









-
Z. japonica is a warm-season turfgrass desirable for both residential and commercial turf use due to its fine leaf texture, low growth habit, low fertility requirements, and wide general tolerance to abiotic stressors such as drought, shade and salinity[1,2]. Although Z. japonica has the best freeze tolerance among warm-season grasses[2], currently, the commercial spread of Z. japonica is still limited to the transition zone and the southern US. In order to expand the species to northern cooler regions, the increased freeze-tolerant cultivars are critically demanded in Z. japonica breeding. Although several cultivars have been tested for cold acclimation and freeze tolerance[3], the lack of knowledge on the molecular mechanisms responsible for cold and freezing response limits breeding progress. Therefore, elucidating the molecular mechanisms and identifying the candidate genes underlying cold stress will accelerate the speed of improvement of freezing tolerance in Z. japonica.
Cold acclimation is a critical process in the development of freezing tolerance, and is defined as exposure to low but above freezing temperatures prior to a freeze stress[4]. Multiple biochemical changes have been identified as associated with cold acclimation, including an increased generation of reactive oxygen species (ROS), an up-regulation of antioxidant enzymes, alteration in plant hormone levels, and accumulation of amino acids with osmotic effects[5]. Meanwhile, cold acclimation and cold tolerance are involved in complicated regulatory pathways. Numerous genes have been found up- or down-regulated during cold acclimation treatment. Among several cold signaling pathways, the CBF/DREB1-dependent cold signaling pathway is well characterized as the key regulatory pathway[6,7]. Three CBF/DREB1s are involved in the regulation of cold response gene (COR) expression and cold tolerance in Arabidopsis and zoysiagrass[8−10]. INDUCER OF CBF EXPRESSION (ICE1), a MYC-type bHLH transcription factor, could activate the expression of CBF genes by directly binding to their promoters under cold stress[8]. LEA proteins are thought to be important for membrane stabilization and to prevent protein aggregation during stress[11]. There are several LEA gene members, COR78/RD29A, COR47, COR15A and COR6.6 in Arabidopsis that are induced by cold stress[12,13]. Although many genes and pathways have been identified in model plant species, the molecular mechanism of cold tolerance in Z. japonica is largely unknown.
Our previous studies have demonstrated that Z. japonica cultivars 'Meyer' and 'Victoria' as freeze tolerant and freeze susceptible genotypes, separately. Holloway et al. reported that 'Victoria' showed more severe winter injury than 'Meyer' in field evaluations[14]. In a lab-based evaluation, 'Meyer' showed less surviving green tissue and regrowth rate than 'Victoria' after freezing without cold acclimation. Meanwhile, the results were reversed if a cold acclimation treatment was added before freezing[15]. In addition, based on LT50 (50% mortality) evaluation, 'Meyer' showed higher freeze tolerance than 'Victoria' after cold acclimation treatment[16]. Meanwhile, genetic analysis identified multiple quantitative trait loci (QTL) regions associated with winter injury, post-freezing surviving green tissue (SGT) and regrowth (RG) using a 'Meyer x Victoria' pseudo-F2 mapping population[14,15]. Several proteins of interest for their association with cold acclimation were identified through a comparative proteomics analysis in 'Meyer' and 'Victoria'[16].
In recent years, the integration of RNA-seq and QTL mapping has been considered to be a reliable method to rapidly identify potential candidate genes, which has been applied in several crop species like rice and maize[17,18]. However, the strategy has not been reported in turfgrass species. The objectives of this study were: (1) to identify differentially expressed genes (DEGs) and functional annotation in response to cold acclimation in Z. japonica, and (2) to identify potential candidate genes crossing transcriptome with previous proteome and QTL mapping studies. The identified potential candidate genes will provide insight into the molecular mechanisms of cold tolerance, and would be helpful for Z. japonica breeding programs.
-
Z. japonica cultivar 'Meyer' and 'Victoria', have been extensively reported in the literature as freeze tolerant and freeze susceptible genotypes, respectively[4,14−16,19], were used in this study. Two-inch plugs of each genetically uniform cultivar were propagated with Fafard 4P potting mix and grown under greenhouse conditions at day/night 27 ± 2 / 22 ± 2 °C for six weeks until fully established. Plants were mowed at 5 cm and fertilized biweekly with 24:8:16 NPK solution of Miracle-Gro Water Soluble All Purpose Plant Food (The Scotts Miracle-Gro® Company, Marysville, OH, USA).
Treatments and experimental design
-
The protocol of the cold acclimation treatment was applied according to Chen et al. with minor modifications[20]. Following establishment in the greenhouse, pots were moved into temperature-controlled Percival plant growth chambers (model I-36LL) for cold acclimation. The growth chambers contained a consistent light emitting diode (LED) which maintained a 10-h photoperiod of 300 µmol·m−2·s−1 active radiation[21]. Plants in the cold acclimation (CA) group were held under 8/2 °C day and night temperatures and 12/12 h light cycles for 48 h, while plants in the non-acclimation (NA) group were maintained at a consistent 22 °C with 12/12 h day and night light cycles. After cold acclimation/non-acclimation, 12 leaf samples of similar maturity status were collected for each 'Meyer' and 'Victoria' with three biological replicates. Leaf samples were frozen in liquid nitrogen immediately and stored in a −80 °C freezer until RNA extraction. The experiment contains four treatments, non-cold acclimated 'Meyer' (NM), cold acclimated 'Meyer' (CM), non-cold acclimated 'Victoria' (NV), and cold acclimated 'Victoria' (CV).
RNA extraction, library preparation and sequencing
-
Total RNA was extracted from frozen leaf samples using the RNeasy Plant Mini Kit (Qiagen, Germany). Only RNA samples that passed quality and quantity checks (RIN value ≥ 7; total amount ≥ 1.5 ug) were used in sequencing library construction. Following this quality control, mRNA was enriched via oligo (dT) beads, and fragmented in fragmentation buffer. First strand cDNA was synthesized using random hexamers and reverse transcriptase, then second strand synthesis buffer (Illumina, San Diego, CA, USA) was added along with dNTPs, RNase H and Escherichia coli polymerase I for second strand synthesis. After a round of purification, terminal repair and poly-A tailing, Illumina PE adapters were ligated and 200 bp cDNA fragments were preferentially selected. Then, cDNA fragments with ligated adapters were enriched using PCR primers. Library quality was assessed by quantification with a Qubit 2.0 fluorometer and qPCR, and insert size was checked on an Agilent 2100. The library was sequenced using an Illumina NovaSeq sequencing platform to generate 150 bp pair-end reads. All the raw sequencing data were deposited into the database of sequence read archive (SRA) in National Center for Biotechnology Information (NCBI) under BioProject ID: PRJNA1040203.
