significant threat to local ecosystems in recent years[46−48]. Our research has identified two primary reasons for its designation as such: firstly, its rapid growth rate, reaching up to 10 cm per day, and secondly, its status as a climbing plant with well-developed roots and vines capable of ascending through vegetation, thereby outcompeting native flora for resources and disrupting local ecological dynamics.
Through comprehensive genomic analysis, we have identified a whole genome triplication (WGT) event in M. boisiana, a process known to generate a substantial number of genes, laying the genetic groundwork for the emergence of novel traits. Our examination of the retained duplicate genes subsequent to this ancient triplication event in M. boisiana uncovered a substantial number of these genes to be associated with the development of key plant organs such as roots, leaves, and stems, as well as processes such as photosynthesis and plant hormone regulation (Fig. 5a). The rapid growth and strong invasiveness of M. boisiana are primarily attributed to its unique plant hormone regulatory mechanisms. The significant expansion of genes related to the biosynthesis of auxin (IAA), gibberellin (GA), and cytokinin (CK) promotes root development and rapid stem elongation, enabling it to efficiently acquire resources and establish its ecological niche. Additionally, abscisic acid (ABA) and jasmonic acid (JA) enhance its stress resistance, allowing M. boisiana to sustain high growth efficiency even under drought or pathogen stress, while ethylene (ETH) strengthens its twining ability, improving its climbing capacity. The interplay of these hormones forms a highly optimized regulatory network, granting M. boisiana a competitive advantage in dynamic ecosystems. This finding underscores the pivotal role that these genes play in the foundation of M. boisiana's evolution into a strangling vine, emphasizing their importance in its adaptative and morphological diversification[49].
Furthermore, our study delved into the physiological characteristics of M. boisiana, particularly focusing on its remarkable growth rate. Through experimental observations, we confirmed that the above-ground stem of M. boisiana can grow at an astonishing rate of 12 to 14 cm per day (Fig. 5b). To unravel the physiological underpinnings of this exceptional growth velocity, we conducted a targeted analysis of the major hormone concentrations within the apical region of its stems.
Our findings revealed a notable abundance of five hormones: SA, ABA, JA, and IAA, along with another hormone (Fig. 5c). Specifically, SA concentrations surpassed 4 ng/mL, while ABA levels exceeded 3 ng/mL, Additionally, JA and IAA concentrations were maintained above 2 and 1 ng/mL, respectively. This hormonal profile suggests a complex interplay among these phytohormones, which likely contributes significantly to the unparalleled growth rate exhibited by M. boisiana.
Phytohormone's crosstalk regulated the rapid growth of shoots
-
To elucidate the relationship between hormones and rapid growth as well as the robust stress tolerance exhibited by M. boisiana, we conducted a comprehensive identification of highly expressed genes involved in the biosynthetic pathways of the four most abundant hormones. Drawing from existing research[50−52], we successfully identified a total of 110 genes, with 22, 56, 13, and 19 genes specifically associated with the synthesis of SA (Fig. 5d), IAA, JA, and ABA, respectively (Supplementary Figs S3−S5).
