









-
MicroRNAs (miRNAs) are an extensive class of 22-nucleotide noncoding RNAs thought to regulate gene expression in post-transcriptional regulation[1]. MiRNAs in plants were first found in Arabidopsis thaliana, and they are involved in many basic biological processes, such as plant growth, morphological formation, fruit development, and biological abiotic stress response[2,3] MiRNAs play an important role in the growth and development of plants. A study by Li found that in regulating plant root development, miR160 downregulates the expression of the auxin response factor (ARF) family genes, thereby controlling plant root differentiation and growth[4]. During the development of floral organs, miR172 promotes flower opening and maturation by inhibiting the expression of the Apetala2 (AP2) gene[5]. MiRNAs also play an important role in plant stress responses. In plant drought stress, miR169 enhances plant drought resistance by downregulating the expression of the nuclear factor Y subunit A (NF-YA) family genes[6]. Under salt stress, miR393 enhances plant salt tolerance by regulating the plant hormone response pathway[7]. In addition, miRNAs are involved in regulating plant virus resistance, climate change, and environmental pollution[8].
MiRNAs in grape, peach, apple, orange, and pear have been studied genome-wide since the development of high-throughput sequencing technology and the completion of fruit tree genome sequencing[3,9−12]. So far, several hundred plant miRNA family species have been found. Wang et al. identified 154 miRNAs from apples belonging to 26 families, some of which were generated by genome duplication during evolution[13]. Zhu et al. identified 47 peach-specific and 47 known miRNAs that targeted 80 genes, among which 3 miRNAs specifically targeted 49 MYBs, of which 19 regulated phenylpropanoid metabolism, a key pathway related to stone hardening and fruit colour development[9]. MiRNAs in apples show tissue-enriched expression patterns. For example, the conserved miRNAs miR156, miR159, miR160, miR167, and miR172 have different expression patterns in different tissues and developmental stages of apples[14]. miRNA172 is a key factor controlling apple fruit size[15].
High expression of miR156 and low expression of miR172 may shorten the juvenile phase, and miR160 and miR393 can promote the transformation of fruit trees at the same time by regulating genes related to auxin signal transduction[16]. Corbsier et al. found that the flowering mechanism of some grafted plants was regulated by miRNAs[17]. Guo found significant differences in the expression of 33 known miRNAs and six newly annotated miRNAs between apple leaf buds and flower buds, mostly showing a negative correlation with the process of flower bud formation[18]. After shoot bending treatment in apples, the differentially expressed miRNAs and their target genes were associated with metabolic pathways such as auxin, abscisic acid, gibberellins, and flowering, and they also participated in various biological processes such as cellular biosynthesis and metabolism[19]. Song et al. found that miR159, miR167, miR396, and their target genes could regulate cell differentiation and internode length, while miR164, miR166, miR171, and their target genes could regulate the growth of the stem apical meristem[20]. Yu et al. found that miRNAs also participate in apple disease resistance[21].
Grafting is a kind of asexual propagation method that can maintain the inherent good properties of scions and is widely used in the production of vegetables, ornamental horticultural crops, fruit trees, and other horticultural crops[22, 23]. Plant grafting has been used for millennia to reduce juvenility and confer biotic and abiotic stress tolerance[24]. Grafting can form a complete vascular bundle between the rootstock and scion, and during the growth and development of grafted plants, large molecules such as proteins, mRNA, and miRNA in the phloem selectively move from the companion cells through plasmodesmata to sieve tubes and are transported over long distances between the rootstock and scion, ultimately reaching the corresponding target cells to regulate the expression of related genes and thereby regulate the development of plant organs. Pant et al. used Arabidopsis thaliana overexpressing miR399 as the scion grafted onto wild-type Arabidopsis and found that the roots of wild-type Arabidopsis accumulated a large amount of mature miR399, while pri-miR399 was barely detected in its roots, indicating that miR399 could move long distances through the phloem[25]. Zhang et al. found in their study that grafting with rootstocks promotes phenolic compound accumulation in grape berry skin during development based on integrative multi-omics analysis[26]. Thieme et al. found that a large number of miRNAs move in specific tissues to regulate plant tissue development[27]. As a signalling molecule, miRNA controls traits such as flower and leaf shape in grafted plants. Wang found differential expression of miR164, miR168, and miR390 in the soybean after drought stress[28]. Buhtz et al. also found differentially expressed miRNAs after grafting in Brassica napus. MiR398, miR399, and miR395 in B. napus may be transmitted from the rootstock phloem to the scion and affect the scion's response to mineral element deficiency[29].
Compared to vegetables and other horticultural crops, there are fewer studies on miRNAs involved in the grafting of fruit trees due to their long life cycle and complex tree structure. In recent years, there has been some progress in miRNA research on the interaction between rootstock and scion in fruit trees, and it has been found that grafting significantly affects the expression levels of many miRNAs. In grafted Citrus seedlings, the expression of miR156, miR157, and miR894 in the leaf petioles is significantly downregulated compared to that in seedlings grown from seeds[30]. Xu found that miRNAs associated with regulating plant growth and development, stress response, and hormone signal transduction were upregulated in grafted Citrus scions, while they were downregulated in the rootstocks[31]. Liu et al. suggested that changes in miRNA expression in grafted watermelon leaves may be one of the reasons why grafting affects plant growth, development, and stress response[32]. Grafting can induce differential expression of miRNAs in Citrus leaf petioles, and compared with seedlings grown from seeds, the expression of miR156, miR157, and miR894 is significantly downregulated in grafted seedlings[30]. An et al. found that grafting altered the expression patterns of miR156 and miR172, which are related to flowering. Specific miRNAs differentially expressed during grafting may be involved in regulating growth, development, and metabolism in fruit trees after grafting[33]. In the grafting process of apples 'Gala' and 'M9', the expression levels of miR172, miR396, miR399, and miR167 changed, which may be related to plant auxin and carbon-nitrogen metabolism processes[18].
There are significant differences in the expression levels of miRNAs in apple fruits grafted with different rootstocks. In 'Fuji' apple fruits, the expression levels of miR156, miR172, miR390, miR397, and miR408 in fruits grafted with dwarfing rootstocks 'M9' and 'B9' were higher, while the expression levels of miR164 and miR167 in fruits grafted with 'MM111' rootstock were higher[34]. Many miRNAs have been discovered in horticultural crops, but the target genes and functional characteristics of most graft-responsive miRNAs are still unclear.
-
The following apple grafting combinations were used in the study: the dwarfing interstock combination M ('Gala'/'Mac 9'/Malus baccata (L.) Borkh) and the standard rootstock combination B ('Gala'/Malus baccata (L.) Borkh). The combinations used in this study were provided by the Research Institute of Pomology of the Chinese Academy of Agricultural Sciences (CAAS), Xingcheng, Liaoning, China (120° 44′ E, 40° 37′ N). For each sample, at least three plants were pooled, and three independent biological replicates were used.