Gene expression quantification and differential expression analysis
-
The raw reads were processed to remove reads containing adapters, uncertain nucleotides (N > 10%), and low-quality reads. The resulting clean reads were evaluated on Q20, Q30, GC content, and used for downstream analyses. Clean reads were aligned to the Zoysia japonica 'Nagirizaki' transcript using the HISAT2[22]. The FPKM (expected number of Fragments Per Kilo bases of transcript sequence per Millions mapped reads) method was used to estimate gene expression levels by counting reads that mapped to genes or exons using HTSeq v0.6.1[23]. Differential gene expression analysis was performed using the DESeq R package[24], and the significant differential expression was filtered using padj ≤ 0.05 and | log2(Fold Change) | ≥ 1.
GO and KEGG enrichment analysis
-
Differentially expressed genes (DEGs) were assigned to Gene Ontology (GO) enrichment analysis via the GOseq R package[25]. GO with corrected p-values < 0.05 are significantly enriched in DEGs. The KOBAS software was used to test the enrichment of DEGs in KEGG pathways, identifying significantly (corrected p-values < 0.05) enriched metabolic or signal transduction pathways associated with the DEGs.
Identification of acclimation response genes in the identified QTL region
-
Eleven overlapping QTL regions were identified in our previous studies that were associated with winter injury in field evaluations and with laboratory-based freezing test traits[14,15,26]. We name these regions as QTL1-11 following the chromosome order. The sequences of QTL regions were extracted from the Zoysia japonica 'Nagirizaki' reference genome based on the position of flanking markers[22]. Then, the sequences were blasted against the Zoysia japonica 'Nagirizaki' transcript to obtain the gene ID[22]. Identified genes that overlapped with the above DEGs were recognized as candidate acclimation response genes.
-
A cDNA library was constructed from total RNA of Z. japonica leaf samples under different cold acclimation treatments. Following Illumina sequencing, the overview of sequencing reads obtained is presented in Table 1. Averaged from three replicates for each sample, there were 50,275,906, 60,245,453, 57,550,037, and 60,264,472 raw reads generated from the NM, CM, NV, CV samples, respectively. The raw reads were processed to remove reads containing adapters, uncertain nucleotides (N > 10%), and low-quality reads. After the quality control, average of 49,055,750, 58,957,880, 56,263,343, and 59,058,380 clean reads were obtained for the NM, CM, NV, CV samples, respectively (Table 1).
Table 1. Summary of reads following Illumina sequencing.
Sample Raw reads Clean reads Clean bases Error rate (%) Q20 (%) Q30 (%) GC content (%) NM1 49,475,254 48,077,382 7.2G 0.03 97.92 94.15 57.25 NM2 51,183,836 50,010,472 7.5G 0.03 97.84 93.99 57.49 NM3 50,168,628 49,079,396 7.4G 0.03 97.88 94.04 57.49 CM1 59,804,056 58,665,446 8.8G 0.03 97.37 92.95 52.47 CM2 67,204,258 65,599,380 9.8G 0.03 97.35 92.94 51.48 CM3 53,728,046 52,608,814 7.9G 0.03 97.11 92.48 52.52 NV1 71,039,902 69,474,296 10.4G 0.03 97.47 93.19 53.68 NV2 46,536,824 45,397,712 6.8G 0.03 97.39 93.03 52.99 NV3 55,073,384 53,918,022 8.1G 0.03 97.08 92.41 53.03 CV1 61,931,458 6,0855,104 9.1G 0.03 97.43 93.12 52.9 CV2 51,018,634 49,819,784 7.5G 0.03 97.28 92.84 52.43 CV3 67,843,324 66,500,252 10.0G 0.03 97.49 93.23 52.78 *NM: 'Meyer' genotype under non-cold acclimated conditions; CM: 'Meyer' genotype under cold acclimated conditions; NV: 'Victoria' genotype under non-cold acclimated conditions; CV: 'Victoria' genotype under cold acclimated conditions. Differentially expressed genes (DEGs) analysis under acclimation treatment
-
Mapping results showed that 92.1% of NM, 85.7% of CM, 85.2% of NV and 83.8% of CV clean reads could be mapped to reference genome, which result in a total of 27,209 genes were identified. We paid particular attention to the DEGs identified for comparison of with/without cold acclimation treatment. The comparisons included non-cold acclimated 'Meyer' vs cold acclimated 'Meyer' (R genotype) (NM vs CM) and non-cold acclimated 'Victoria' vs cold acclimated 'Victoria' (S genotype) (NV vs CV). For NM vs CM, 4,609 DEGs were up-regulated and 3,605 DEGs were down-regulated after treatment. For NV vs CV, 3,758 DEGs were up-regulated and 3,516 DEGs were down-regulated in response to cold acclimation (Fig. 1a, Supplemental File 1).
