









-
All species within the genus Fragaria produce white petals. The well-known strawberry (F. × ananassa) is derived from hybridization between two octoploid wild species, F. virginiana and F. chiloensis[1−3]. In contrast, the pink-flowered strawberry originates from a distant cross between the white-flowered cultivated strawberry (F. × ananassa, 2n = 8x = 56), and the red-flowered Potentilla palustris (2n = 6x = 42)[4,5]. In China, the pink-flowered strawberry cultivar can bloom year-round in the southern regions under suitable temperatures, and from May to October in open fields in northern areas. The pink-flowered strawberry exhibits strong adaptability, thriving in full sunlight while also tolerating shade, and demonstrates robust stress resistance and vigorous growth. Flower color, the most prominent ornamental trait of the pink-flowered strawberry, is characterized by a diverse range of petal hues, making it an ideal material for landscaping and potted displays with considerable economic value[6,7]. Given the economic and ornamental value of the pink-flowered strawberry, understanding the molecular mechanisms underlying its flower development is crucial.
Flower development is a critical phase in the life cycle of higher plants, marking the transition from vegetative to reproductive growth. This process is tightly coordinated through the integration of endogenous and environmental signals, including phytohormone signaling, photoperiod, and temperature fluctuations[8]. Plant hormones play indispensable roles in both the initiation and progression of flowering. Key regulatory pathways, such as those involving gibberellin signaling and photoperiodic responses, have been elucidated using functional genomic approaches. For instance, Wang et al. employed transcriptomic profiling to decode the genetic regulatory network underlying flowering transition in Rosa rugosa, examining dynamics in sucrose/starch metabolism, phytohormone signaling, and environmental response pathways related to photoperiod and vernalization[9]. Multi-omics (e.g., transcriptomics + hormone profiling) deciphers regulatory networks, bridges molecular and physiological phenotypes, and advances understanding of floral development in plants. For example, to explore the molecular mechanisms of floral bud differentiation in Litsea cubeba, He et al. performed transcriptomic analyses of female and male floral buds across three developmental stages using RNA-seq. This study yielded 160.88 Gbp of clean data, assembled 100,072 unigenes (38,658 annotated), and integrated hormone content dynamics with differential expression analysis. Key hormone-related genes were identified, and stage-specific transcription factors (e.g., upregulated MADS-box, downregulated bZIP genes) were highlighted[10]. Similarly, Xu et al. investigated flowering regulation in Camellia sinensis via RNA-seq, identifying a core transcriptome of 92 genes and hormone-associated differentially expressed genes. Correlating these with endogenous hormone levels during floral development allowed constructing a hormonal regulatory network involving SOC1, LFY, FT, and MYC as integrators[11].
Plant microRNAs (miRNAs), typically 20–24 nucleotides in length, function as key post-transcriptional regulators of gene expression. They mediate mRNA cleavage, translation inhibition, or DNA methylation, thereby modulating diverse biological processes including organ morphogenesis, growth and development, hormone signaling, and stress responses[12−14]. miRNAs play particularly crucial roles in flower development[15−17]. For example, Silva et al. revealed that the core mechanism underlying meristem maturation and floral transition in cultivated tomato involves miR156-SBPs (SQUAMOSA PROMOTER BINDING PROTEIN-LIKE) recruiting age-related cues, their integration with gibberellin (GA) signaling, and the miR319-TCP4 homolog LANCEOLATE (LA), and the subsequent regulation of SFT and floral identity genes through this integrated pathway[18]. Sun et al. identified four members of the VvmiR172 family in grapevine and characterized their expression patterns during flower development, establishing VvmiR172c as a key regulator through its targeting of AP2[19]. Additionally, certain miRNAs trigger the production of phased secondary small interfering RNAs (phasiRNAs), which further expand their regulatory roles in stress responses and developmental processes[20,21].
To investigate post-transcriptional regulation during petal coloration in pink-flowered strawberry, our research group previously identified and validated four miRNA-target pairs involved in this process: FamiR156a-FaSPL13A, FamiR396e-FabHLH79, FamiR858_R-2-FaMyb308, and FamiR828a-FaMYB114[22]. Building on these findings, the present study re-analyzed small RNA and degradome sequencing data and integrated them with transcriptome profiles to identify novel miRNA-target interactions. Additionally, during flower development in pink-flowered strawberry, miRNA triggers were identified and PHAS loci were annotated. To further explore the post-transcriptional regulatory mechanisms in flower development and petal coloration of pink-flowered strawberries, the sequencing data were re-examined and phasiRNAs were identified. These analyses enhance our understanding of post-transcriptional regulatory mechanisms in pink-flowered strawberry, and provide valuable insights for genetic engineering-based breeding strategies.
-
The pink-flowered strawberry cultivar 'Sijihong' was used in the study, which was bred by our research group and grown in the open field of the Strawberry Resource Nursery of Shenyang Agricultural University (Shenyang, China). According to the opening degree of sepals, and the opening degree and the color change of petals, the flower bud development is divided into five stages: young bud stage (PF_L), coloration beginning stage (PF_Z), big bud stage (PF_D), half-opening stage (PF_B), and blooming stage (PF_S)[23]. To explore the molecular mechanism of flower development and petal color variation during the flower bud development in pink-flowered strawberry, the flower bud at the front three developmental stages with significantly different petal color, PF_L, PF_Z, and PF_D, were selected as high-throughput sequencing in this study (Fig. 1). The a* of the fresh petals were measured with a colorimeter (CR-400 Koniea Minolta, Japan) to distinguish the colors and the diameter was measured using a vernier caliper. The petals of 'Sijihong' at three developmental stages were collected, quickly frozen in liquid nitrogen, and stored in a refrigerator at −80 °C for RNA extraction. Three biological replicates were set up for each treatment.

Figure 1.
Petal colors at different developmental bud stages (PF_L: young bud stage; PF_Z: coloration beginning stage; PF_D: big bud stage) of pink-flowered strawberry cv. 'Sijihong'.
The modified CTAB method was used to extract total RNA from the petals of 'Sijihong' at different development stages[24], and whether it was degraded or not was identified by 1% agarose gel electrophoresis. The Nanodrop 2000c spectrophotometer (Thermo Scientific, Waltham, the United States) was used to detect the concentration and quality, resulting in determining the integrity of RNA. The extracted RNA was used to construct the library of transcriptome, small RNA, and degradation sequencing. The library construction and sequencing were carried out by Lianchuan Biological Technology Co., Ltd. (Hangzhou, China). The steps were as follows: High-quality total RNA (RIN > 7.0) was used for strand-specific library construction. mRNA was purified using Dynabeads™ Oligo (dT) and fragmented. cDNA was synthesized with reverse transcription followed by second-strand synthesis incorporating dUTP. After end repair, A-tailing, and adapter ligation, size selection was performed, and UDG enzyme treatment was applied for strand marking. Libraries were amplified with PCR (eight cycles) and sequenced on an Illumina NovaSeq 6000 platform (2 × 150 bp).
