









-
Watercress (Nasturtium officinale R. Br.), originated from European Mediterranean area, distributed in south and north America and Asia, belongs to the cruciferous herbs. Watercress is widely cultivated as a horticultural crop, with 124 acres of cultivation in the UK and 638 acres in the US[1,2]. It has been used as vegetable to make salads and soups throughout a long history and contains abundant nutrition including vitamin C, minerals, and bioactive compounds[3,4]. Since watercress is rich in polyphenols and glucosinolates[5,6], it is helpful for preventing diabetes and gastrointestinal disease[7]. Due to its cultivation area near rivers and streams, watercress manifests robust endurance to extreme flooding stress. In addition, the regeneration capacity of watercress is strong and it can be reproduced from cuttings. Even though many genes and gene families involved in the flooding stress response and regeneration capacity have been identified in various plants, it is still an open question how watercress adapted to flooding stress and has a strong regenerative capacity from the perspective of evolutionary genomics.
About 5.6 million years ago (Mya), the Mediterranean Sea became disconnected from the world's oceans, and desiccated during the Messinian salinity crisis[8]. About 5.33 Mya, the Atlantic waters rapidly refilled the Mediterranean through the present Gibraltar Strait, which is known as the Zanclean flood. The flood may have lasted for up to several thousand years. The Mediterranean Sea level rose more than 10 m per day during this extremely abrupt flood. This long term flood put enormous flooding pressure on plants near the Mediterranean Sea.
Polyploidy arising from whole-genome duplication (WGD) is an important mechanism for plants in adapting to harsh environments. Cruciferae is a large family containing more than 3,700 species[9]. At present, more than 30 species in the cruciferous family have been sequenced. The first acquired plant genome, the Arabidopsis thaliana reference genome, offers convenience for researchers[10]. In the genome research of cruciferous WGD, it is found that cruciferous species share the core eudicot common hexaploidization (ECH), which occurred approximately 140 Mya[11]. The Aethionema genus diverged the earliest in the family. After separation of the genus Aethionema, the remaining species share a mutual WGD which occurred in Arabidopsis thaliana, Cardamine hirsuta[12], and Capsella rubella[13], etc. approximately 47 Mya[14]. Meanwhile, Brassica species experienced an extra whole genome triplication (WGT) which causes a difference involving gene loss in three subgenomes and subsequent genomic rearrangement[15,16]. Under environmental pressure, genome polyploidization leads to variations in the plant genome's size, structure, and function[17−19]. It plays a crucial role in morphological characteristics, ecological habits, and promoting evolution.
At present, most of the studies in watercress generally concentrate on the application of functional compounds. Germplasm innovation and molecular breeding in watercress are still slow. It is still an open question that how watercress adapted to flooding stress and acquired a strong regenerative capacity from the perspective of evolutionary genomics. With the improvement of high-throughput sequencing technology and reducing costs, it is common to obtain reference genomes via sequencing. Reference genomes play important roles in research and can provide the gene positions and gene sequences. Therefore, it is necessary to obtain the watercress genome to lay a foundation for subsequent research. In our study, we present a high-quality and chromosome-scale genome assembly of watercress using a combination of PacBio long reads, Illumina HiSeq, optical mapping, and Hi-C chromatin interaction maps. Our assembled watercress genome achieves a high level of continuity and completeness. We explain how watercress adapts to the aquatic environment from the perspective of genomics. This research not only provides valuable resources for comparative analyses of Cruciferae genomes, but also facilitates the genomics-assisted breeding of watercress in the future.
-
The genomes of Aethionema arabicum, Leavenworthia alabamica, Arabidopsis thaliana, Arabidopsis lyrata, and Carica papaya were downloaded from Phytozome v13 (
https://phytozome-next.jgi.doe.gov/ ) and the genomes of Brassica rapa (v3.0), Brassica olercea (v1.1), Thellungiella halophila, Thellugiella parvula, Capsella rubella, and Cardamine hirsuta were downloaded from the Brassicacea Database (http://brassicadb.org/brad/ ). The genome of Malus domestica (GDDH13V1.1) was downloaded from the Rosaceae database (https://iris.angers.inra.fr/gddh13/the-apple-genome-downloads.html ), and the genome of Nelumbo nucifera was downloaded from the lotus database (http://lotus-db.wbgcas.cn ).Genome sequence and assembly
-
The plant material was provided by Seed Needs company (
www.myseedneeds.com ). Stems derived from the same watercress plant were used for cuttings planted into perlite, and the root tissues were collected for genomic DNA isolation and library construction. Genomic DNA was extracted using the cetrimonium bromide (CTAB) method, size fractionated with BluePipin (Sage Science, Inc, MA, USA), used for library construction following the PacBio SMRT library construction protocol, and sequenced on the PacBio Sequel platform (Pacific Biosciences, CA, USA). After passing Nanodrop and Qubit quantitative testing, the library for sequencing was constructed using large (> 20 kb) DNA fragments and sequenced using the PacBio Sequel platform. The data obtained by sequencing were subjected to quality control to obtain subreads and was used to assemble the genome de novo. BLASR[20] was used to align the subreads sequenced by PacBio to the assembly results and then correct the assembly results with Arrow. Contamination by bacterial and fungal sequences was filtered through the three rules of control GC content, PacBio subreads' coverage depth, and Illumina reads' coverage depth. The Illumina second-generation sequence data were aligned to the assembly results after decontamination treatment using BWA-MEM software[21]. Pilon-1.20 was used to further correct the assembly results. BioNano optical spectra assisted the assembly. According to the BioNano company's methods, genomic DNA with high molecular content was extracted from leaf tissues and digested using Nt.BspQI. The fluorescent label was made. Finally, the labeled DNA molecules wereimaged by the Irys imaging system. The molecules that satisfied the following conditions were filtered out and subjected to data cleaning: (1) molecules < 150 kb; (2) molecule signal to noise ratio (SNR) < 2.75 and label SNR < 2.75; (3) label intensity > 0.8. The results of the PacBio assembly were mapped to the BioNano optical spectra and used to construct the scaffold. HiC-Pro[22] was used to evaluate the quality of Hi-C data. Finally, the final assembly result was evaluated using BUSCO[23].Genome size estimation
-
A sequencing library with an insert length of 400 bp was constructed and sequenced using the Illumina Hiseq X Ten platform. Sequenced data were compared with plant chloroplast and mitochondrial databases using BWA-MEM software for quality control and then were used for k-mer analysis. The genome size was calculated according to the following formula: Genome size = k-mer number/k-mer peak depth.