Qualitative and quantification of miRNAs
-
On June 21, 2016, six healthy trees without pests or diseases were selected for B and M. Each sample obtained 2 cm long and wide bark slices, and two samples (BS and BR) in B, three samples (MS, MI, and MR) were collected in M, with three biological replicates for each tree. Before transferring the bark samples to a −80 °C ultra-low-temperature freezer, they were briefly stored in liquid nitrogen. First, total RNA needs to be extracted from the bark samples. Then, the extracted RNA samples were sent to Biomarker Technologies for small RNA sequencing. After passing quality control, the library was constructed. To obtain RNA with a length of 18–30 nt, PAGE gel electrophoresis was used for gel cutting and separation. Subsequently, the Clean Reads, which were trimmed for adapter sequences, low-quality bases, and contaminants, were aligned to the Rfam13 database for annotation.
The expression of miRNAs in each sample was calculated using the TPM algorithm and normalised. The equation for TPM normalisation is as follows:
$ T P M=\dfrac{Read\;count*\mathrm{1,000,000}}{Mapped\;Reads} $ In the equation, readcount represents the number of reads that correspond to the miRNA, and Mapped Reads represent the number of reads that are mapped to all miRNAs. Known and novel miRNAs were identified using the miRDeep2 software.
Analysis of differentially expressed miRNAs (DEMs)
-
After normalization of expression levels using the TPM algorithm, the influence of sequencing depth differences between different samples can be eliminated, enabling a scientific and accurate comparison of differential expression of miRNAs among samples. To investigate the differential expression of miRNAs between two biological conditions, DESeq was used to perform differential expression analysis between sample groups. The fold change (FC) and false discovery rate (FDR) were used as filtering criteria. In the original hypothesis test, the recognized Benjamini Hochberg correction method was used to correct the significance p-values, and the FDR was ultimately used as the key indicator for filtering differentially expressed miRNAs. We compared the miRNAs of B and M to determine whether there were differences under the intermediate rootstock grafting. In this study, the threshold for defining differentially expressed miRNAs was set at |log2(FC)| ≥ 0.58 and p-value ≤ 0.05.
Prediction of miRNA target genes
-
For the sequencing data of this study, we used the Target Finder software to predict the target genes of miRNAs, using known miRNAs and newly predicted miRNAs with corresponding gene sequence information of the species. After completing the prediction of target genes, we used the BLAST software to compare the predicted target genes with the nr, Swiss Prot, GO, COG, KEGG, KOG, and Pfam databases to obtain annotation information for the target genes.
Analysis of co-expression trends of DEMs
-
DESeq2_edgeR p value = 0.05, FC = 1.5. Analysis of the different patterns of changes in miRNA expression abundance in two different parts of the bark, and grouping mRNAs with the same expression trend into a dataset, creating an expression pattern graph for the dataset. The distance measurement method used is Euclidean distance, and the clustering method is K-means clustering. Finally, the expression pattern will be divided into K categories.
Joint analysis of miRNA and mRNA
-
Based on the miRNA sequencing data in this study and the transcriptome sequencing in early research using differentially expressed miRNAs as the screening criteria, we searched for mRNAs regulated by these differentially expressed miRNAs, and focused on analyzing the negative regulatory relationship between miRNAs and mRNAs[35]. At the same time, functional enrichment analysis was performed on the resulting mRNAs. The targeting relationship between miRNAs and mRNAs can be visualized using a Sankey diagram.
Expression analysis by reverse-transcription quantitative PCR
-
Total RNA was extracted from phloem of rootstock, interstock, and scion of 'Gala'/'Mac 9'/Malus baccata (L.) Borkh and 'Gala'/Malus baccata (L.) Borkh using the Polysaccharide and Polyphenol RNA Extraction Kit (Tiangen, China). Real-time quantitative PCR (qRT-PCR) was carried out to validate the levels of DEGs from phloem of B and M combinations according to the instructions of the Prime ScriptTM RT reagent Kit with gDNA Eraser (TaKaRa, Code No. RR036A), 1,000 ng of the total RNA was reverse-transcribed with random primers. The reaction system included 1,000 ng of RNA, 2 µL of 5 × PrimeScript RT Master Mix (Perfect Real Time), and RNase-free dH2O was added to obtain a 20 µL volume; the reaction conditions were 37 °C for 15 min and 85 °C for 5 s. Four target genes with miRNA were selected from the transcriptome data. Primers were designed with primer premier 5.0 software and relevant primers were synthesized by Sangon Biotech (Shanghai, China). The qRT-PCR was performed using TB Green Premix Ex Taq II (TaKaRa, Code No. RR820B) in a Light Cycler 96 quantitative PCR detection system (Roche, Switzerland). The qRT-PCR aliquot contained 10 µL TB Green Premix Ex Taq II (Tli RNaseH Plus) (2 ×), 6 µL of ddH2O, 2 µL of cDNA, 0.8 µL of each of the forward and reverse primers (10 μM), and 0.4 µL of ROX Reference Day II (50 ×). The reaction conditions included preincubation at 95 °C for 30 s, followed by 40 cycles at 95 °C for 7 s, 60 °C for 30 s, and 72 °C for 30 s, with melting (65~97 °C, +1 °C/cycle; holding time: 1 s). The q-Actin gene of malus was used as the reference to correct expression levels of related genes. All the PCR assays were performed with three biological replicates. The relative expression levels were calculated with the 2−ΔΔCᴛ method[36].
The reverse transcription of miRNA was usually performed using the stem-loop method, and miRNA primers were designed using Shanghai Biotech's online primer design tool (
https://www.sangon.com/class_mirna_stemloop.html ) which was a relatively complex process. First, total RNA was extracted, and then miRNA first-strand cDNA was synthesised. The reaction system included 10 l of miRNA L-RT Solution mix, 1.5 l of miRNA L-RT Enzyme mix, 2 l of total RNA, 1 l of stem-loop primer (10 M), and RNase-free water, for a total volume of 20 l. The reaction mixture was incubated at 16 °C for 30 min, then at 37 °C for 30 min, and finally at 85 °C for 5 min to inactivate the enzyme. The resulting cDNA could be used for miRNA qPCR. miRNA qPCR used a miRNA fluorescent quantification PCR kit (dye method), which was similar to the target gene qPCR method. Specific miRNA primers were used, and typically three miRNAs were selected for detection, with 5s rRNA serving as an internal reference gene. The reaction system included miRNA primers, cDNA, 2 miRNA qPCR master mix, ROX Reference dye (L) or (H), and water. The miRNA qPCR reaction was usually pre-treated at 95 °C, followed by 40 cycles, each consisting of annealing at 95 °C for 30 s, annealing at 60 °C for 30 s, and extension at 72 °C for 30 s. After qPCR detection, the relative expression level of miRNA was calculated using the 2−ΔΔCᴛ method by determining the Ct value and comparing it to the expression level of the internal reference gene, thus inferring the expression level of miRNA. All primers used in reverse-transcription quantitative PCR (qRT-PCR) are listed in Supplemental Table S1. We selected two miRNAs, namely mdm-miR156a, and mdm-miR396a, and their four target genes (At3g21360, CYP707A2, EIX2, and SPL9). -
The study used 15 phloem samples from five different combinations to create a library. The clean data obtained after quality control processing amounted to a total of 366.87 M, with a minimum of 16.64 M clean reads in each sample. The clean reads from each sample were aligned to the Malus x domestica GDDH13 v1.1 reference genome using Bowtie software, with alignment efficiency ranging from 44.28% to 61.14% (Supplemental Table S2). By comparing mapped reads with mature miRNA in the miRbase (v22) database, the study identified 291 known miRNAs (Supplemental Table S3). The majority of these known miRNAs (72.16%) were 21 nt in length (Fig. 1a). The reads that were not mapped to known apple miRNAs in miRbase (v22) were used to predict novel miRNAs using the miRDeep2 package. As a result, 159 novel miRNAs were identified in the study's materials. Their precursors were folded into the secondary hairpin structure by RNAfold, such as Novel miR6, Novel miR16, and Novel miR158 (refer to Fig. 1b for more information). The majority of these novel miRNAs (88.05%) were 24 nt in length. The study also conducted miRNA family analysis using the search software and identified 281 miRNAs from 80 miRNA families (Supplemental Table S4). Among these families, the mdm-MIR171-1, mdm-MIR159, mdm-MIR172, and mdm-MIR395 families accounted for approximately 8.5%, 7.1%, 5.7%, and 5%, respectively (Fig. 1c).