Transcriptome data analysis, and identification of differentially expressed transcripts
-
The high-quality total RNA (RIN > 7.0) was used for transcriptome library construction by adopting the fr-firstrand method according to the manufacturer's instructions. The transcriptome data have been uploaded to the NCBI GEO database under Accession No. GSE125777. The differentially expressed transcripts in different developmental stages were screened and counted, which were enriched and analyzed by KEGG using the Lianchuan Bio-cloud platform online tools (
www.omicstudio.cn/tool ). Key TFs were further screened for correlation analysis between the three comparisons using Sytoscape software. Subsequently, TBtools-II was employed to perform cluster heatmap analysis on the differentially expressed transcripts[25], and the differentially expressed genes were identified and annotated. Among them, the main parameters for constructing a heat map were Row Scale and 0–1 distribution (Zero To One).Small RNA, degradome sequencing, and data preprocessing
-
Small RNA sequencing can quickly identify the small RNA groups of the whole spectrum of species and predict novel miRNAs. Degradome sequencing conducted for the identification and functional analysis of the target genes of small RNA. Nine small RNA libraries (PF_L1, PF_L2, PF_L3, PF_Z1, PF_Z2, PF_Z3, PF_D1, PF_D2, PF_D3), and one degradation library were constructed. The 18–30 nt small RNA sequences in the samples were identified and analyzed using high-throughput sequencing technology, relying on the sequencing analysis software ACGT101-miR and ACGT301-DEG101 combined with CleaveLand. Sequencing was carried out on the Illumina HiSeq 2500 platform of Lianchuan Biological Technology Co., Ltd. (Hangzhou, China) according to standard operating procedures. The sRNAome sequencing data have been uploaded to the NCBI Gene Expression Omnibus (GEO) database (
www.ncbi.nlm.nih.gov ) under the Accession No. GSE193522.The raw sequencing data were preprocessed using the software sRNAminer[26]. In brief, the process consisted of the following steps: removing the adapters of the raw data, checking the sRNA length distribution, using Seq Collapser to merge repetitive sequences, removing rRNA and plasmid contamination, using Alignment to convert the format of bowtie files, and sort the file based on the position of reads mapped on the Fragaria_vesca_v4.0.a1 genome, and finally generating. bam files that could be manually viewed for subsequent analysis.
Identification of miRNA and PHAS loci, and prediction of miRNA triggers
-
Using the One Step sRNAminer function in the sRNAminer software, miRNAs and PHAS loci were identified. Relevant file information, including genome files, raw sequencing FASTQ files, rRNA databases, and organelle databases, were input into the functional interface. Subsequent bioinformatics analyses were conducted using the generated result files. In this study, known miRNAs were named based on sequence homology to previously characterized miRNAs[27], and novel miRNAs were named with the prefix 'miRN'. Furthermore, given that the biogenesis of phasiRNAs is predominantly initiated by 22-nt miRNAs, the predicted potential miRNA triggers of PHAS loci were incorporated into the analysis. The phasiRNA trigger function in the sRNAminer software offers a convenient and rapid analytical approach for PHAS trigger identification, enabling the prediction of miRNAs that might induce the generation of a specific PHAS locus.
Identifying target genes of miRNAs
-
The adapter sequences were removed from the degradation data in the same way as the sRNA data, and then the clean data was analyzed through CleaveLand. The Degradome Analysis function of sRNAminer software was used to analyze the degradation library with clean data, and somewhere along the way, it was necessary to prepare the genome file, the genome annotation file, and the miRNA file. The annotation information of miRNAs target genes was obtained by combining the analysis results with the transcriptome results.
qRT-PCR and statistical analysis
-
To verify the high-throughput sequencing results, qRT-PCR was used to verify the expression trends of differentially expressed miRNAs and their target genes at three stages during the flower bud development of 'Sijihong' with significantly different petal colors. The miRNA First Strand cDNA Synthesis Kit (by stem-loop) (Nazyme, MR101), and the HiScript IV All-in-One Ultra RT SuperMix for qPCR (Nazyme, R433) were used to reverse transcribe miRNAs and their target genes, respectively. The real-time fluorescence quantitative kits for miRNAs and target genes were miRNA Unimodal SYBR qPCR Mastter Mix (Nazyme, MQ102), and ChamQ Universal SYBR qPCR Mastter Mix (Nazyme, Q711), respectively. U6 and FaDBP were used as the internal reference genes of miRNAs and target genes, respectively[28]. The real-time fluorescence quantitative instrument was QuantStudio 6 real-time PCR system (Life, USA), three repeats were set for qRT-PCR, and the petals of the young bud stage were used as the control, the relative expression was calculated via the 2−ΔΔCt method[29], and the related primer sequences involved are shown in Supplementary Table S1. The results are reported as mean standard deviation (SD). Statistical analyses were conducted using a two-way analysis of variance (ANOVA) combined with Tukey's post-hoc test (* p < 0.5, ** p < 0.01, *** p < 0.001, **** p < 0.0001).
-
Flower buds and petal sizes of 'Sijihong' were investigated across three developmental stages, characterized by a gradual darkening of flower color. The three stages included young bud stage (PF_L) with fully closed sepals and pale white petals (0 < a* < 10), the bud size is 0.5–0.7 cm, and the petal size is 0.35–0.5 cm, coloration beginning stage (PF_Z) with sepals slightly open, petals appear and begin to stain (10 < a* < 35), the bud size is 0.7–0.75 cm, and the petal size is 0.45–0.6 cm, and big bud stage (PF_D) with open sepals and closed petals but the deepest color (50 < a* < 60), the bud size is 0.75–0.9 cm, and the petal size is 0.55–0.7 cm (Fig. 1).
In this study, transcriptome sequencing was performed on the PF_L, PF_Z, and PF_D petals during the flower development of pink-flowered strawberry. There have been reports about clean data in nine samples (PF_L1, PF_L2, PF_L3, PF_Z1, PF_Z2, PF_Z3, PF_D1, PF_D2, PF_D3) obtained after transcriptome sequencing. According to the genome data of the octoploid strawberry, the RNA-seq data were reanalyzed, and 68.68 GB of illumina RNA-seq high-quality clean reads were obtained after removing the low-quality reads[30]. In the pairwise comparison groups, there were 2,081 differentially expressed genes in PF_Z vs PF_L, of which 962 were up-regulated, and 1,119 were down-regulated. There were 1,491 up-regulated and 2,783 down-regulated differential gene IDs in PF_D vs PF_Z. The most differentially expressed gene IDs in PF_ D vs PF_L were observed, with 5,554 genes, of which 1,838 were up-regulated, and 3,716 were down-regulated (Fig. 2a). The results of Venn analysis showed that 457 of these differentially expressed gene IDs were differentially expressed only in PF_Z vs PF_L, 995 in PF_D vs PF_Z, and 1,889 gene IDs were differentially expressed only in PF_D vs PF_L (Fig. 2b).
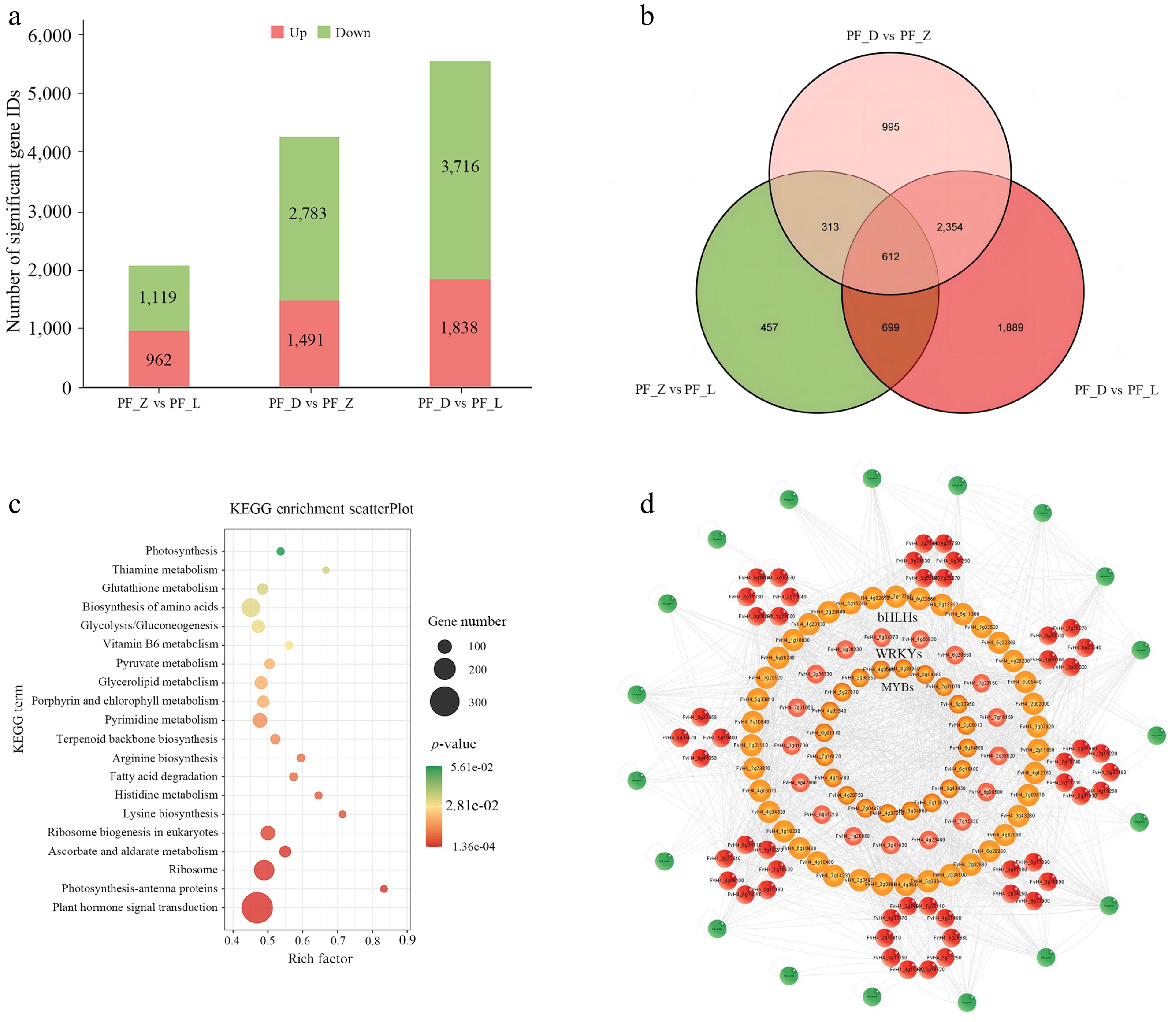
Figure 2.
