









-
Lagerstroemia indica is among the most widely used ornamental species within the Lagerstroemia genus (Lythraceae), valued for its vibrant summer blossoms, extended flowering season, striking colors, smooth bark, and pollution resistance[1]. However, while L. indica flowers for a prolonged season, each individual bloom lasts only 1−2 d from opening to peak display, making the flower's ornamental period brief[2]. Additionally, although it blooms throughout summer, L. indica has generally passed its peak display by China's National Day, limiting its potential as a showpiece for that occasion. Therefore, prolonging the flowering period of L. indica and improving its ornamental and aesthetic values have become one of the important objectives of the study.
Petal expansion is a key process in the life cycle of flowering plants, involving cell expansion and cell wall relaxation[3]. Many studies have shown that cell wall relaxation is one of the central mechanisms of petal expansion, and expansins play an important role in this process[4]. Expansins promote cell wall expansion by relaxing cellulose-half-cellulose complexes in the cell wall. The expansin gene family in plants is usually categorized into four subfamilies: EXPA (α-Expansins), EXPB (β-Expansins), EXLA (Expansin class A), and EXLB (Expansin class B)[5]. The EXPA gene family has been shown to play an important role in petal expansion in several plant species[6]. Although there have been studies on the EXP gene family in several species, there is still a lack of research on the molecular mechanism of petal expansion and related genes in L. indica. In particular, it is not clear which genes are closely related to cell wall relaxation and petal expansion in L. indica. Therefore, it is of great significance to study the EXP gene family of L. indica and reveal its role in petal expansion to prolong the flowering period and enhance the ornamental value.
Current research on flower opening across different species indicates that this process is influenced by a combination of carbohydrate metabolism, cell wall expansion, water transport, and hormonal regulation[7]. Studies have shown that flower opening is often accompanied by carbohydrate metabolism, including starch and polysaccharide breakdown, suggesting that sugar absorption and polysaccharide degradation play critical roles in petal expansion[8]. In support of this, detached cut flowers often fail to open without an external sugar source, underscoring the importance of carbohydrate availability[9]. Research on species such as Gladiolus gandavensis, Antirrhinum majus, and Eustoma grandiflorum further reveals that supplementing with glucose or sucrose can promote flower opening, reinforcing the role of carbohydrates in this process[10−12].
Water availability also plays a crucial role in flower opening, as water uptake into buds increases cell turgor and drives petal expansion, even under conditions where leaves may appear wilted[13]. Hormones such as ethylene, auxin, and gibberellin are also fundamental regulators in flower opening, influencing petal expansion through coordinated hormonal signaling[14]. Additionally, the loosening of cell walls-mediated by proteins such as expansins and enzymes like xyloglucan endotransglycosidase/hydrolase (XTH) - is essential for petal cell expansion[15].
The aim of this study was to systematically excavate the role of the EXP gene family during petal expansion in L. indica through transcriptome analysis. Transcriptome sequencing was first performed on L. indica flower buds and petals at four flowering stages to identify genes that were differentially expressed during petal expansion. Through functional validation, especially the expression patterns of LiEXLA1 and LiEXLA2 genes and their potential roles in petal expansion, this study provides new perspectives on the molecular mechanisms of petal expansion in L. indica. In addition, the regulatory role of hormone signaling pathways in petal expansion was explored, revealing how hormones such as growth hormone and gibberellin synergistically regulate the role of the EXP gene family in petal expansion.
-
Materials were obtained from robust and uniformly growing L. indica 'Lan Zi' variety from Zhejiang Agricultural Forestry University, Zhejiang Province, China. Observations and collections were made in early July 2022 during the growth of new shoots. Flowering is classified according to morphological characteristics and time course. Stage 3 has petals that are not yet fully expanded, but the size and shape of the petals are basically the same; therefore, it is divided into four stages: tight bud closure (S1), bud expansion (S2), first bloom (S3), and full bloom (S4). Buds or flowers were collected from each of the four periods (Fig. 1). Each sample was repeated three times and frozen in liquid nitrogen. RNA was extracted from these samples using TRIzol reagent (TaKaRa Inc., Dalian, China), and the RNA samples were detected using Nano Drop and Agilent 2100 Bioanalyzer (Thermo Fisher Scientific, Waltham, USA).
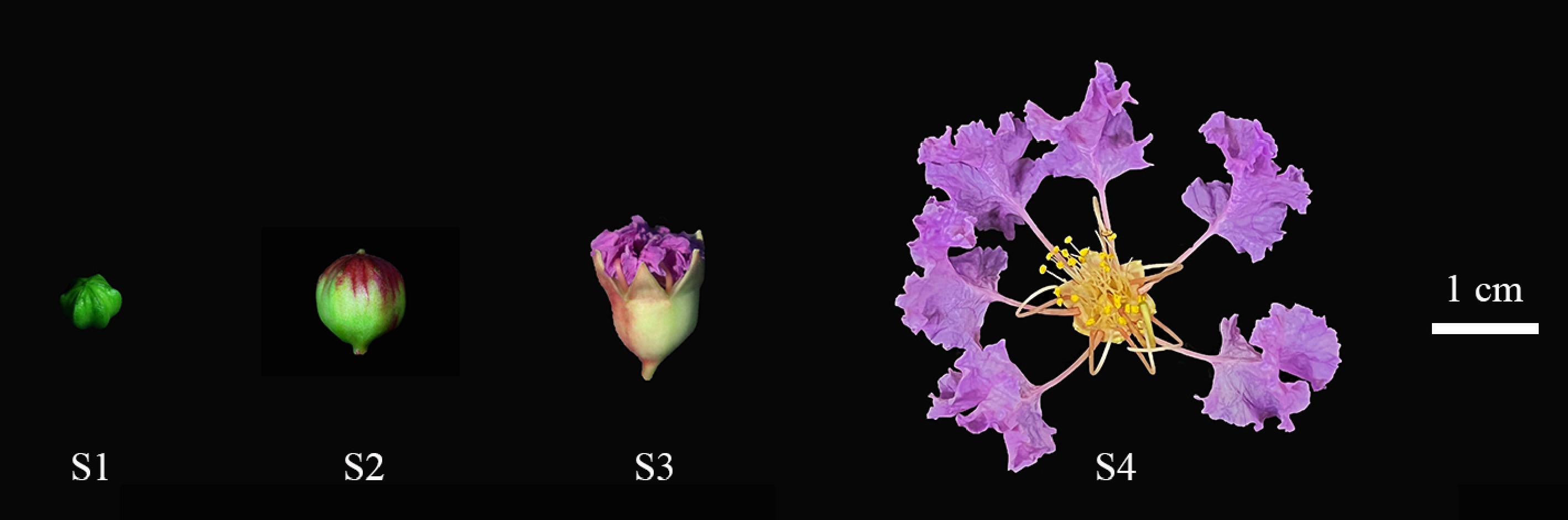
Figure 1.
Four stages of L. indica flower opening. (S1) When the bud is closed and the petals do not show color. (S2) Petals begin to show color. (S3) Petals gradually split, petals and sepals gradually spread, stigma elongated. (S4) Petals and sepals fully extended, anthers tightly closed, stigma green and mucous.
cDNA library construction and sequencing
-
After total RNA was extracted from L. indica buds or flowers, the sampling time point consisted of four phases with three biological replicates per phase. A cDNA library was constructed and sequenced from 12 samples. Total RNA was treated by either mRNA enrichment or rRNA removal. The obtained RNA is then fragmented by interrupting the buffer. A cDNA strand was synthesized using the interrupted mRNA as a template, and then the double-stranded cDNA was synthesized by configuring a double-stranded synthesis reaction system. The cDNA fragment obtained in the previous step was amplified by PCR. The PCR product was heated and denatured into a single strand, and then the single strand DNA was cycled with a bridge primer to obtain a single-strand circular DNA library. Finally, the RNA was sequenced after identification. Sequencing was performed using the DNBSEQ sequencing platform with a sequencing depth of 30X to ensure that each sample had sufficient coverage to detect low-abundance transcripts[16]. Sample data were quality controlled by SOAPnuke and assembled using Trinity software[17]. Transcriptome data filtering was based on the following criteria: an error rate below 0.01 and a quality score Q30 greater than 90%[18]. Data normalization between samples was performed using the TPM method to ensure consistency of gene expression[19]. Biological replicates (n = 3) were assessed for Differences among treatments were tested using the ANOVA method and followed by the Duncan test for the difference test between average treatment pairs with adjusted p value[1].
De novo assembly, functional annotation, and classification
-
SOAPnuke and Trinity software were used for filtering and assembly, and Tgicl was used to cluster transcripts to remove redundancy to obtain Unigene. The assembled Unigene was annotated by seven functional databases (KEGG, GO, NR, NT, SwissProt, Pfam, and KOG) to obtain the protein functional annotation and metabolic pathway annotation of Unigene. Blastx annotated the uniform gene in NR, KOG, KEGG, and SwissProt. Blastn was used to annotate genes in NT, and Blastx was used to annotate uniform genes in NR, KOG, KEGG, and Swiss-Prot. Blast2Go and NR were used to annotate GO, and InterProScan5 was used to annotate InterPro.
Analysis of differentially expressed genes (DEGs)
-
Clean reads and unigenes were compared using Bowtie2, and RSEM was used to determine the gene expression levels of each sample. Differential gene expression level (FPKM) provided a measure of the expression abundance of unigenes. In DEG analysis, all gene expression differences were corrected for false discovery rate (FDR) (Benjamini-Hochberg method) to control for multiple testing errors. The criteria for genes to be considered significantly differentially expressed (DEGs) were |log2FC| > 1 and p-value < 0.05. DEGs underwent GO function categorization and KEGG pathway enrichment analysis. The significance threshold for the GO and KEGG pathways was p-value < 0.05.
Expression analysis of LiEXP gene in L. indica vulgare blooms
-
Total RNA was extracted from L. indica sinensis, and cDNA synthesis was performed using HiScript® III All-in-One RT SuperMix (Vazyme, Nanjing, China). Primers for differentially expressed genes associated with the flowering process were designed by Primer 5.0 software, and Li18S (Accession: MG704137) was used as a reference gene[20]. The primer sequences of the candidate genes are shown in Supplementary Table S1, and were synthesized by Hangzhou Youkang Biotechnology Co. (Hangzhou, China). RT-qPCR was performed using an ABI 7300 real-time PCR instrument (Applied Biosystems, Foster City, CA, USA) and SYBR® Premix ExTaq™ (TaKaRa, Dalian, China), and the experiment consisted of three replicates, and the data were analyzed by the 2−ΔΔCᴛ method. To analyze the expression pattern of the LiEXP gene during flowering, bud and flower samples were collected at four flowering stages (S1, S2, S3, and S4), and the samples were rapidly frozen in liquid nitrogen and stored at −80 °C. RNA extraction and cDNA synthesis were carried out as described above.