RNA-seq
-
Total RNA was isolated separately from the blades, flowers, petioles, roots, and siliques of three uniformly growing N. officinale individuals using a QIAGEN RNeasy plant mini kit (QIAGEN, Hilden, Germany). Thereafter, RNA-seq libraries were constructed with the TruSeq Sample Preparation (Illumina Inc, CA, USA) and IsoSeq Library Construction kits (Pacific Biosciences, CA, USA). Paired-end sequencing with 150 bp was conducted on the HiSeq X Ten platform (Illumina Inc, CA, USA). In addition, full-length transcriptome sequencing was conducted for mixed samples by using the PacBio Sequel platform.
Gene prediction and annotation
-
The methods of homology prediction, transcriptome-based sequencing, and de novo annotation were used to predict the genes of the watercress genome. The protein sequence of watercress was aligned with the protein sequences of Aethionema arabicum, Arabidopsis thaliana, Brassica rapa, Brassica oleracea, Capsella rubella, Malus domestica, Nelumbo nucifera, and Thellugiella parvula. The gene set used for predicting homology was obtained with GeneWise software. Based on the root, stem, leaf, flower, petiole, and silique tissues of watercress, Iso-seq (mixed sequencing) and RNA-seq (each tissue is sequenced separately) data were used for transcriptome prediction using PASA[24], and 1,000 genes were randomly selected from the results of transcriptome prediction and were used to test the model parameters using Augustus[25], Geneid[26], and SNAP software. The results of the test were used for de novo annotation. Finally, the EvidenceModeler (EVM) was used for integration. The genome of the watercress was aligned with the Rfam database (release 13.0) through BLASTN (E-value ≤ 1e-5) to identify the rRNA, snRNA, and miRNA genes in the genome. tRNAscan-SE v1.3.1 software[27] was used to annotate the tRNA gene, and RNAmmer v 1.2 software[28] was used to predict rRNA and subunits. Predicted protein sequences were aligned with the SwissProt and TrEMBL databases (E-value ≤ 1e-5), and the best result was used for functional annotation. KAAS[29] was used to analyze the metabolic pathways that the protein-coding genes may participate in InterProScan v5.24[30] was used to align the data with public databases such as Pfam, SMRT, ProDom[31], PANTHER, PRINTS, and PROSITE[32], and predict gene functions through protein domains and motifs. The Gene Ontology (GO) functional annotation of each gene was obtained through the corresponding InterPro entry.
Repetitive element prediction
-
Simple sequence repeats (SSRs) were identified using MISA[33], and the repeat sequences were identified using homologous prediction and de novo annotation. The genome was aligned with the Repbase transposon database and the Mips-REdat database. RepeatMasker software[34] was used for the identification of repeated sequences at the DNA level. Extensive de-novo TE Annotator (EDTA) was used for de novo annotation, and two results were combined to obtain the final repeated sequence.
Gene family clustering and GO enrichment analysis
-
Gene families were clustered into watercress, Aethionema arabicum, Arabidopsis lyrata, Arabidopsis thaliana, Brassica rapa, Brassica oleracea, Capsella rubella, Cardamine hirsuta, Leavenworthia alabamica, Thellungiella halophila, Thellugiella parvula, and Carica papaya using OrthoMCL software[35]. The longest transcripts were retained. BLASTP software was used to align them with protein sequences of the selected species (E-value ≤ 1e-5), and then the Markov Cluster Algorithm (MCL) was used to identify homologous gene families in each species.
Ks analysis
-
The coding sequences (CDSs) from watercress were aligned with those of other species using LAST (v 2.27.1). The tandem repeats and low scores were filtered to obtain high-quality aligned results. JCVI mcscan[36] (Python version) was used to identify collinear blocks within a genome or between different genomes, each containing at least five collinear gene pairs. The MCScanX script was used to calculate the Ks value between gene pairs.
Analysis of WGD
-
The syntenic orthologs of watercress and other Brassicaceae species were found using LAST (v 2.27.1) with watercress as the query genome and other Brassicaceae species as the subject genome. The dot plot was painted by graphica dot plot in the JCVI mcscan program. The syntenic blocks between Aethionema arabicum and Arabidopsis thaliana, Cardamine hirsuta were identified using JCVI mcscan (Python version). We selected the doubling genes and highlighted them in four colors.
Gene retention after WGD
-
The syntenic blocks within watercress were identified using JCVI and the tandem repeats were filtered. The recently doubled genes were aligned with Arabidopsis thaliana using the BLAST program blastp (E-value ≤1e-5). The homologous genes from Arabidopsis thaliana were analyzed by AgriGO v2.0 (
http://systemsbiology.cau.edu.cn/agriGOv2/index.php ). Brassicaceae group analysis was selected. The analysis tool selected was singular enrichment analysis (SEA). Hypergeometric distribution was used for the statistical test. The significance level was 0.05. The minimum number of mapping entries was 5. The gene ontology type was complete GO. Pfam-A was downloaded from the Pfam database (http://pfam.xfam.org/ ). Genes related to leaf development were selected. HMM scan[37] and PlantTFDB (http://planttfdb.gao-lab.org/ ) were used to search the transcription factors. The expression patterns were constructed with the ggplot package (R).Positively selected gene analysis
-
We calculated the Ka and Ks values by using the collinear gene pairs obtained by alignment within watercress and constructed the dot plot with an R script. The genes (Ka/Ks > 1) were selected and aligned with Arabidopsis thaliana using BLASTP (E-value ≤ 1e-5). We annotated the gene with hmmer and BLASTP, and identified the transcription factors with PlantTFDB and BLASTP. The expression patterns were constructed with the ggplot package (R).