Annotation and pathway analysis of differentially expressed genes. (a) The number of differentially expressed genes in the three pairwise comparison groups. (b) Venn diagram showing the intersections of differentially expressed genes in three pairwise comparison groups. (c) The top 20 KEGG significant enrichment pathways of differentially expressed genes in the PF_D vs PF_Z vs PF_L comparison group. (d) The correlation network diagrams of differentially expressed transcription factors in the three comparison groups.
In addition, KEGG enrichment analysis of PF_D vs PF_Z vs PF_L was performed for further analysis of biological function. A total of 136 pathways were enriched and according to p value screening, the enrichment results of the first 20 pathways are shown in Fig. 2c. The differentially expressed genes in PF_D vs PF_Z vs PF_L participated in plant hormone signal transduction, photosynthesis-antenna proteins, ribosome and ascorbate and aldarate metabolism, etc., in which the plant hormone signal transduction pathway enriched the most transcription factors (323). Transcription factor families were involved, such as bHLHs (19), MYBs (29), WRKYs (35), NACs (21), ARFs (13), ERFs (25), and SPLs (6) (Supplementary Table S2), which indicated that transcription factors play an important role in flower development and petal coloration in pink-flowered strawberry.
Screening of key transcription factors related to flower development
-
Transcription factors differentially expressed in the three comparisons were screened, and correlation analysis was performed with a threshold set to p ≤ 0.01, rho ≥ 0.95, or rho ≤ −0.95 (Fig. 2d). The results showed that most of the correlations among different transcription factors were positive. Among them, the number of bHLH family transcription factors was the most, with 39, followed by MYBs and WRKYs, with 20 and 17, respectively.
In the constructed correlation network, only 47 pairs were negatively correlated, and the remaining 592 pairs were all positively correlated, including ERF, MYB, bHLH, WRKY, bZIP, etc. Among them, the AP2-like ethylene-responsive transcription factor AIL1 (FvH4_1g18270), and transcription factor bHLH93-like (FvH4_1g18930) had the most correlation pairs (25 and 24). Moreover, they were co-regulators of many transcription factors, suggesting that these transcription factors play important roles in the development of petals in pink-flowered strawberry. Therefore, heatmaps of transcription factors were constructed based on the FPKM values, and their expression patterns were analyzed. The results showed that most of them showed a down-regulated expression trend compared with the control PF_L (Supplementary Fig. S1), and the remaining fraction showed an up-regulated expression, and up-down expression trend, which can provide an effective reference for studying at the post-transcriptional level.
miRNAs directed post-transcriptional regulation of flower development in pink-flowered strawberry
-
To investigate the post-transcriptional regulation of petal coloration in pink-flowered strawberry, small RNA sequencing and degradome sequencing were performed on the petals of 'Sijihong' in three stages (PF_L, PF_Z, PF_D) with significantly different flower colors during flower development. Raw reads were reported in a previous study[21], which was obtained from nine small RNA libraries and a mixed degradation library. The raw data obtained by the sRNAminer software was re-analyzed.
151 known miRNAs from 55 families were identified, and most of them are 21 nt. Using the genome of Fragaria vesca as the reference genome, chromosomal location analysis of all identified miRNAs was performed by using TBtools-II (Fig. 3a). The number of miRNAs located by Fvb1-Fvb7 was 17, 19, 45, 23, 27, 35, and 23, respectively. Among them, the largest number of known miRNAs were located on chromosome Fvb3, with 41 miRNAs from 21 families, and chromosome Fvb6 has been located to the most novel miRNAs, with 14. It is worth noting that most of the known identified miRNAs from the same family are not located on the same chromosome; for example, FvmiR156 family members are widely distributed on Fvb1, Fvb2, Fvb3, Fvb4, and Fvb5. In addition, the number of known miRNA family members was counted, among them, FvmiR169 family (12 members), FvmiR156 family (11 members), FvmiR171 family (8 members), and FvmiR2275 family (8 members) have more family members (Fig. 3b). Thirty eight novel miRNAs have been identified, which are mainly 21 nt and 22 nt, only miRN10 is 24 nt, without novel miRNA is 20 nt. Most families have only one member; only the miRN17 family, and the miRN36 family have two members (Fig. 3b).
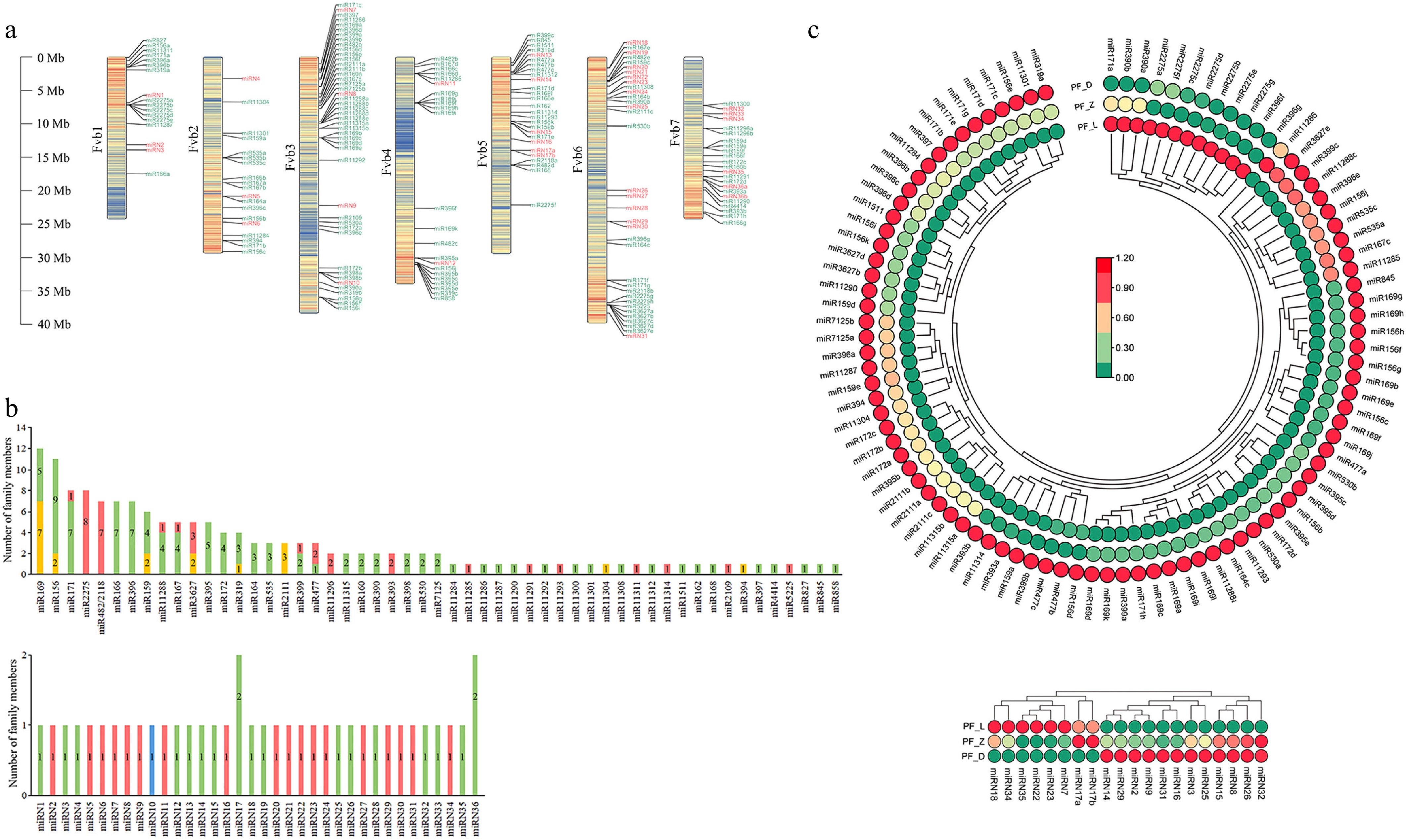
Figure 3.