Identification and physicochemical properties analysis of LiEXP gene family
-
Based on the hidden Markov model of the Pfam database (
http://pfam.xfam.org/ ), the transcriptome was searched for genes containing the DPBB_1 domain (PF03330) and the Pollen_allerg_1 domain (PF01357). The HMMER program was used to screen out the candidate genes with high confidence. At the same time, from the TAIR database (www.arabidopsis.org/index.jsp ) to download Arabidopsis thaliana EXP protein sequences, using Blast analysis tool executables (http://ncbi.nlm.nih.gov/blast/ ), A. thaliana proteins will be collected with the transcriptome sequences compared, and according to the threshold conditions for E-value < 1×10−10 to credibility, higher genetic screening. Finally, the intersection of the identification results of the two methods was used as the final EXPs candidate gene of L. indica. And through the batch of NCBI Web CD-Search Tool (www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi ), validation of candidate genes of conservative domain structure using online tools such as ExPASy ProtParam (https://web.expasy.org/protparam/ ) to calculate LiEXP family proteins, amino acids, isoelectric point, and the basic parameters such as molecular weight and hydrophobic Plant-mPloc uses an online site (www.csbio.sjtu.edu.cn/bioinf/plant-multi/# ) for LiEXP protein subcellular localization prediction.Analysis of conserved domain and motif of the LiEXP gene family
-
The Batch Web CD-Search Tool was used to search the sequential conservative domain, and then TBtools (Chen, C.J.; Guangzhou, China) software was used for graphical display. The conserved motifs of EXP gene family protein sequences of L. indica were identified by MEME (
https://meme-suite.org/meme/ ) and visualized by TBtools.Multiple sequence alignment and phylogenetic analysis of LiEXP
-
The amino acid sequences of 27 LiEXP genes were compared by Jalview. By comparing the gene sequences with those of A. thaliana, Nicotiana tabacum, and Oryza sativa, as well as those related to flower opening in O. fragrans, R. hybrida, D. caryophyllus, G. gandavensis, and C. praecox, the gene sequences were classified into four subfamilies. Phylogenetic tree construction was performed by the maximum likelihood (ML) method with MEGA X software. We used the WAG evolutionary model and the 1,000 times self-help method (Bootstrap) for stability verification of the tree. Sequence alignment was performed using the ClustalW algorithm to ensure the accuracy of sequence alignment.
Gene cloning and identification of transgenic plants
-
Using the full-length sequences of the known genes LiEXLA1 and LiEXLA2, Takara, through its online website (
https://www.takarabio.com/learning-centers/cloning/primer-design-and-other-tools ), designed and synthesized specific primers from Hangzhou Youkang Biological Technology Co., Ltd. (Hangzhou, China). The region involved in the primer contains the complete ORF sequence and is used to clone the full length of the target gene. The gene sequence was constructed into the pORE_R4 vector by homologous recombination, and A. thaliana was transformed by flower impregnation. The T3 seeds were seeded, and a number of wild A. thaliana were planted as controls. The plants growing for 45 d were photographed, and data such as stem diameter (2 cm away from the stem base), plant height, and petal length were recorded. -
Separate transcriptomes were obtained from four phases of L. indica bloom, and three biological replicates were performed in each phase, for a total of 12 samples. A total of 77.09 GB of raw data was obtained during transcriptome sequencing. The original data is uploaded to NCBI with the accession PRJNA970535. After the sequence splicing and de-redundancy processing, 90,717 Unigene sequences were obtained in the transcriptome data, respectively. For the length distribution, see Supplementary Fig. S1. After excluding unqualified reads from the original data, the proportion of filtered reads was higher than 79.17%, and the Q20 value was higher than 96.91%. On the whole, the proportion of bases with low quality < 20 was low, indicating that the sequencing quality was good (Supplementary Table S2). High-quality data from 12 samples was reassembled using Trinaty software, and 90,717 single genes were obtained. The total length of a single gene was 143,564,327 bp, and the average length was 1,582 bp. The length of N50 is 2,160 bp, the length of N90 is 876 bp, and the number of GC is 47.12% to 48.12% (Supplementary Table S3).
Gene functional annotation
-
By comparing Unigene sequences with the NR, Swiss-Prot, GO, COG, KOG, eggNOG, and KEGG databases, 79,423 annotated Unigene sequences were obtained, accounting for 87.55% of the total number. Supplementary Table S4 presented statistical results for genetic annotations, which amounted to 77,590 (NR: 85.53%), 66,981 (NT: 73.84%), 62,000 (Swiss Prot: 68.34%), 63,065 (KOG: 69.52%), 61,459 (KEGG: 67.75%), 61,136 (GO: 67.39%), and 63,095 (Pfam: 69.55%). Unigenes were annotated, and 37,711 (41.57%) unigenes were annotated simultaneously by all databases. Figure 2a shows the Venn diagram of the number of unigenes annotated by KEGG, GO, NR, SwissProt, and KOG. The unigenes sequence was compared with the NR database, and corresponding functional annotations were obtained. According to the NR functional annotation results, the proportion of different species on the unigenes annotation was counted, and the species distribution map was drawn (Fig. 2b). The unigenes of the L. indica samples were highly similar to those of five plants. They were Punica granatum (83.88%), Syzygium oleosum (0.96%), Eucalyptus grandis (0.95%), Corymbia citriodora subsp. variegata (0.93%), and Rhodamnia argentea (0.87%). Unigenes were annotated in the KOG database and compared with 25 functional groups in the KOG database for functional classification statistics (Fig. 2c). Among them, the maximum number of unigenes in the category of 'General function prediction only' is 11,720. Followed by 8,849 types of 'signal transduction mechanisms' and 5,459 types of 'function unknown'. The number of unigenes added to the 'Cell motility' category is at least seven.
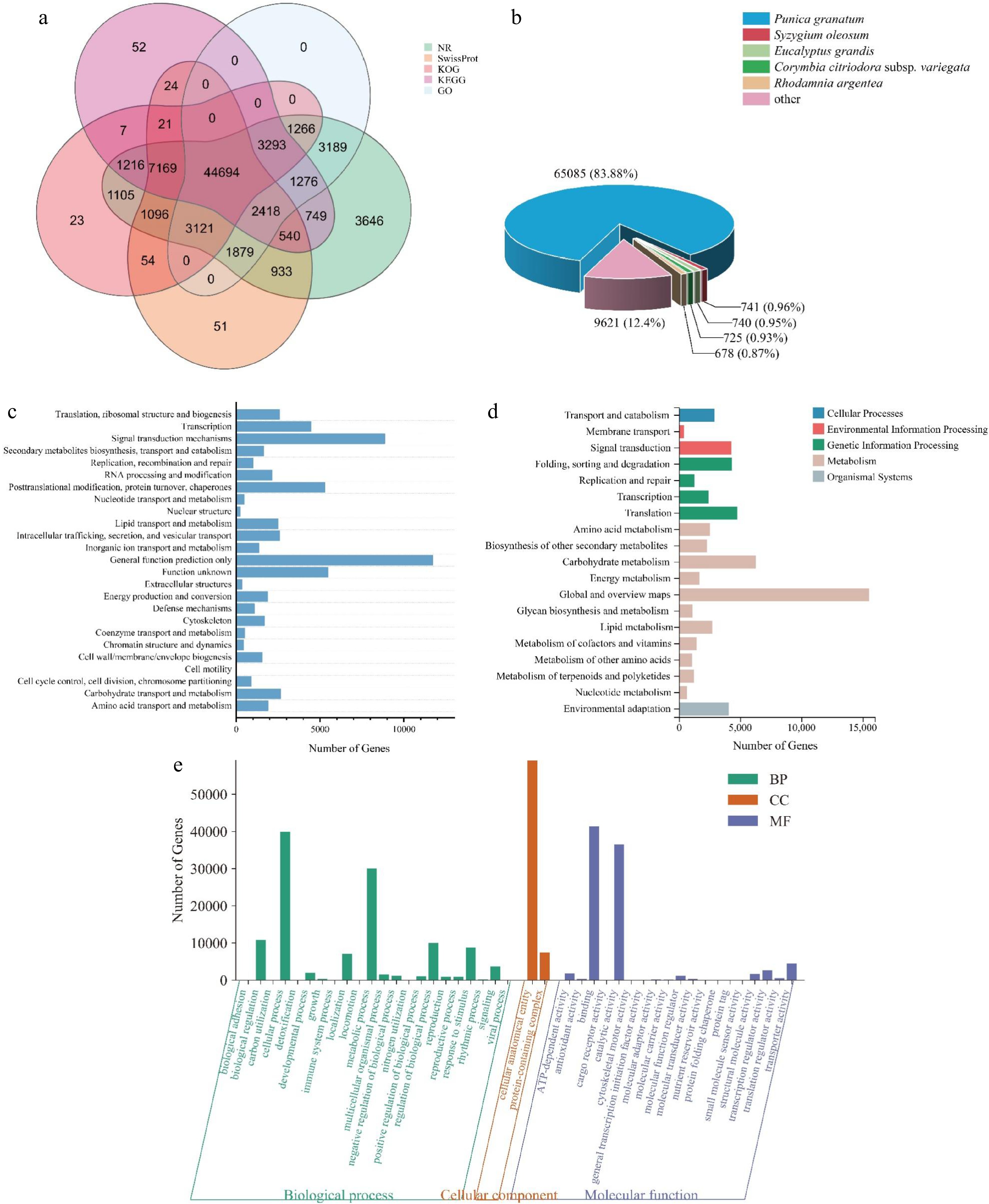
Figure 2.
Gene functional annotation. (a) Venn diagram of the number of unigenes annotated in different public databases. (b) Species distribution of NR annotation. (c) Functional classification of KOG annotation. The x-axis represents the corresponding number of unigenes, the y-axis represents the KOG function classification name. (d) Functional classification and pathway unigenes assembled by KEGG. (e) GO enrichment with three primary classifications of biological process, cellular component, and molecular function.