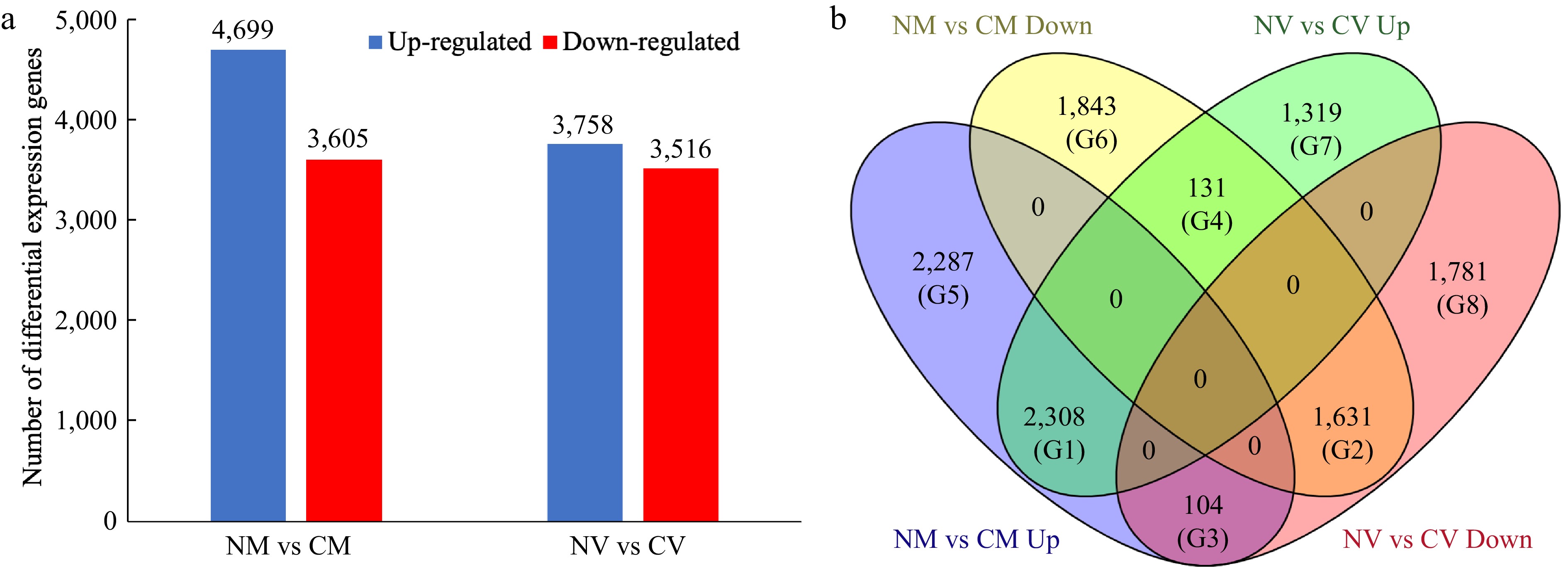
Figure 1.
Differentially expressed genes (DEGs) in Z. japonica in response to cold acclimation. (a) Number of DEGs identified in the 'Meyer' (resistant) and 'Victoria' (susceptible) genotypes. (b) Venn diagram of differentially expressed genes, showing the unique and overlapping genes expressed among comparison groups. NM vs CM: non-cold acclimated 'Meyer' vs cold acclimated 'Meyer' (R genotype); NV vs CV: non-cold acclimated 'Victoria' vs cold acclimated 'Victoria' (S genotype). G1−G8 indicated eight groups.
To further evaluate DEGs patterns, a Venn diagram was used to show the unique and overlapping genes expressed among comparison groups (Fig. 1b). Accordingly, the DEGs were categorized into eight groups (G1-G8): G1, 2,308 DEGs were up-regulated in both R and S genotypes; G2, 1,631 DEGs were down-regulated in both genotypes; G3, 104 DEGs were up-regulated in R but down-regulated in S genotype; G4, 131 DEGs were up-regulated in S but down-regulated in R genotype; G5, 2,287 DEGs were only up-regulated in R genotype; G6, 1,843 DEGs were only down-regulated in R genotype; G7, 1,319 DEGs were only up-regulated in S genotype; G8, 1,781 DEGs were only down-regulated in S genotype.
Gene Ontology (GO) classification of differentially expressed genes
-
The DEGs were assigned to enriched GO terms to annotate the gene function in terms of molecular function, cellular component, and biological process. Within cultivar expression differences, CM vs NM and CV vs NV, were the primary comparisons of interest. For up-regulated genes in CM vs NM, the major categories within molecular function were nucleoside phosphate binding (GO:1901265), carbohydrate derivative binding (GO:0097367) and purine nucleotide binding (GO:0017076). The top GO terms in cellular component were intracellular (GO:0005622) and membrane (GO:0016020), while single-organism process (GO:0044699), single-organism cellular process (GO:0044763) and biosynthetic process (GO:0009058) were identified as major biological process. For down-regulated genes in 'Meyer', the enriched biological processes were photosynthesis (GO:0015979) and alpha-amino acid biosynthetic process (GO:1901607), while enriched cellular components were membrane protein complex (GO:0098796) and thylakoid (GO:0009579). In 'Victoria', the up-regulated genes were mainly assigned to single-organism process (GO:0044699) and biosynthetic process (GO:0009058) in terms of biological process, intracellular (GO:0005622) and membrane (GO:0016020) in terms of cellular component, and ion binding (GO:0043167) in terms of molecular function. The down-regulated genes were enriched in biological process of transmembrane transport (GO:0098656, GO:1903825, GO:0003333), cellular component of photosystem I (GO:0009522, GO:0009538) and molecular function of transmembrane transporter activity (GO:0008514, GO:0005342, GO:0046943). The GO analysis of up-regulated DEGs is shown in Fig. 2, and more detail can be found in Supplemental File 2.
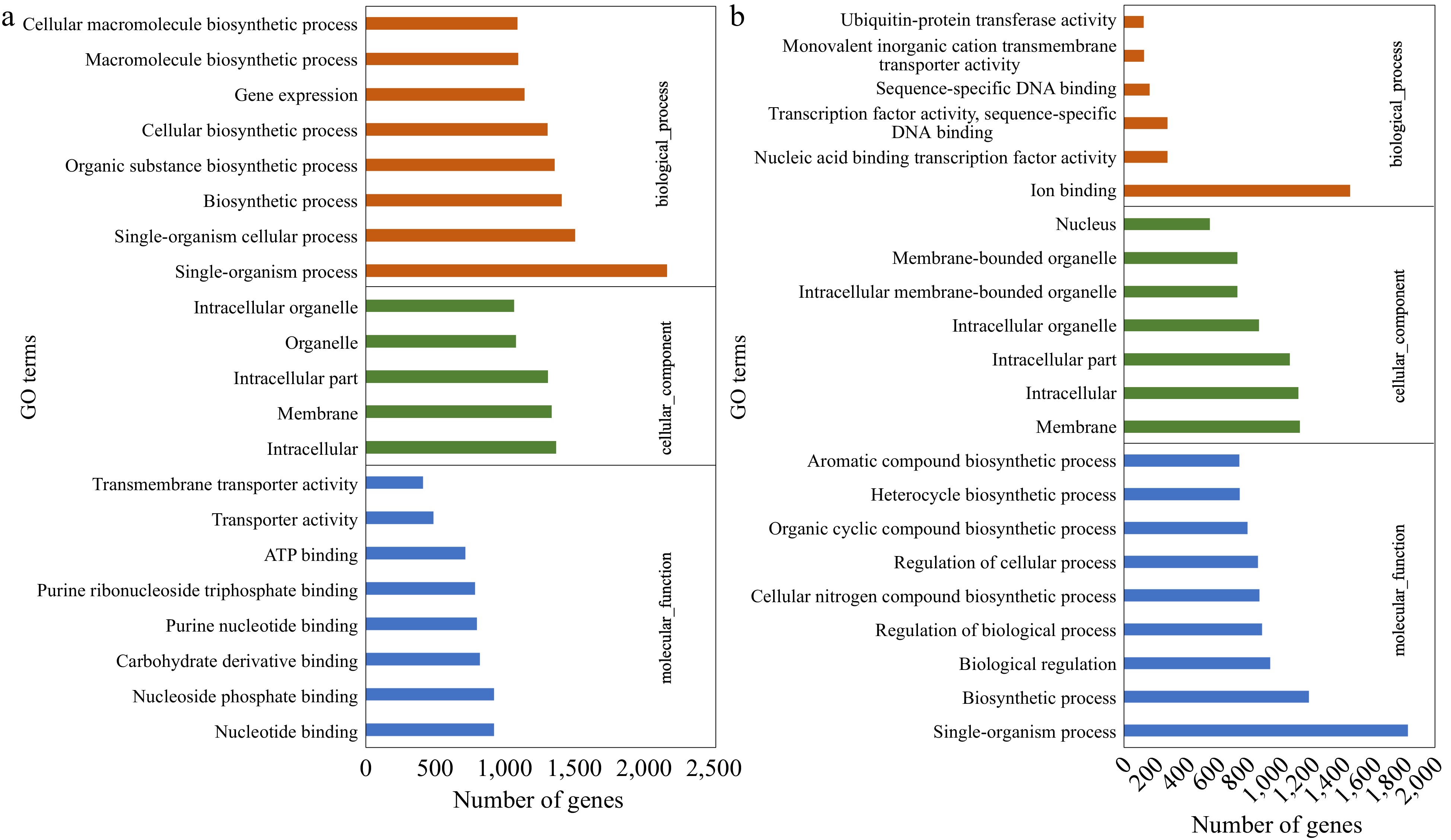
Figure 2.