Figure 1.
The length distribution and family of miRNAs identified in apple. (a) Length distribution of known and novel miRNAs. (b) Secondary structures of some novel miRNAs (Novel miR6, Novel miR16, and Novel miR158). (c) Analysis of the miRNA family.
Dicer and DCL enzymes were known to have strong sequence cleavage preference for 5'U. Analysis on miRNA base bias was used to compare that of identified miRNA with typical miRNA. First base preference of miRNA and base preference on all sites were shown in Fig. 2. First base preference of miRNA has a strong preference for U and tenth base preference for A, which complied with the base preference rule of miRNA, indicating that the miRNA obtained through sequencing was high quality and confidence.
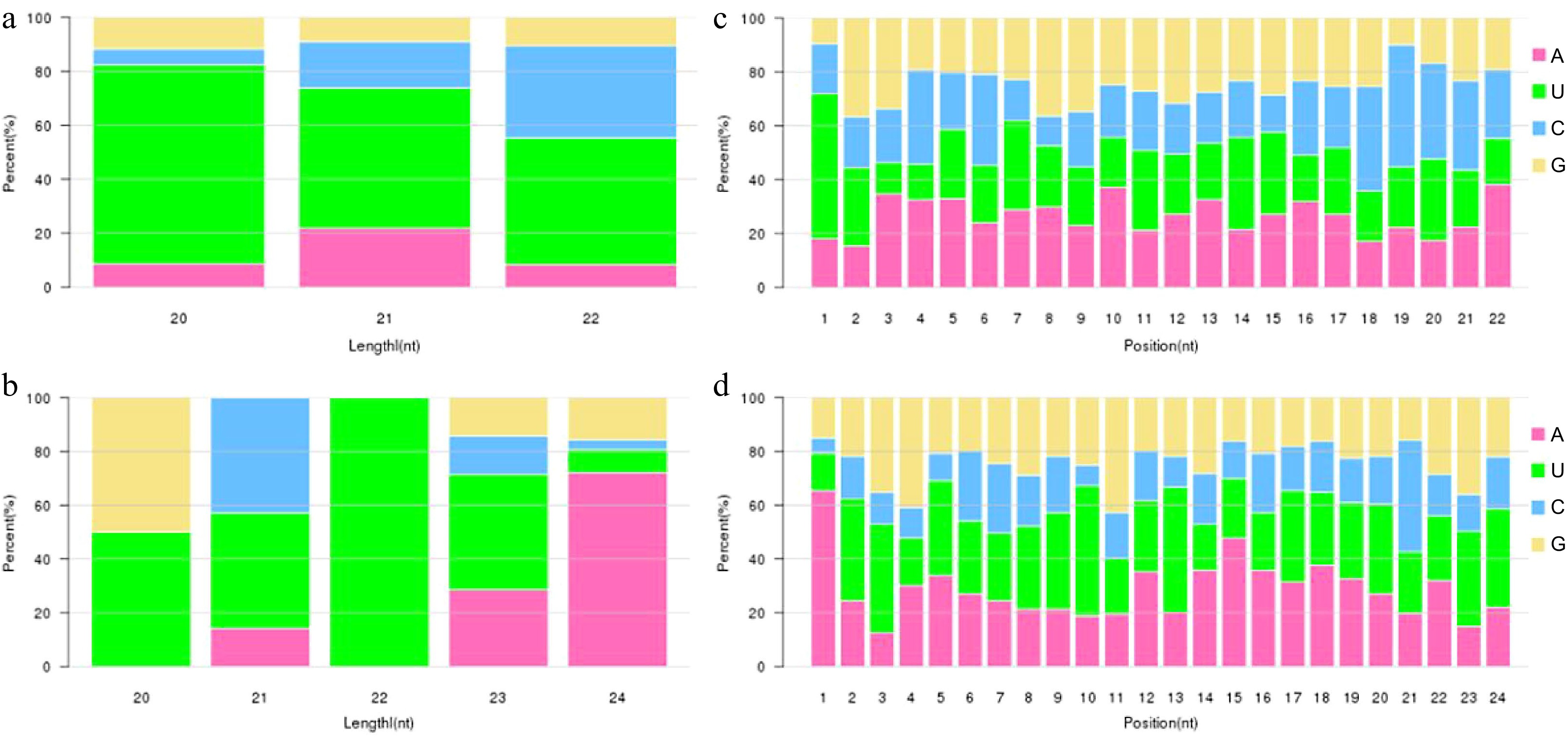
Figure 2.
Analysis of the miRNA nucleotide bias. (a), (b) First base preference of known and novel miRNA in different length, respectively. (c), (d) Base preference on known and novel miRNA at each position, respectively.
Quantitative analysis of the miRNA
-
Expression of miRNAs in each sample was calculated and normalized by TPM algorithm. All miRNA expression in each sample is listed in Supplemental Table S3. Distribution of miRNA expression described overall miRNA expression pattern in each sample. Distribution of TPM density in each sample is shown in Fig. 3a, b. In each sample, the miRNA’s log10TPM was mainly between 1 and 3. The miRNA counts from each sample with ranging from 353 to 411 (Fig. 3c). The expression levels of the miRNAs in the sample’s three replicates tended to be roughly consistent, indicating that biological reproducibility was reliable. We analyzed co-expressed and specifically expressed miRNAs in different samples with Venn plots, and a total 358 miRNAs were expressed together in all combinations (Fig. 3d).