KEGG annotation results (Fig. 2d) showed that the most representative metabolic pathways were global and overview maps (15,540, 25.46%). The main metabolic pathways include carbohydrate metabolism (6,291, 10.31%), lipid metabolism (2,720, 4.46%), and amino acid metabolism (2,548, 4.17%). Blast2GO software was used to analyze GO enrichment (Fig. 2e). GO functions were mainly enriched in three aspects: biological processes, cell components, and molecular functions. The GO functional distribution involved 43 functional subclasses, and biological processes included 22 functional subclasses. The cellular process and metabolic process had the most functions, with 39,989 (14.43%) and 30,068 (10.85%), respectively. Among them, the GO function subclasses in response to flower opening are mainly related to cellular processes, catalytic activity, metabolic processes, and biological regulation.
Analysis of differentially expressed genes
-
In this study, three stages were used to represent the opening process of L. indica. S1 vs S2 represents the process by which the green petal is driven primarily by cell division until pigment synthesis begins. S2 vs S3 represents the process by which petal cells grow slowly and their pigment content increases rapidly until maximum pigmentation is achieved. S3 vs S4 represents the process of petal growth, mainly through cell expansion to bloom. For the three phases, 41,639, 33,396, and 34,079 DEGs were obtained, respectively (Fig. 3a; Supplementary Table S5). Between S1 and S2, 13,330 and 12,697 transcripts were up-regulated and down-regulated, respectively. In comparison with S3 and S2, 9,388 and 8,814 transcripts were up-regulated and down-regulated, respectively (Fig. 3b). There were fewer up-regulated transcripts (8,417) than down-regulated transcripts (11,424) in the S3 vs S4 comparison. This indicated that the number of DEGs decreased and the number of up-regulated genes decreased during flower opening.
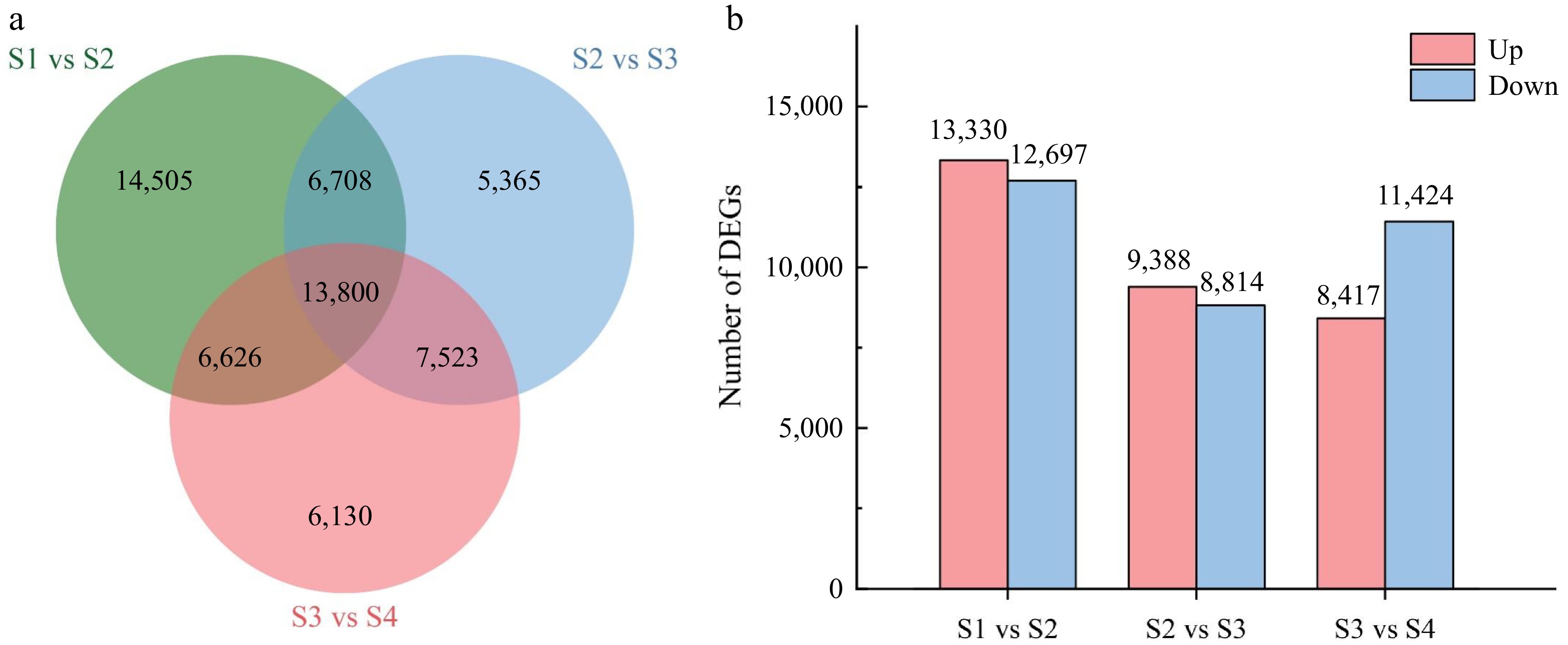
Figure 3.
Comparative analysis of differentially expressed genes (DEGs) between the four stages of flower opening. (a) Venn diagram of the number of DEGs between the stage comparisons. (b) The number of up-regulated and down-regulated DEGs between the three comparisons.
The GO database was used to analyze the three comparison DEGs, and all DEGs were annotated with GO. It is divided into three functional categories: molecular function, cell component, and biological process. Fourteen biological processes (Fig. 4a) were enriched in DEGs. Of these, six terms are enriched in each comparison. Moreover, genes in the group of proton export across the plasma membrane were enriched in all comparison groups. The 'pectin catabolic process' group was enriched in S2 vs S3 and S3 vs S4. Interestingly, the 'cell component' category showed significant changes across all analysis groups. Moreover, DEGs associated with the cell wall were significantly enriched when flowers opened. These data suggest that complex gene regulatory mechanisms underlie petal development and that cell wall dilation plays a potentially important role in L. indica flower opening.
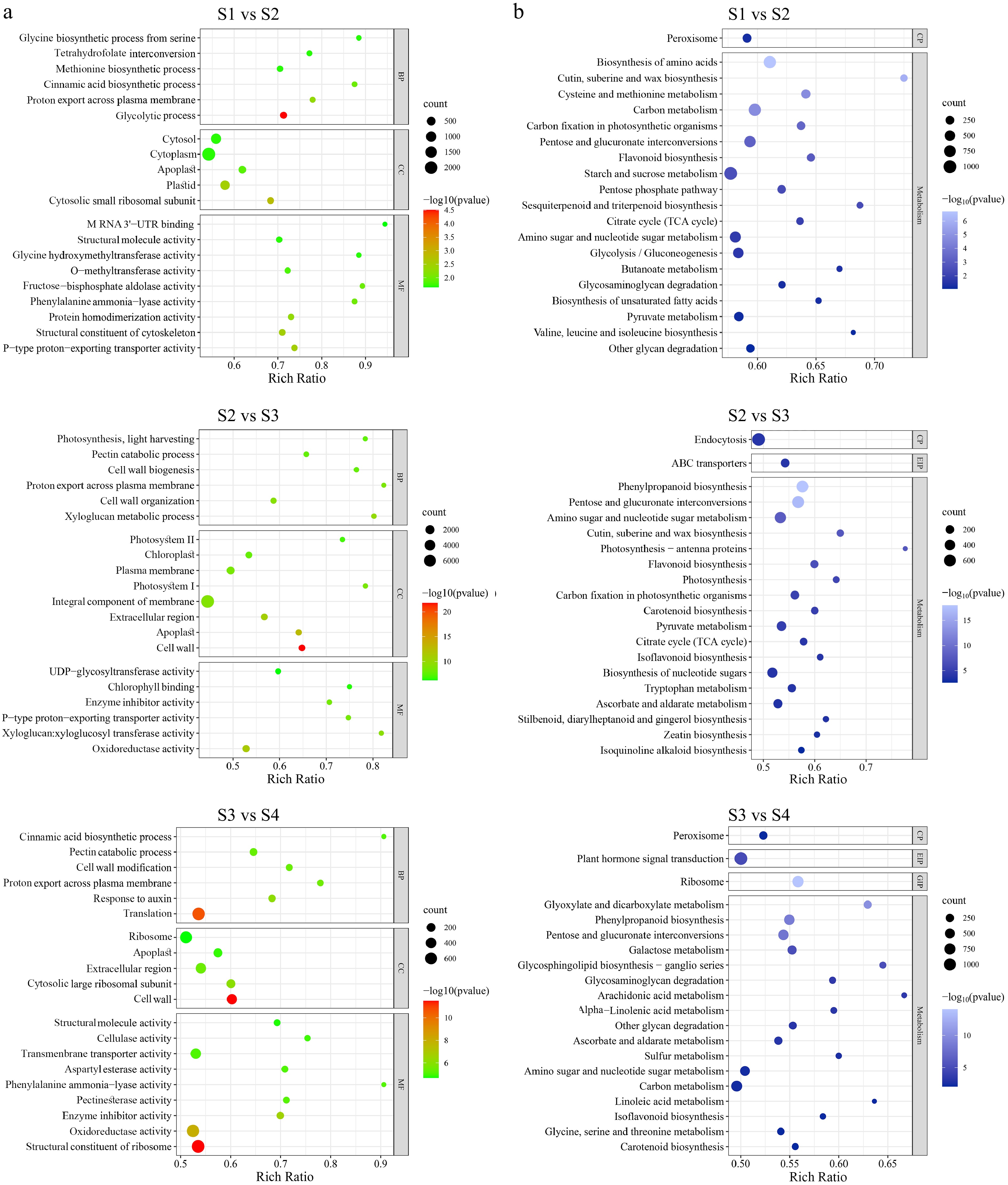
Figure 4.
Enrichment analysis of differentially expressed genes (DEGs) in four stages of flower opening. (a) GO enrichment with DEGs between different flower opening stages. BP, CC, and MC represent biological process, cellular component and molecular function respectively. (b) KEGG pathway enrichment with DEGs between different flower opening stages. CP, EIP, and GIP respectively stand for cellular processes, environmental information processing, and genetic information processing. The number of genes in each category is equal to the dot size. The color of the dot represents the p-value.
The KEGG Homology Annotation System (KOBAS) was used for further functional classification and pathway assignment of the three comparison DEGs. Of the seven official classifications of the KEGG metabolic pathway, the three stages of L. indica samples were annotated to the four branches: cellular processes, environmental information processing, genetic information processing, and metabolism. The enrichment bubble diagram showed the enrichment of the KEGG pathway in three dimensions, and Fig. 4b showed the first 20 GO terms with the lowest p-value. The KEGG pathway enrichment results of differential genes showed that among the top 20 metabolic pathways with the lowest p-value, two metabolic pathways were co-enriched. Are 'pentose and glucuronate interconversions' and 'amino sugar and nucleotide sugar metabolism'. All of them were significantly enriched (p-value < 0.05).