Regeneration-related gene analysis
-
Some genes such as WOX transcription factors and the SERK gene family promote the regeneration of side branches. The protein domains of homeobox and leucine-rich repeat (LRR) in watercress, Arabidopsis thaliana, and Brassica rapa were identified using Pfam and hmmer 3 software. WOX transcription factors were detected by the accession number 'PF00046', and SERK genes were detected by the accession number 'PF13306' of the Pfam annotation. We counted the distribution of the four kinds of genes and calculated the gene expression levels in different organs. Mafft (v7.271)[38] was used for multiple alignment, and Fasttree[39] was used to construct the phylogenetic tree with maximum likelihood. MEGA software and ITOL (
https://itol.embl.de/ ) were used to modify and illustrate the results. The expression patterns were constructed with the ggplot package (R). -
The genome size of watercress was ~346.0 Mb, as estimated by the k-mer analysis (k = 17; peak depth = 21×) based on Illumina paired-end short reads (Supplementary Fig. S1). To achieve a chromosome-level assembly of the watercress genome (Fig. 1a), we further generated 141× coverage of PacBio SMRT long reads (48.7 Gb with an N50 length of 14.7 kb), a 263× optical map data (90.9 Gb with an N50 length of 191.3 kb), and 338× Hi-C data (117.0 Gb). The initial assembly was performed based on PacBio-only data and then polished with PacBio long reads and Illumina short reads. After potential contamination sequences were eliminated, the total length of the assembled genome was 330.0 Mb, with a contig number of 488 and an N50 size of 2.45 Mb. Subsequently, the contigs were scaffolded using a BioNano-based consensus map, which resulted in 361 scaffolds with an N50 length of 5.85 Mb. The total length of the final assembled scaffolds was 337.51 Mb, corresponding to 97.5% of the genome size estimated by the genome survey (Supplementary Tables S1–S4). Assembly quality assessment showed that 97.9% of complete genes could be identified in BUSCO (Supplementary Table S5). Lastly, 96.6% of the assembly was anchored onto 16 pseudo-chromosomes using Hi-C data (Supplementary Fig. S2; Fig. 1b; Supplementary Table S6).
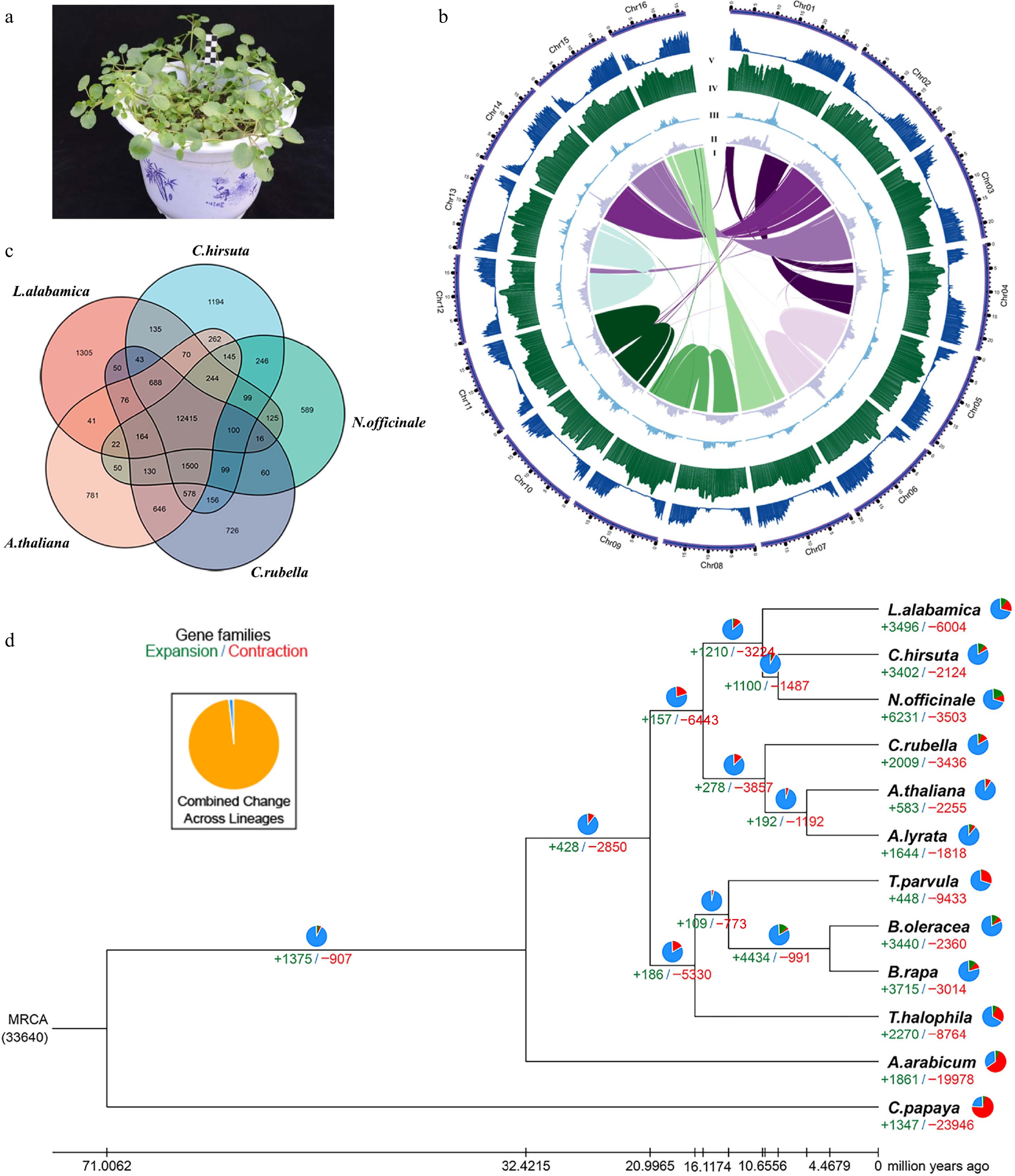
Figure 1.
Plant morphology, genome assembly, and genome features of watercress. (a) the watercress cultivar used in this study. (b) The landscape of the watercress genome: syntenic blocks (I), copia density (II), gyspy density (III), GC content (IV), and gene density (V). (c) Venn diagram representing the shared and unique gene families among watercress, A. thaliana, C. hirsuta, C. rubella, L. alabamica, and N. officinale. (d) Phylogenetic tree showing the divergence time and the evolution of gene family size for 12 species including watercress. The numbers at the branches represent gained (green) and lost (red) gene families. The small pie charts represent the percentage of expanded (green), contracted (red), and unchanged (blue) gene families.
By combining ab initio, homology-based, and transcriptome data, a total of 38,945 protein-coding genes were annotated with an average length of 2,040 bp, an average CDS length of 1,189 bp, and a mean number of 5.6 exons per gene (Supplementary Table S7). Among the predicted protein-coding genes, 37,868 (97.2%) received known functional annotation in a public database (Supplementary Table S8). In addition, annotation of noncoding RNA genes yielded 2,043 rRNAs, 558 snRNAs, 320 miRNAs, and 884 tRNAs (Supplementary Table S9). We annotated repetitive sequences by combining homology-based and de novo prediction. Approximately 49.15% (165.88 Mb) of the assembled genome was annotated as repetitive sequences (Supplementary Table S10). Long terminal repeats (LTRs) are the most abundant in the watercress genome, representing 41.96%. A total of 129,308 SSRs were identified in the watercress genome, most of which are mono-nucleotide repeats (61.22%) and di-nucleotide repeats (27.76%) (Supplementary Table S11).