Identification of miRNAs and expression analysis of differentially expressed miRNAs during the flower development of pink-flowered strawberry. (a) Location of miRNAs on chromosomes, with known miRNAs in green font, and novel miRNAs in red font. (b) The statistics of the number of identified miRNAs family members. (c) Heatmaps showing expression patterns of the different expression miRNAs during flower development.
In this study, it is considered that if PF_D.exp/PF_L.exp > 2 or PF_D.exp/PF_L.exp < 0.5, it was characterized by differential expression. Based on this, the heatmap analysis of differentially expressed miRNAs (96 known miRNAs, and 20 novel miRNAs) was performed (Fig. 3c). Heatmap analysis showed that most of the known miRNAs showed an up-regulated expression trend with the deepening of petals color in pink-flowered strawberry, while the rest showed a down-regulated expression trend (FvmiR171a, FvmiR390a, FvmiR390b, FvmiR2275b, FvmiR2275e, FvmiR2275g, FvmiR396f, and FvmiR396g), first downward and then upward (FvmiR2275a, FvmiR2275f, FvmiR2275c, and FvmiR2275d), and first upward and then downward (FvmiR11286). Heat map analysis of novel miRNAs shows that, with the deepening of the color of pink-flowered strawberry petals, 12 of them (miRN32, miRN26, FamiRN8, miRN15, miRN25, miRN3, miRN16, miRN31, miRN9, miRN2, miRN29, miRN14) show a trend of gradual rise, six of them (miRN7, miRN23, miRN22, miRN35, FamiRN34, miRN18) show a trend of gradual downward. Two novel miRNAs (miRN17a and miRN17b) show the trend of first upward and then downward, and no novel miRNAs show the trend of first downward and then upward.
Integrating sequencing data, and screening key miRNAs-targets pairs
-
To more accurately determine the regulatory relationship between key miRNAs and their target mRNAs during petal coloration in 'Sijihong', degradome sequencing results were also re-analyzed, with a comprehensive analysis conducted by integrating transcriptome and small RNA sequencing data. Based on degradome sequencing results, 552 miRNA-target pairs were identified. Combined with expression level data and transcriptome annotations, 83 of these pairs exhibited differential expression with opposite expression trends (Supplementary Table S3). The partial T-plot diagrams of miRNAs cleaving the target genes in the degradation library is shown in Supplementary Fig. S2, and the red line represents the predicted cleavage site of the corresponding miRNAs. Pathway enrichment analysis was performed for these targeted differentially expressed genes (Fig. 4a), with results indicating that these genes were mainly enriched in the following pathways, 00190 (Oxidative phosphorylation), 00500 (Starch and sucrose metabolism), 00564 (Glycerophospholipid metabolism), 00730 (Thiamine metabolism), 00910 (Nitrogen metabolism), 03022 (Basal transcription factors), 03040 (Spliceosome), 04075 (Plant hormone signal transduction), 04120 (Ubiquitin mediated proteolysis), 04141 (Protein processing in endoplasmic reticulum), and 04626 (Plant-pathogen interaction). Among them, the most genes were enriched in the 04075 (Plant hormone signal transduction) pathway (four genes), namely FaSPL13A (FvH4_5g08580), FaSPL1 (FvH4_6g26530, FvH4_3g19650), and FaSPL12 (FvH4_4g23650).
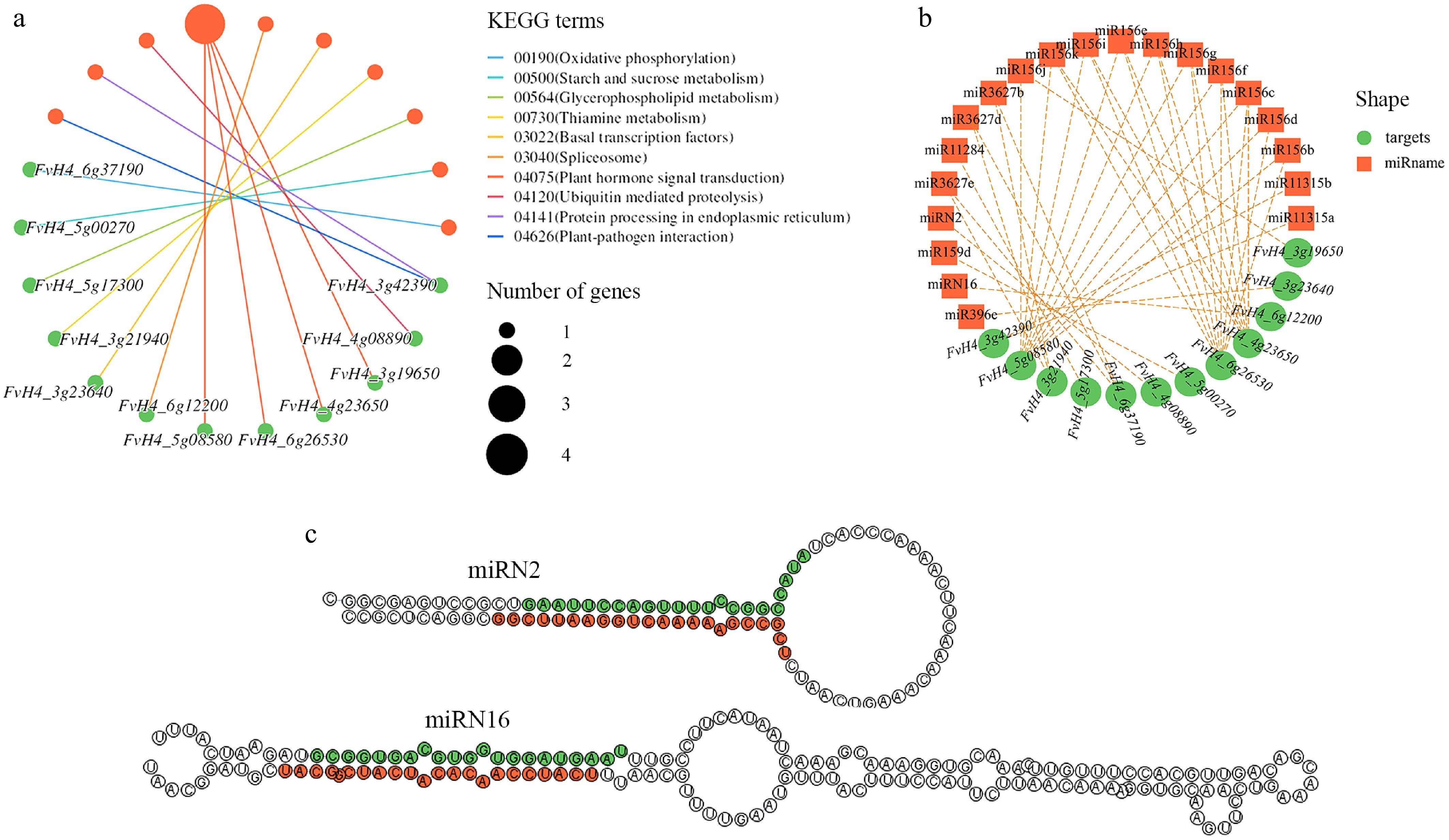
Figure 4.