Transcriptome analysis of key genes during flower opening
-
In order to explore the key genes regulating the opening process of the L. indica flower and the molecular mechanism of flower opening regulation, the common DEGs in the four stages were analyzed in terms of carbohydrate, hormone pathway, water, and cell wall expansion.
Plant growth is regulated by hormones such as auxin (AUX), abscisic acid, cytokine (CK), gibberellin (GA), and ethylene (ETH)[14,21]. Exogenous AUX, ABA, GA, and ETH affected petal expansion and opening. Supplementary Table S6 shows the single genes involved in the known plant hormone biosynthesis and signaling pathways for L. indica flower opening, and the DEGs in the flower opening process are shown in Fig. 5. Single genes that have been annotated as participating in GA biosynthesis or signaling pathways are also differentially expressed during flower opening. The expression of gibberellin receptors (GID: CL3355.Contig1_All, CL4650.Contig4_All, and Unigene24568_All) in the GA pathway increased continuously during flower opening. The results showed that GA played a role in the opening process of L. indica (Fig. 5a). Many ABA biosynthesis genes were up-regulated during flower opening (Fig. 5b). In the auxin signaling pathway, the expression of most genes increases with flower opening. For example, AUX1, AUX/IAA, SAUR, and ARF-related genes (Fig. 5c). The results indicated that the balance of auxin biosynthesis and catabolism was strictly controlled during the opening process of L. indica. The expression of many single genes in the CK biosynthetic pathway is down-regulated in flower opening, such as AHK, ARR-B, ARR-A, and CKX. In addition, transcriptome-level changes in other important plant hormones occur during flower opening.
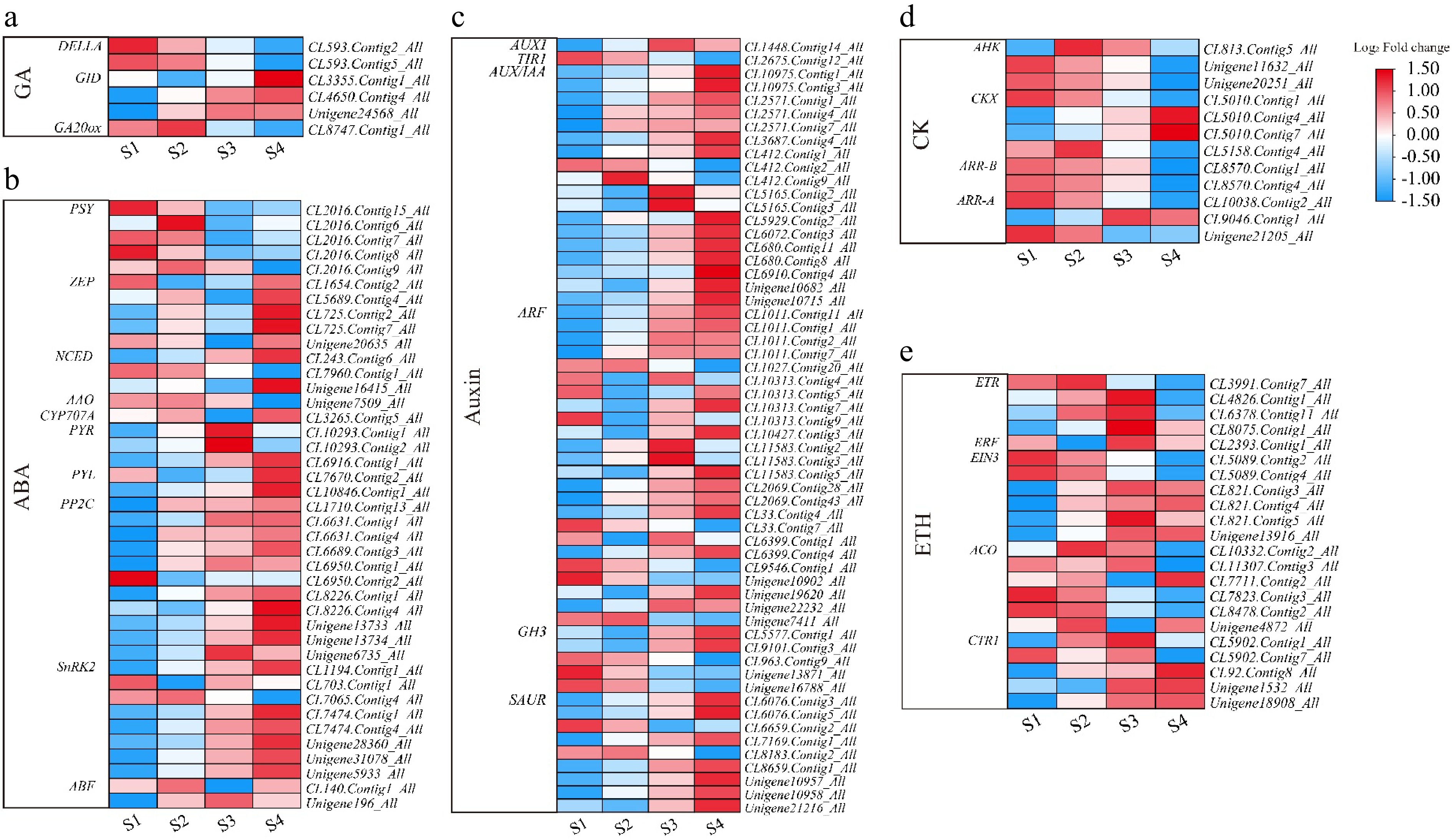
Figure 5.
Heatmap of plant hormone-related genes during flowering of L. indica. Heatmap of major genes involved in the biosynthesis and signaling pathways of (a) gibberellic acid, (b) abscisic acid, (c) auxin, (d) cytokinin, and (e) ethylene. After homogenization, the value of expression appears in the heatmap, with red representing high expression and blue representing low expression.
During flower opening, cell wall expansion factors regulate cell wall expansion to promote the expansion of petal cells[22]. According to research reports, xyloglucan endotransglycosylase/hydrolase (XTH) is a kind of cell wall relaxation factor. It is involved in the formation and opening of petals by catalyzing the cleavage and reconnection of xyloglucan molecules, thereby changing the structure of cell walls[23]. During the opening process of L. indica, the expression of most XTH family genes gradually increased and reached its highest value in the S4 period (Fig. 6, Supplementary Table S7). This indicates that the XTH gene is involved in flower opening and plays an important role in the opening of L. indica. In addition, the expansin (EXP) gene can also promote cell growth and petal elongation[24]. Among the differentially expressed genes, Unigene22997_All and Unigene4839_All are up-regulated, but Unigene6210_All and Unigene23132_All are down-regulated. It is speculated that different EXP proteins have different regulatory effects. Most genes in the XYL gene family were downregulated, suggesting that the XYL gene may regulate the growth and development of other plants. In addition, the expression of genes involved in cell wall synthesis, modification, or hydrolysis, such as cellulose synthetase (CES), pectinesterase (PE), polygalacturonase (PG), and pectin lyase (PL), was assessed, as these genes may be involved in the process of L. indica blossom opening (Fig. 6; Supplementary Table S7).
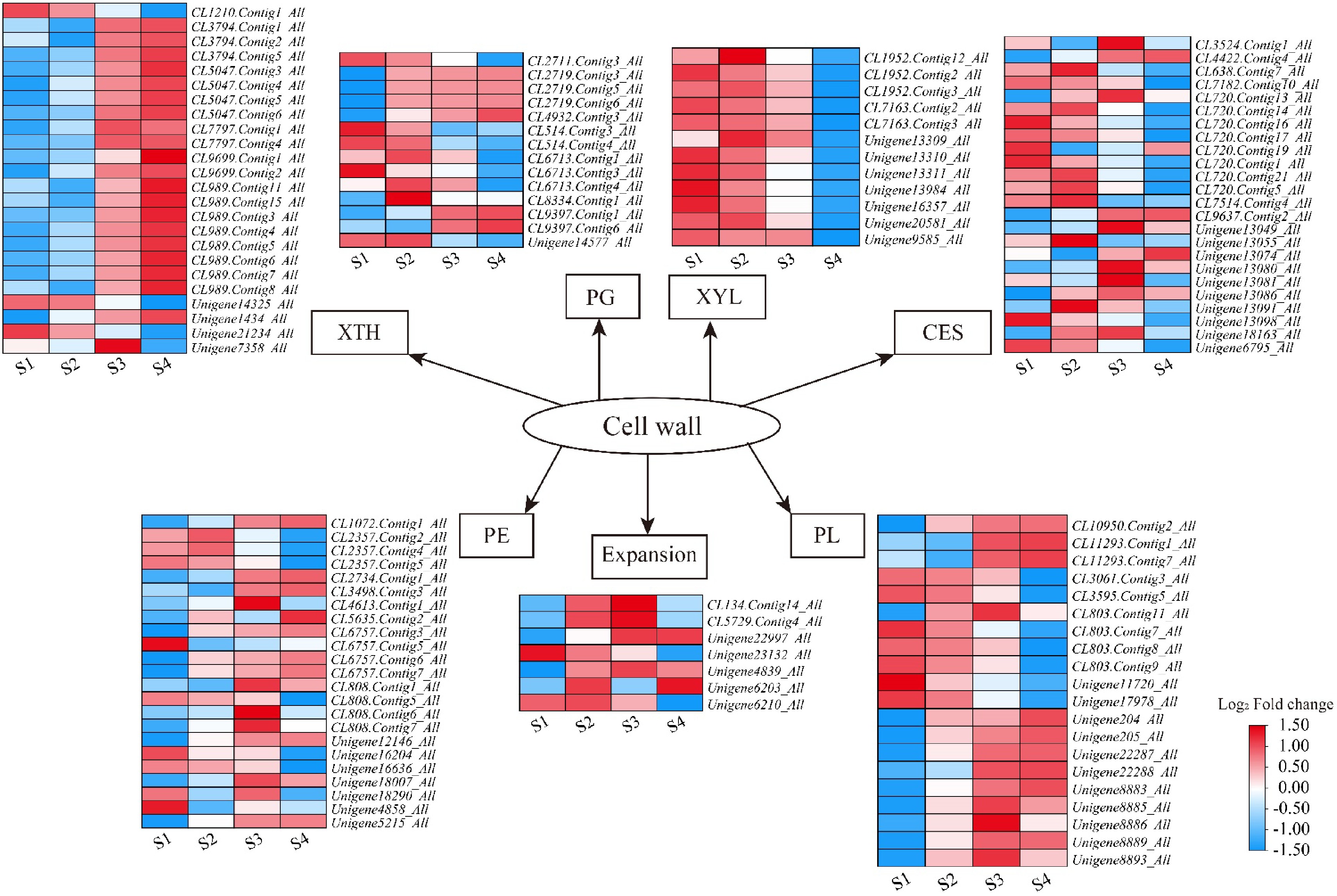
Figure 6.