Gene family evolution and phylogenetic analysis
-
OrthoMCL gene family clustering was conducted, based on the predicted proteomes of watercress and 11 other sequenced species. Consequently, 32,248 watercress genes were clustered into 16,004 gene families, including 315 watercress-unique families, whereas 6,697 genes were unclustered (Supplementary Table S12). Among all the studied species, B. oleracea has the largest number of gene families (21,034) and unique gene families (1,205). On the other hand, ortholog analysis revealed that watercress, A. thaliana, C. hirsuta, C. rubella, L. alabamica, and N. officinale shared a core set of 12,415 gene families (Fig. 1c).
To gain insight into the evolution of watercress, a maximum likelihood phylogenetic tree was constructed for the 11 Brassicaceae species and one outgroup species (Carica papaya) based on 675 single-copy gene families. Molecular dating suggested that watercress diverged from C. hirsuta around 9.2 Mya, following the divergence of the most recent common ancestor of the two species and L. alabamica around 10.7 Mya (Fig. 1d). Analyses of gene family expansion and contraction found that 6,231 gene families expanded and 3,503 families became smaller in the watercress genome.
In order to further predict their biological functions, the genes in the expansion gene family and the specific gene family were analyzed by GO enrichment. It was found that the genes in the expanded and specific gene families were significantly enriched in the processes of auxin transport and regulation of hormone levels (Supplementary Fig. S3).
Watercress-specific WGD occurred about 4.7 to 12.6 Mya
-
To estimate the WGD events during the evolutionary course of watercress, we first plotted the distributions of Ks (synonymous substitution rate) for paralogous genes within the genomes of Arabidopsis thaliana, Carica papaya, and Nasturtium officinale. The Ks distributions of orthologous genes between Nasturtium officinale and four species (Aethionema arabicum, Arabidopsis thaliana, Brassica rapa, and Cardamine hirsuta) were also plotted (Fig. 2a). The Ks distributions of paralogs show that the Ks peak of the WGD event (β) is obvious in C. papaya, but it is slightly lower in watercress. Two additional obvious Ks peaks representing the WGD for watercress were identified: one peak between 0.5 and 1 shared by A. thaliana (α event) and another peak specific to watercress between 0 and 0.5. Moreover, the Ks peak distributions of orthologs were used to infer the divergence time. Watercress first diverged from A. arabicum and then from B. rapa, A. thaliana, and C. hirsuta successively. A comparison of the Ks peak of the watercress WGD with Ks peaks of the orthologs among species indicated that a recent independent WGD event occurred in watercress after the divergence from A. thaliana and C. hirsuta. The time of the watercress-specific WGD was estimated to be at around 4.7 to 12.6 Mya[40], coinciding with Zanclean flood at about 5.33 Mya. Therefore, we hypothesised that the independent WGD in watercress could be related to the Zanclean flood.
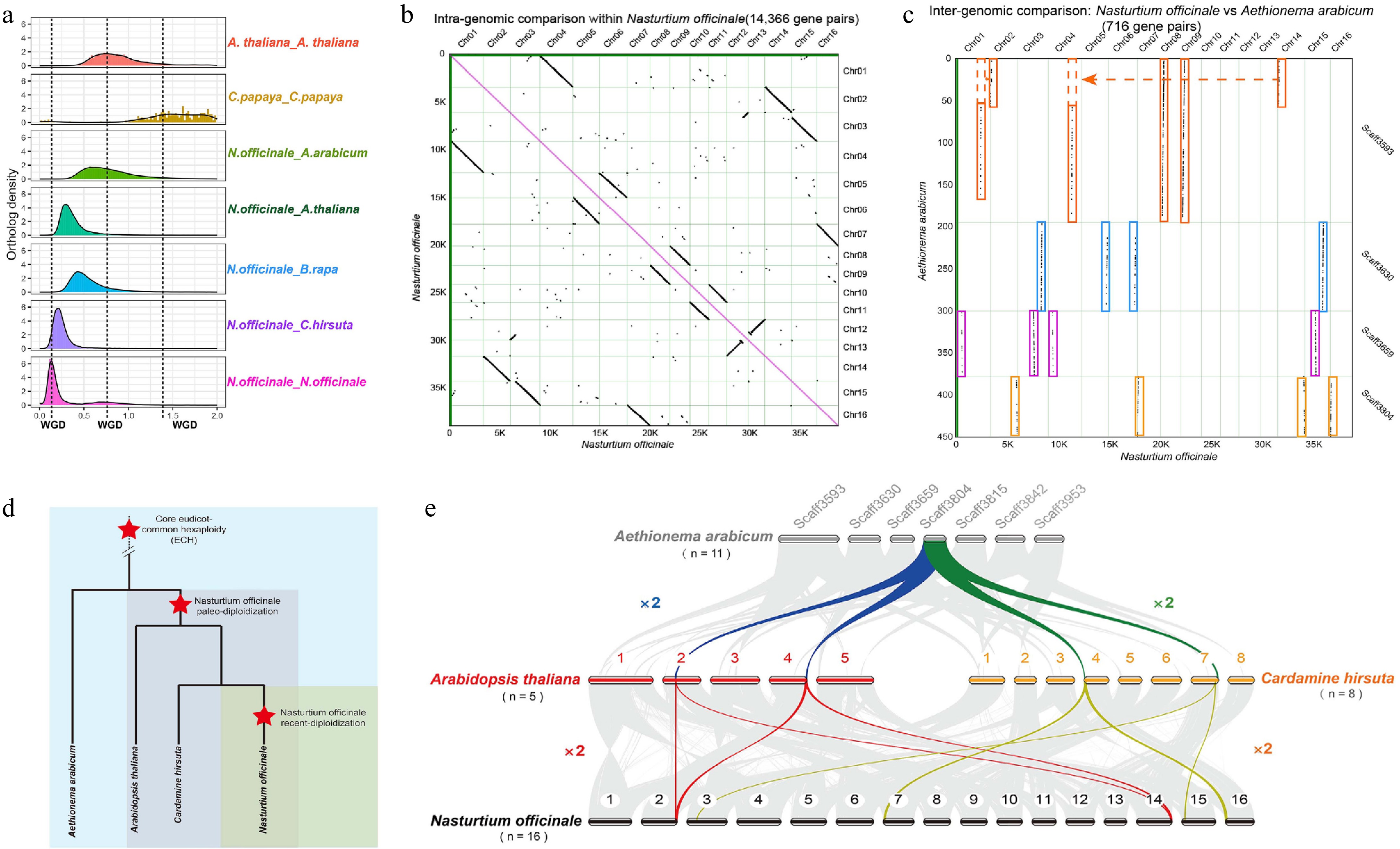
Figure 2.