Gene Ontology (GO) classification of the up-regulated DEGs identified in (a) CM vs NM and (b) CV vs NV comparisons. DEGs were annotated in three categories: biological process, cellular component, and molecular function.
KEGG classification of differentially expressed genes
-
DEGs above were annotated using the KEGG database. Among the up-regulated genes in 'Meyer', 23 significant pathways were identified, with metabolic pathways, biosynthesis of secondary metabolites, and ribosome being the top three. Among down-regulated genes in 'Meyer', 13 pathways were identified, and top three were metabolic pathways, biosynthesis of secondary metabolites, and carbon metabolism. In the S genotype 'Victoria', up-regulated DEGs were assigned to 16 pathways, in which the top two pathways were the same as the R genotype, followed by plant hormone signal transduction. Meanwhile, the down-regulated DEGs were only assigned to two pathways: metabolic pathways and N-Glycan biosynthesis. The KEGG analysis of up-regulated DEGs is shown in Fig. 3, and more detail can be found in Supplemental File 3.
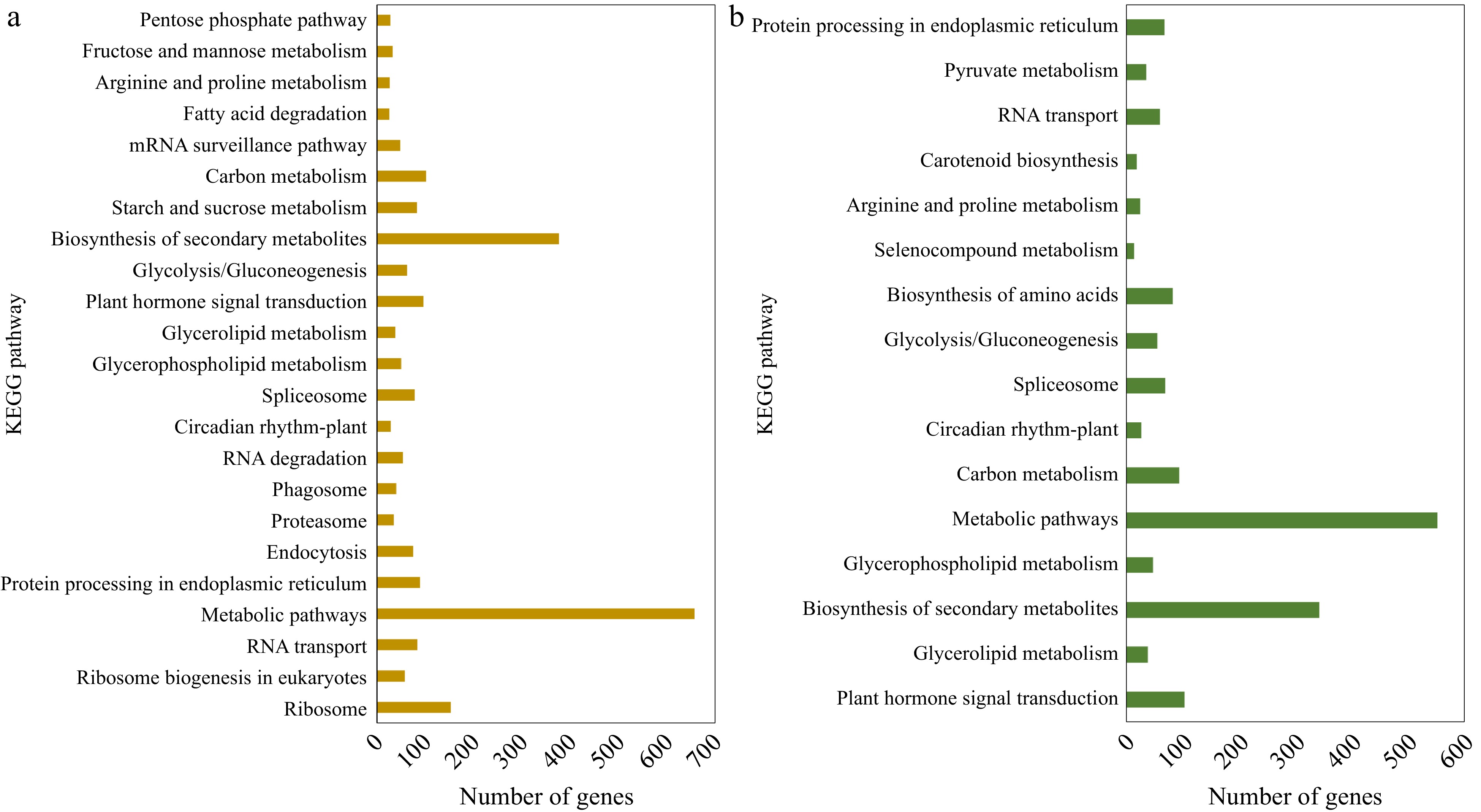
Figure 3.
