









-
Whole Genome Sequencing (WGS) utilizes high-throughput sequencing platforms to decode the complete genomic content of cells or tissues. As advanced sequencing technology has developed in recent years, WGS enables the parallel sequencing of thousands to millions of DNA molecules[1]. Applying WGS provides a vital bioinformatics foundation for research in strawberry genomics research, facilitating comprehensive studies of genome structure and function, as well as the identification of functionally relevant genes[2].
The main cultivated strawberry species, Fragaria × ananassa (2n = 8x = 56), presents challenges for exploring their molecular mechanisms due to high heterozygosity and complex chromosome structures[3]. In contrast, the diploid woodland strawberry (Fragaria vesca, 2n = 2x = 14) has emerged as the primary model species for gene functional research within the Rosaceae family. This is primarily attributed to its compact plant size, low heterozygosity, small genome, and efficient in vitro regeneration and transformation[3]. Among the diploid strawberry accessions frequently employed in experimental studies, three are particularly prominent: 'Hawaii 4', 'Ruegen', and 'Yellow Wonder'.
The genome sequencing and assembly of 'Hawaii 4' (H4) was first released in 2011 (version 1.0). This initial sequencing was conducted using Roche 454, Illumina Solexa, and SOLID technologies[4]. Since then, advancements in sequencing technology have led to continuous improvements in the H4 genome. By 2023, a telomere-to-telomere, gap-free genome of H4 was successfully assembled[5]. In 2022, a high-quality genome for 'Yellow Wonder' (YW) was assembled and annotated[6]. However, the genome of 'Ruegen' (RG), another commonly used accession for genetic transformation, remains unavailable. Although H4, YW, and RG share many similar morphological traits, they also exhibit distinct characteristics. For example, H4 produces runners and yellow fruits, while YW bears yellow fruits but lacks runners. In contrast, RG is characterized by its red fruits and non-running habit. A comparative analysis between RG and YW could provide valuable insights into the anthocyanin synthesis pathway. Thus, assembling the genome of RG will serve as a solid foundation for studying the mechanisms underlying strawberry color formation and supporting molecular breeding programs.
Transcriptome sequencing based on Illumina high-throughput sequencing platforms offers several key advantages: high information yield, rapid sequencing speed, low data redundancy, and nucleotide-level resolution for profiling transcriptional activity across species. These characteristics make high-throughput sequencing technology serves as an efficient tool for exploring functional genes in non-model plants, leading to its widespread adoption in horticultural plant research[7]. When a reference genome is unavailable, de novo transcriptome assembly from RNA sequencing data enables gene expression quantification. However, challenges such as fragmentation and incorrect assembly of RNA sequencing data can lead to issues like low sequencing accuracy and incomplete gene coverage. These issues may negatively affect the accuracy and reproducibility of gene expression levels[8]. The availability of a high-quality reference genome (such as the newly assembled RG genome) significantly improves transcriptome studies by enabling genome-guided analysis. This approach enhances both the detection sensitivity and quantification accuracy of transcriptomic data, providing more reliable results for downstream applications.
In this study, third-generation sequencing was integrated with Hi-C-assisted assembly to generate a high-quality genome assembly for the RG strawberry. The assembled genome spans 221.31 Mb with a contig N50 of 8.35 Mb, representing a significant genomic resource. Additionally, comparative transcriptome analysis of fruits from the diploid RG and octoploid 'Yanli' varieties was performed across multiple developmental stages, with a particular focus on expression profiling of key anthocyanin biosynthesis genes. These findings provide valuable insights into the genetic regulation of anthocyanin accumulation in strawberry fruits, offering a molecular foundation for breeding improved strawberry varieties with enhanced quality traits.
-
Fragaria vesca, 'Ruegen' (RG) were grown in pots and maintained at Shenyang Agricultural University (Shenyang, China). Two biological replicates were collected from the RG fruit and sent to Biomarker Technologies Company (Beijing, China) for DNA library preparation and sequencing. The genomic DNA of RG was extracted using a DNAsecure Plant Kit (TIANGEN, Beijing, China), after verifying the quality and concentration of the DNA library by 1% agarose gel electrophoresis and Qubit 2.0 Fluorometer (Life Technologies, USA). The construction of 220-bp paired-end (PE) libraries was carried out utilizing the NEBNext® Ultra™ DNA Library Prep Kit. Subsequently, these libraries underwent sequencing on the Illumina HiSeq X Ten platform at the Biomarker Technologies Company, Beijing (China). To eliminate adapters and leading/trailing ambiguous or low-quality bases, the raw Illumina sequencing reads were processed using Trimmomatic version 0.33[9] and Cutadapt version 1.13[10]. Ultimately, 21.37 Gb of clean reads were obtained and employed for the assembly evaluation and error correction of the genome assembly.
Genome size and heterozygosity analysis
-
By utilizing k-mer analysis, the genome size was determined, and the heterozygosity of RG was assessed. High-quality sequencing reads amounting to 21.37 Gb was processed to construct a depth distribution curve for k-mers (k = 19). This was accomplished with the assistance of the 'kmer freq stat' software, which was crafted by Biomarker Technologies Company, Beijing, China. The genome size (GS) of RG was then calculated using the formula GS = k-mer number/average k-mer depth[11]. The heterozygosity was estimated based on the formula[12].