Screening of key miRNAs-targets. (a) Pathway enrichment analysis of key differentially expressed genes. (b) Correlation network of key miRNAs-targets. (c) The secondary structure prediction of two novel identified miRNAs.
In addition, the corresponding network map of miRNAs and these targeted genes was constructed (Fig. 4b), and a total of 40 miRNA-targets pairs were screened, including the classic miR156s-SPLs pairs. Their crucial regulatory roles in the process of plant flower development have been reported in many studies. Moreover, two novel identified miRNAs showed the secondary structure prediction results in Fig. 4c, which both have the stem-loop structure typical of miRNA. The candidate target gene of miRN2 was FvH4_4g08890, and the transcriptome annotation was uncharacterized protein LOC101302346 [Fragaria vesca subsp. vesca]. The candidate target gene of miRN16 was FvH4_6g12200, and the transcriptome annotation was putative disease resistance protein RGA3 [Fragaria vesca subsp. vesca]. Gene sequences targeted by two novel miRNAs were retrieved and blastx using NCBI (
https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastx&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome ), the results showed that the miRN2 candidate target gene FvH4_4g08890 was also annotated as an unknown protein in other plants of Rosaceae, and the alignment of miRN16 candidate target gene FvH4_6g12200 in other plants of Rosaceae was consistent with the transcriptome annotation, a putative disease resistance protein.Subsequently, the expression patterns of these miRNA-targets were analyzed. Quantitative real-time PCR (qRT-PCR) validated that the expression trends were consistent with the sequencing results. During flower development in pink-flowered strawberry, FamiR156s showed an upregulated expression trend, whereas its target genes FaSPLs exhibited an opposite expression pattern (Fig. 5a−c). Similarly, FamiR396e and its target genes, transcription initiation factor TFIID subunit 6-like (FaTAF6, FvH4_3g23640) (Fig. 5d), as well as FamiR11315 and its target F-box protein CPR30-like isoform X3 (FaCPR30, FvH4_3g42390) (Fig. 5e), displayed analogous expression dynamics. Additionally, the novel FamiR16 was highly accumulated at the large bud stage, while its target gene FaRGA3 (FvH4_6g12200) showed negligible expression during the same stage (Fig. 5f).
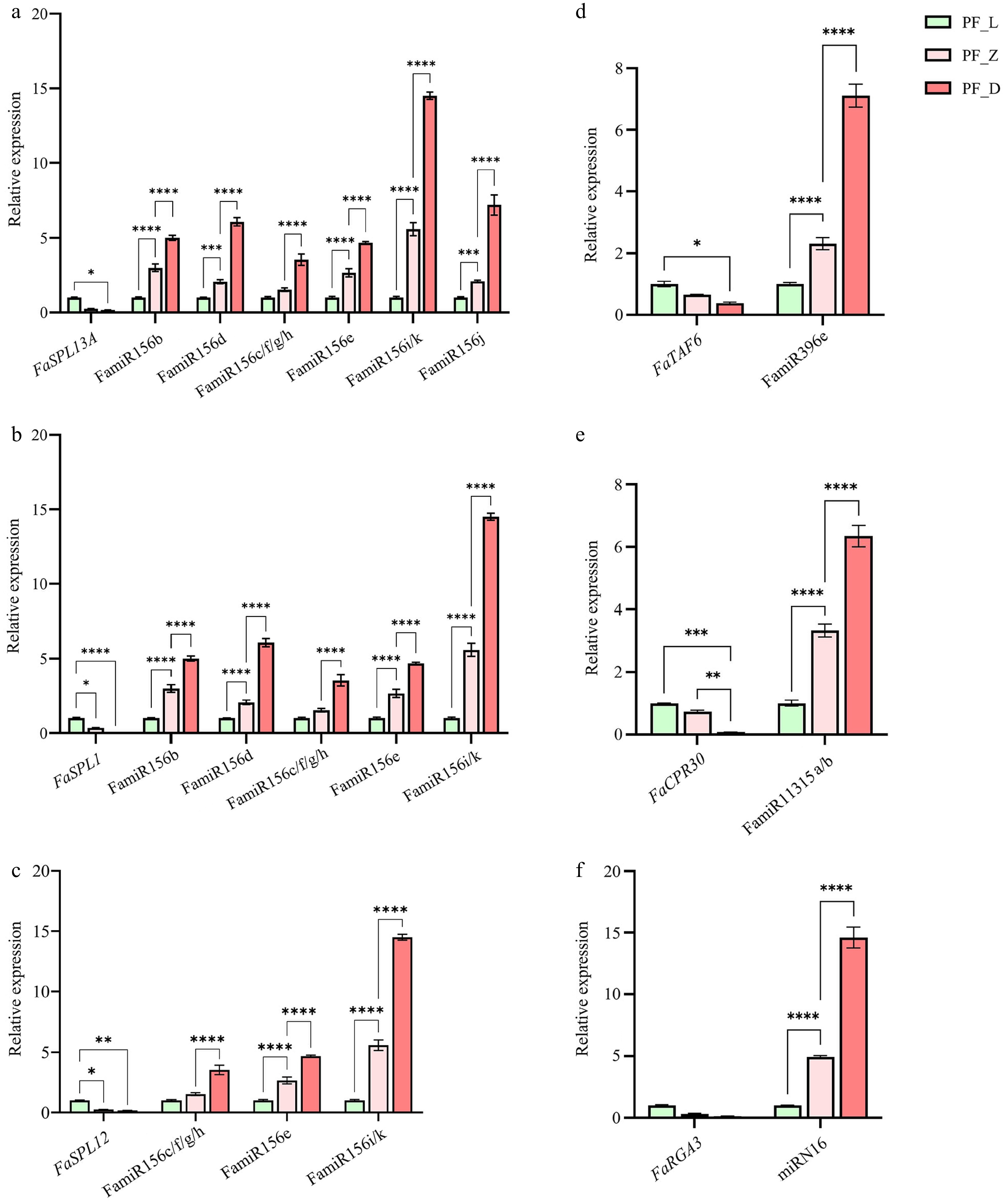
Figure 5.
Expression pattern analysis of key miRNAs-targets in three developmental stages of 'Sijihong'. (a)–(c) Expression patterns of FamiR156s and their target genes FaSPLs. (d) The expression pattern of FamiR396e and its target gene, transcription initiation factor TFIID subunit 6-like (FaTAF6, FvH4_3g23640). (e) The expression pattern of FamiR11315 and its target gene F-box protein CPR 30-like isoform X3 (FaCPR30, FvH4_3g42390). (f) The expression pattern of novel miR16 and its target gene FaRGA3 (FvH4_6g12200).
Identification and expression patterns of PHAS loci of flower development in pink-flowered strawberry
-
To investigate the regulatory role of phasiRNAs in the petal coloration process of 'Sijihong', PHAS loci were additionally identified using sRNAminer software during the re-analysis of small RNA sequencing data. A total of 237 21-nt PHAS loci, and 48 24-nt PHAS loci were identified by setting Phasing Score to 10 and p-value to 0.01 (Supplementary Table S4). It was found through chromosomal location analysis of all identified PHAS loci that 31, 27, 69, 24, 37, 54, and 43 PHAS loci were located on chromosomes Fvb1 to Fvb7 in turn (Fig. 6a). Among them, 21-nt PHAS loci and 24-nt PHAS loci were the most located on chromosome Fvb3, with 58 and 11 respectively.