Heatmaps of petal expansion-related genes in L. indica during flower opening. XTH: xyloglucan endotransglucosylase/hydrolase; PG: polygalacturonase; XYL: xylosidase; CES: cellulose synthase; PE: pectinesterase; PL: pectate lyase. After homogenization, the value of expression appears in the heatmap, with red representing high expression and blue representing low expression.
Aquaporins (AQPs) are the main channels mediating water transmembrane transport in plants, which can promote water transmembrane diffusion and osmotic pressure formation. It is widely found in plant cell membranes and plays a key role in plant growth[25]. A total of 28 aquaporins were annotated by the transcriptome. There were 12 plasma membrane intrinsic proteins (PIPs), nine vacuolar in vivo proteins (TIPs), and seven nodule intrinsic membrane proteins (NIPs) (Fig. 7; Supplementary Table S8). The expression levels of most PIPs, TIPs, and one NIP (Unigene16644_All) continued to increase during the flower opening process. In the process of flower opening, the expression of most NIPs reaches its maximum in the S2 and S3 stages. Carbohydrates provide energy and the foundation for plant growth and development and regulate plant growth and development. Figure 4b shows the metabolic pathway of common enrichment in L. indica samples at four periods. Among them, 11 DEGs were involved in the 'Pentose and glucuronate interconversions' with elevated expression, and eight were continuously downregulated (Fig. 7; Supplementary Table S8). Seven DEGs were up-regulated and seven were down-regulated in the 'Amino sugar and nucleotide sugar metabolism' pathways.
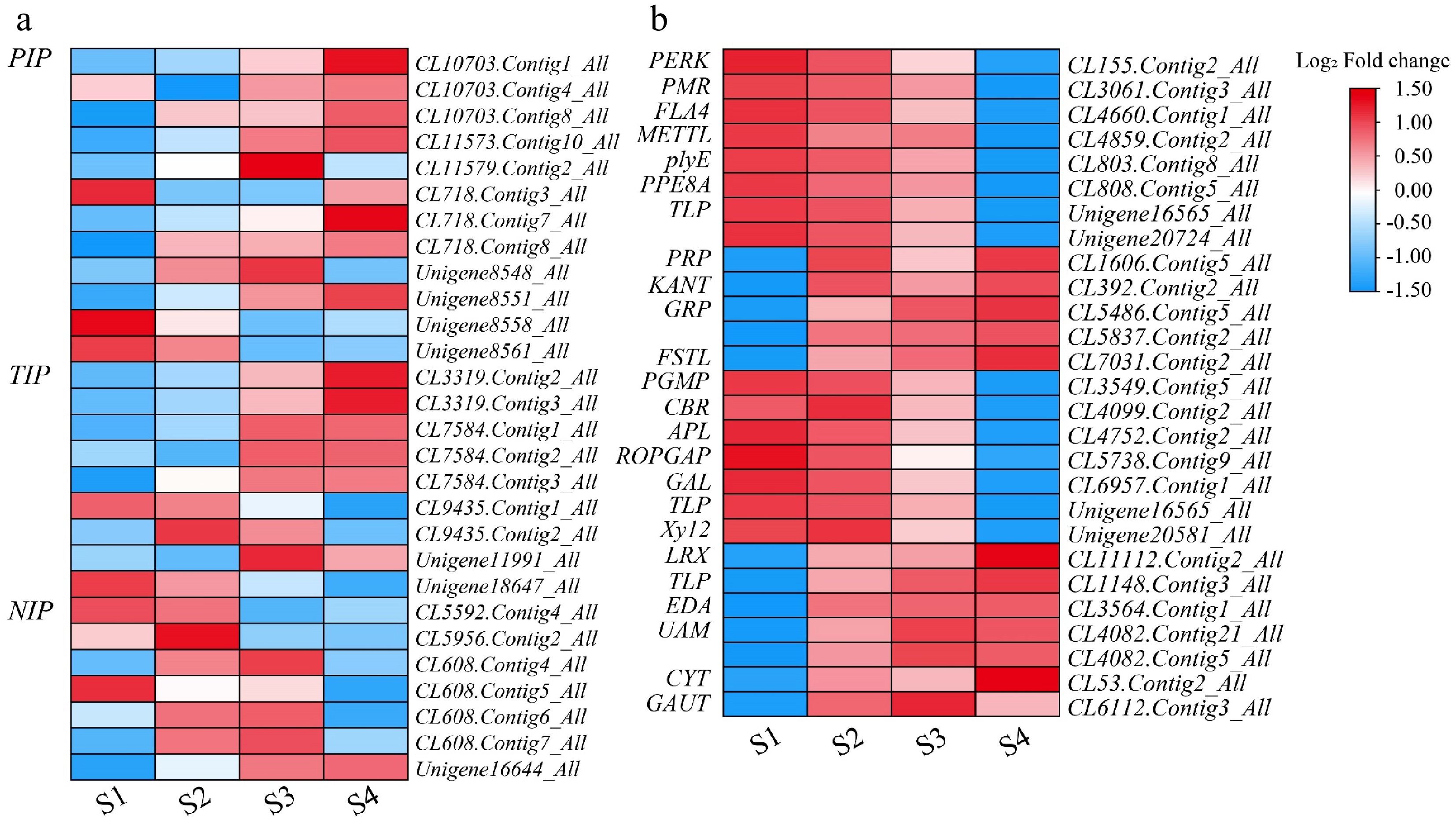
Figure 7.
Heatmap of genes related to water and carbohydrate during flower opening in L. indica. Heatmaps of major genes involved in (a) water and (b) carbohydrates After homogenization, the value of expression appears in the heatmap, with red representing high expression and blue representing low expression.
Verification of transcriptome reliability by RT-qPCR
-
In order to test the authenticity of the transcriptome sequencing results, 16 candidate genes were randomly selected from the key genes in Supplementary Table S1 for RT-qPCR validation. They are AUX/IAA (CL3687.Contig4_All), ARF (CL33.Contig4_All), GH3 (CL5577.Contig1_All), ETR (CL8075.Contig1_All), AHK (Unigene20251_All), CKX (CL5010.Contig7_All), DELLA (CL593.Contig5_All), Expansin (CL2478.Contig1_All), XTH (CL5047.Contig5_All), PIP (CL10703.Contig1_All), TIP (CL7584.Contig1_All), ARR-A(CL10038.Contig2_All), AUX1(CL1448.Contig14_All), TIR1(CL2675.Contig12_All), ERF(CL2393.Contig1_All), and EIN3(CL5089.Contig2_All). A correlation analysis was conducted between RT-qPCR verification results and RNA-seq data. It can be seen that the correlation coefficient between the two is high, and the expression trend is basically the same (Supplementary Fig. S2). This indicates that the results of RNA-seq analysis are reliable.
Identification and physicochemical properties of LiEXP gene family members
-
Through a single comparison and Pfam and HMMER searches, the compared candidate genes were submitted to the NCBI-CDD online website for structural verification in the conserved domain. Finally, 27 LiEXP genes (22 LiEXPA genes, two LiEXPB genes, two LiEXLA genes, and one LiEXLB gene) were obtained and named. In addition, the reported families of extensor protein genes in plants are summarized, along with their number and distribution among the four subfamilies, as shown in Supplementary Table S9. The physicochemical properties of the proteins encoded by the LiEXP gene were further analyzed (Supplementary Table S10). The results showed that the encoded protein was composed of 249−323 amino acid residues with a molecular weight of 26,538.67−35,651.22 Da. The isoelectric point values of LiEXPA6 and LiEXPB2 were less than 7, which were acidic proteins, and the rest were basic proteins. The instability index of the LiEXPs protein family ranged from 60.65 to 80.57, all of which were unstable proteins. The average hydrophobicity of all the LiEXPs proteins was lower than 0, indicating that the LiEXPs protein family was hydrophilic. Subcellular localization prediction analysis showed that LiEXPs were located in the cell wall. The results showed that LiEXPs mainly consisted of 14.86% α-helix, 25.64% β-folding, 6.73% random curling, and 52.76% extended chains. Therefore, proteins encoded by the LiEXPs gene have certain differences in amino acid sequence length and protein characteristics. The protein family is mainly composed of alkaline, hydrophilic, and unstable proteins that are located in the cell wall.
Phylogenetic analysis of plant EXP protein
-
According to an amino acid sequence comparison analysis (Supplementary Fig. S3), there were eight conserved cysteine residues in the N-terminal of L. indica expansin, four conserved tryptophan residues in the C-terminal, and one conserved HFD motif in the EXPA subfamily. MEGA11 software was used to construct a phylogenetic tree for the amino acid sequences of some plant extended protein families. It includes amino acid sequences of L. indica expansin and A. thaliana, N. tabacum, and O. sativa expansin family genes. And amino acid sequences of other species retrieved by BLAST, such as G. grandifloras, R. hybrida, C. praecox, C. praecox, and O. fragrans, are associated with flower opening. According to the evolutionary tree analysis, the 27 members of the L. indica expansin gene family can be divided into four subfamilies: LiEXPA, LiEXPB, LiEXLA, and LiEXLB (Fig. 8). The LiEXPA subfamily accounted for the highest proportion, accounting for about 81.5% in 22 of 27 expansin genes. The proportion of LiEXLB subfamily genes was the smallest, with only one subfamily member gene. LiEXPA7, LiEXPA8, and LiEXPA9 are closely related to DcEXPA1, DcEXPA2, GgEXPA1, OfEXPA2, and RhEXPA1. LiEXLA1 and LiEXLA2 are closely related to OfEXLA1. LiEXPA19 is closely related to CpEXPA1 and OfEXPA4.