PacBio SMRT sequencing and assembly
-
The SMRT Bell library was prepared using a DNA Template Prep Kit 1.0, and 20-kb SMRTbell libraries were constructed. De novo assembly was performed using Canu v1.7[13] with the parameters 'genomeSize = 250000000', and Falcon v3.0 with the parameters 'length_cutoff = 3000, length_cutoff_pr=8000', and wtdbg v1.1.006 (
https://github.com/ruanjue/wtdbg ). Quickmerge[14] was employed to optimize the genome and remove redundant sequences. Subsequently, the Illumina data were aligned to the assembly contigs from Quickmerge pipeline using bwa mem v0.7.12[15]. Finally, the optimized genome was then refined using Pilon version 1.22[16] to enhance assembly efficiency.Hi-C library construction and scaffolding
-
To anchor the scaffolds onto chromosomes, a Hi-C library was constructed utilizing the Illumina HiSeq X Ten platform. The trimmed reads from the RG genome were aligned to the assembly using BWA version 0.7.12 software[15], with specific parameters set as follows: bwa index -a bwtsw fasta; bwa aln -M 3 -O 11 -E 4 -t 2 fq1; bwa aln -M 3 -O 11 -E 4 -t 2 fq2. For filtering low-quality reads and conducting quality assessments, the HiC-Pro pipeline was employed[17], using the parameters specified as 2hic fragments.py -v -S -s 100 -l 1000 -a -f -r -o for mapping. Subsequently, LACHESIS[18] was utilized to parse and model the genomic sequence locations, with the following parameters: cluster min re sites = 48; cluster max link density = 2, cluster noninformative ratio = 2, order min n res in trun = 14, order min n res in shreds = 15. Finally, to enhance the accuracy and completeness of the genome assembly, the gaps in the LACHESIS-based assembly were filled using PBjelly[19]. The completeness of the genome assembly was evaluated by BUSCO version 3.0.2[20] based on a benchmark of 1,440 conserved plant genes.
RNA sequencing (RNA-seq)
-
Isolate total RNA using an improved CTAB method as described by Wang et al.[21]. The concentration of RNA sample was tested by a NanoDrop spectrophotometer (Thermo Fisher Scientific Inc., USA) and a Qubit 2.0 Fluorometer (Life Technologies, USA). Assess the integrity of RNA samples with an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, USA). The developmental stages of the fruits can be categorized based on color and size as green (G), white (W), turning (T), and red (R). Samples were collected from the fruits of RG and 'Yanli' at various stages and subsequently ground into powder in liquid nitrogen. A single RNA library, featuring an average insert size ranging from 250 to 300 base pairs (bp), was constructed utilizing the NEBNext UltraTM RNA Library Prep Kit, adhering to the instructions provided by the manufacturer, Illumina (NEB, USA). The quality of this library was subsequently evaluated using the Agilent Bioanalyzer 2100 system. Following this, RNA samples from the developmental stages of the fruits were subjected to sequencing on the Illumina HiSeq X Ten platform (Illumina, USA).
-