Whole-genome duplication in the watercress genome. (a) The density distribution of Ks for paralogs and orthologs within and between species. (b) Dot plot of syntenic genes within the watercress genome. (c) Dot plot of syntenic genes between the genomes of A. arabicum and watercress. (d) Model of species divergence. (e) Model of species WGD.
To further elucidate the extra WGD of the watercress genome, dot plots of paralogs within watercress (Fig. 2b) as well as orthologs between watercress and A. arabicum (Fig. 2c) were constructed. In detail, a total of 127 syntenic blocks (with no fewer than five orthologs) including 14,366 gene pairs were identified with the watercress genome (Supplementary Table S13). We also identified 21 blocks including 716 collinear gene pairs between watercress and four scaffolds of A. arabicum (Supplementary Table S13). The dot plot between watercress and A. arabicum provided a clear 1:4 syntenic depth ratio. Furthermore, a syntenic depth ratio of 1:2 was inferred between watercress and A. thaliana (Fig. 2e). The analysis of syntenic relationships within and between species collectively indicated that watercress underwent an extra WGD event after its divergence from Arabidopsis thaliana. The first differentiation was from Aethionema arabicum, then from Arabidopsis thaliana, and the final differentiation was from Cardamine hirsuta (Fig. 2d, e). The watercress-specific WGD might provide ample genomic resources for the evolutionary adaptation to the flooded environment in the Mediterranean area.
Evolution of genes involved in regeneration capacity in the watercress genome
-
Strong regenerative ability is one of the mechanisms of plants' adaptation to aquatic environments. The regeneration-related genes mainly include the WOX and SERK gene families, LEC2, and BBM[41]. We identified 29 WOX and 9 SERK genes in watercress, while 28 WOX and 8 SERK genes were found in Brassica rapa (Fig. 3a–c; Supplementary Tables S14 & S15). Brassica rapa has fewer WOX and SERK genes than watercress. We did not identify SERK2 in Brassica rapa. Watercress retained 26 WOX and 6 SERK genes after WGD, accounting for 89.66% and 66.67% of each gene family, respectively. Furthermore, WOX6 was identified as a positively selected effect gene and was retained after WGD. Watercress RNA-seq data showed high expression of most SERK genes (Fig. 3d; Supplementary Table S16), and these might play an important role in the regeneration of watercress.
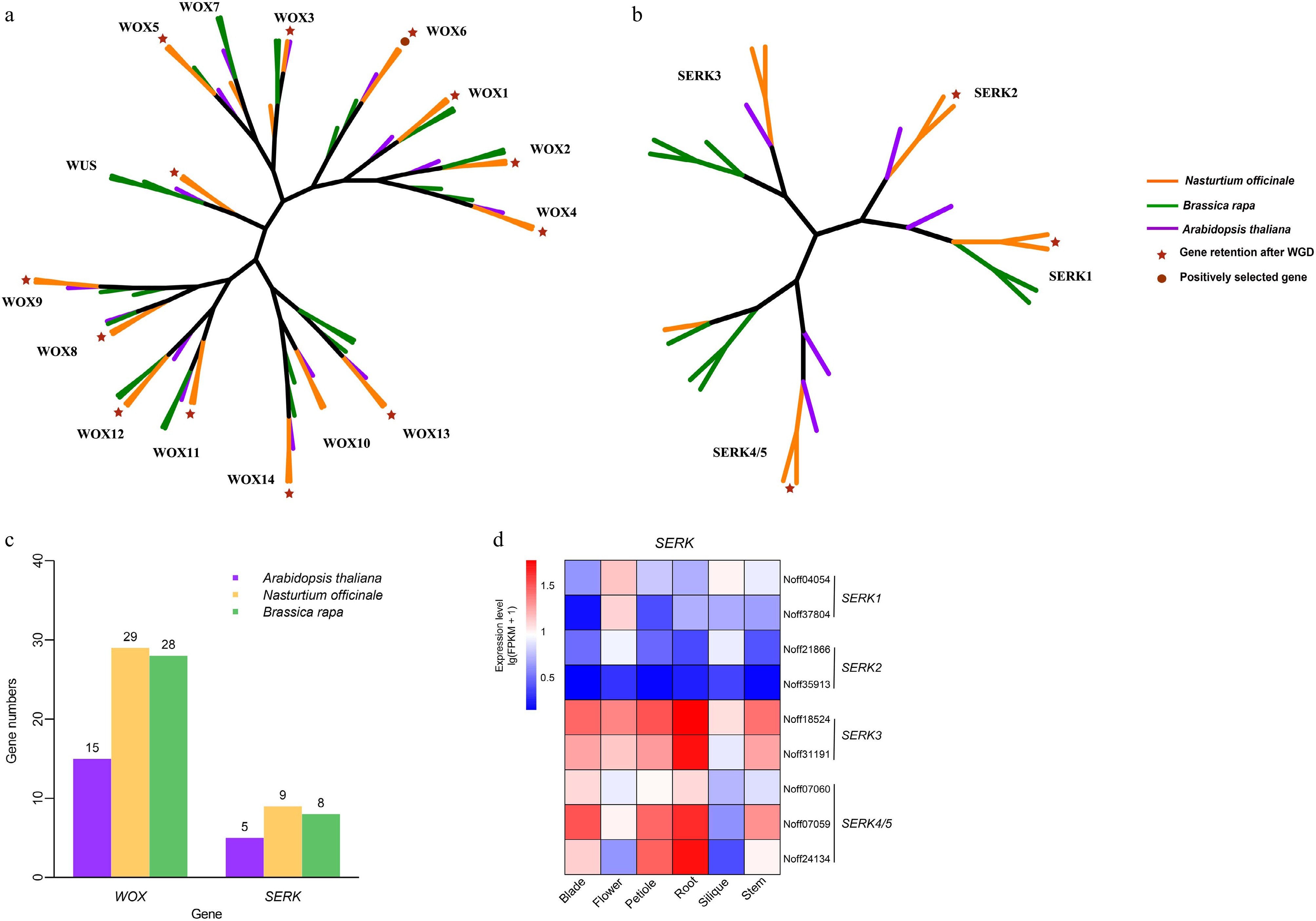
Figure 3.