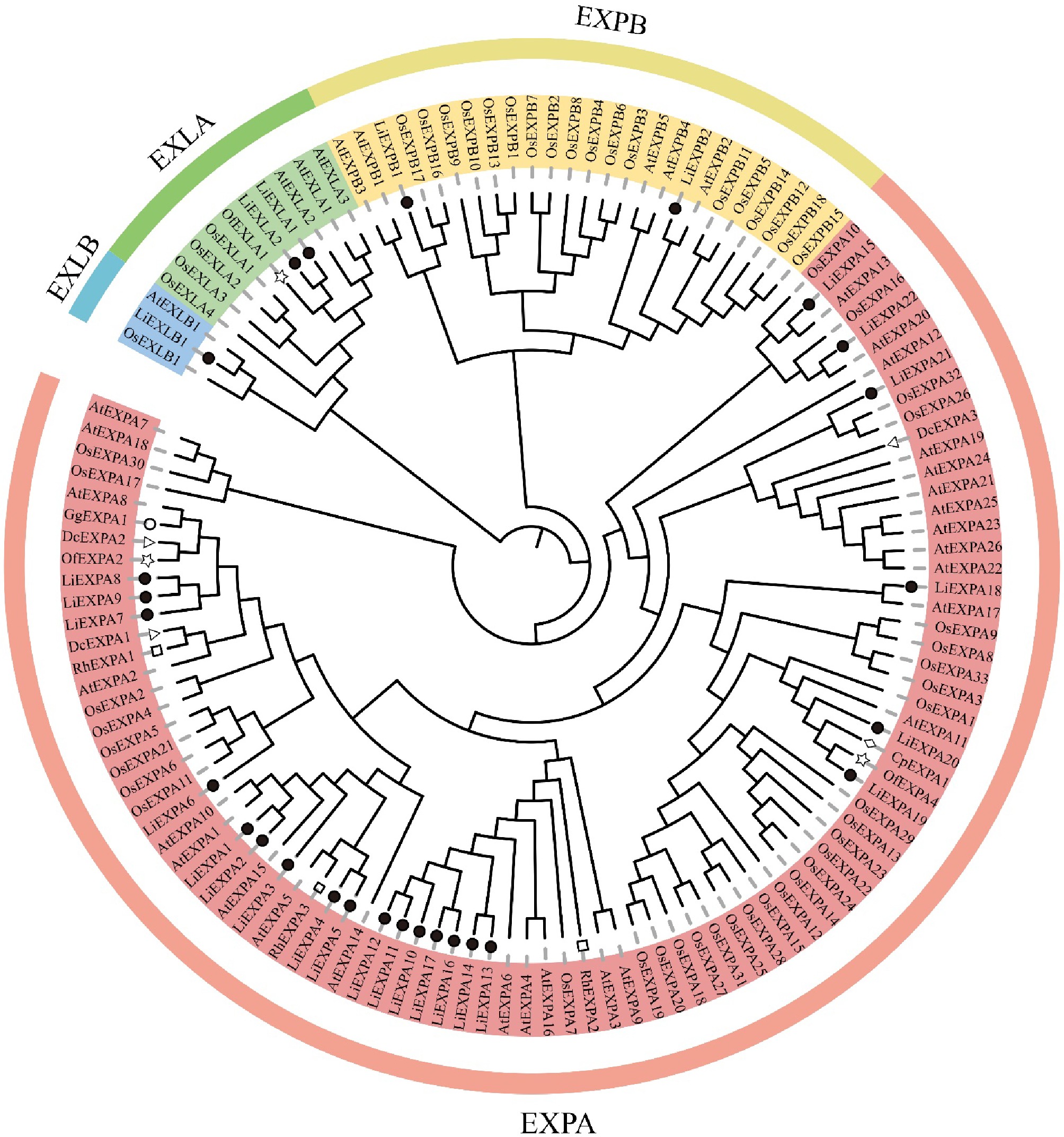
Figure 8.
Plant expansin phylogenetic tree. Each subgroup is distinguished by a different color. At: A. thaliana; Os: O. sativa; Nt: N. tabacum; Li: L. indica; Of: O. fragrans; Rh: R. hybrida; Dc: D. caryophyllus; Gg: G. grandifloras; Cp: C. praecox.
Analysis of conserved domain and motif of LiEXP gene family
-
The protein domain analysis of L. indica expansin showed that DPBB1 (Pfam ID: PF03330) and Pollen allerg1 (Pfam ID: PF01357) were the only two domains in all L. indica expansin family members (Fig. 9b). A total of 20 different conserved motifs (Fig. 9c) were found in the amino acid sequence analysis of the extended protein of L. indica using MEME software, with an average of four to nine conserved motifs per extended protein gene. As can be seen from the Fig. 9 the types of conserved motifs of expansin genes are stable within the same subfamily, while there are large differences among subfamilies. The conservative motif combination of the LiEXPA subfamily was 5/2/7/3/1/6/4, and the conservative motif combination of the LiEXPB subfamily was 16/13/8/11. All LiEXLA subfamilies are 15/2/14/8/11/7/13/20 combinations, while LiEXLB subfamilies are mostly 16/13/8/11 combinations. It is noteworthy that Motif 5, Motif 6, Motif 1, and Motif 10 are unique to the LiEXPA subfamily. Motif 18 is unique to the LiEXPB subfamily, and Motifs 15, 14, and 20 are unique to the LiEXLA subfamily.
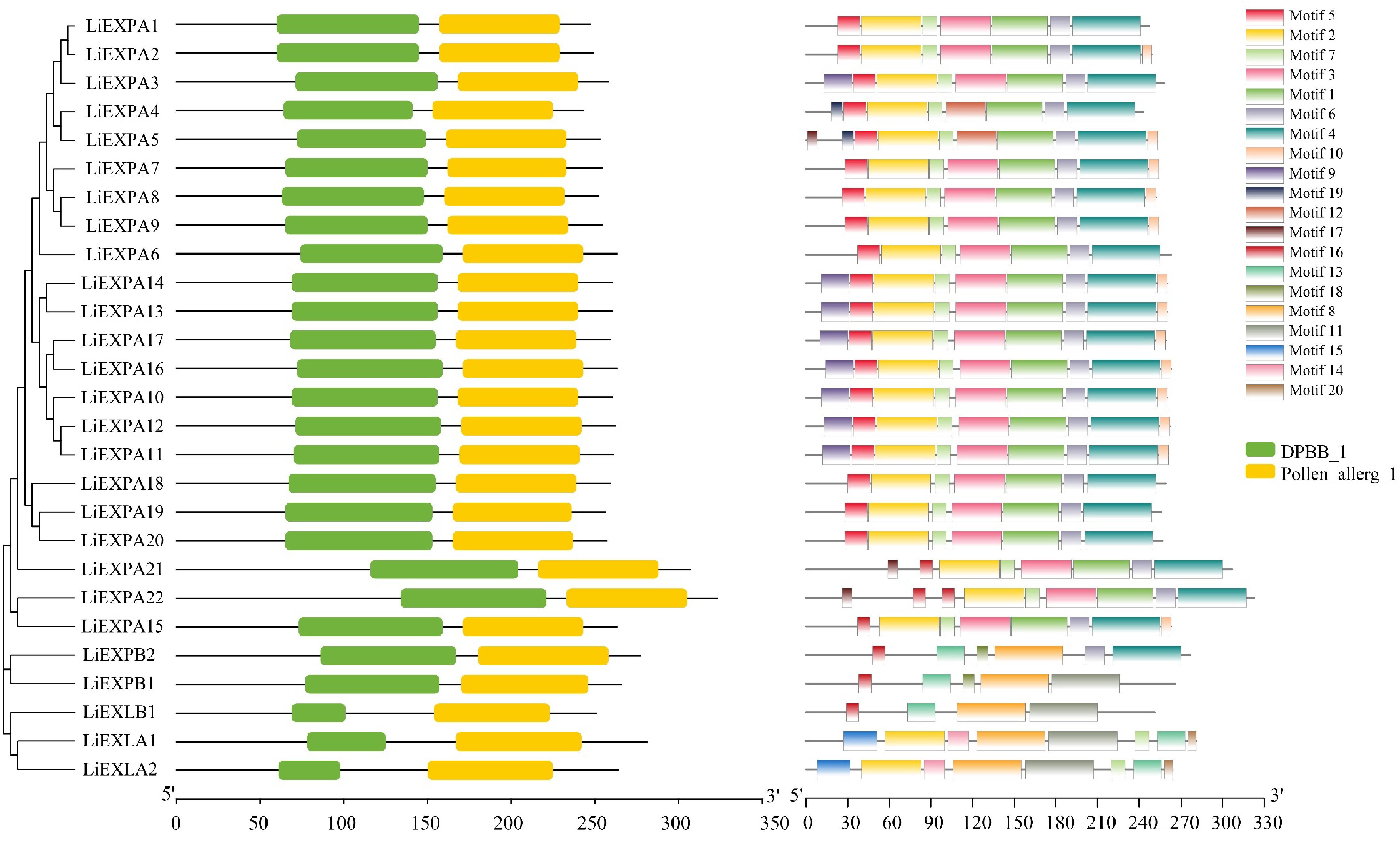
Figure 9.
Analysis of the (a) evolutionary tree, (b) protein motif structure, and (c) domain of the LiEXPs gene family. Different colored modules represent different domains or motifs.
Expression patterns of LiEXP gene during flower opening
-
RT-qPCR analysis of the expression pattern of LiEXPs in the opening process of L. indica (Fig. 10) showed that the relative expression levels of LiEXPA17, LiEXPA22, LiEXPB1, LiEXPB2, and LiEXLB1 genes were significantly higher in the early stage (S1−S2) than in the late stage (S3−S4). The relative expression levels of LiEXP22, LiEXPB1, LiEXPB2, and LiEXLB1 were at a high level in the first two periods (S1−S2) of the opening process of L. indica. The expression trend was relatively stable, and it was significantly down-regulated in the S3 stage, suggesting that it may play a certain role in the opening of L. indica flower buds. The expression of the LiEXPA17 gene was downregulated gradually with the process of flower opening. However, the expression levels of LiEXPA19, LiEXPA20, LiEXPA21, LiEXLA1, and LiEXLA2 genes in the early flowering period (S1−S2) were relatively stable, and the relative expression levels of LiEXPA19, LiEXPA20, LiEXPA21, LiEXLA1, and LiEXLA2 were gradually up-regulated in the late flowering period (S3−S4). The expressions of LiEXPA19, LiEXLA1, and LiEXLA2 increased gradually during the opening period and reached their highest levels in the S4 period. The expression levels of LiEXPA20 and LiEXPA21 were low in the early flowering period, reached their highest value in the S3 period, and gradually decreased in the S4 period. In addition, LiEXPA1-10 and LiEXPA11-16 showed stable expression in the whole flower opening process, which may have little relationship with flower opening.