KEGG pathway classification of the up-regulated DEGs identified in (a) CM vs NM and (b) CV vs NV comparisons.
Identification of acclimation response genes in the identified QTL regions
-
By searching against the reference genome and transcript, we identified a total of 674 genes from QTL regions previously identified as associated with freezing and winter injury traits, while 168 of them were overlapped with above DEGs list (Fig. 4; Table 2; Supplemental File 4). The DEGs were identified in all the 11 QTL regions except in QTL5 on chromosome 7. There were 74 genes identified in QTL1, which was the one with the highest number of genes, 16 genes in QTL2, six genes in QTL3, 19 genes in QTL4, nine genes in QTL6, 15 genes in QTL7, 12 genes in QTL8, 10 genes in QTL9, six genes in QTL10, and one gene in QTL11. We found that a lot of these genes have been reported to be involved in plant stress response, such as Late embryogenesis abundant protein, Heat stress transcription factor, and other transcription factors including NAC, WRKY, MYB, and ethylene-responsive transcription factors. There were also several signal transduction related genes like CBL-interacting protein kinase, auxin-responsive protein, and ABSCISIC ACID-INSENSITIVE 5-like protein. More details of these genes are shown in Supplemental File 4.
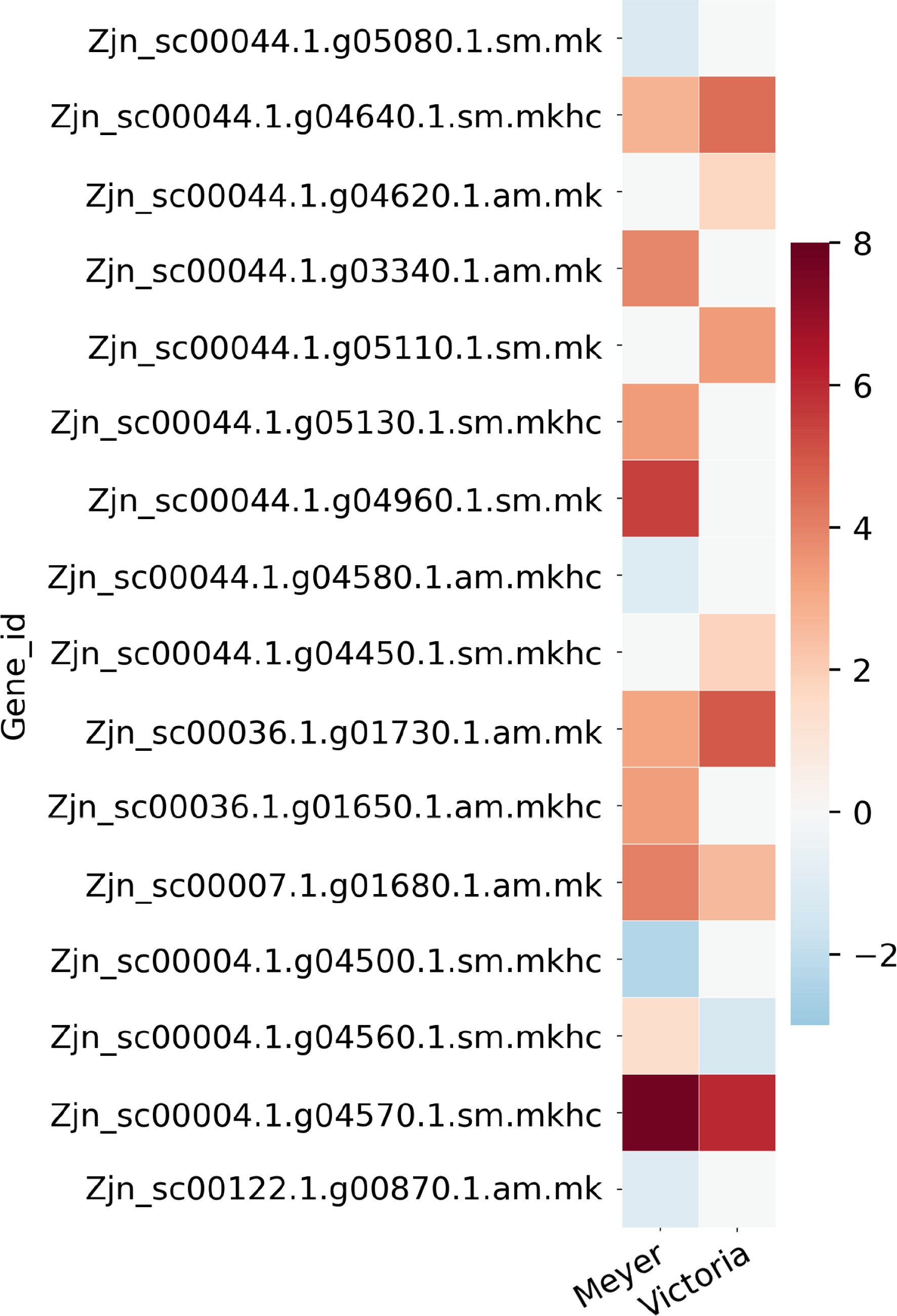
Figure 4.
Heat map of partial candidate acclimation response genes that were identified both in the QTL regions previously reported in Brown et al.[15] and the differentially expressed genes reported here. Color bar shows the log2(Fold Change) value, red color indicates up-regulation, blue color indicates down-regulation, and white color indicates no significant change.
Table 2. Partial list of candidate acclimation response genes that were identified both in the QTL regions previously reported in Brown et al.[15] and differential expression genes reported in this study. Full gene list and QTL information are provided in Supplemental File 4.