Remarkably, we observed that the genes encoding chorismate mutase (CM) and phenylalanine ammonia-lyase (PAL) (Fig. 5e, f), which are crucial for the biosynthesis of SA (Fig. 5g), displayed high expression levels across all organs of M. boisiana. This ubiquitous high expression pattern of the PAL gene indicates that SA production is a fundamental and consistently active process throughout the plant, likely contributing to a sustained hormonal milieu that fosters not only rapid growth but also resilience against adverse conditions. The pervasive nature of this high expression underscores the significance of SA in the overall physiology and adaptative strategies of M. boisiana. Furthermore, our analysis revealed that M. boisiana harbors a greater number of genes involved in auxin synthesis, including tryptophan aminotransferase of Arabidopsis (TAA), Toll/interleukin-1 receptor (TIR), cytochrome P450 79B (CYP79B), Trichostatin A (TSA), and YUCCA (YUC), as well as chorismate mutase (CM) genes related to SA synthesis, EPSIN1 (EPS1) transcription factors, neoxanthin synthase, chloroplastic (NSY) genes, and oxophytodienoate reductase 2 (OPR2) transcription factors. All of these genes were more abundant in M. boisiana compared to A. thaliana, I. cairica, and Ipomoea trifida. This finding suggests that M. boisiana possesses an enhanced genetic repertoire for hormone biosynthesis, potentially conferring unique growth and adaptive advantages. Notably, members of the YUC family, particularly YUC10 and YUC11, exhibited the most pronounced differences compared to other plant species, highlighting their potential significance in the rapid growth and development of M. boisiana. Based on our observations, we proposed that the EPS1 transcription factor involved in SA synthesis, along with YUC11 and TIR2 transcription factors participating in auxin synthesis, and the OPR2 transcription factor associated with ABA synthesis, collectively contributed to the enhanced root growth of M. boisiana through mechanisms such as gene expansion, gene dosage and specific expression in roots. These transcriptional regulators likely orchestrate complex hormonal signaling networks, enabling the plant to achieve its remarkable growth rates and stress tolerance (Fig. 5h).
-
The genomic landscape of the Convolvulaceae family is currently limited, encompassing only a few low-quality genome sequences from species such as C. australis[42], C. campestris[53], C. epithymum[54], I. aquatica[55], I. cairica (morning glory), and a handful of sweet potato species[56]. This scarcity of genomic data restricts our in-depth understanding of this plant family. Members of the Convolvulaceae family generally exhibit climbing and vine-like growth habits. Among these, sweet potatoes not only possess significant economic value but also face challenges in pest and disease control, as well as genetic improvement[57]. In addressing these challenges, some research has turned its focus to M. boisiana[58,59]. Although classified as a wild species, M. boisiana holds important economic significance and potential value for the genetic improvement of sweet potatoes. However, current research on M. boisiana remains relatively scarce, lacking corresponding genomic studies and genetic information.
This study unveils the genomic characteristics of M boisiana, enriching the genomic research of the Convolvulaceae family and providing a valuable reference for constructing a more comprehensive evolutionary framework for sweet potatoes and their relatives. I. batatas is a vital food and economic crop, yet there remains room for improvement in growth rate, stress resistance, and root system development. Given M. boisiana's exceptional performance in these aspects, its genomic data can serve as a valuable resource for identifying key genes involved in root development, disease resistance, and growth regulation. Notably, the root development-related genes in M. boisiana may provide novel genetic resources for enhancing sweet potatoes' nutrient and water uptake efficiency, ultimately improving yield and adaptability. The intricate interplay of plant hormones within M. boisiana reveals a highly optimized regulatory network that governs its rapid growth and environmental adaptability. These findings not only deepen our understanding of hormone crosstalk in plant growth and adaptation but also offer new strategies for genetic improvement in sweet potatoes. Furthermore, this study elucidates whole-genome triplication (WGT) and chromosomal rearrangements in Convolvulaceae, shedding light on how plants in this family have leveraged gene expansion and neofunctionalization to enhance growth potential and stress tolerance. From an evolutionary perspective, these insights can guide researchers in optimizing sweet potato genome design to enhance its environmental resilience and disease resistance.
In the future, by utilizing multi-omics approaches, we are committed to constructing a more comprehensive and in-depth genetic research framework[60,61]. Our objective is to achieve a more mature and thorough understanding of the genetic characteristics of Convolvulaceae plants. This effort will not only enrich our knowledge of these fascinating plants but also will contribute significantly to the sustainable development of agriculture and the conservation of ecosystems.
-
Studies have shown that hormone biosynthesis pathways play a crucial role in the rapid growth and strong stress tolerance of M. boisiana. Among the 110 identified related genes, CM and PAL genes exhibit high expression levels across all organs, highlighting the essential role of SA synthesis in the plant's growth and adaptability. Compared to other plants, M. boisiana demonstrates a significant advantage in genes related to the synthesis of IAA and SA. Notably, key genes such as YUC10, YUC11, EPS1, and OPR2 contribute to its development and the coordination of hormone signaling networks through gene expansion and specific expression, endowing M. boisiana with exceptional growth rates and stress resilience.