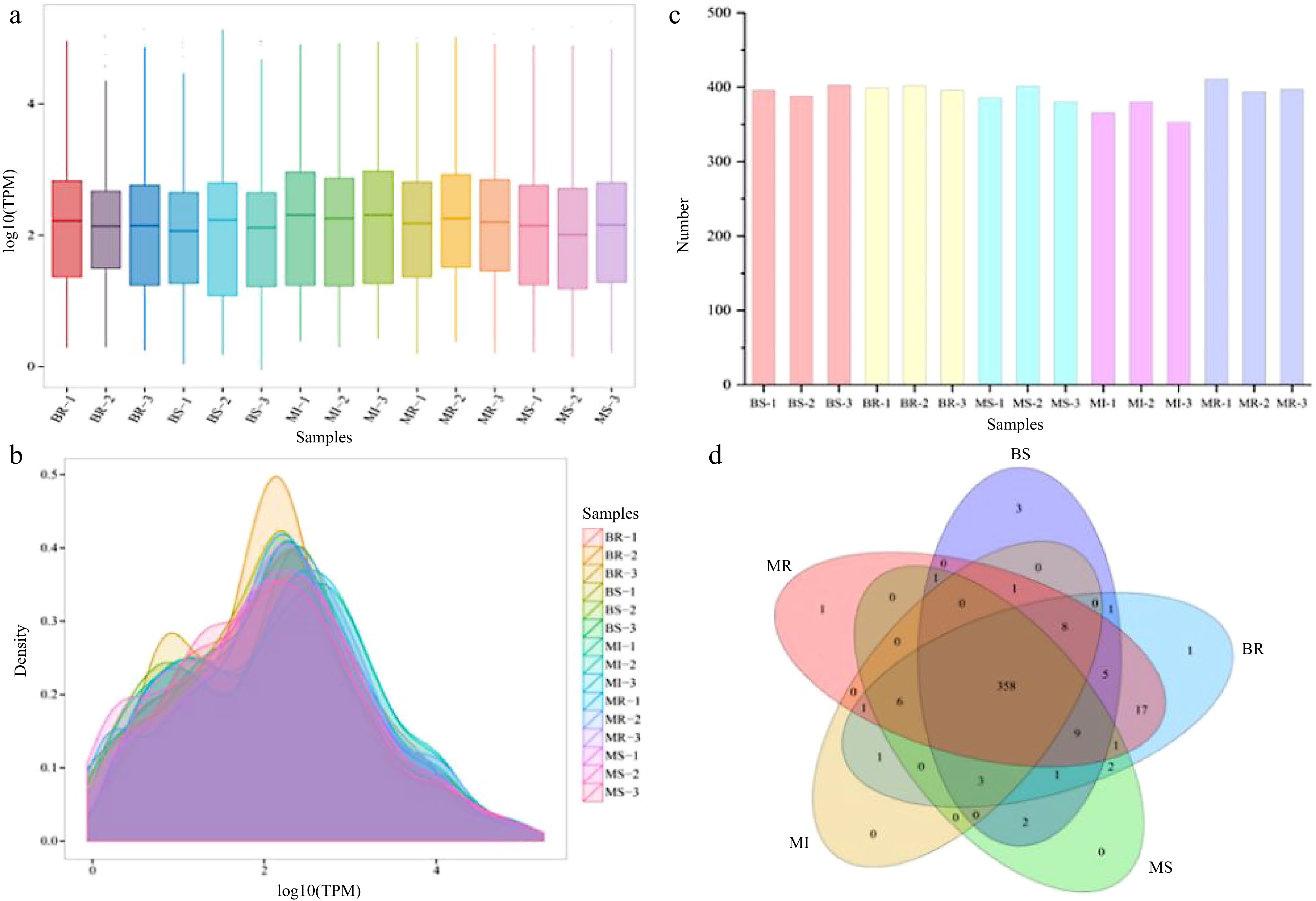
Figure 3.
Analysis of the miRNA expression status in each sample. (a) Boxplot of the overall distribution of miRNA expression in each sample. (b) TPM density distribution (the curves with different colors in the figure represent different samples, the abscissa of the point on the curve represents the logarithm value of the corresponding sample TPM, and the ordinate of the point represents the probability density). (c) Number of miRNA statistics in each sample. (d) Venn analysis of miRNAs in each combination (the number of miRNAs in each combination were taken as three repeated union sets).
Target gene analysis for miRNAs
-
MiRNA target genes were predicted based on sequences of known miRNAs, novel miRNAs and gene sequences of corresponding species. The Target Finder software (v1.6) was employed for target gene prediction in this study. Summary of miRNA target gene prediction was shown in Table 1. The results demonstrated that 4,792 genes were predicted to be targeted by 436 miRNAs.
Table 1. Predicted known and novel miRNAs with targets.
Types All miRNA miRNA with target Target gene Known miRNA 291 291 4,194 Novel miRNA 159 145 783 Total 450 436 4,792 In 4,792 target gene, with 4,648 of them annotated in various databases (Fig. 4), including Non-redundant protein sequence database (nr) (4,641), Kyoto encyclopedia of genes and genomes (KEGG) (3,561), Gene ontology database (GO) (3,964), and Clusters of orthologous groups (COG) (1,675). A total of 4,125 target genes were annotated in Malus domestica, and they accounted for 88.9%. In GO enrichment analyses, the major GO terms for target genes were membrane, membrane part, cell, cell part, and organelle for cellular components (CCs); the major GO terms in the binding and catalytic activity were molecular functions (MFs); and the major GO terms in biological processes (BPs) were cellular process and metabolic process. The top three KEGG pathways were plant-pathogen interaction (177), plant hormone signal transduction (143), and starch and sucrose metabolism (119). The top three COG function classes were general function prediction only (256), signal transduction mechanisms (216), and carbohydrate transport and metabolism (164). In general, these target genes were mostly related to plant signal transduction and material transport.

Figure 4.
The analysis of the miRNA’s target genes. (a) NR homologous species distribution. (b) KEGG pathway enrichment analysis. (Y-axis: name of KEGG pathway annotation; X-axis: number and percentage of genes involved in this pathway over counts of all annotated genes). (c) GO annotation classification statistics. (d) COG function classification of consensus sequence (X-axis: the COG functional classifications; Y-axis: the functional categories corresponding to each classification).
Differential expression of miRNAs (DEMs) under grafting interstock
-
We compared the miRNAs of B and M to determine whether there was a difference under grafting interstock. During the detection of DEMs, In this project, threshold for defining DEMs was set as |log2(FC)| ≥ 0.58; p-value ≤ 0.05. Fold change (FC) refers to ratio in expression between two samples (groups). P-value represents significancy of difference in expression. We detected DEMs by comparing five pairs of samples (Fig. 5a). A total of 291 DEMs were identified (Supplemental Table S5). The comparison of BS vs MS (B scion vs M scion) revealed 36 upregulated miRNAs and 43 downregulated miRNAs. BR vs MR (B rootstock vs M rootstock), in contrast, had 57 DEMs, of which 22 miRNAs were upregulated and 35 miRNAs were downregulated. We analyzed the DEMs of the BS vs MS and BR vs MR combinations using Venn diagrams (Fig. 5b). A total of 10 miRNAs were upregulated in the scions but downregulated in the rootstocks. A total of six miRNAs were downregulated in the scions but upregulated in the rootstocks. Additionally, after grafting the interstock, there were one DEM that were commonly upregulated in the BS vs MS and BR vs MR groups and three DEMs that were commonly downregulated in these groups (Fig. 5b). The DEMs were subjected to cluster analysis, and a cluster heat map was created (Fig. 5c, d). After grafting the intermediate rootstock, there will be differential expression of tree miRNAs, with a decrease in the expression levels of mdm-miR156 in the scion and mdm-miR172 in the rootstock. The expression levels of the miRNAs in the group’s three replicates tended to be roughly consistent, indicating that biological reproducibility was reliable.

Figure 5.
The analysis of the DEMs in each sample. (a) Number of DEMs in each group. (b) Venn diagram analysis of DEMs (different colors represent different comparison combinations). (c) Heat map of DEMs with BR vs MR. (d) Heat map of DEMs with BS vs MS.