'Ruegen' (RG) is a diploid woodland strawberry characterized by its red fruit and non-running growth habit. The polymerase reads were filtered to obtain subreads. Statistical analysis indicated that the original subread obtained from the PacBio sequencing platform had an N50 of 11,158 bp and an average length of 6,448 bp. Using HiC-Pro[17], the raw data was processed through alignment, interaction classification, and quality control. Our results (Supplementary Table S1) demonstrated high library quality, with 79.73% valid cis-interaction pairs (25.40 million) and 100% uniquely aligned read pairs (31.86 million), indicating efficient genome-wide alignment. Hi-C scaffolding generated a high-quality chromosome-scale assembly, successfully anchoring 221.31 Mb (99.11% of the genome) onto seven pseudochromosomes (Supplementary Table S2). The anchored sequences demonstrated outstanding continuity, with 219.34 Mb (99.99%) represented by gap-free, oriented contigs (ATCG length), achieving nearly complete chromosomal representation with all major arms correctly oriented. Based on a k-mer analysis (k = 19), the genome was evaluated with heterozygosity of 0.09%, repetitive sequences of 36.13%, and GC content of 38.48%.
To improve the genome assembly accuracy, scaffolds were fragmented into 50 Kb segments and reassembled using Hi-C sequencing. Regions that deviated from the original assembly sequence were flagged as potential error regions, with positions showing low Hi-C coverage depth further identified as error points. This approach facilitated systematic error correction in the initial assembly. After the correction, the assembly achieved a contig N50 of 8.35 Mb. The Hi-C sequencing yielded a total of 13.79 Gb of clean data (62.39 X coverage). Following the Hi-C assembly, gap filling was conducted using PBjelly[19], yielding a high-quality genome assembly with a total size of 221.31 Mb. The final assembly exhibited excellent continuity, with a contig N50 of 8.35 Mb, N90 of 1.04 Mb, and an overall GC content of 38.48% (Table 1). Genome information was visualized through a comprehensive circular plot (Fig. 1).
Table 1. Statistical information on genome assembly.
Assembly feature Number Scaffold number 50 Scaffold length 221,321,825 Scaffold N50 (bp) 33,167,685 Scaffold N90 (bp) 23,655,317 Scaffold max (bp) 40,042,862 Gap total length (bp) 3,010 Contig number 142 Contig length (bp) 221,318,815 Contig N50 (bp) 8,351,392 Contig N90 (bp) 1,040,108 Contig max (bp) 18,184,794 GC content (%) 38.48 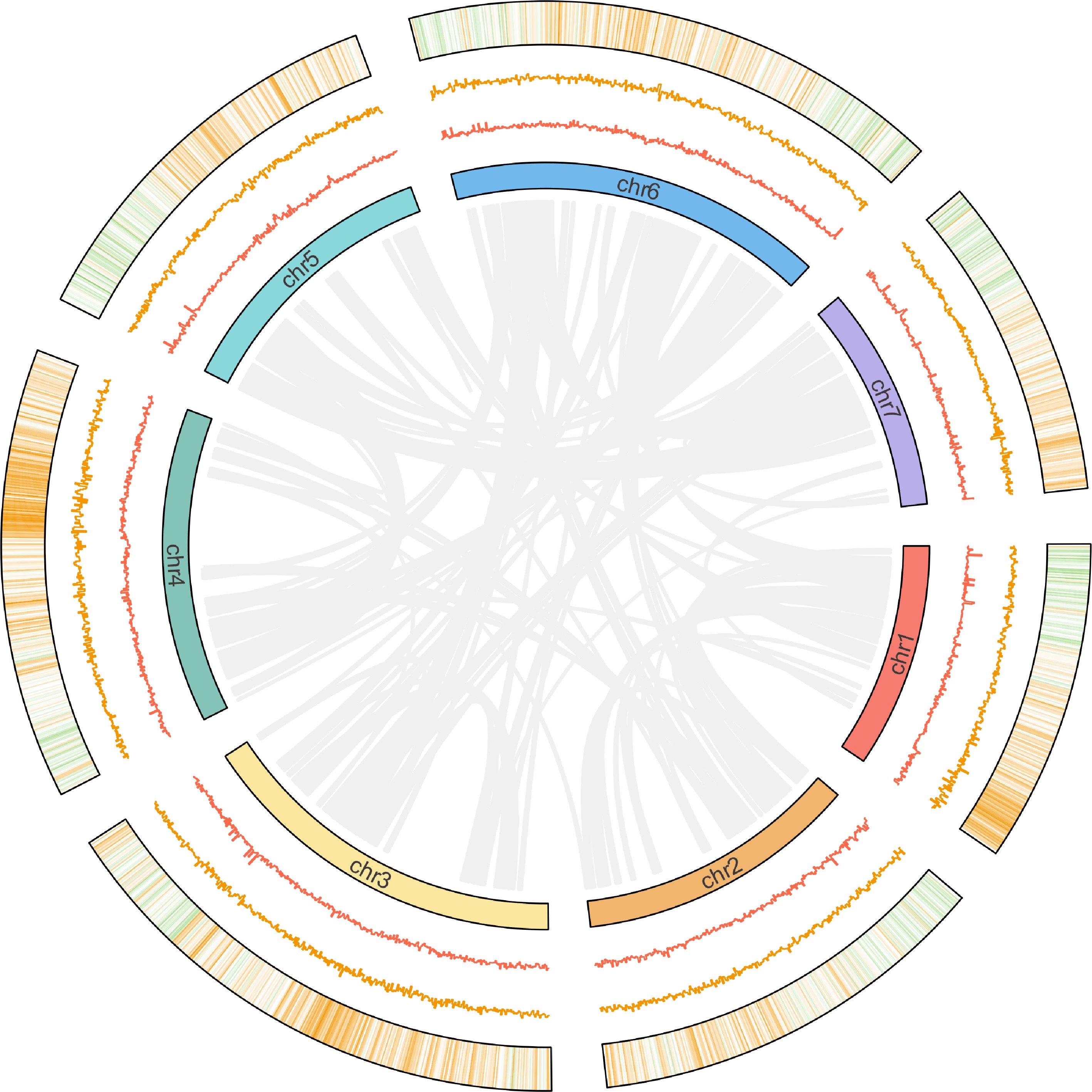
Figure 1.
Circular visualization of 'Ruegen' genome. The circular diagram displays (from inner to outer rings): (1) chromosomal homologous relationships; (2) GC content distribution (100-kb windows); (3) GC skew patterns (100-kb windows); (4) gene density (100-kb windows).
Chromosome-scale scaffolding validation and BUSCO assessment
-
To visualize chromatin interactions, the genome was partitioned into 100-kb bins, and the density of read pairs between bins was plotted as a heatmap (Fig. 2). The heatmap revealed that seven distinct chromosomal blocks, each showing stronger intra-chromosomal (diagonal) than inter-chromosomal (off-diagonal) interaction signals and a clear distance-dependent decay of interaction frequency, consistent with expected Hi-C contact patterns. In addition, minimal off-diagonal noise suggests high scaffolding accuracy with no large-scale misassemblies.
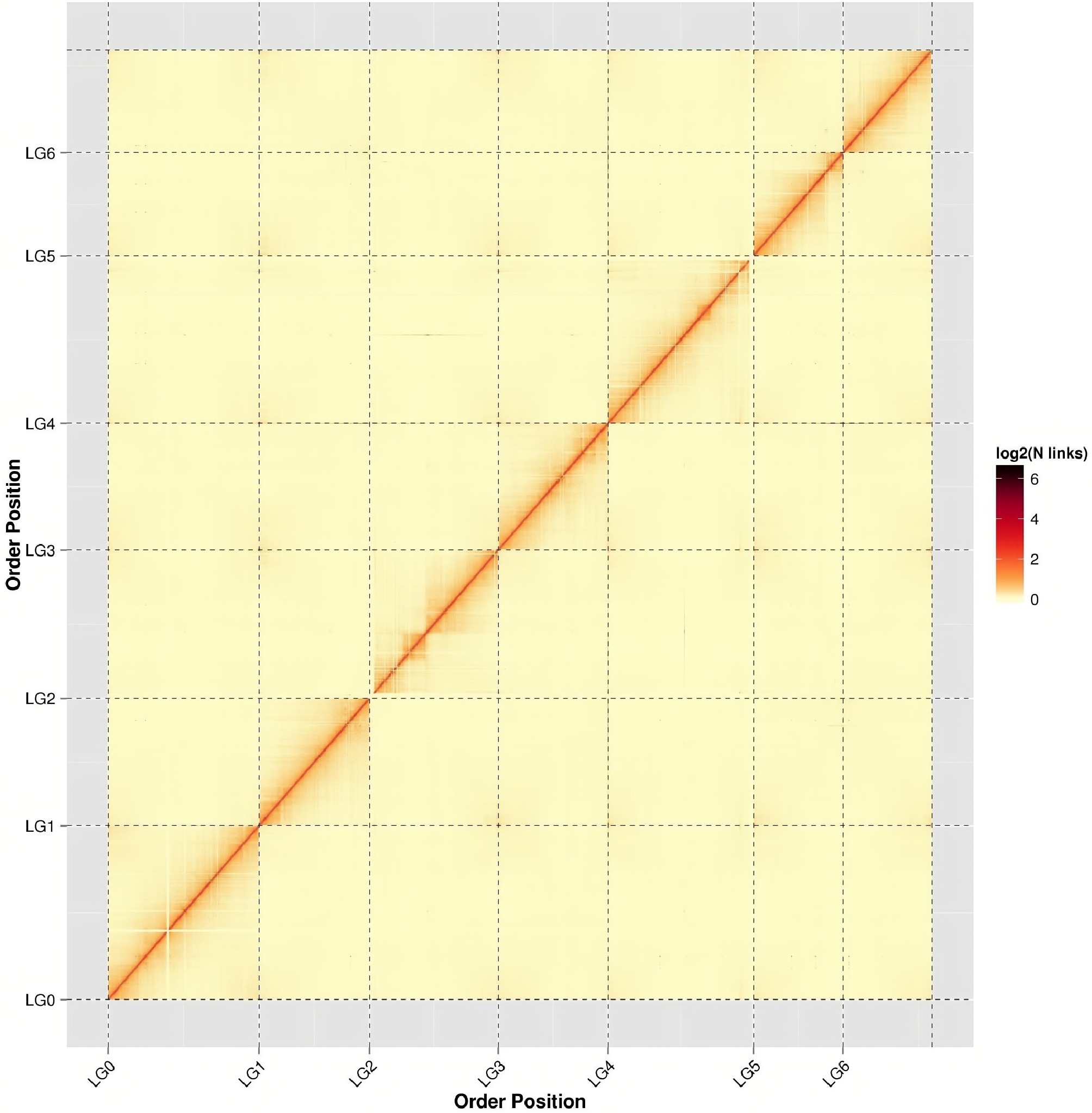
Figure 2.