Analysis of genes related to regeneration. (a) Phylogenetic tree of the WOX gene family. (b) Phylogenetic tree of the SERK gene family. (c) The distribution of WOX and SERK gene family members in Arabidopsis thaliana, Brassica rapa, and Nasturtium officinale. (d) The expression pattern of SERK gene family members in different organs of Nasturtium officinale.
Evolution of genes involved in leaf morphogenesis in the watercress genome
-
Decreasing leaf surface area is one of the plant's strategies to adapt to various abiotic stresses[42]. Research has shown that the TCP4 and TCP10 genes regulate plant leaf shape and size[43,44], and overexpression of the TCP4 and TCP10 genes results in a reduction in leaf surface area. The TCP4 and TCP10 genes could be silenced by the antagonist miRNA gene, MIR319[43]. In the watercress genome, WGD yielded two homologous copies each for TCP4 and TCP10. RNA-seq data showed that the TCP4 and TCP10 genes were highly expressed in watercress leaves (Fig. 4a; Supplementary Table S17). Interestingly, MIR319 is missing in the watercress genome. High expression of TCP4 and TCP10, combined with the deletion of MIR319, might contribute to the reduction in leaf area in watercress.
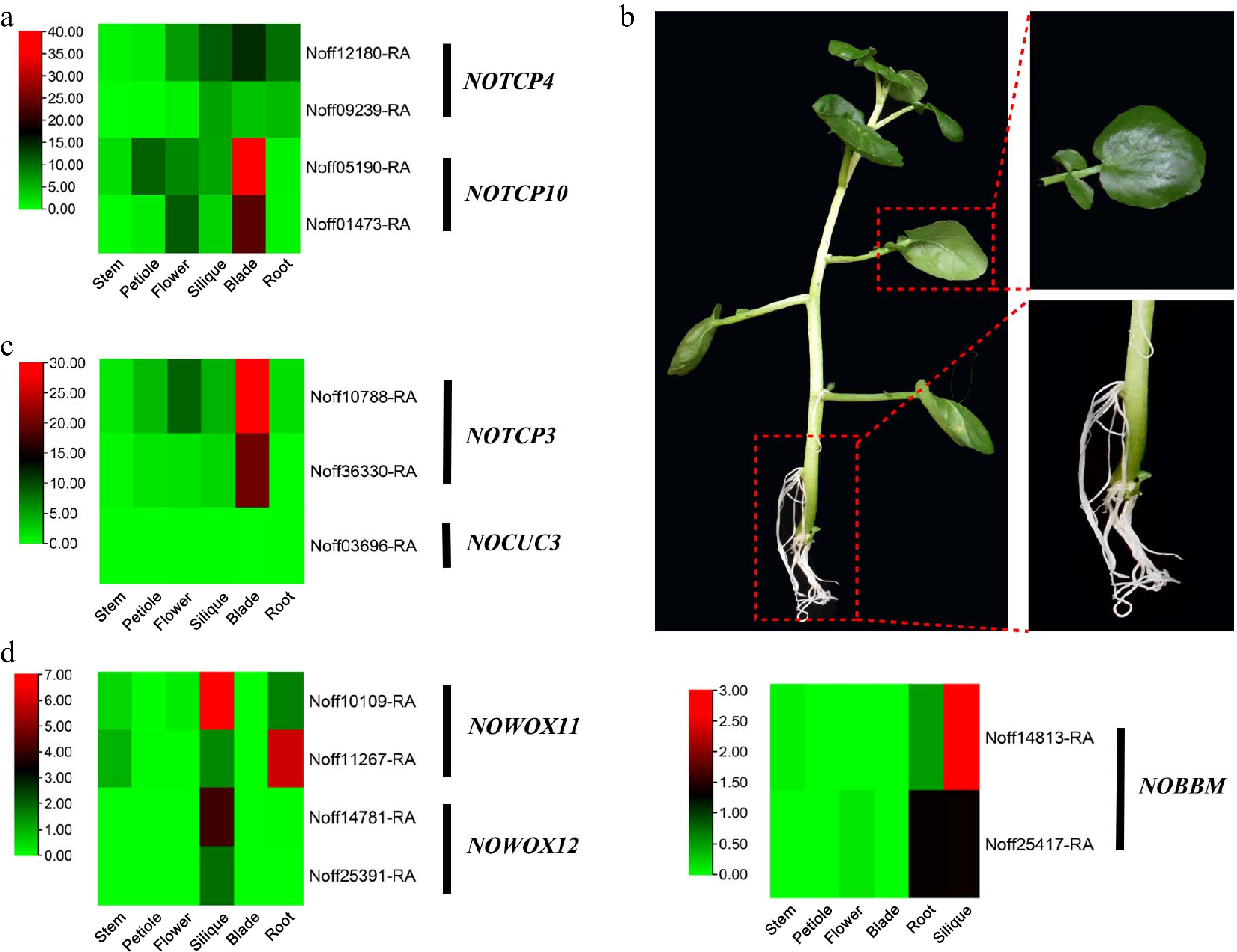
Figure 4.
Evolution of genes involved in leaf and root morphogenesis in the watercress genome. (a) Heatmap of the expression levels of NoTCP4 and NoTCP10 in different organs. (b) Leaves and aerial adventitious roots of watercress. (c) Heatmap of the expression levels of NoTCP3 and NoCUC3 in different organs. (d) Heatmap of the expression levels of NoBBM, NoWOX11, and NoWOX12 in different organs.
Watercress has broad leaves (Fig. 4b). Studies have shown that WOX1 and WOX3 are the key genes involved in leaf width[45,46]. Plant leaves appear as narrow strips if WOX1 and WOX3 genes are missing or broken. WGD yielded two homologous copies each for WOX1 and WOX3 in the watercress genome (Fig. 3a; Supplementary Table S14), which might help watercress to form broad leaves that float in the water.
The leaf margin of watercress is entire or nearly entire (Fig. 4b). The CUC2 and CUC3 genes both regulate the formation of serrated leaf edges[47]. In A. thaliana, increasing the expression level of CUC2 resulted in leaves with deep and large serrations[48]. The CUC gene could be silenced by TCP3[49]. It is noteworthy that CUC2 is absent from the watercress genome. RNA-seq data demonstrated that TCP3 genes were highly expressed in the watercress blade, which inhibited the expression of CUC3 (Fig. 4c; Supplementary Table S17).
Evolution of genes involved in root morphogenesis in the watercress genome
-
Watercress has many aerial adventitious roots (Fig. 4b). The key genes involved in aerial adventitious root formation mainly include BBM, WOX11, and WOX12[50,51]. In the watercress genome, WGD yielded two homologous copies each for BBM, WOX11, and WOX12. The watercress RNA-seq data revealed that most of the BBM, WOX11, and WOX12 genes exhibit high expression in roots and siliques (Fig. 4d; Supplementary Table S18), suggesting that they play important roles in the formation of aerial adventitious roots to adapt to hypoxia stress.