This work was supported by the startup funds for the double first-class disciplines of crop science in Hainan University (RZ2100003362), the National Natural Science Foundation of China (32172614), Hainan Province Science and Technology Special Fund (ZDYF2023XDNY050), Hainan Provincial Natural Science Foundation of China (324RC452), and the Project of National Key Laboratory for Tropical Crop Breeding (No. NKLTCB202337). We thank the editor and anonymous reviewers for their insightful comments and suggestions.
-
The authors confirm contribution to the paper as follows: study conception and design, project leading: Wang W, Chen F; plant materials collection: Guo G; data analyses: Guo G, Garcia-Caparross P, Zhang J, Zhang J, He S, Li Y, Xue J. All authors reviewed the results and approved the final version of the manuscript.
-
The raw data of genome sequencing, transcriptome raw reads, as well as genome assembly, genome annotation files, have been submitted to the database National Genomics Data Center (PRJCA020455 and PRJCA032116).
-
The authors declare that they have no conflict of interest.
-
accompanies this paper at (https://www.maxapress.com/article/doi/10.48130/tp-0025-0007)
-
Received 10 December 2024; Accepted 21 February 2025; Published online 24 March 2025
-
A high-quality M. boisiana reference genome was constructed through a combination of Nanopore sequencing.
M. boisiana and 61 other plant species were used to analyze the evolutionary relationships, divergence and gene family expansion and contraction.
Integrated transcriptome analysis to investigate the role of plant hormones in the invasion mechanism of M. boisiana.
- Supplementary Fig. S1 The Sequencing flowchart of M. boisiana.
- Supplementary Fig. S2 The chloroplast genome of M. boisiana.
- Supplementary Fig. S3 Genes specifically associated with the synthesis of IAA.
- Supplementary Fig. S4 Genes specifically associated with the synthesis of JA.
- Supplementary Fig. S5 Genes specifically associated with the synthesis of ABA.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Guo G, García-Caparros P, Zhang J, Zhang J, Li Y, et al. 2025. Chromosomal reference genome of Merremia boisiana: unveiling the secrets of the tropical rainforest's killer plant. Tropical Plants 4: e012 doi: 10.48130/tp-0025-0007 

Chromosomal reference genome of Merremia boisiana: unveiling the secrets of the tropical rainforest's killer plant
- Received: 10 December 2024
- Revised: 20 February 2025
- Accepted: 21 February 2025
- Published online: 24 March 2025
Abstract: Merremia boisiana, a captivating species endemic to tropical rainforest habitats, belongs to the esteemed Convolvulaceae family. Renowned for its dazzling golden flowers and exceptional growth rate. This plant rapidly expands, covering other vegetation, suppressing the growth of native species, and altering light availability and nutrient distribution within the forest, thereby impacting ecological balance. Here, we report the first high-quality M. boisiana genome assembly, comprising 510 Mb with a contig N50 of 21 Mb and an assembly completeness of 98.7%. This assembly includes the identification of 15 chromosomes and the annotation of 37,389 protein-coding genes, with a high annotation rate of 99.2%. By integrating genomic data from other Convolvulaceae species, we analyzed the karyotype evolution of M. boisiana and uncovered the fundamental ploidy level of the Convolvulaceae family. Based on existing research, we identified 110 highly expressed genes involved in the biosynthesis of SA, IAA, JA, and ABA, all of which play essential roles in plant growth. The EPS1 transcription factor, involved in SA synthesis, along with YUC11 and TIR2, which participate in auxin biosynthesis, and OPR2, associated with ABA biosynthesis, collectively contribute to the enhanced root growth of M. boisiana through mechanisms such as gene expansion, gene dosage, and root-specific expression. This study not only sheds light on the genetic complexity of M. boisiana but also provides a promising direction for improving stress resistance in sweet potatoes and advancing ecological research. Our findings promote the sustainable utilization of this species while broadening our understanding of tropical plant genomics.
-
Key words:
- Rain forest killer /
- Tropical plant /
- Reference genome /
- Phytohormone crosstalk