Hi-C assembly chromosome interaction heatmap. chr1−chr7 represents chromosome groups 1−7. The horizontal and vertical axes represent the order of each bin on the corresponding chromosome group.
Next, BUSCO analysis confirmed exceptional assembly completeness. 98.6% (1,591/1,614 BUSCOs) complete, including 96.6% single-copy and only 1.1% missing genes. Gene annotation achieved 94.6% completeness (Supplementary Table S3), with an 86.99% functional annotation rate (Table 2). These results collectively validate the assembly's robustness for downstream functional genomics applications.
Table 2. Functional annotation statistics results.
Description Reference databases Number Percent (%) Total 36,071 Annotated 31,377 86.99 InterPro 23,870 66.18 GO 16,366 45.37 KEGG_ALL 30,574 84.76 KEGG_KO 10,674 29.59 Swissprot 19,054 52.82 TrEMBL 30,140 83.56 TF 1,736 4.81 Pfam 23,481 65.10 NR 30,947 85.79 KOG 23,055 63.92 Unannotated 4,694 13.01 Repeats annotation
-
Chromosomes harbor substantial amounts of transcriptionally inactive repetitive DNA, which constitutes a major genomic component. These sequences are broadly classified into two categories: tandem repeats and interspersed repeats. For comprehensive annotation, we employed a multi-tiered approach: database-curated TEs, de novo prediction, and integrated annotation. This strategy identified 79.97 Mb of repetitive sequences, representing 36.13% of the genome (Table 3). Among these repetitive sequences, LTR retrotransposons dominated (35.12 Mb, 15.87%), followed by DNA transposons (29.62 Mb, 13.39%). De novo prediction revealed 10.93 Mb (4.94%) of novel repeats absent in RepBase, underscoring its utility for uncovering uncharacterized elements. The analysis highlights that ~1/3 of the genome comprises diverse repetitive elements, with retrotransposons being the most prevalent class.
Table 3. Statistics of the repeated sequences.
RepBase TEs TE proteins De novo Combined TEs Length (bp) % in genome Length (bp) % in genome Length (bp) % in genome Length (bp) % in genome DNA 22,861,785 10.33 4,802,499 2.17 19,895,368 8.99 29,624,293 13.39 LINE 2,837,652 1.28 1,789,441 0.81 3,417,282 1.54 4,676,541 2.11 SINE 10,031 0 0 0 8,265 0 18,296 0.01 LTR 26,068,846 11.78 9,354,199 4.23 22,028,459 9.95 35,119,957 15.87 Satellite 352,115 0.16 0 0 257,026 0.12 549,061 0.25 Simple_repeat 0 0 0 0 274,771 0.12 274,771 0.12 Other 299 0 747 0 0 0 1,046 0 Unknown 393,519 0.18 18,210 0.01 10,928,536 4.94 11,128,498 5.03 Total 51,904,045 23.45 15,956,836 7.21 56,499,821 25.53 79,971,177 36.13 Gene annotation and comparative analysis
-
Gene annotation was performed using an evidence-integration approach through the EVidenceModeler pipeline[22−24]. This incorporated: Ab initio predictions, homology-based search, and transcriptome support from RNA-Seq data. The analysis identified 36,071 high-confidence protein-coding genes with a mean gene length of 2,826 bp and an average CDS length of 1,019 bp. Gene structure analysis revealed an average of 4.67 exons per gene, with a mean exon length of 325.46 bp. Comparative analysis revealed that conserved exon-intron architectures across four accessions, similar CDS length distributions in RG, H4, and CFRA2339, and increased short CDS proportion (< 800 bp) in YW (Fig. 3). Gene functional annotation was performed through comprehensive BLAST alignments against multiple reference databases, including NCBI NR, KOG, KEGG, etc. (Table 2). Functional annotation achieved: 86.99% annotation rate (31,377 genes), high coverage in NR (85.79%) and TrEMBL (83.56%), 84.76% KEGG pathway annotation (29.59% with KO assignments), and 13% (4,694 genes) remaining unannotated.