QTL
regionGene_id log2FoldChange 'Meyer' log2FoldChange 'Victoria' Blast gene accession Blast gene name QTL1 Zjn_sc00044.1.g05080.1.sm.mk −1.2035 NS Q9C5Q2 AI5L3_ARATH ABSCISIC ACID-INSENSITIVE 5-like protein 3 QTL1 Zjn_sc00044.1.g04640.1.sm.mkhc 2.7812 4.4685 Q7X996 CIPK2_ORYSJ CBL-interacting protein kinase 2 QTL1 Zjn_sc00044.1.g04620.1.am.mk NS 1.7587 Q7XIW5 CIPKT_ORYSJ CBL-interacting protein kinase 29 QTL1 Zjn_sc00044.1.g03340.1.am.mk 3.9076 NS Q7XHZ0 HFB4B_ORYSJ Heat stress transcription factor B-4b QTL1 Zjn_sc00044.1.g05110.1.sm.mk NS 3.422 A3AHG5 LEA17_ORYSJ Late embryogenesis abundant protein 17 QTL1 Zjn_sc00044.1.g05130.1.sm.mkhc 3.3773 NS Q10MB4 MYB2_ORYSJ Transcription factor MYB2 QTL1 Zjn_sc00044.1.g04960.1.sm.mk 5.4299 NS A0SPJ6 NAMB2_TRITD NAC transcription factor NAM-B2 QTL1 Zjn_sc00044.1.g04580.1.am.mkhc −1.0768 NS P42736 RAP23_ARATH Ethylene-responsive transcription factor RAP2-3 QTL1 Zjn_sc00044.1.g04450.1.sm.mkhc NS 1.8656 Q9SW70 SRP_VITRI Stress-related protein QTL2 Zjn_sc00036.1.g01730.1.am.mk 3.1617 4.869 Q6VBA4 HFC1A_ORYSJ Heat stress transcription factor C-1a QTL2 Zjn_sc00036.1.g01650.1.am.mkhc 3.3333 NS Q93WV4 WRK71_ARATH WRKY transcription factor 71 QTL6 Zjn_sc00007.1.g01680.1.am.mk 4.0415 2.6034 Q6K846 IAA9_ORYSJ Auxin-responsive protein IAA9 QTL8 Zjn_sc00004.1.g04500.1.sm.mkhc −2.3169 NS Q7XBH4 MYB4_ORYSJ Transcription factor MYB4 QTL8 Zjn_sc00004.1.g04560.1.sm.mkhc 1.3851 −1.3166 P14717 PAL1_ORYSJ Phenylalanine ammonia-lyase QTL8 Zjn_sc00004.1.g04570.1.sm.mkhc 7.7281 5.9998 A2X7F7 PAL2_ORYSI Phenylalanine ammonia-lyase QTL10 Zjn_sc00122.1.g00870.1.am.mk −1.0261 NS O80341 EF102_ARATH Ethylene-responsive transcription factor 5 *log2FoldChange, positive value indicates up-regulation after cold acclimation, negative value indicates down-regulation after cold acclimation, NS indicates non-significant. Identification of acclimation response genes that overlapped with previous proteomics study
-
We previously identified 62 protein spots differentially accumulated in abundance under cold acclimation using 'Meyer' and 'Victoria'[16]. Five of them were repeatedly identified in the above DEGs, including Late embryogenesis abundant protein, Zinc finger protein, MAP kinase, Phospholipase D, and Photosystem II protein. There were 14 DEGs that belong to the Late embryogenesis abundant protein gene family, 16 to the MAP kinase gene family, 17 to the Zinc finger protein gene family, nine to the Phospholipase D gene family, and 12 to the Photosystem II protein gene family (Fig. 5; Table 3; Supplemental File 5). The gene Zjn_sc00044.1.g05110.1.sm.mk, which encodes a Late embryogenesis abundant protein (LEA17), was also found to be present in the QTL1 region listed above.
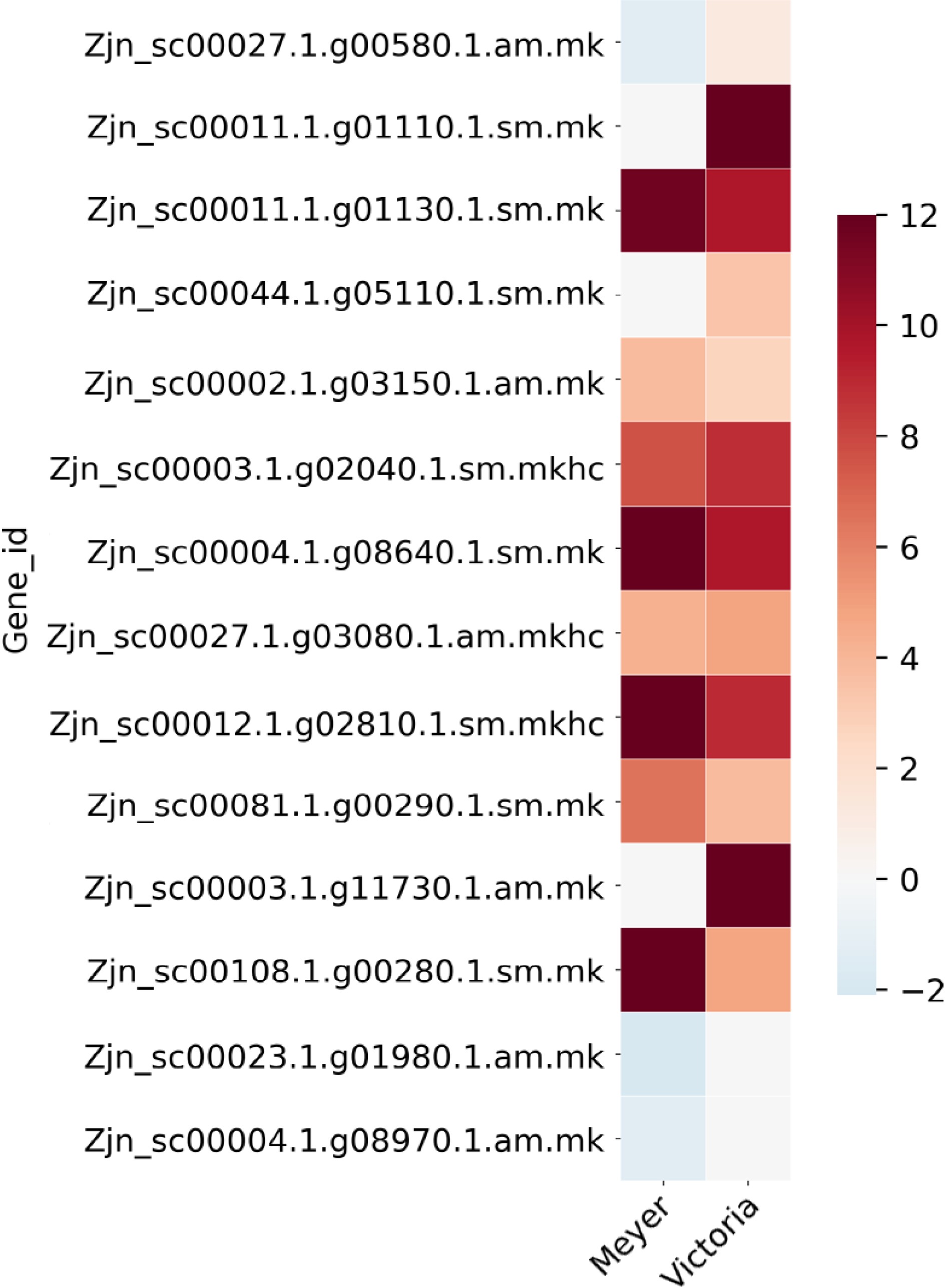
Figure 5.