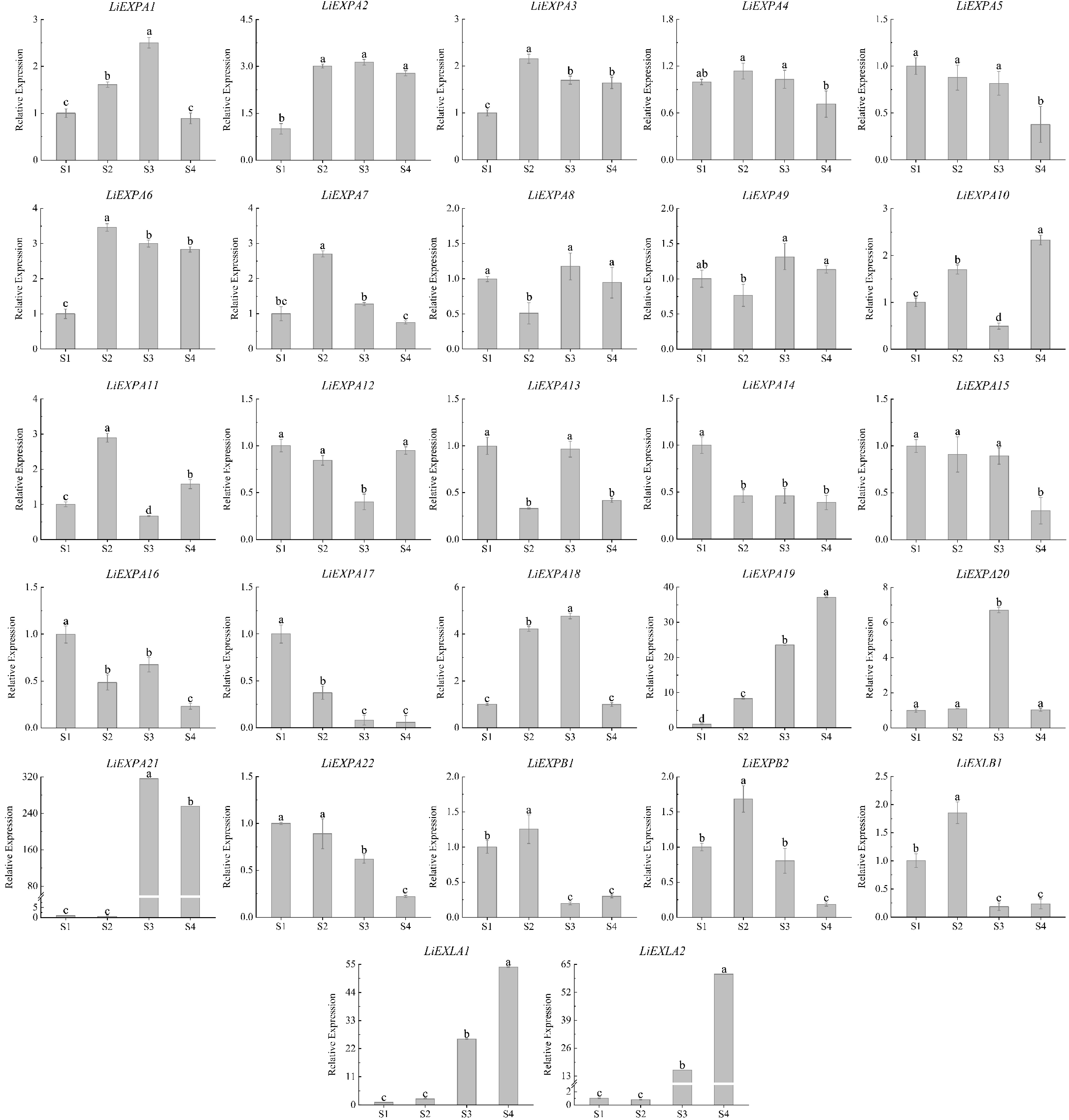
Figure 10.
Expression analysis of LiEXP gene family during flower opening. Significant differences are using ANOVA and Duncan's test (p < 0.05) and are represented by different letters above the error bars.
Identification and analysis of transgenic plants
-
The two genes, LiEXLA1 and LiEXLA2, were significantly upregulated in expression at the later stages of flowering (S3−S4), and phylogenetic analyses showed that they were highly homologous to related genes involved in petal expansion (e.g., OfEXLA1). In addition, the expression patterns of these two genes were consistent with the key stages of petal expansion, and thus they were selected as validation candidates. The effects of the LiEXLA1 and LiEXLA2 genes on the stem size of A. thaliana were observed and analyzed (Figs 11, 12). In this study, wild-type A. thaliana that grew normally for 45 d was compared with T3-generation transgenic A. thaliana. The results showed that the stem diameter of LiEXLA1 and LiEXLA2 transgenic A. thaliana was significantly different from that of the wild type, and the stem diameter of LiEXLA1 and LiEXLA2 transgenic A. thaliana was significantly higher than that of the wild type. In addition, we also observed the petal area of A. thaliana and found that the petal area of wild-type A. thaliana was significantly lower than that of the transgenic strain. The flowering time of wild A. thaliana was also significantly longer than that of transgenic strains.

Figure 11.
Wild-type and transgenic LiEXLA1 A. thaliana growth 45 d.
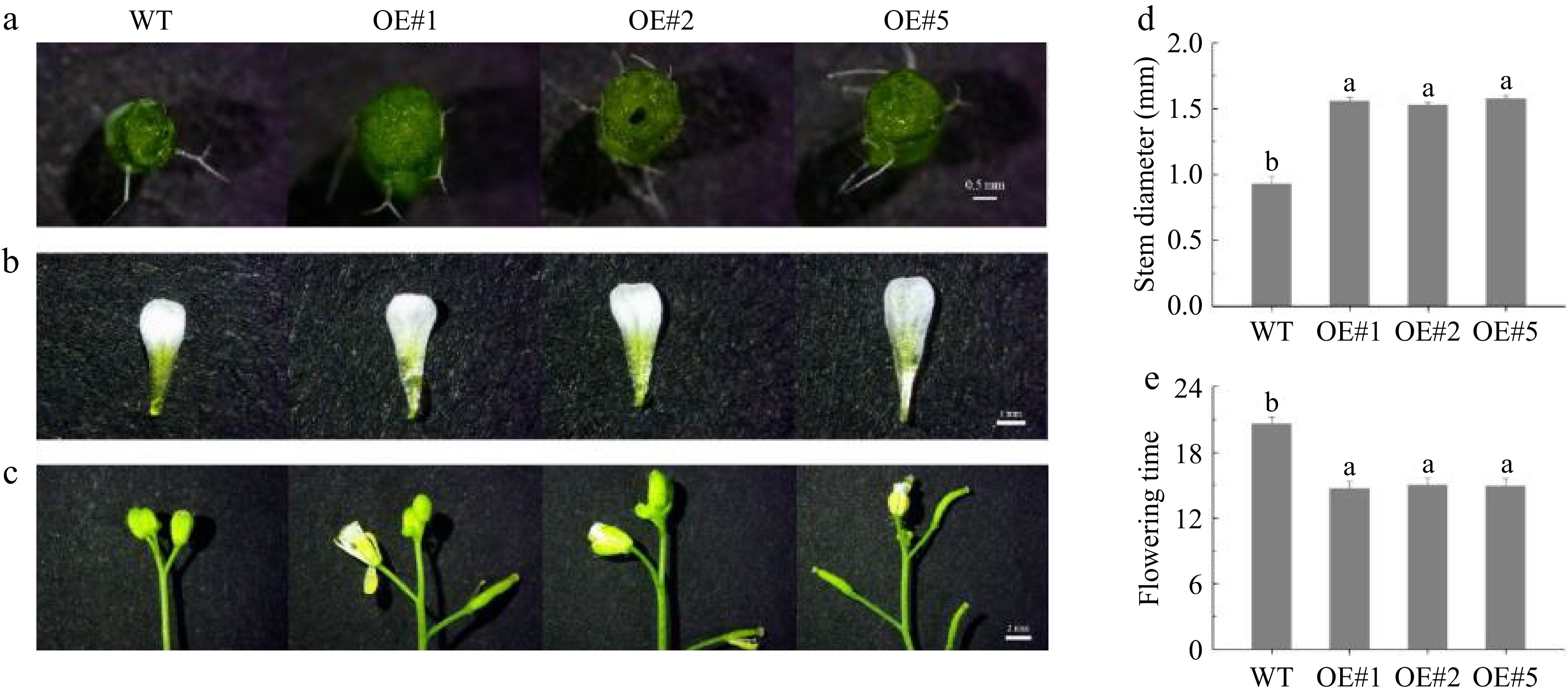
Figure 12.
Wild-type and transgenic LiEXLA2 A. thaliana growth 45 d.
-
Petal expansion is a crucial step in the plant flowering process, and cell wall relaxation is considered to be one of the key factors in this process[26]. The present study revealed the high expression of LiEXPA and LiEXLA family genes, especially LiEXLA1 and LiEXLA2, during petal flowering in L. indica, suggesting that these genes play an important role in cell wall relaxation and expansion. These findings are consistent with findings in other species, such as in flowers such as Rosa rugosa, Moso Bamboo, and Syringa oblata, where the EXP gene family has also been shown to play a central role in petal expansion[27−29]. During cell wall relaxation, extension proteins promote cell wall expansion by relaxing the noncovalent bonds of cellulose-hemicellulose complexes[30]. The results of this study showed that the expression of LiEXPA19 and LiEXLA1/2 genes was significantly up-regulated at the critical stage of petal expansion, suggesting that these genes may promote petal growth and expansion by regulating cell wall relaxation.
Petal expansion in relation to water transport
-
Water plays an important role in petal expansion, driving petal unfolding by promoting cell swelling and maintaining cell expansion pressure[31]. In this study, RNA-seq analysis revealed that several genes related to water transport showed differential expression during petal expansion in L. indica, especially PIP (plasma membrane intrinsic protein) and TIP (tissue intrinsic protein). These water channel proteins play a key role in petal expansion by regulating water transport across membranes. For example, the expression of PIP family members is consistently elevated during petal expansion, suggesting their important role in water regulation and cell expansion. Previous studies have shown that efficient water transport is an important factor in plant petal expansion, and the present study further supports this hypothesis[12,32].
The role of sugar metabolism in petal expansion
-
Sugar metabolism plays a crucial role in petal expansion, mainly through the provision of energy and the building blocks of cell walls[33,34]. Analysis of transcriptome data showed that several sugar metabolism-related genes showed significant expression changes at all stages of Zoysia petal flowering, especially at the initial flowering stage, where the metabolism and distribution of sugars are crucial for petal expansion. Especially at the S2 stage, the expression of related genes in the sugar xenobiotic pathway and polysaccharide degradation pathway rose, suggesting a close relationship between sugar utilization and petal expansion. The supply and utilization of sugar sources are crucial in petal expansion and flowering, and cut flowers lacking sugar sources lead to flowering disorders, a phenomenon that has been verified in other plants[35,36]. In addition, this study further demonstrated the central role of sugar metabolism in petal expansion, especially through the pathway of glycolysis and starch catabolism to provide energy and precursors for rapid petal expansion.