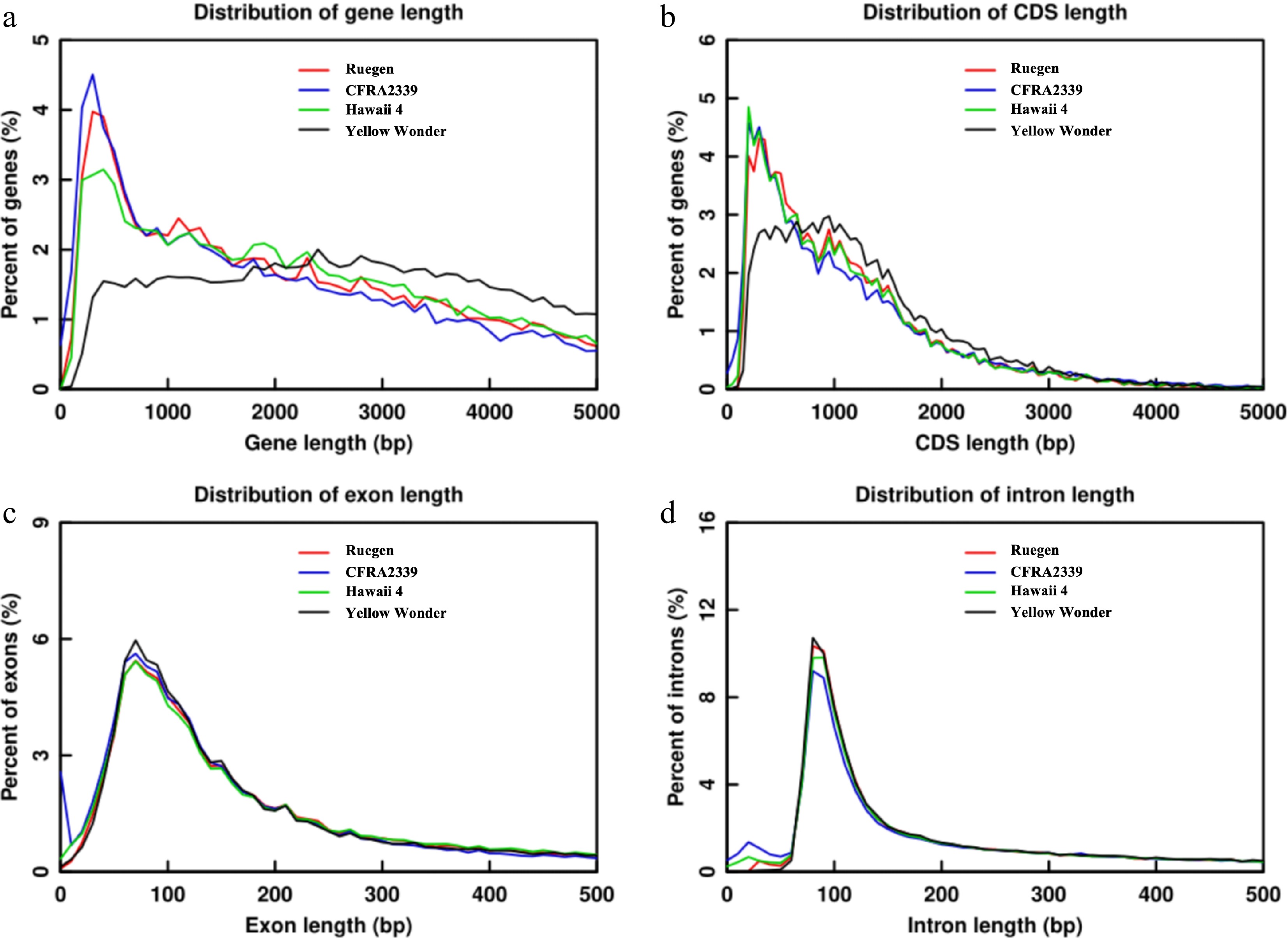
Figure 3.
Comparative distribution of gene structural elements in 'Ruegen', 'CFRA2339', 'Hawaii 4', and 'Yellow Wonder'. (a) Gene length distribution (bp), showing the percentage of genes in each size bin. (b) CDS length distribution (bp). (c) Exon length distribution (bp). (d) Intron length distribution (bp).
Non-coding RNA annotation
-
Non-coding RNAs (ncRNAs) including rRNA, tRNA, snRNA, miRNA, and others, represent functionally important RNA molecules that are not translated into proteins but they play important biological roles. Our annotation identified several major classes of ncRNAs. One hundred and seven miRNAs (0.006% of the genome) were identified, 496 tRNAs (0.017%, Supplementary Table S4). It was also found snRNAs were the most abundant (0.022%, Supplementary Table S4). The comprehensive identification of these ncRNA classes, particularly the complete rRNA complement and splicing-related snRNAs, confirms the high assembly quality of regulatory genomic regions.
Comparative transcriptomic analysis of anthocyanin biosynthesis in diploid and octoploid strawberries
-
Using the RG genome as a reference, transcriptome analysis was performed to investigate anthocyanin biosynthesis pathways. For the diploid RG (red-fruited F. vesca), fruits were collected at four developmental stages (green, white, turning, red, Supplementary Fig. S1a) with three biological replicates per stage. RNA-seq generated 89.18 Gb of high-quality clean data (average 5.21 Gb/sample, Q30 ≥ 90.65%), with alignment rates of 93.87%−95.86% to the RG genome (Supplementary Table S5). To compare with octoploid strawberries, 'Yanli' (a red-fruited cultivated variety) was analyzed using its published genome (phased into two haplotypes, Hap1 and Hap2)[25]. Similar developmental stages (Supplementary Fig. S1b) were sequenced, yielding 5.82 Gb/sample (Q30 ≥ 92.93%) with 84.84%−88.19% alignment rates (Supplementary Table S5). This analysis identified 12,036 novel genes (6,335 functionally annotated), confirming data reliability.
Phenylpropanoid-derived flavonoids, particularly anthocyanins[26,27], were the main focus. Twenty-seven genes related to anthocyanin biosynthesis and transport were screened[28−31]. The corresponding gene IDs and FPKM values for RG and all alleles in 'Yanli' were listed in Supplementary Table S6. The naming convention for the gene IDs of RG and 'Yanli' is provided in Supplementary Table S6. The gene expression levels of these genes were compared at different developmental stages of RG and 'Yanli' fruit, and the gene expression levels were plotted as heat maps. Significant differences in the expression patterns of these genes during fruit development of RG and 'Yanli'. Interestingly, it was found that 12 genes showed distinct expression patterns between RG and all 'Yanli' alleles (Supplementary Fig. S2). Among the 15 genes exhibiting partial allelic similarity between RG and 'Yanli', the study categorized them into two functional groups for analysis, early biosynthetic genes (EBGs, n = 7) and late biosynthetic genes (LBGs, n = 8) (Figs 4, 5). Five genes were found in EBGs (Fig. 4a) and three genes in LBGs (Fig. 5a) had consistent allele expression in 'Yanli' but differed from RG. Among them, MYB1 exhibited opposing trends: decreasing in RG but increasing in 'Yanli' during ripening (Fig. 5a). While not all alleles in 'Yanli' showed expression patterns consistent with RG, we identified several that exhibited conserved regulation (Figs 4b & 5c). Notably, two FaLAR alleles (FxaYL_411g0810410 and FxaYL_412g0808700) demonstrated a gradual decrease in expression during fruit development, mirroring the expression pattern observed for FvLAR (FvRG_4g0010500) in the diploid RG (Fig. 5c). In addition, AHA10 showed identical expression patterns between RG and 'Yanli' (Fig. 5b). These findings suggest potential functional conservation of this gene between diploid and octoploid strawberries. Our comparative transcriptomic analysis of fruit development stages not only reveals ploidy-specific differences in anthocyanin biosynthesis pathways but also provides a comprehensive framework for elucidating the molecular mechanisms underlying anthocyanin synthesis regulation in Fragaria species.