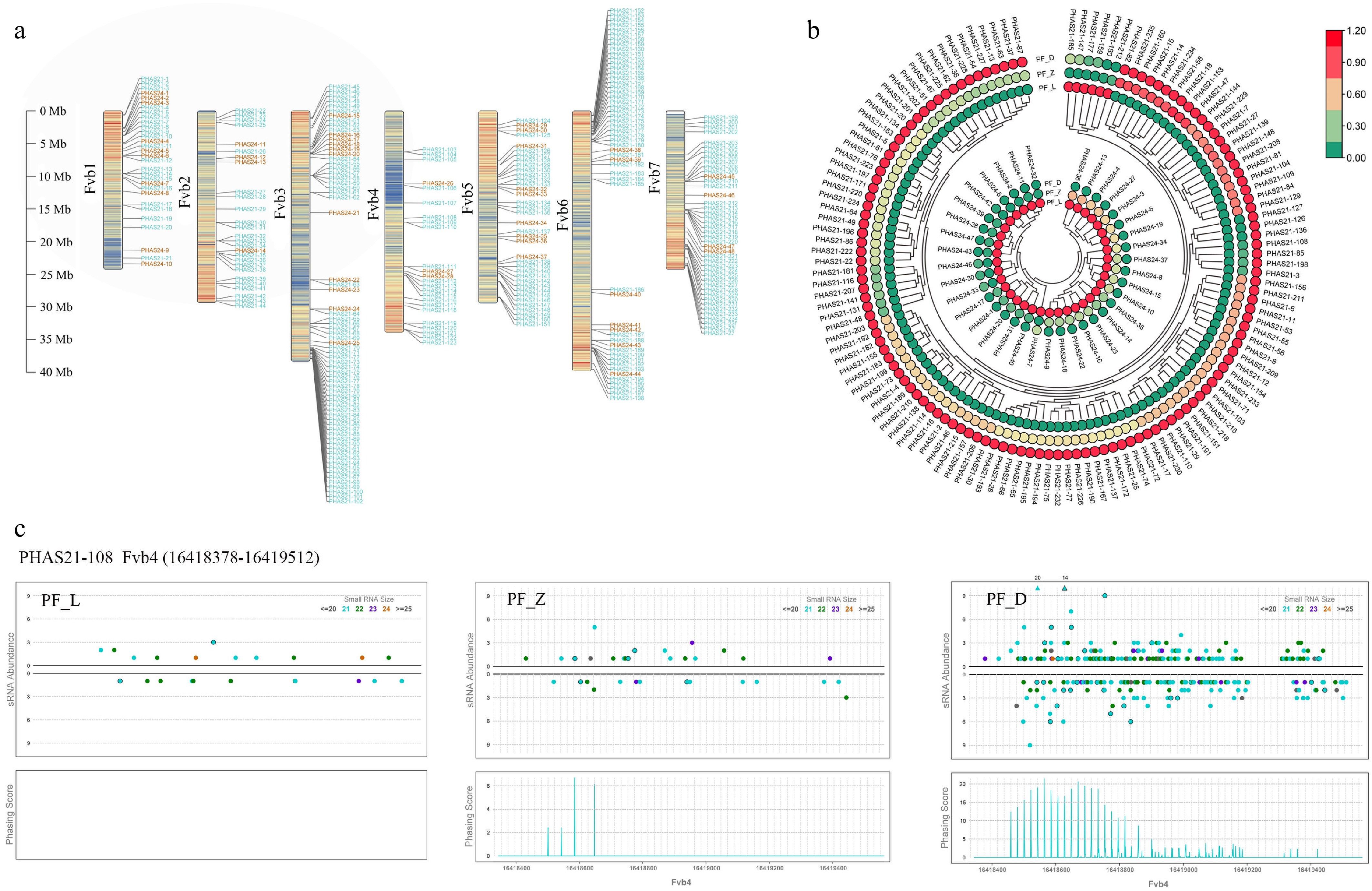
Figure 6.
Identification of PHAS loci and expression analysis of differentially expressed PHAS loci during the flower development of pink-flowered strawberry. (a) Location of PHAS loci on chromosomes, with 21-nt PHAS loci in blue font and 24-nt PHAS loci in orange font. (b) Heatmaps showing expression patterns of the different expression PHAS loci during flower development. (c) PHAS2-108 located on Fvb4 was annotated as transcription factor bHLH77-like, and the results of PHASList to Graph in three developmental stages.
Furthermore, the expression levels of the differentially expressed PHAS loci were also identified (Fig. 6b). Among all 21-nt PHAS loci, 130 loci showed differential expression characteristics with the deepening of petal color, among which PHAS21-147, PHAS21-159, PHAS21-177, PHAS21-180, PHAS21-185, and PHAS21-212 loci were down-regulated, and the rest were up-regulated. In contrast, 37 differentially expressed 24-nt PHAS loci showed down-regulated expression trend. In other words, most of the differentially expressed 21-nt PHAS loci accumulated at higher levels in PF_D, while differentially expressed 24-nt PHAS loci accumulated at higher levels in PF_L.
To evaluate the coding potential of these loci, coding capacity was calculated via CPC2[30], identifying 72 PHAS loci (Supplementary Table S5), 70 with 21-nt PHAS loci and only two 24-nt PHAS loci with coding capabilities, PHAS24-10, and PHAS24-34. To determine the possible functions of these sites, functional annotation and classification were performed, and the results showed that most of these sites were associated with resistance. Notably, PHAS21-108 was annotated as transcription factor bHLH77-like, and the results of PHASList to Graph showed that the expression level was gradually enhanced with the flower developed from 'Sijihong', and the specific expression was in the big bud stage (Fig. 6c). The annotation results of PHAS21-216, PHAS21-217, and PHAS21-219 were NAC domain-containing proteins, while these three loci were not specifically expressed in the three key stages during flower development in 'Sijihong' (Supplementary Fig. S3). Two 24-nt PHAS loci, PHAS24-10, and PHAS24-34 were annotated as unknown proteins with no specific expression in the three developmental stages (Supplementary Fig. S4).
Identification of miRNA triggers, and annotation for PHAS loci during the petal coloration process
-
The nine known miRNA families and six novel miRNA families were found to trigger phasiRNA production from PHAS loci, and most miRNAs were 22 nt, including conserved family (FvmiR2275, FvmiR11288, FvmiR482/2118, FvmiR11285, FvmiR2109, FvmiR390, FvmiR168, FvmiR393, FvmiR5225), and novel family (miRN7, miRN10, miRN11, miRN16, miRN20, miRN23) (Supplementary Table S6). Among them, the FvmiR482/2118 super miRNA family triggered the most 21 nt phasiRNAs. All 24-nt-PHAS loci were triggered by the miR2275 family. The number of miRNA triggers within best regions for PHAS loci varied across chromosomes, with counts of 13, 4, 21, 10, 10, 21, and 13 for chromosomes Fvb1 to Fvb7, respectively. It is worth mentioning that with the development of pink-flowered strawberry and the deepening of petal color, the 21-nt PHAS loci triggered by miRNAs also increased, with 21, 31, and 37 loci in PF_L, PF_Z, and PF_D stages, respectively. However, the best samples of miRNAs triggering 24-nt PHAS loci were all from the young bud stage.