Analysis of the DEMs co-expression trends
-
We conducted a co-expression trend analysis of the DEMs from the BR vs MR and BS vs MS combinations to study the expression of these DEMs at different samples, with a total of nine expression patterns (Fig. 6 & Supplemental Table S6). Most of the gene trends a and b indicate that the miRNA expression of scion and rootstock will be affected by the dwarfing interstock. mdm-miR172 was Cluster 9. mdm-miR156 was Cluster 6, as well as mdm-miR167 and mdm-miR395 was Cluster 6.
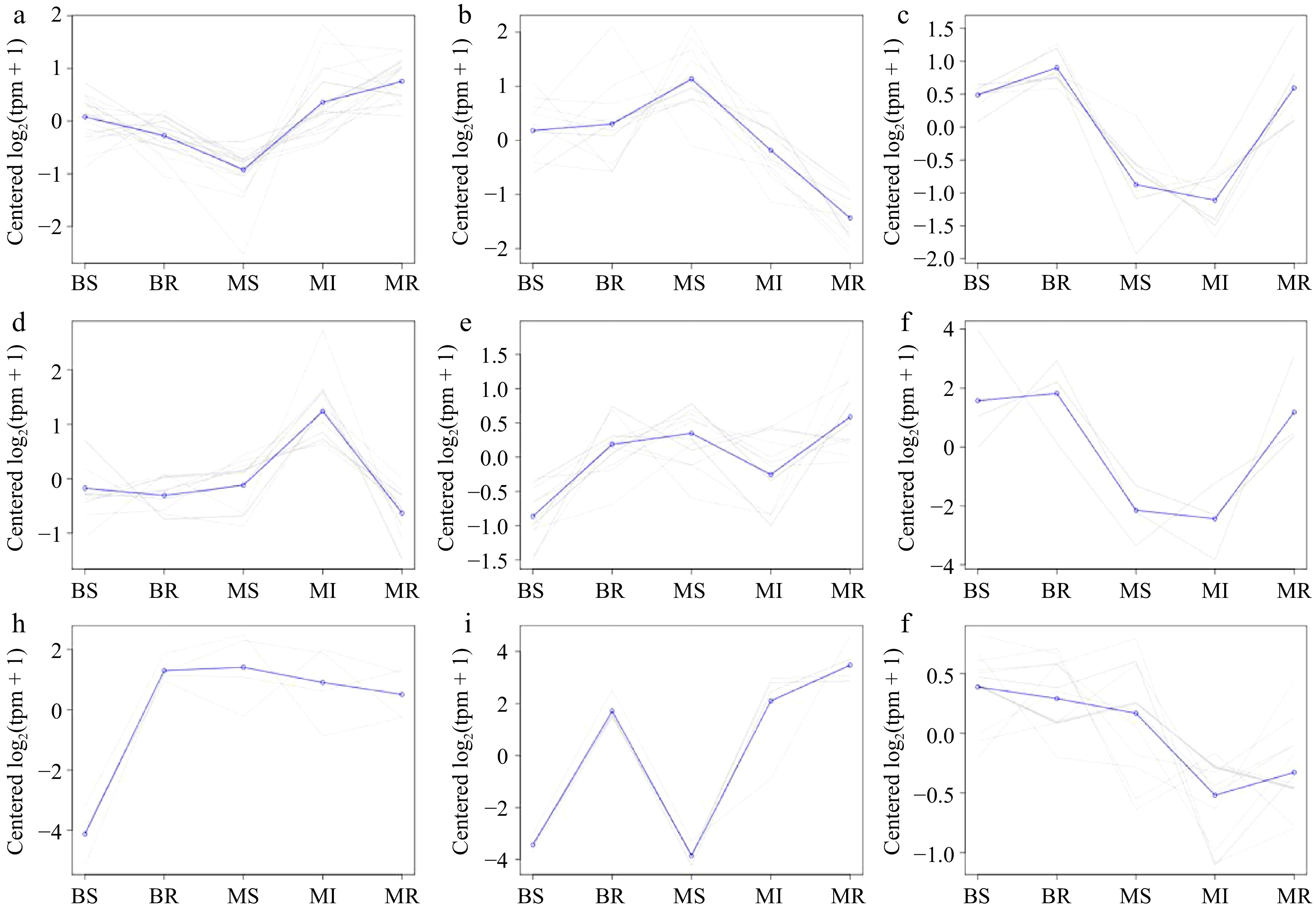
Figure 6.
A co-expression trend analysis of the miRNAs in each sample. (a)−(j) Results plots for each class of the co-expression trend clustering in all DEMs with Cluster 1−9, respectively.
In this research, we explored the effects of interstock in grafting dwarfing on mRNA and miRNA in the cambium. Generally, individual miRNA not only has no function but is also easily degraded. Only when it forms RISC with Ago protein can it play an effective role, where miRNA plays a guiding role, guiding RISC to specific mRNA regions and exerting its function[37]. Typically, plant miRNA can form complete complementary pairing with the coding region of mRNA and play a role in inhibiting gene expression by inducing mRNA degradation[38]. We used the DEGs from our previously published article and the DEMs from this experiment for joint analysis[35]. Using DEMs as the screening standard, we searched for the mRNA regulated by these DEMs, and focused on analyzing the negative regulatory relationship between miRNA and mRNA (Fig. 7). Meanwhile, KEGG functional enrichment analysis was performed on the resulting mRNA. The relative expression levels of miRNA and target genes are shown in Supplemental Figs S1, S2.
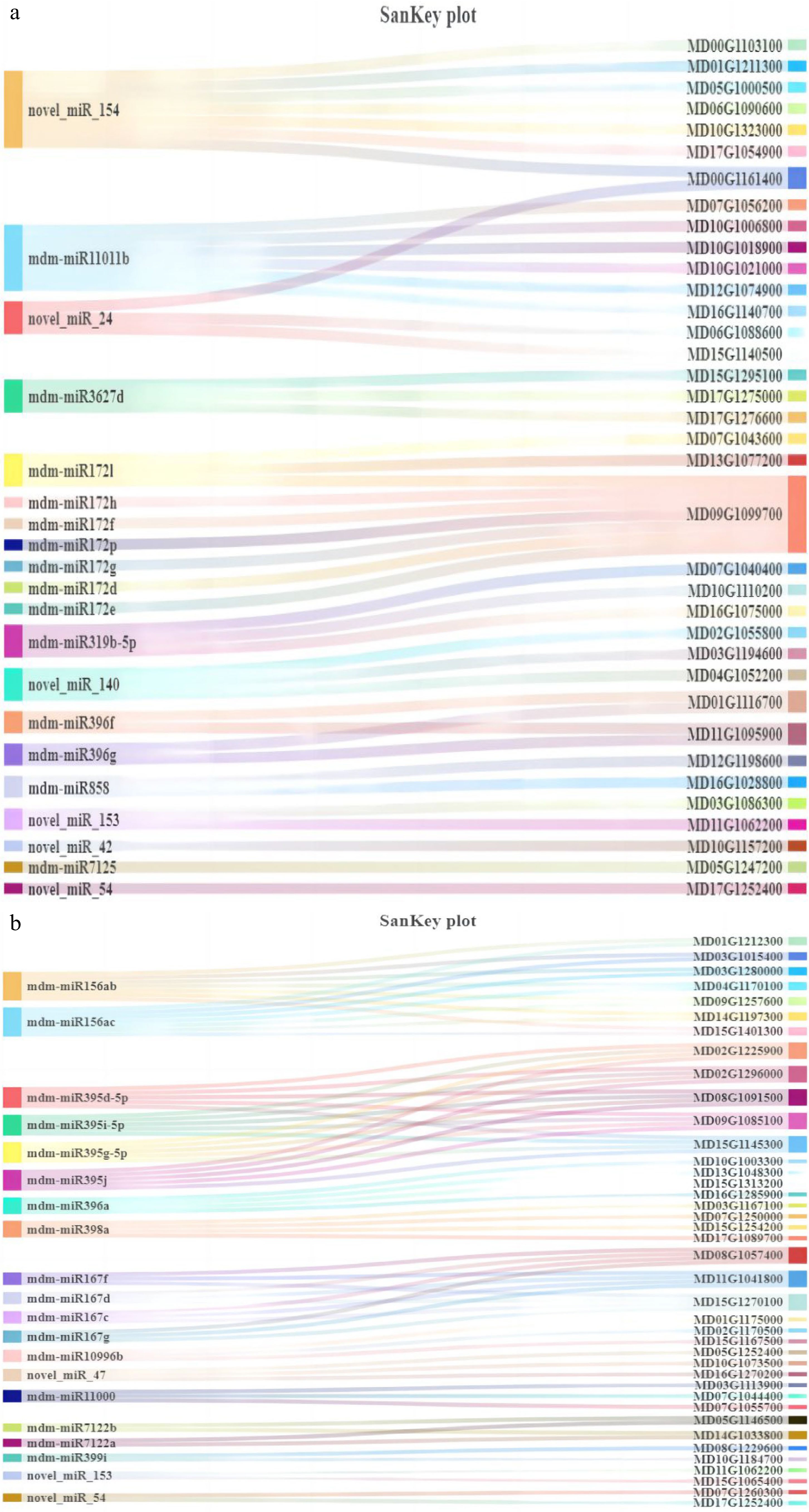
Figure 7.