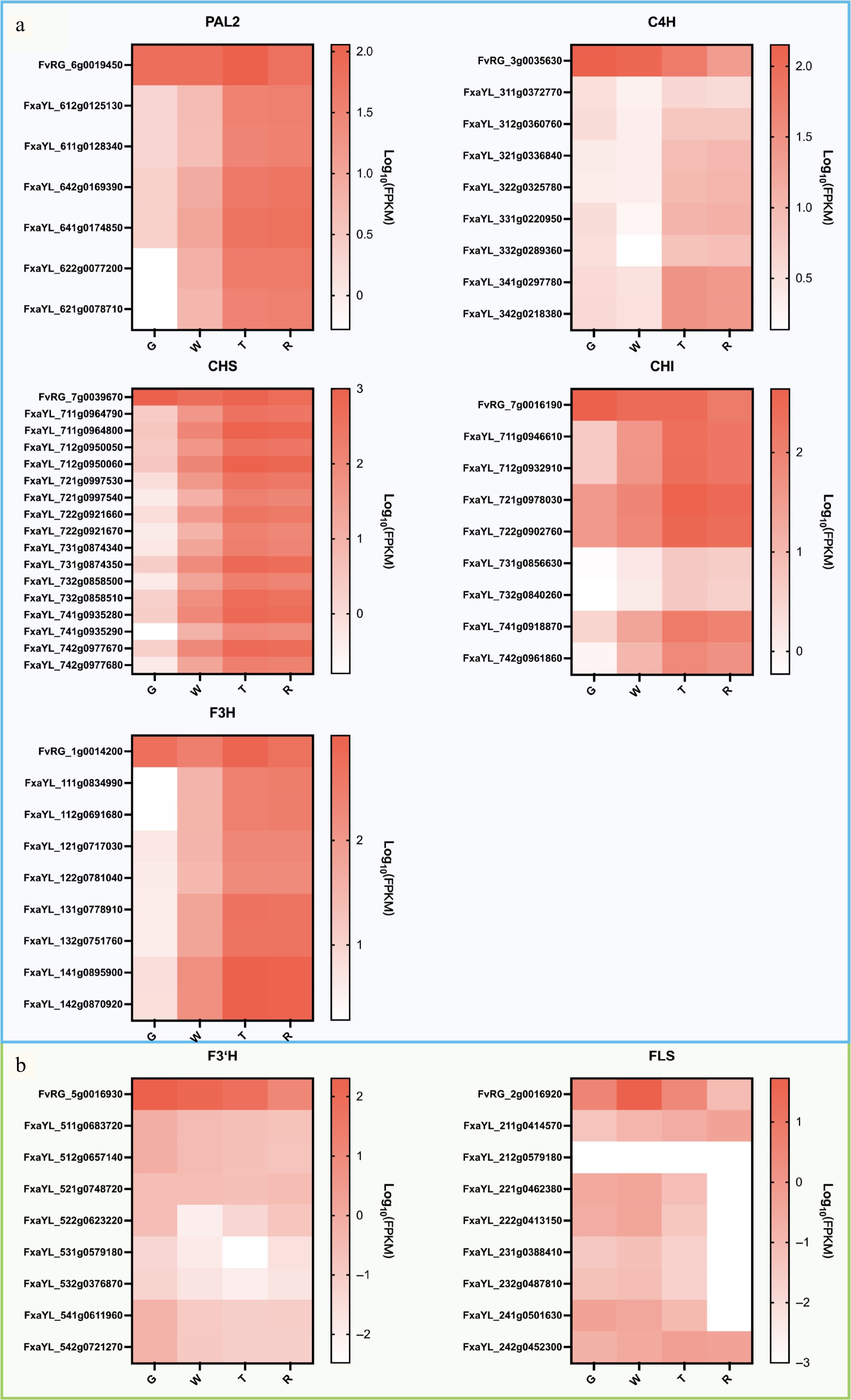
Figure 4.
Heat map of expression levels of EBGs during fruit development in 'Ruegen' (RG) and 'YanLi' (YL). (a) Five EBGs show divergent expression patterns between 'Yanli' (YL) and RG while maintaining consistent allelic expression within YL. (b) F3'H and FLS exhibiting expression patterns concordant with specific YL alleles. The color scale represents log10(FPKM + 0.0001) normalized expression values.