Heat map of Late embryogenesis abundant protein (LEA) genes that were identified as differentially expressed after cold acclimation in Z. japonica, which were also differentially expressed in protein abundance in a previous study[16]. Color bar shows the log2(Fold Change) value, red color indicates up-regulation, blue color indicates down-regulation, and white color indicates no significant change.
Table 3. List of Late embryogenesis abundant protein (LEA) genes that were identified as differentially expressed after cold acclimation in Z. japonica, which were also differentially expressed in protein abundance in a previous study[16].
Gene_id log2FoldChange
'Meyer'log2FoldChange
'Victoria'Blast gene
accessionBlast gene name Protein spot 64[16] 6.7 NS Late embryogenesis abundant protein 3 (LEA3) Zjn_sc00027.1.g00580.1.am.mk −1.3435 1.2295 P46518 LEA14_GOSHI Late embryogenesis abundant protein Lea14-A Zjn_sc00011.1.g01110.1.sm.mk NS Inf Q94JF2 LEA14_ORYSJ Late embryogenesis abundant protein 14 Zjn_sc00011.1.g01130.1.sm.mk 11.646 9.6458 Q94JF2 LEA14_ORYSJ Late embryogenesis abundant protein 14 Zjn_sc00044.1.g05110.1.sm.mk NS 3.422 A3AHG5 LEA17_ORYSJ Late embryogenesis abundant protein 17 Zjn_sc00002.1.g03150.1.am.mk 3.8076 2.7007 A3AHG5 LEA17_ORYSJ Late embryogenesis abundant protein 17 Zjn_sc00003.1.g02040.1.sm.mkhc 7.597 8.8459 A3AHG5 LEA17_ORYSJ Late embryogenesis abundant protein 17 Zjn_sc00004.1.g08640.1.sm.mk Inf 9.7227 Q96273 LEA18_ARATH Late embryogenesis abundant protein 18 Zjn_sc00027.1.g03080.1.am.mkhc 4.2847 4.8089 Q42376 LEA3_MAIZE Late embryogenesis abundant protein, group 3 Zjn_sc00012.1.g02810.1.sm.mkhc Inf 8.9382 Q42376 LEA3_MAIZE Late embryogenesis abundant protein, group 3 Zjn_sc00081.1.g00290.1.sm.mk 6.5069 3.7991 Q9LJ97 LEA31_ARATH Late embryogenesis abundant protein 31 Zjn_sc00003.1.g11730.1.am.mk NS Inf P09444 LEA34_GOSHI Late embryogenesis abundant protein D-34 Zjn_sc00108.1.g00280.1.sm.mk Inf 4.7263 P09444 LEA34_GOSHI Late embryogenesis abundant protein D-34 Zjn_sc00023.1.g01980.1.am.mk −2.0172 NS F4KFM8 LEA65_ARATH Late embryogenesis abundant protein At5g17165 Zjn_sc00004.1.g08970.1.am.mk −1.2806 NS F4KFM8 LEA65_ARATH Late embryogenesis abundant protein At5g17165 * log2FoldChange, positive value indicates up-regulation after cold acclimation, negative value indicates down-regulation after cold acclimation, NS indicates non-significant, Inf indicates infinite value. -
Cold acclimation, a low but above freezing temperature treatment, is a critical process to the development of freezing tolerance. The process directly inhibits metabolic reactions, and indirectly inhibits cold-induced osmotic stress, oxidative stress as well as numerous other genes and pathways by affecting lipid composition of membranes, sugars, and proline molecules, and increasing total soluble proteins[13,27]. To further disentangle the differences in metabolic response to freezing, the present study compared the transcriptomes of 'Meyer', a freeze-tolerant cultivar, and 'Victoria', a freeze-susceptible cultivar, under cold acclimated and non-cold acclimated conditions. The study identified abundant genes and pathways in response to cold acclimation, which provides insights into the molecular mechanisms underlying cold acclimation response in Z. japonica. In addition, the findings provide an opportunity to validate our previous studies on QTL mapping and proteomics, with the overlapped genes showing great value to be used for selection in breeding for freeze tolerance in Z. japonica.
Different response to cold acclimation in 'Meyer' and 'Victoria'
-
As mentioned above, our previous studies have demonstrated that 'Meyer' and 'Victoria' perform differently when exposed to cold acclimation and freezing[14−16]. The findings of this study provide evidence to explain these differences from the gene regulation mechanisms activated in response to cold acclimation. Cold response in plants is a very complex trait, which involves many different gene regulation and metabolic pathways[28]. The molecular basis of cold acclimation and acquired freezing tolerance in plants has been extensively studied, especially in model plant Arabidopsis. The CBF/DREB1-dependent cold signaling pathway has been characterized as the key cold signaling pathway[8,9]. In Arabidopsis, several CBF/DREB1s have been reported to be involved in the regulation of COR gene expression and cold tolerance[8,9]. In addition, the CBF/DREB1 (mainly CBF3/DREB1A) pathway was found to be controlled by transcription factor ICE1 (inducer of CBF expression1)[11]. In zoysiagrass, six DREB1 and two DREB2 unigenes were up-regulated significantly at the 2h-cold time point, and one DREB2 unigene was up-regulated at the 72h-cold time point using 'Meyer'[10]. DREB and ICE transcription factors were also identified in our study. There were two ICE1 transcription factors were found, Zjn_sc00034.1.g02330.1.am.mkhc was up-regulated in both cultivars, while Zjn_sc00094.1.g00080.1.am.mk was found in the G3 group (up-regulated in 'Meyer' but down-regulated in 'Victoria'), which indicates it might contribute to the higher cold tolerance in 'Meyer' (Supplemental File 1). There were 17 DREB transcription factors found, two of them in G1, two in G2, two in G5, six in G6, one in G7, and one in G8 (Supplemental File 1). Despite the common change trend in G1 and G2, more DREB genes were changed in 'Meyer' (G5, G6) than in 'Victoria' (G7, G8), indicating the two cultivars might have adopted different DREB members in response to cold stress.