Sankey diagram of the top 20 miRNA and their target genes. (a) BR vs MR’s top 20 miRNA and their target genes. (b) BS vs MS’s top 20 miRNA and their target genes.
As shown in Fig. 8, after the joint analysis of differential miRNA and transcriptome sequencing in the cambium of two grafted combinations, 401 target genes were annotated in KEGG, involving a total of 100 pathways. Among them, the most significant enrichment was the starch and sucrose metabolism pathway (ko00500), which involved 42 related genes. In the scion, a total of 371 target genes were annotated in KEGG, involving 97 pathways, mainly enriched in plant hormone signal transduction (ko04075, 30 target genes). The KEGG enrichment results indicate that the interstock may affect the expression of related genes in the root starch and sucrose metabolism pathway and the scion plant hormone signal transduction pathway, thereby affecting the growth and development of the plant, resulting in a dwarf phenotype.
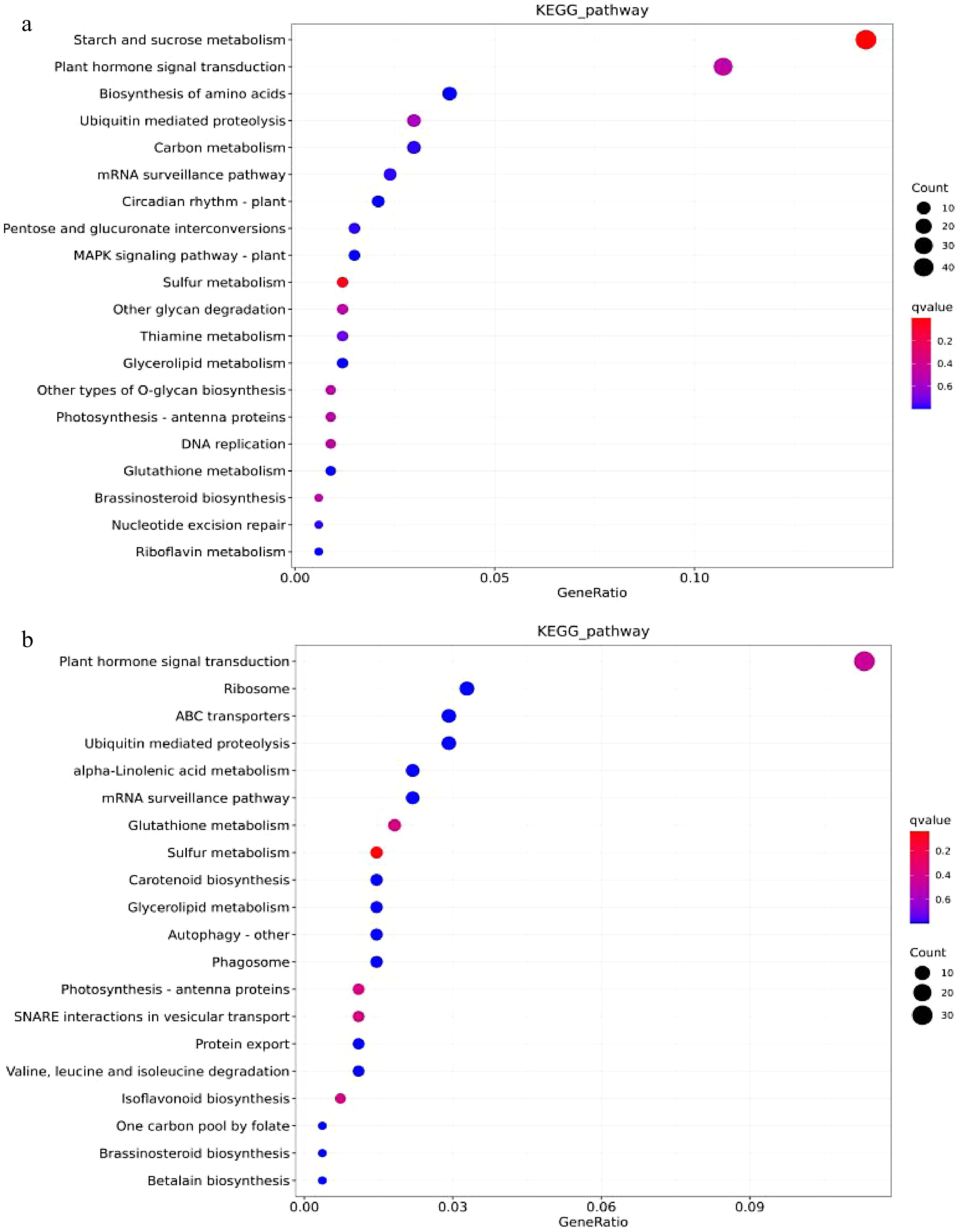
Figure 8.
The KEGG enrichment analysis of DEM’s target genes. (a) BR vs. MR’s DEMs enrichment bubble plots for top 20 KEGG pathways (b) BS vs. MS’s DEMs enrichment bubble plots for top 20 KEGG pathways.
Verify miRNA-seq results using qRT- PCR
-
To verify miRNA-seq results, we selected three miRNAs, namely mdm-miR156a and mdm-miR396a, and their four target genes (EIX2, CYP707A2, At3g21360, and SPL9) at B and M stages were selected for qRT-PCR. The targeting relationships between these two miRNAs and four genes are shown in Supplemental Table S7. Expression patterns of the three miRNAs and four target genes in B and M were consistent with results of the RNA-seq analysis (Fig. 9). Thus, the transcriptome data were correct and reliable and truly reflected the transcription levels of genes involved in phloem development after grafting dwarfing interstock.