Regulation of petal expansion by hormone signaling pathways
-
The role of hormone signaling pathways, especially growth hormone (auxin) and gibberellin (GA), in petal expansion has received widespread attention[37]. In this study, we showed that several genes related to growth hormone and gibberellin exhibited significant differential expression during petal expansion in L. indica. Specifically, the expression of GA receptor genes was significantly up-regulated from S3 to S4 stages, suggesting that gibberellins play an active role in promoting petal flowering and expansion. Changes in growth hormone levels also had a significant effect on petal expansion, especially during the early petal development stage, and the expression of genes related to growth hormone signaling, such as AUX/IAA and SAUR, increased during petal expansion[38]. In addition, ABA (abscisic acid) and ethylene play regulatory roles during petal aging and decline. Our data suggest that the cross-talk of these hormones may form a complex regulatory network on the petal expansion process in L. indica.
Identification and bioinformatics analysis of the expansin gene family of L. indica
-
In order to better understand the function and evolution of the LiEXP gene family, this study analyzed the LiEXPA gene family of L. indica in comparison with the EXP genes of other species, including R. hybrida, O. fragrans, and A. thaliana. The results showed that the LiEXPA genes of R. indica shared significant structural consistency in conserved motifs with the EXP genes of these species, especially in the distribution of cysteine and tryptophan residues, reflecting the evolutionary conservation of these genes in function. These key loci are thought to play important roles in cell wall relaxation and petal expansion. Among the LiEXPA gene family in L. indica, this study found that the conserved motifs of some specific genes, such as LiEXPA19, were highly consistent with RhEXPA1 in R. hybrida. Previous studies have shown that RhEXPA1 has an important function in petal expansion in R. hybrida[39]. This high degree of homology suggests that LiEXPA19 may have a similar function in L. indica to promote rapid petal expansion by relaxing the cellulose-half-cellulose network in the cell wall. In addition, the positions of the LiEXLA1 and LiEXLA2 genes in L. indica in the phylogenetic tree were closely clustered with OfEXLA1 in O. fragrans, and both of these genes were significantly expressed at the critical stage of petal expansion[40]. Related studies in O. fragrans suggest that OfEXLA1 may adapt to environmental changes by regulating the flexibility of the cell wall, thus ensuring normal petal expansion. This evolutionary functional conservation further supports a critical role for LiEXLA1 and LiEXLA2 in L. indica petal expansion. Notably, although the AtEXP gene family of A. thaliana is mainly involved in cell wall relaxation in leaves and roots, certain genes such as AtEXPA8 are also associated with petal development[41]. Comparison of the conserved motifs of L. indica and A. thaliana genes revealed that certain EXP genes of L. indica may be functionally more specialized, especially in petal expansion and floral organ development. From an evolutionary perspective, the existence of such conserved motifs suggests that the functions of EXP genes in angiosperms may have originated from common ancestral genes and have been further specialized to meet different adaptive needs as the species diversified. LiEXP genes in L. indica may act synergistically with biological processes, such as sugar metabolism and hormone signaling pathways, through a specific regulatory network of gene expression to shape its unique petal expansion mechanism. In addition, since the whole genome sequence of L. indica is publicly available, fine-grained analysis of the LiEXP gene family can be performed in the future by combining the genome with the transcriptome, e.g., by integrative analyses to identify regulatory elements in the promoter regions of genes as well as the global regulatory network of gene expression[42].
Analysis of expression pattern of expansin of L. indica
-
The expression patterns of the LiEXPA19, LiEXLA1, and LiEXLA2 genes were similar to those of the GgEXPA1 and RhEXPA1 genes by RT-qPCR. That is, the expression level was low in the bud stage, increased sharply in the initial opening stage, and reached its peak in the blooming stage. Therefore, we speculated that LiEXPA19, LiEXLA1, and LiEXLA2 have similar functions to GgEXPA1 and RhEXPA1, participate in petal extension, and are key EXP gene family members in the opening process of L. indica. Moreover, the evolutionary tree (Fig. 8) shows that LiEXPA19, LiEXLA1, and LiEXLA2 are close to OfEXPA4 and OfEXLA1, respectively, thus further clarifying our inference. And functional validation of the genes LiEXLA1 and LiEXLA2 in transgenic A. thaliana yielded that the above two genes can promote flower opening[41]. In addition to LiEXLA1 and LiEXLA2, we also preliminarily screened genes such as LiEXPA10 and LiEXPA19, which showed significant expression changes during the early stages of flowering. Although not functionally validated in the current study, we plan to further validate their potential roles in petal expansion in future studies. In addition, LiEXPA20 and LiEXPA21 also showed similar expression patterns, suggesting that LiEXPA20 and LiEXPA21 are also key members of the EXP gene family in the opening process of L. indica. In addition, LiEXPA1-10 and LiEXPA11-16 showed stable expression in the whole flower opening process, which may have little relationship with flower opening. The other four LiEXP genes (LiEXP22, LiEXPB1, LiEXPB2, and LiEXLB1) showed a higher expression level in the early stage of flower opening, while the expression level decreased significantly in the petal expansion stage, which was similar to the expression pattern of RhEXPA3 in R. hybrida[39]. RhEXPA3 was mainly involved in xylem development, suggesting that these four LiEXP genes are not the key members of the EXP gene family in the opening process of L. indica.
The aim of this study was to reveal the role of the EXP gene family in the petal expansion of L. indica through transcriptome analysis and to deeply analyze how factors such as hormone signaling, sugar metabolism, and water transport interact with each other in this process. These results not only provide a theoretical basis for the floral regulation of L. indica but also provide a new direction for ornamental improvement and genetic breeding of related plants.
-
In this study, transcriptome analysis revealed the key roles of the LiEXPA and LiEXLA gene families in petal expansion in L. indica. LiEXLA1 and LiEXLA2 were significantly upregulated at the later stages of petal expansion (S3−S4), and functional validation demonstrated that they significantly promoted petal expansion and accelerated flowering time, confirming their importance in cell wall relaxation. In addition, this study also found that L. indica petal expansion is closely related to hormone signaling pathways, sugar metabolism, and water transport. The expression of hormones such as growth hormone and gibberellin, as well as sugar metabolism and water channel proteins, play important roles in petal expansion. This study provides a new perspective on the molecular mechanism of petal expansion in L. indica and provides a theoretical basis for floral regulation and breeding of L. indica and other ornamental plants. In the future, other potential genes (e.g., LiEXPA10 and LiEXPA19) can be further validated, and the regulatory network of L. indica petal expansion can be improved by combining genomic data.
This work was supported by grants from the Zhejiang Provincial Natural Science Foundation of China (Grant No. LY21C160001), and the Zhejiang Science and Technology Major Program on Agricultural New Variety Breeding (Grant No. 2021C02071-4).
-
The authors confirm contribution to the paper as follows: study conception and design: Zhang G, Zhao Y, Gu C; data collection: Li Z; analysis and interpretation of results: Zhang G, Wu Z, Yang L; draft manuscript preparation: Zhang G, Zhao Y, Zong J. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Guozhe Zhang, Yu Zhao, Zhiqiang Wu
- Supplementary Table S1 Primer sequences for candidate genes.
- Supplementary Table S2 Clean reads quality statistics.
- Supplementary Table S3 Quality metrics of unigenes of L. indica.
- Supplementary Table S4 The statistical result of unigene functional annotation.
- Supplementary Table S5 Statistics of differential genes quantity.
- Supplementary Table S6 Phytohormone-related genes during flowering in L. indica.
- Supplementary Table S7 Genes associated with petal expansion during flower opening in L. indica.
- Supplementary Table S8 Genes related to water and carbohydrates during flower opening in L. indica.
- Supplementary Table S9 The categories of expansion in different species.
- Supplementary Table S10 Physicochemical properties of the LiEXP gene.
- Supplementary Fig. S1 Plot of the length distribution of Unigene sequences.
- Supplementary Fig. S2 qRT-PCR analysis plot of random candidate genes.
- Supplementary Fig. S3 Amino acid sequence comparison of members of the LiEXPs gene family.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang G, Zhao Y, Wu Z, Li Z, Zong J, et al. 2025. Deciphering the genetic basis of flower opening in Lagerstroemia indica: transcriptomic and functional insights into expansin-mediated petal expansion. Ornamental Plant Research 5: e018 doi: 10.48130/opr-0025-0013 

Deciphering the genetic basis of flower opening in Lagerstroemia indica: transcriptomic and functional insights into expansin-mediated petal expansion
- Received: 05 November 2024
- Revised: 11 January 2025
- Accepted: 12 February 2025
- Published online: 06 May 2025
Abstract: Lagerstroemia indica is popular for its bright flower colors and long bloom period. However, although L. indica has a long flowering period, the flowering time of a single flower is short, lasting only 1−2 d. Petal expansion is a key process that affects the length and ornamental quality of the flowering period. However, the molecular mechanism of petal expansion in L. indica remains unclear. The molecular mechanisms underlying flower opening in L. indica were investigated through transcriptome sequencing of flower buds and blooms at four developmental stages. Analysis of differentially expressed genes (DEGs) indicated enrichment in cellular processes, metabolic regulation, and biological signaling pathways. KEGG pathway analysis revealed significant roles for carbohydrate, lipid, and amino acid metabolism in the flowering process. Additional pathway analysis identified key genes and processes related to carbohydrate utilization, hormone signaling, water transport, and cell wall expansion that contribute to petal opening regulation. A comprehensive examination of the expansin gene family proteins, known for promoting cell wall loosening and extension, identified 27 expansin genes in L. indica, which were categorized into four subfamilies with conserved structures and motifs. Of these, LiEXPA10, LiEXPA19, LiEXLA1, and LiEXLA2 showed heightened expression in the later stages of flowering (S3−S4), suggesting a central role in petal expansion. Functional validation in Arabidopsis thaliana demonstrated that LiEXLA1 and LiEXLA2 promote accelerated flowering and enhanced petal expansion in transgenic lines. These findings offer new insights into the genetic and molecular basis of flower opening in L. indica and provide a foundation for breeding strategies aimed at improving ornamental traits.
-
Key words:
- Lagerstroemia indica /
- Flower opening /
- Expansin gene family /
- Hormone signaling /
- Functional validation