Meanwhile, to clarify the annotation information and expression patterns of genes encoded by PHAS loci, the sequence information of all identified PHAS loci was aligned and integrated with genomic information and transcriptome results. The annotation information of genes with the highest bit score was retained, the results obtained are shown in the Supplementary Table S7. There were 82 21-nt PHAS loci and five 24-nt PHAS loci with encoding capabilities, most of them were annotated as resistance proteins. Combined with genome annotation and transcriptome differential expression analysis, it was found that 31 PHAS loci triggered by miRNAs were differentially expressed during the flower development of 'Sijihong'. Supplementary Table S8 lists the PHAS loci triggered by miRNA, while satisfying the conditions that were differentially expressed in the PF_D vs PF_Z vs PF_L comparison group. Notably, PHAS21-198 was triggered by FvmiR393, and the PHAS locus was annotated as auxin signaling F-Box 2 (FvAFB 2) gene. Figure 7 showed FvmiR393-triggered phasiRNAs produced from the FvAFB2 locus.
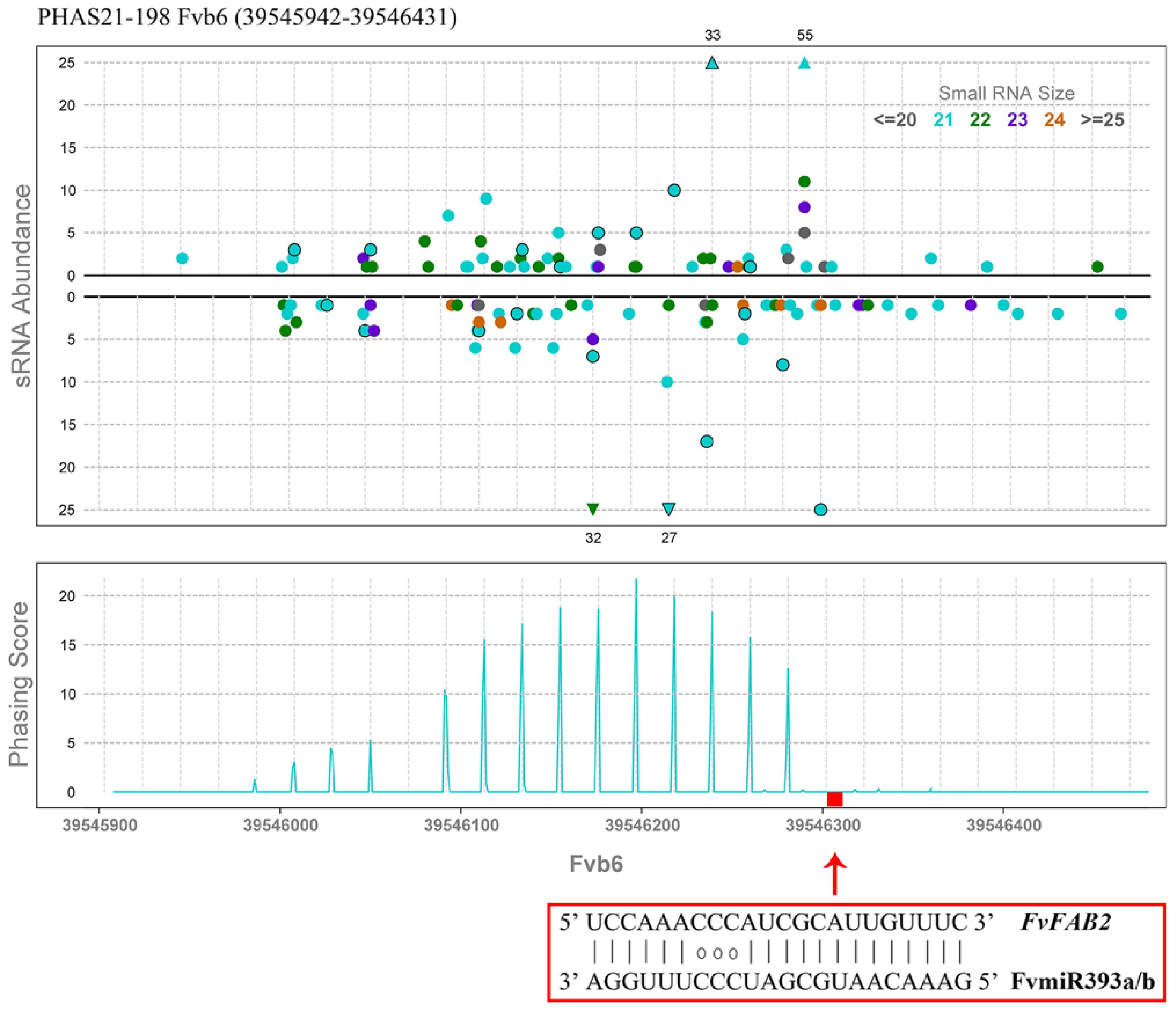
Figure 7.
FvmiR393a/b triggered phasiRNAs produced from the auxin signaling F-Box 2 (FvFAB2) locus in 'Sijihong'.
-
Flower development in angiosperms is a tightly spatiotemporally coordinated process that not only governs reproductive success but also determines the economic value of many crop and horticultural plants. This complex process is orchestrated by endogenous hormonal signals, which integrate diverse environmental cues to precisely regulate a series of key developmental events. Notably, transcriptional reprogramming serves as a fundamental molecular mechanism that underpins the specificity and robustness of plant responses to these hormonal signals[31,32]. Phytohormones act as central regulators, and their functional divergence across species and developmental stages is increasingly being unraveled through transcriptomic approaches, which enable the identification of hormone-responsive genes and regulatory networks[33−35]. The present transcriptomic analysis underscores the dominance of hormone signaling in pink-flowered strawberry: KEGG enrichment revealed that the 'Plant hormone signal transduction' pathway harbored the largest number of differentially expressed transcription factors (TFs, 323 in total) across PF_L, PF_Z, and PF_D stages (Fig. 2c). This enrichment is consistent with observations in Rosa rugosa and Litsea cubeba, where hormone signaling cascades were identified as core drivers of floral transition[9,10], highlighting the evolutionary conservation of hormone-centric regulation in angiosperm flower development. Within this pathway, key TF families—including ERFs, bHLHs, MYBs, NACs, and SPLs—emerged as critical transducers of hormone signals. Wang et al. found that TFs of bHLH, ERF, MYB, bZIP, and NAC families changed significantly during the flower growth[36]. In addition, Liu et al. comprehensively summarized that NAC transcription factors play a central role in the process of flowering and ripening of plants, and are crucial for the yield and quality of fruits[37]. In the present study, correlation network analysis further identified AIL1 (FvH4_1g18270), and bHLH93-like (FvH4_1g18930) as central regulatory hubs, each interacting with over 20 co-regulators (Fig. 2d). These TFs are known to mediate crosstalk between ethylene, auxin, and gibberellin signaling[36,37], suggesting their role in integrating multiple hormone pathways to coordinate petal cell expansion, pigment biosynthesis, and sepal opening—traits that define the phenotypic transition from PF_L to PF_D, where TFs such as SPLs and ARFs likely mediate crosstalk between gibberellin signaling and floral transition, analogous to regulatory modules in Rosa rugosa[9] and Petunia[38].