Gene retention after WGD in the watercress genome
-
After WGD, a total of 14,353 homologous gene pairs including 22,476 genes were retained, which account for 57.71% of all genes in the watercress genome. The functions of homologous Arabidopsis thaliana genes were annotated (Supplementary Fig. S4a). There were 221 homologous Arabidopsis thaliana genes associated with leaf development found in the annotation and 454 genes related to leaf development were identified in watercress (Supplementary Fig. S4b), accounting for 2.01% of all retained genes. Seven transcription factor families including MYB, TCP, AP2, B3, WRKY, SBP, and bZIP were found, including 82 genes, accounting for 18.06% of the total number of genes related to leaf development (Supplementary Table S19). The MYB transcription factor has the largest number of genes, containing 23 genes, followed by TCP transcription factors, with 18 genes (Supplementary Fig. S4c, d). To further understand the expression of transcription factors, the gene expression patterns were analyzed. In the TCP transcription factors' expression pattern (Supplementary Fig. S4e; SupplementaryTable S17), the expression level of TCP3, TCP4, and TCP10 was high in the blades. The retention of leaf development-related genes within the watercress genome provided ample genomic resources for the formation of the leaf with entire or nearly entire margins, and broad and small surface area, which might play an important role in escaping various abiotic stresses caused by the Zanclean flood.
Positively selected genes in the watercress genome
-
The Ka/Ks value of collinear gene pairs within the watercress genome was calculated. The dot blot showed 95 gene pairs (Ka/Ks > 1) including 174 genes that were positively selected (Supplementary Fig. S5a). Among these genes, 15 were transcription factors belonging to B3, MYB, bZIP, and Pcc1 (Supplementary Fig. S5b; Supplementary Table S20), accounting for 8.62% of the positively selected genes. In particular, the B3 transcription factor has nine genes.
-
Watercress, a leafy vegetable containing high nutrition, is consumed globally. The chromosome-level reference genome of watercress was explored in this study (genome size 337.51 Mb), and 96.6% of the sequences were assembled. The assembly results were evaluated with BUSCO. The reliability of the genome was high. Through gene prediction and functional annotation, we identified 38,945 protein-coding genes and also obtained 2,043 rRNA genes, 558 snRNA genes, 320 miRNA genes, and 884 tRNA genes. A total of 129,308 SSRs were identified. The repeated sequences accounted for 49.15% of the genome size, which indicated that the number of repeated sequences may have expanded the genome size of watercress. Studies have shown that some hormones play an important role in the adaptation of plants to flooding stresses[52]. We found that the genes in the expanded and specific gene families were significantly enriched in the process of auxin transport and regulation of hormone levels, which may be related to the physiological characteristics of the rapid growth of watercress and adaptation to flooding stress.
Watercress originates from the European Mediterranean area. About 5.33 Mya, the Zanclean flood put enormous flooding pressure on plants near the Mediterranean Sea. Flooding poses huge challenges to plant growth and reproduction. Rice uses the 'Quiescence' and 'Escape' strategies to adapt to flooding, involving genes such as SUB1A, SK1, and SK2[53]. We did not find their homologous genes in the watercress genome. A previous study has shown that NoNAC36a and NoMOB1a work together to regulate plant height increases in response to flooding stress in watercress[54]. Some plants, such as rice and water lilies, have evolved well-developed aeration tissues to adapt to flooding[55]. Watercress has well-developed adventitious roots, and aerated tissues were found in both its roots and stems, which increase its flood tolerance[55,56]. From the perspective of the watercress genome, we found that watercress might have adapted to various abiotic stresses caused by the Zanclean flood through two strategies. One is to enhance the tolerance to various abiotic stresses; the other is to escape various abiotic stresses caused by flooding through floating growth.
The regeneration capacity of watercress is strong and is one of its mechanisms of adaptation to the aquatic environment. Polyploidy arising from WGD is an important mechanism for plants in adapting to harsh environments[57]. We found 26 WOX genes and 6 SERK genes were retained after WGD, accounting for 89.66% and 66.67% of each gene family, respectively. The multiplication and retention in the watercress genome of regeneration-related genes provided ample genomic resources for the evolutionary adaptation to the flooded environment. Decreased leaf surface area is one of the strategies used by plants to adapt to various abiotic stresses[42]. There are two homologous copies each for TCP4 and TCP10 in the watercress genome after the watercress WGD events. The TCP4 and TCP10 genes were highly expressed in the watercress blade. Interestingly, the MIR319 gene, which can silence TCP4 and TCP10, is missing in the watercress genome, which might result in the reduction in leaf surface area in watercress. It is helpful for watercress to enhance its tolerance to various abiotic stresses caused by the Zanclean flood. The key genes involved in aerial adventitious root formation mainly include BBM, WOX11, and WOX12. There are two homologous copies each for BBM, WOX11, and WOX12 in the watercress genome after the WGD events. Most of the BBM, WOX11, and WOX12 genes were highly expressed in the roots and siliques. The multiplication and retention in the watercress genome of genes associated with aerial adventitious root formation could provide ample genomic resources for the evolutionary adaptation to the flooded environment. Many transcription factors help plants to survive stressful conditions. Some MYB transcription factors have been reported to regulate plants' responses to cold, salt, drought, and oxidative stresses[58]. Pcc1 plays a role in responding to pathogenic stresses[59]. Certain bZIP genes help plants defend against biotic and abiotic stresses[58]. Members of the B3 superfamily are involved in the response to salt, drought, and temperature stresses[60]. Fifteen positively selected genes belonging to B3, MYB, bZIP, and Pcc1 might have helped watercress endure extreme living conditions.
Floating growth helps watercress escape flooding stress. According to previous reports, WOX1 and WOX3 are the key genes involved in leaf width. There are two homologous copies each for WOX1 and WOX3 in the watercress genome after WGD events. The multiplication and retention in the watercress genome of WOX1 and WOX3 genes may play an important role in broad leaf formation. Wider leaves were helpful for watercress floating in the water to escape the hypoxic stress caused by the Zanclean flood. The CUC2 and CUC3 genes both regulate the formation of serrated leaf edges. CUC2 is absent from the watercress genome, which might inhibit the formation of serrated leaf edges in the watercress. We also found that TCP3 genes were highly expressed in the watercress blade. The high expression of TCP3 may suppress the expression of CUC3, favoring the formation of leaves with entire or nearly entire margins. Watercress leaves are characterized by broad and entire or nearly entire margins, which helped watercress floating in the water to escape the hypoxic stress caused by the Zanclean flood.