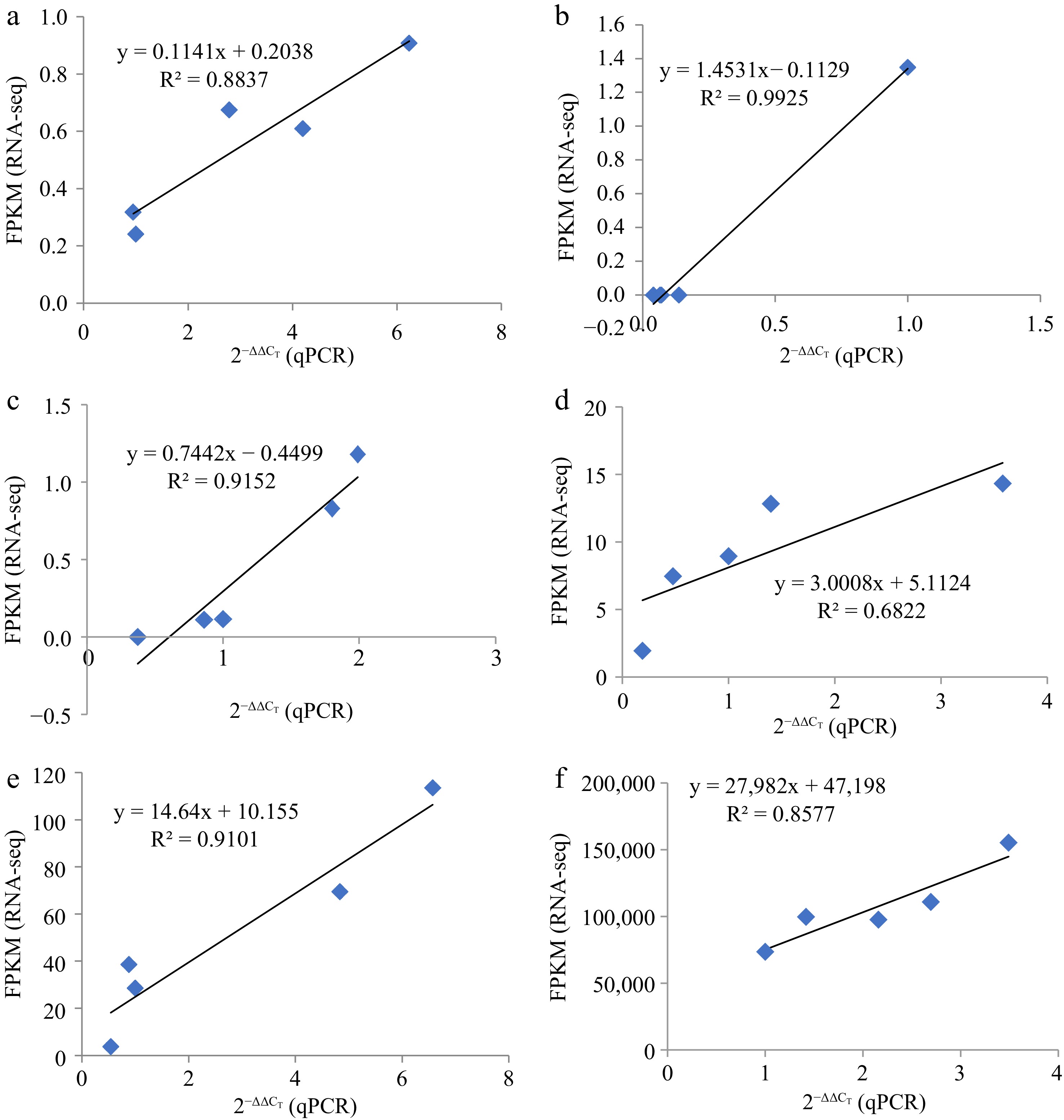
Figure 9.
The linear relationship between RNA-seq and qRT-PCR data. Represented by the linear relationship between RNA-seq and qRT-PCR Data. (a) miR156a, (b) miR396a, (c)–(f) were EIX2, CYP707A2, At3g21360, and SPL9, respectively.
-
MiRNA is a type of small single-stranded endogenous non-coding RNA, usually about 20 to 24 nucleotides long, that regulates gene expression levels by inducing inactivation or degradation of target mRNAs[33]. Through high-throughput sequencing technology, researchers have successfully identified many miRNAs in grafted apples. miRNAs affect the growth and development of grafted apple scions and rootstocks by regulating the expression of target genes[39]. Wang et al. identified 96 miRNAs in the apple rootstock 'M26' through small RNA library construction and Illumina high-throughput sequencing, of which the expression of 23 miRNAs was influenced by grafting[6]. Wang et al. compared the miRNA libraries of apple rootstocks 'M26' and 'JM2', and identified a total of 277 miRNAs, of which 23 miRNAs showed significantly altered expression during grafting[39]. In addition, researchers have also identified some miRNA target genes in grafted apples through target prediction and validation[39−41]. For example, the miR156 a/b and the Squamosa promoter binding protein like (SPL) gene family exhibited opposite expression patterns in the rootstock 'M26'. Meanwhile, the study also found that miR395 was expressed at a low level in the rootstock 'M26', possibly because its target gene ATP Sulfurylase 3 (ATPS3) was inhibited during grafting. Li & Mao further identified miRNA target genes in the apple rootstock Malus hupehensis, including transcription factors, hormone synthesis, and signal transduction-related genes, among others[42].
Grafting can shorten the juvenile phase of fruit trees and improve their stress resistance. It was found that miR156, miR172, and miR395 can transport long distances in grafted plants, and the expression of many miRNAs changed significantly after grafting[37, 43−47]. miR156 is an important regulatory factor that participates in the transition from vegetative growth to reproductive growth in plants. It is the main regulatory factor for the transition from the juvenile phase to the adult phase, and has been shown to be the most conserved miRNA in Arabidopsis, maize, rice, and other plants[43]. Overexpression of miR156 under long-day conditions can cause delayed flowering and a decrease in the expression of SPL family members[44, 48]. In this experiment, the expression level of mdm-miR156 decreased in the interstock combination scion, which may promote the transition of the plant to the adult phase and obtain early flowering characteristics. Low expression of miR172 may shorten the juvenile phase[16]. In this study, mdm-miR172 was downregulated in the interstock of the grafting. miR395 is a key factor that regulates plant sulfur metabolism. It targets three ATP sulfurylase genes, ASP1, ASP3, and ASP4. When miR395 is induced by low sulfur stress, the transcription level of ASP1 decreases. When sulfur supply is normal, the transcription level of ASP1 increases, and the expression of mdm-miR395 is completely suppressed[49].