Beyond coding genes, small RNA (sRNA) transcriptomics has expanded our understanding of hormonal-post-transcriptional crosstalk[39]. The evolutionary conservative role of miR156 in controlling flowering is supported by the fact that the overexpression lines of Arabidopsis and Nicotiana tabacum show delayed flowering phenotype and prolonged juvenile stage[40−42]. In Coffea arabica, sRNA-seq across eight floral developmental stages identified 557 miRNA precursors and 173 21-nucleotide phased siRNA (phasiRNA) loci, with miR156 (targeting SPL) and miR172 (targeting AP2) interacting with ethylene signaling to regulate meristem dormancy[31]. Moreover, 24-nt phasiRNAs derived from disease resistance genes were preferentially accumulated in latent floral buds, and their downregulation upon rehydration coincided with ethylene-induced anthesis—revealing a novel layer of hormonal-sRNA regulatory crosstalk[31]. The present sRNAomic and degradomic analyses identified 151 conserved miRNAs (55 families), and 38 novel miRNAs, with 552 miRNA-target pairs validated by degradome sequencing (Fig. 3a, b). FamiR156s exhibited sustained upregulation from PF_L to PF_D (Figs 3c, 5a−c), while their target FaSPLs (FaSPL13A, FaSPL1, FaSPL12) were downregulated. This inverse relationship is consistent with the conserved role of miR156-SPL in mediating vegetative-to-reproductive transition[40−42]; however, the present study extends this function to petal coloration by linking FaSPLs to hormone signaling pathways (Fig. 4a). This suggests that miR156-SPL not only controls developmental timing but also integrates hormone signals to regulate pigment biosynthesis—a trait unique to ornamental strawberries. miR393, a conserved regulator of auxin signaling[43−45], was found to target the auxin receptor gene AFB2 (auxin-signaling F-box 2). Degradome analysis confirmed FvmiR393-mediated cleavage of FvAFB2, which was further associated with phasiRNA production (PHAS21-198, Fig. 7). This targeted relationship underscores the role of miRNAs in shaping auxin signaling dynamics—critical for petal cell polarity and expansion[44]—and bridges post-transcriptional regulation to phasiRNA-mediated amplification. Novel miRNAs further expanded the regulatory repertoire of pink-flowered strawberry. For example, FamiRN16 was highly expressed in PF_D, while its target FaRGA3 (a putative disease resistance protein) was repressed (Fig. 5f). This suggests a unique trade-off between flower development and stress defense, where novel miRNAs prioritize reproductive growth by suppressing stress-responsive genes—a likely adaptation to the hybrid's environmental resilience.
In addition to miRNAs, phasiRNAs have emerged as essential regulators in flowering plants, orchestrating developmental trajectories and mediating stress responses through their precise gene-silencing activities[46,47]. PHAS loci, genomic regions that generate phased small interfering RNAs (phasiRNAs) upon cleavage by trigger microRNAs (miRNAs), are key post-transcriptional regulators of plant floral development, governing processes from floral transition to gametophyte maturation via species-conserved and lineage-specific pathways[48,49]. The present analysis identified 285 PHAS loci (237 21-nt, 48 24-nt), with 31 loci showing differential expression across developmental stages (Supplementary Table S8). These PHAS loci play two critical roles in integrating hormone signaling, transcriptional, and post-transcriptional regulation: the 21-nt PHAS locus PHAS21-198 (annotated as AFB2) was triggered by miR393 cleavage (Fig. 7), generating phasiRNAs that likely reinforce the suppression of AFB2 mRNA. This 'miRNA-initiated, phasiRNA-amplified' silencing cascade enhances the precision of auxin signaling regulation—essential for maintaining the balance between cell proliferation and differentiation during petal maturation[45]. Similar cascades have been reported in Arabidopsis for hormone receptor genes[47], confirming the conservation of this mechanism in strawberry. Chromosomal mapping of PHAS loci revealed stage-dependent accumulation: 21-nt PHAS loci (e.g., PHAS21-108) were upregulated from PF_L to PF_D, while 24-nt PHAS loci were enriched in PF_L (Fig. 6a, b). PHAS21-108, annotated as a bHLH77-like TF, exhibited PF_D-specific expression (Fig. 6c), directly linking phasiRNA production to transcriptional regulation. This suggests that phasiRNAs not only amplify miRNA signals but also encode functional TFs—closing the loop between post-transcriptional and transcriptional regulation. Notably, all 24-nt PHAS loci were triggered by the miR2275 family, which was most active in PF_L (Supplementary Table S6). This stage specificity aligns with the role of 24-nt phasiRNAs in suppressing transposable elements[46], suggesting that pink-flowered strawberry prioritizes genome stability during early bud development (PF_L) before shifting to 21-nt phasiRNA-mediated developmental regulation in later stages (PF_Z, PF_D). Evolutionarily, the identification of species-specific miRNAs and PHAS loci reflects the regulatory innovations underlying the success of the Fragaria × Potentilla hybrid, which combines the ornamental traits of P. palustris with the horticultural adaptability of F. × ananassa. These innovations highlight the role of non-coding RNAs in driving phenotypic diversity in hybrid species.
In summary, the present integrated multi-omics analysis reveals that flower development in pink-flowered strawberry is governed by a tightly coordinated network where hormone signaling, transcriptional/post-transcriptional regulation, and phasiRNA-mediated amplification are inextricably linked. This network ensures the precise timing and execution of petal coloration and bud maturation, while also incorporating species-specific regulatory elements to adapt to environmental and horticultural demands. These findings not only serve as a foundation for future studies on ornamental strawberry biology, but also provide a paradigm for dissecting complex developmental processes in other horticultural crops.
This study was supported by the National Natural Science Foundation of China (31701964), and Shenyang Science and Technology Plan Project - Seed Industry Innovation Special (Grant No. 23-410-2-07).
-
The authors confirm contributions to the paper as follows: study conception and design: Yue J, Lei J, Xue L; sample collection: Xue L; analysis of transcriptome data: Zheng Y, Zhao J; analysis of the sRNAsome and degradome data: Bi M, Dai H; combined analysis of sequencing data and experimentation: Yue J; draft manuscript preparation: Yue J; feedback on the analysis and manuscript: Xue L, Lei J. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 The primers used for qRT-PCR analysis.
- Supplementary Table S2 Gene IDs and expression changes for TFs in hormone signaling pathways.
- Supplementary Table S3 The differentially expressed target genes of the differentially expressed known and novel miRNAs by degradome sequencing.
- Supplementary Table S4 Identification of PHAS loci during flower development of pink-flowered strawberry.
- Supplementary Table S5 Prediction of coding ability of PHAS loci.
- Supplementary Table S6 Identifcation of miRNA triggers.
- Supplementary Table S7 Annotation information of genes encoded by PHAS loci.
- Supplementary Table S8 The PHAS loci triggered by miRNAs were differentially expressed in the comparison group PF_D vs PF_Z vs PF_L.
- Supplementary Fig. S1 Heatmaps showing the expression patterns of the key differentially expressed transcription factors related to flower development.
- Supplementary Fig. S2 T-plots of the targets cleaved by the corresponding miRNAs from the degradome library.
- Supplementary Fig. S3 PHAS21-216, PHAS21-217 and PHAS21-219 in three developmental stages.
- Supplementary Fig. S4 PHAS24-10 and PHAS24-34 in three developmental stages.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yue J, Zheng Y, Zhao J, Bi M, Dai H, et al. 2025. Integrated high-throughput sequencing approach provides insights into the mechanism of flower development in pink-flowered strawberry. Fruit Research 5: e040 doi: 10.48130/frures-0025-0031 

Integrated high-throughput sequencing approach provides insights into the mechanism of flower development in pink-flowered strawberry
- Received: 29 June 2025
- Revised: 23 August 2025
- Accepted: 08 September 2025
- Published online: 01 November 2025
Abstract: Pink-flowered strawberry possesses significant ornamental value; however, the molecular mechanisms underlying its flower development remain poorly understood. In this study, an integrated multi-omics strategy—combining transcriptome, sRNAome, and degradome sequencing—was employed to analyze three key stages of petal coloration (young bud, coloration initiation, and big bud) in the pink-flowered strawberry cultivar 'Sijihong'. The present analysis revealed dynamic transcriptional reprogramming primarily governed by hormone-signaling transcription factors (TFs), among which ERFs (AIL1), and bHLHs (bHLH93-like) were identified as central regulatory components. Importantly, extensive post-transcriptional regulation was uncovered, which is mediated by 151 conserved, and 38 novel miRNAs, and contributes to petal development. Among these regulatory mechanisms, the repression of SPL transcription factors by FamiR156 and the triggering of phasiRNA production from the auxin receptor gene FvAFB2 by FvmiR393 are included. Additionally, 237 phasiRNA-producing loci (PHAS) were mapped, and these loci exhibit stage-specific expression patterns during floral maturation. This study delineates a hierarchical regulatory network that integrates transcriptional control, miRNA-guided silencing, and phasiRNA-mediated amplification to coordinate flower development, thereby offering valuable mechanistic insights and genetic resources for future horticultural improvement.
-
Key words:
- Pink-flowered strawberry /
- Flower development /
- Transcription factors /
- miRNAs /
- PHAS