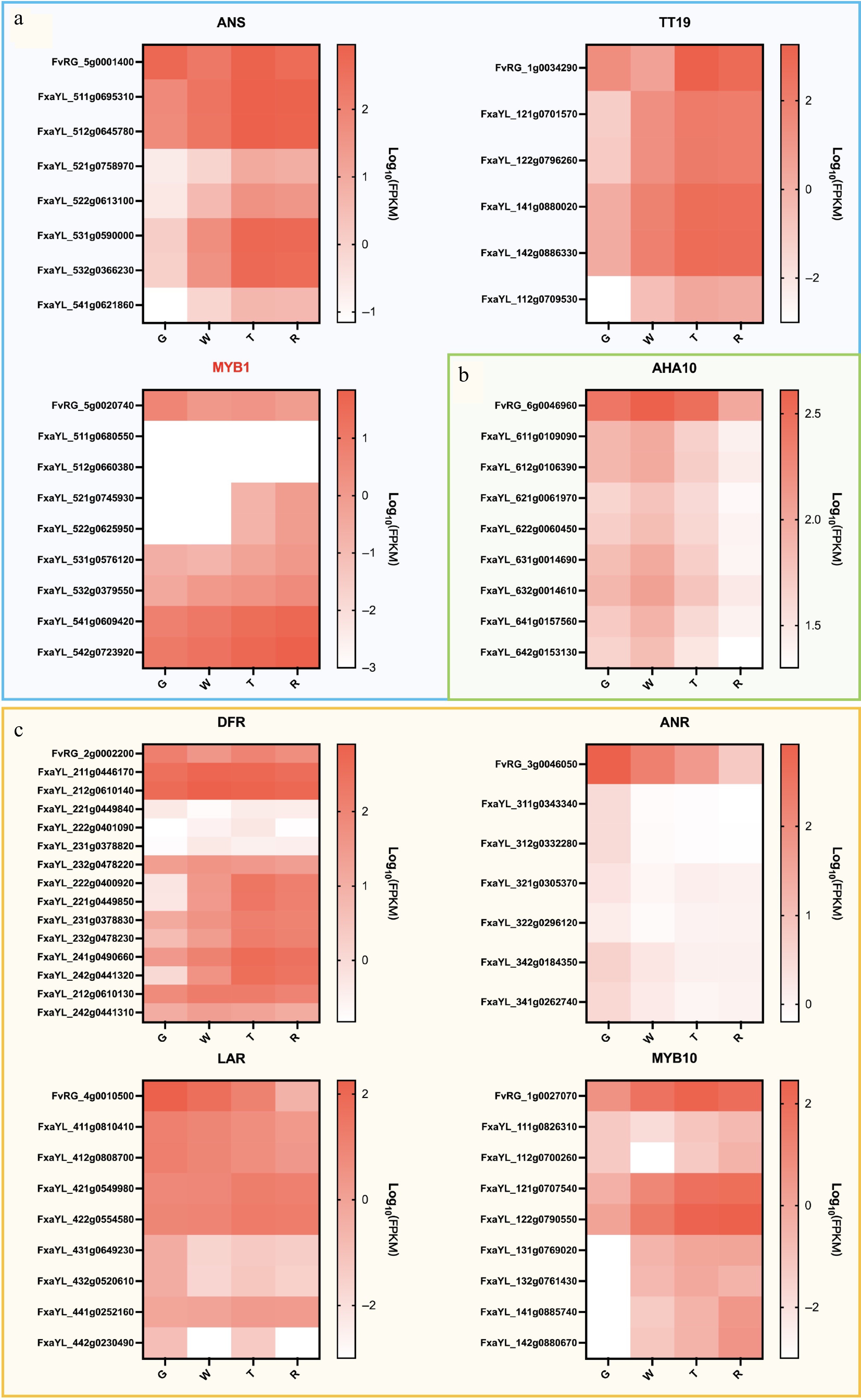
Figure 5.
Heat map of expression levels of LBGs during fruit development in 'Ruegen' (RG) and 'YanLi' (YL). (a) ANS, TT19, and MYB1 show divergent expression patterns between 'Yanli' (YL) and RG while maintaining consistent allelic expression within YL. (b) The expression pattern of AHA10 gene is similar in YL and RG. (c) Four RG LBGs exhibiting expression patterns concordant with specific YL alleles. The color scale represents log10(FPKM + 0.0001) normalized expression values.
-
Current genomic resources for Fragaria vesca feature high-quality assemblies across major accessions. Notably, the 'Yellow Wonder' genome assembly spans 220 Mb with an exceptional scaffold N50 of 34 Mb[6]. The 'Hawaii 4' accession, recognized as the first sequenced strawberry variety, has recently been enhanced through telomere-to-telomere (T2T) technology, yielding an upgraded version 6.0 assembly of 220.8 Mb with a scaffold N50 of 34.34 Mb[5]. In this study, the chromosome-scale assembly of the 'Ruegen' strawberry was reported, comprising 221.31 Mb with a contig N50 of 8.35 Mb. This resource substantially enriches genomic tools for this widely utilized diploid strawberry model. Collectively, these assemblies provide comprehensive coverage of principal F. vesca research accessions, establishing a robust foundation for comparative genomic analyses within the species.
The 'Ruegen' accession, a key model system for strawberry genetic transformation studies, has historically utilized the reference genomes of 'Hawaii 4' and 'Yellow Wonder' as genomic proxies. However, this approach presents significant constraints for anthocyanin research due to fundamental differences in pigmentation phenotypes among these accessions. Recent studies have focused on elucidating the molecular mechanisms underlying fruit color variation in strawberry cultivars. The R2R3-MYB transcription factor MYB10 is a well-characterized activator of structural genes in the strawberry anthocyanin biosynthesis pathway. Mutations in MYB10 have been identified as the primary cause of variations in anthocyanin accumulation and distribution in both diploid (F. vesca) and octoploid (F. × ananassa) strawberries[32]. For instance, in the white-fruited cultivar 'Snow Princess', an 8-bp insertion at the C-terminus of FaMYB10-2 leads to premature translation termination[33]. Recent telomere-to-telomere genome sequencing of the white-fruited 'Chulian' strawberry revealed two critical mutations in FaMYB10, an 8-bp insertion in FaMYB10 on chr1-2-1, and a C→A point mutation in FaMYB10 on chr1-2-2[34]. In the white-flesh mutant 'Xiaobai', a frameshift insertion mutation (FaMYB10AG-insert) of FaMYB10 caused the low expression of FaUFGT and low anthocyanin content in ripe flesh, which is key to producing white flesh and fruit[35]. Additionally, A missense mutation in FvF3H impaired the encoded enzyme's catalytic efficiency, disrupting the anthocyanin biosynthesis pathway and resulting in pink strawberry fruit pigmentation[36].