In this study, it was found that after grafting the interstock, the expression level of mdm-miR395 was downregulated, and its effect on plant growth and development was not clear. According to the regulatory mechanism between miRNA and target genes, miRNA and their corresponding target genes show a negative correlation in expression regulation. This study found that the expression of most miRNAs and their target genes showed a negative correlation after grafting, while a small number of miRNAs and target genes showed a positive correlation or no correlation. Generally, gene expression is influenced by more than one factor, and the expression of miRNA target genes is also regulated by factors other than miRNA. To date, the complex regulation of miRNA on target genes has not been fully elucidated[50]. This result preliminarily reveals the expression patterns of miRNA and their target genes between grafted apple rootstocks. Through KEGG enrichment analysis, the effects of these differentially expressed genes on plant physiology and growth and development mainly manifested in the pathways of root starch and sucrose metabolism and the plant hormone signaling pathways in the scion. The problems of how to regulate gene expression and transcription need further research. In this study, to clarify the genetic interaction mechanism between the grafted rootstocks and to explore miRNAs that play important roles in plant growth, stress resistance, and anti-aging, are of great practical significance for improving grafting resistance, disease resistance, yield, and quality through genetic improvement of rootstocks and the interaction between rootstocks and scions.
Compared with the control, there were 79 differentially expressed miRNAs in the scion and 57 differentially expressed miRNAs in the interstock, including 36 upregulated miRNAs and 43 downregulated miRNAs in the scion and 22 upregulated miRNAs and 35 downregulated miRNAs in the interstock. Through KEGG enrichment analysis, the interstock may affect the expression of related genes in the root starch and sucrose metabolism pathways and the scion plant hormone signaling pathways, thereby affecting plant growth and development and causing plant dwarfing. The downregulation of mdm-miR156 in the scion and mdm-miR172 in the interstock may promote the plant's transition to the adult phase and obtain early flowering characteristics. This study not only provides responsive miRNAs and target genes to apple grafting, but also enriches and supplements the mechanism of apple grafting. It also provides candidate miRNAs and target genes that respond to apple grafting-induced dwarfing and early flowering, enriches the research on the mechanism of apple grafting-induced dwarfing and early flowering, and provides a new theoretical basis for the creation of new apple interstock varieties.
-
Grafting is a common technique in horticulture for improving fruit tree performance. A total of 281 miRNAs were categorized into 80 families, including 159 newly discovered ones. Grafting with an interstock led to 79 differentially expressed miRNAs (DEMs) in the scion, with 36 up-regulated and 43 down-regulated miRNAs, and 57 DEMs in the rootstock, with 22 up-regulated and 35 down-regulated miRNAs, when compared to the control. Grafting using a dwarfing interstock influenced DEMs in the entire plant, causing a decrease in mdm-miR156 expression in the scion and mdm-miR172 in the rootstock. Analysis of DEMs and their target genes indicated that miRNAs influence scion growth by affecting aspects such as plant hormone synthesis, signal transduction, and nutrient balance. Combined miRNA and mRNA analysis revealed that the dwarfing effect of the interstock might impact genes related to starch and sucrose metabolism in the rootstock and plant hormone signaling in the scion. KEGG enrichment analysis identified pathways affecting plant physiology and growth, including root starch and sucrose metabolism, and hormone signaling. Notably, differentially expressed miRNAs in the scion and interstock influence related genes and contribute to plant dwarfing. The downregulation of mdm-miR156 and mdm-miR172 could promote the transition to the adult phase and early flowering. This research sheds light on miRNA regulation in grafted apples, enhancing our grasp of grafting-induced changes and providing a foundation for creating new apple interstock varieties.
-
The authors confirm contribution to the paper as follows: study conception and design: Gao Y, Feng J, Wang D; data collection: Gao Y, Wang K, Li L; analysis and interpretation of results: Li Q, Liu Z, Li Z, Wang G, Wang L, Lu X; draft manuscript preparation: Li Q, Feng J, Sun S, Lu X, Guo H. All authors reviewed the results and approved the final version of the manuscript.
-
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA008505) that are publicly accessible at
https://ngdc.cncb.ac.cn/gsa [51, 52]. We would like to thank OmicShare tools, a free online platform for plotting (
https://www.omicshare.com/tools , accessed on 13 February 2023), the ImageGP, an easy-to-use data visualization web server for scientific researchers (http://www.ehbio.com/ImageGP/ , accessed on 26 February 2023), and the Hiplot team (https://hiplot.com.cn , accessed on 2 February 2023) for providing technical assistance and valuable tools for data analysis and visualization. This work was supported by the Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2021-RIP-02) and Fundamental Research Funds for Central Non-profit Scientific Institution, grant number 161018201614.-
The authors declare that they have no conflict of interest.
- Supplemental Table S1 Primer sequences for real-time fluorescence quantitative PCR.
- Supplemental Table S2 Statistics on clean reads mapping against reference genome.
- Supplemental Table S3 Expression of miRNAs in each sample (TPM).
- Supplemental Table S4 The miRNA family analysis.
- Supplemental Table S5 All DE-miRNA.
- Supplemental Table S6 Analysis of the DEMs co-expression trends.
- Supplemental Table S7 Targeting relationship between miRNA and target genes.
- Supplemental Fig. S1 BR vs MR's miRNAs and target genes expression levels and targeting relationships.
- Supplemental Fig. S2 BS vs MS′s miRNAs and target genes expression levels and targeting relationships.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Li Q, Gao Y, Wang K, Sun S, Lu X, et al. 2023. Identification of microRNAs and target genes in apple (Malus domestica) scion and rootstock with grafted interstock. Fruit Research 3:34 doi: 10.48130/FruRes-2023-0034 

Identification of microRNAs and target genes in apple (Malus domestica) scion and rootstock with grafted interstock
- Received: 02 August 2023
- Accepted: 22 September 2023
- Published online: 05 December 2023
Abstract: Apple (Malus domestica) is an important multi-functional horticultural crop. However, thus far, there have been few reports concerning the molecular mechanism by which interstock regulates rootstock and scion growth. MicroRNAs (miRNAs) are a type of post-transcriptional regulator that regulates apple growth and development, fruit quality, hormone signal transduction, and stress response. Interstock grafting resulted in differences in miRNA distribution between apple tissues. The regulatory roles of miRNAs in the grafting of apples are not yet clear. In this study, 15 libraries were constructed using the phloem of an apple grafted on two types of interstock and rootstock by high-throughput sequencing. A total of 281 miRNAs were catalogued into 80 families, and 159 novel ones were identified. Compared with the control, the grafting combination with the interstock resulted in 79 differentially expressed miRNAs (DEMs) in the scion, with 36 up-regulated and 43 down-regulated miRNAs, and 57 DEMs in the rootstock, including 22 up-regulated and 35 down-regulated miRNAs. Grafting with the dwarfing interstock led to DEMs in the whole plant, including a decrease in the expression of mdm-miR156 in the scion and mdm-miR172 in the rootstock. Predictive analysis of the DEMs and their target genes suggested that miRNAs mediate scion growth through several aspects, such as plant hormone synthesis and signal transduction, and nutrient absorption and balance. Through combined miRNA and mRNA analysis, the dwarfing effect of the interstock may affect the expression of genes related to the starch and sucrose metabolism pathways in the rootstock and the plant hormone signal transduction pathway in the scion. The available evidence facilitates a better understanding of the role of miRNAs in the response of apples after grafting interstock. One possible reason for the stunting of apple trees and the promotion of early flowering caused by the dwarfing interstock is the decrease in expression levels of mdm-miR156 and mdm-miR172.
-
Key words:
- Apple /
- miRNA /
- Grafting /
- Interstock /
- Target genes