-
In conclusion, this study provides a reference genome of watercress and proved that watercress has undergone three WGD events in its evolution. The time of the watercress-specific WGD was estimated to be around 4.7 to 12.6 Mya, coinciding with Zanclean flood at about 5.33 Mya. From the perspective of the watercress genome, we found that watercress may have adapted to various abiotic stresses caused by the Zanclean flood through two strategies. One is to enhance the tolerance to various abiotic stresses; the other is to escape various abiotic stresses by floating growth. All results provided useful information for understanding how watercress adapted to flooding stress and lay the foundation for future watercress production and molecular breeding.
We thank Yi Li from the University of Connecticut for scientific advice. This work was supported by the Jiangsu Seed Industry Revitalization Project [JBGS(2021)015, JBGS(2021)064], the Fundamental Research Funds for the Central Universities (KYPT2024001), a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, the Nanjing Science and technology planning project (202109022), and the China Agriculture Research System (CARS-23-A-16).
-
The authors confirm their contributions to the paper as follows: study conception and design: Hou X, Li Y; data collection: Yan C, Ma X, Ran J; analysis and interpretation of results: Deng S, Li Z, Bai A; draft manuscript preparation: Yan C, Li Z, Bai A, Chen F; review and editing: Hou X, Li Y, Deng S. All authors reviewed the results and approved the final version of the manuscript.
-
The Illumina and PacBio sequencing data have been deposited in the NCBI database with the accession code PRJNA1272309.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Chao Yan, Suchong Deng, Zhidong Li
- Supplementary Table S1 Statistics of the watercress genome assembly after correction using PacBio subreads.
- Supplementary Table S2 Statistics of the watercress genome assembly after decontamination and further correction using Illumina sequencing data.
- Supplementary Table S3 The statistics of raw data from Hi-C sequencing.
- Supplementary Table S4 Statistics of the final genome assembly of watercress.
- Supplementary Table S5 Assessment of the completeness of the watercress genome assembly by BUSCO.
- Supplementary Table S6 The statistics of the pseudochromosomes of watercress.
- Supplementary Table S7 Predicted protein-coding genes in the watercress genome.
- Supplementary Table S8 The statistics of gene function annotations.
- Supplementary Table S9 Summary of predicted ncRNA genes in the watercress genome.
- Supplementary Table S10 The statistics of repeat contents in the watercress genome.
- Supplementary Table S11 Statistics of the identified SSRs in the watercress genome.
- Supplementary Table S12 The statistics of clustered gene families in 12 species.
- Supplementary Table S13 The statistics of homologous blocks and collinear gene pairs.
- Supplementary Table S14 The summary in WOX gene family.
- Supplementary Table S15 The summary in SERK gene family.
- Supplementary Table S16 The statistics of SERK gene expression data.
- Supplementary Table S17 The statistics of gene expression data involved in leaf morphogenesis.
- Supplementary Table S18 The statistics of gene expression data involved in root morphogenesis.
- Supplementary Table S19 The summary of transcription factor involved in leaf development retained after WGD.
- Supplementary Table S20 Positively selected genes that may respond to abiotic stresses.
- Supplementary Fig. S1 The genome size of watercress estimated by 17-mer analysis.
- Supplementary Fig. S2 Interchromosomal Hi-C contact map of the N. officinale genome.
- Supplementary Fig. S3 The GO enrichment of N. officinale gene family. (a) GO functional enrichment of the watercress-expanded genes. (b) GO functional enrichment of the watercress-specific genes.
- Supplementary Fig. S4 The analysis of gene retention of WGD. (a) The functional pathway annotation plot of homologous Arabidopsis thaliana gene aligned with retained gene after WGD in Nasturtium officinale. (b) The specific presentation of homologous Arabidopsis thaliana gene related to leaf development. (c) The distribution of transcription factor numbers related to leaf development in Nasturtium officinale. (d) The example of gene retention after WGD (TCP10). (e) The expression patterns of TCP transcription factors.
- Supplementary Fig. S5 The identification of positively selected genes and expression patterns of positively selected transcription factors in the watercress genome. (a) The Ka/Ks distribution dot plot of homologous gene pair. (b) The positively selected transcription factors numbers.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yan C, Deng S, Li Z, Bai A, Ma X, et al. 2025. Genome of plant watercress (Nasturtium officinale R. Br.) illuminates genomic basis for marine-incursions adaptation in the Mediterranean area. Vegetable Research 5: e031 doi: 10.48130/vegres-0025-0025 

Genome of plant watercress (Nasturtium officinale R. Br.) illuminates genomic basis for marine-incursions adaptation in the Mediterranean area
- Received: 10 May 2025
- Revised: 07 June 2025
- Accepted: 01 July 2025
- Published online: 02 September 2025
Abstract: Watercress (Nasturtium officinale R. Br.), an herbaceous plant in the cruciferous family, has a long history of use as a vegetable. In this report, we present a high-quality assembly of the watercress genome, based primarily on PacBio and Hi-C sequencing data. The assembled genome of watercress was 337.51 Mb in size, with a contig N50 length of 3.26 Mb and a scaffold N50 length of 5.85 Mb. Approximately 49.15% (165.88 Mb) of the assembled genome was annotated as repetitive sequences, with long terminal repeats (LTRs) being the most abundant, representing 41.96% of the genome. About 96.6% of the assembly was anchored onto 16 pseudo-chromosomes using Hi-C data. Analyses of the syntenic relationships within and between species collectively indicated that watercress underwent an additional whole-genome duplication (WGD) event after divergence from Arabidopsis. The time of the watercress-specific WGD was estimated to be around 4.7 to 12.6 million years ago (Mya), which coincided with the Zanclean flood about 5.33 Mya. After the WGD, a total of 14,353 homologous gene pairs, including 22,476 genes were retained, accounting for 57.71% of all genes in the watercress genome. From the perspective of the watercress genome, we found that watercress might adapt to various abiotic stresses caused by the Zanclean flood through two mechanisms: one is enhancing tolerance to various abiotic stresses, and the other is escaping various abiotic stresses by floating growth. The watercress genome and transcriptome presented here provide useful information for subsequent molecular breeding and understanding how watercress adapted to an aquatic environment.
-
Key words:
- Watercress /
- Whole-genome duplication /
- Genome evolution /
- Regeneration /
- Aquatic adaptation