Comparative transcriptome analysis across successive developmental stages provides critical insights into the dynamic physiological and biochemical regulation of fruit maturation. To enable cross-ploidy comparisons, this approach was extended through parallel RNA-seq of octoploid strawberry cultivars. Comparative transcriptomic analysis of anthocyanin-related genes between diploid and octoploid strawberries reveals ploidy-dependent variation in anthocyanin biosynthesis pathways. The genes can be clustered with similar expression patterns across different samples, which may help infer their potential functions based on known gene roles. For example, when studying anthocyanin-related genes, it was found that MYB1 exhibited completely opposite expression patterns during fruit development in both RG and 'Yanli' strawberry varieties. MYB1 has been established as a critical negative regulator of anthocyanin biosynthesis in multiple Fragaria species[37,38]. Functional analyses reveal that silencing FcMYB1 expression partially restores anthocyanin biosynthesis in white-fruited varieties, leading to red pigmentation development[39]. Temporal expression profiling shows that MYB1 homologs in both F. chiloensis and F. × ananassa reach peak transcript levels during late fruit ripening stages, consistent with our observations of FaMYB1 expression in the octoploid 'Yanli' cultivar. Notably, the diploid F. vesca 'Ruegen' exhibits an opposite FvMYB1 expression pattern compared to octoploid varieties. This functional divergence suggests that MYB1 serves as a key regulator of ploidy-dependent anthocyanin accumulation patterns in Fragaria. The observed differences likely reflect fundamental variations in anthocyanin pathway regulation between diploid and octoploid strawberries. Future investigations should characterize MYB1 regulatory networks across ploidy levels and identify interacting partners that modulate its activity.
In addition, a comparative analysis of gene expression patterns between diploid and octoploid strawberries during fruit development could provide valuable insights into the genetic evolutionary mechanisms underlying cultivated strawberry domestication. Current genomic evidence suggests that modern octoploid strawberries (Fragaria × ananassa) originated through polyploidization events involving at least four diploid ancestors[40]. Specifically, the F. vesca-derived subgenome demonstrates predominant influence[41]. These analyses (Figs 4 & 5; Supplementary Fig S2) reveal that in the 'Yanli' cultivar, most genes exhibit dominant expression from the A-subgenome (F. vesca). However, we identified several notable exceptions to this pattern. For example, two A-subgenome genes (FxaYL_511g0680550 and FxaYL_512g0660380) from the FaMYB1 cluster showed no detectable expression (FPKM = 0; Supplementary Table S6). In contrast, two B-subgenome genes (FxaYL_121g0707540 and FxaYL_122g0790550), representing functional components of FaMYB10, displayed significant expression levels. Modern sequencing technologies enable deeper comparative genomic and transcriptomic analyses across species. In this study, a high-quality chromosome-scale assembly of the Fragaria vesca 'Ruegen' genome was successfully generated using Hi-C scaffolding. The completed genome spans 221.31 Mb with contig N50 = 8.35 Mb, representing the first comprehensive reference genome for the widely used 'Ruegen' transformation model. This high-precision genomic resource will facilitate advanced molecular research in strawberry.
This work was supported by the National Natural Science Foundation of China (Grant No. 32130092) and the project of Liaoning Province Germplasm Innovation Grain Storage and Technology Special Program (Grant No. 2023020525-JH1/102-04).
-
The authors confirm their contributions to the paper as follows: experiments design, manuscript writing and revision: Li X, Zhang J, Zhang Z; conducting bioinformatic analysis, data analysis: Li X, Liu S. All authors reviewed the results and approved the final version of the manuscript.
-
The whole genome sequence data reported in this paper have been deposited in the Genome Warehouse in the National Genomics Data Center[42,43], Beijing Institute of Genomics, Chinese Academy of Sciences/China National Center for Bioinformation, under accession number GWHFPXR00000000.1, which is publicly accessible at https://ngdc.cncb.ac.cn/gwh.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 Hi-C data of various types.
- Supplementary Table S2 Hi-C assembly data.
- Supplementary Table S3 BUSCO assessment.
- Supplementary Table S4 Statistics of non-coding RNA annotation.
- Supplementary Table S5 Table of sequence alignment results between sample sequencing data and selected reference genome of RG and 'Yanli'.
- Supplementary Table S6 Gene ID and FPKM of anthocyanin synthesis-related genes.
- Supplementary Fig. S1 Fruit developmental stages of (a) 'Ruegen' and (b) 'Yanli' cultivars.
- Supplementary Fig. S2 The expression patterns of all alleles in 'Yanli' are different from those of RG genes. The color represents the log10 (FPKM+0.0001) gene expression level in the sample.
- Copyright: © 2025 by the author(s). Published byMaximum Academic Press, Fayetteville, GA. This articleis an open access article distributed under Creative CommonsAttribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Li X, Liu S, Zhang J, Zhang Z. 2025. Genome assembly and transcriptome profiling of the woodland strawberry (Fragaria vesca) 'Ruegen'. Fruit Research 5: e031 doi: 10.48130/frures-0025-0022 

Genome assembly and transcriptome profiling of the woodland strawberry (Fragaria vesca) 'Ruegen'
- Received: 26 April 2025
- Revised: 25 May 2025
- Accepted: 30 May 2025
- Published online: 11 August 2025
Abstract: The diploid woodland strawberry, Fragaria vesca (2n = 2x = 14), has emerged as a premier model system for Rosaceae functional genomics due to its compact genome, low heterozygosity, and tractable genetic transformation. Commonly used research accessions for woodland strawberry include 'Ruegen' with red fruits, 'Yellow Wonder', and 'Hawaii 4' with yellow fruits. While reference genomes are available for 'Yellow Wonder' and 'Hawaii 4', 'Ruegen' has remained lacking in this regard. In this study, SMRT and Hi-C technology were employed to assemble the 'Ruegen' genome, yielding a total size of 221.31 Mb with a contig N50 of 8.35 Mb. Gene prediction identified 36,071 protein-coding genes, of which 85.79% exhibited significant homology to sequences in the NCBI NR database. RNA-seq of fruits from the diploid 'Ruegen' and octoploid 'Yanli' varieties was conducted across multiple developmental stages. Comparative transcriptomic analysis revealed divergence in anthocyanin-related gene expression patterns. Notably, MYB1 homologs exhibited diametrically opposed expression profiles during fruit development between 'Ruegen' and 'Yanli'. These integrated genomic and transcriptomic resources significantly advance strawberry functional genomics research.
-
Key words:
- Fragaria vesca /
- Ruegen /
- Genome /
- RNA-seq