Expression of CORs (cold-regulated genes) regulated by CBF/DREB1s has been shown to be critical in plants for cold and freezing stress[29]. The cold-responsive protein kinase 1 (CRPK1), a plasma membrane protein which could phosphorylate 14-3-3 proteins, was proved to be a negative regulator of freezing tolerance. The crpk1 Arabidopsis mutant showed enhanced freezing tolerance[30]. There were two CRPK1 genes found in our study, Zjn_sc00002.1.g07100.1.sm.mkhc was up-regulated in both cultivars, while Zjn_sc00068.1.g00990.1.sm.mkhc was only down-regulated in 'Meyer' (Supplemental File 1). Considering CRPK1 gene is a negative regulator, decreased expression of Zjn_sc00068.1.g00990.1.sm.mkhc might enhance cold tolerance in 'Meyer'.
To alleviate the oxidative damage in plant cells under cold stress, plants exhibit enhanced activities of antioxidant enzymes, such as superoxide dismutase (SOD), peroxidase (POD), catalase (CAT), glutathione peroxidase (GPX), and ascorbic acid peroxidase (APX) and eliminate excess reactive oxygen species (ROS)[31]. In this study, one, two, three and 34 genes were annotated as GPX, CAT, SOD and POD. The GPX gene was up-regulated in both cultivars. The two CAT genes were only changed in 'Victoria', one up-regulated and one down-regulated. The two SOD genes were up-regulated in both cultivars, while one SOD gene was only up-regulated in 'Meyer'. Five POD genes were down-regulated in both cultivars, three genes were up-regulated in both cultivars, three genes were up-regulated in 'Meyer' but down-regulated in 'Victoria', six genes only up-regulated in 'Meyer', four genes were only down-regulated in 'Meyer', one gene was only up-regulated in 'Victoria', while 12 POD genes were only down-regulated in 'Victoria' (Supplemental File 1). These findings indicated that more antioxidant genes were up-regulated in 'Meyer', and down-regulated in 'Victoria', suggesting that 'Victoria' might suffer more severe oxidative damage than 'Meyer' during cold stress.
Validated and overlapping findings with previous studies
-
Integrating QTL information and gene expression variation is a common strategy for candidate gene screening[32−34]. In previous studies, we have identified multiple QTL associated with field winter injury, and lab-based post-freeze surviving green tissue and regrowth using a 'Meyer x Victoria' mapping population, and 11 QTL regions of interest were found that overlapped across different traits and under different environments. By searching genome sequences of these regions and integrating the DEGs identified in this study, we found a total of 168 genes spread across 10 of 11 QTL regions (Table 2; Supplemental File 4). Some of these have been proved to be involved in response to cold acclimation and freezing stress. Two CIPK (CBL-interacting protein kinase) genes were found, one was up-regulated in both cultivars, another one was only up-regulated in 'Victoria'. Arabidopsis CIPK3 and CIPK7 were reported to be involved in response to cold stress[35,36]. Zjn_sc00044.1.g04610.1.am.mk, annotated as a POD gene discussed above, was only down-regulated in 'Victoria', which might impair the freezing tolerance of 'Victoria'. Heat stress transcription factor (HSF) act as components of signal transduction that induce the expression of heat shock protein (HSP) in response to a broad range of abiotic stresses[37]. In this study, two HSF genes and a HSP gene located in QTL regions were found up-regulated by cold acclimation, while one of HSF gene, Zjn_sc00044.1.g03340.1.am.mk, only changed in 'Meyer' (Table 2). In addition, there were several transcription factors found in QTL regions, including MYB, NAC, ERF, WRKY, which function in regulating downstream cold response genes, and trigger a series of physiological and biochemical responses.
Several genes were found overlapped with our previous comparative proteomic study between 'Meyer' and 'Victoria' under cold acclimation, including LEA, Zinc finger protein, MPK, Photosystem II (PSII) protein and Phospholipase (Table 3, Supplemental File 5). LEA proteins were identified in different plant species and proved that they are important factors in regulating plant freezing tolerance[12,38]. There were 14 LEA genes identified in DEGs, most of them were up-regulated in both cultivars, while three of them were down-regulated in 'Meyer'. It was noted that one LEA gene, Zjn_sc00044.1.g05110.1.sm.mk, was also found in the above QTL region, which shows great value to be used for cold tolerance improvement in Z. japonica. In plants, the cold response involves decreased photosynthetic capacity, and inhibition of photosystem II (PSII) activity is one of the most notable indicators of cold stress[39]. Recently, Zhang et al. reported that the expression of several PSII related genes were altered in Zoysia japonica during cold stress[40]. We identified a PSII protein in the proteomics study, which shows higher abundance in 'Meyer' but lower abundance in 'Victoria'[16]. In the present study, 12 genes encoding the PSII complex protein were found to be differentially expressed after cold acclimation. Most of the PSII genes were decreased during cold stress, while four of them were up-regulated in 'Victoria', and only one of them was up-regulated in 'Meyer'. The inconsistency between protein and transcript level might be due to post-translational modifications. Mitogen-activated protein kinases (MPK) were identified in both proteomics and transcriptomics levels in response to cold acclimation. MPK cascades are important signaling modules that convert environmental stimuli into cellular responses. Recent studies show that MPK3, MPK4, and MPK6 are rapidly activated after cold treatment[41]. Zhao et al. discovered that the MKK5-MPK3/MPK6 cascade negatively regulates cold response by promoting the degradation of ICE[42]. In contrast, the MEKK1-MKK2-MPK4 cascade positively regulates cold response, indicating the different roles of two MAP kinase cascades in the regulation of COR gene expression and freezing tolerance[43]. In this study, there were 16 MPK genes in DEGs, five of them were up-regulated and one was down-regulated in both cultivars. Expression of the rest of MPK genes showed different trends: three of them were only down-regulated in 'Meyer', and two genes were only down-regulated and five genes were only up-regulated in 'Victoria' (Supplemental File 5). These results indicated several different MPK pathways function in cold stress response in Z. japonica. Further studies are, however, necessary to demonstrate the function of these overlapped genes in Z. japonica response to cold acclimation and freezing stress.
-
This study examined the differentially expressed genes between freeze-susceptible 'Victoria' and freeze-tolerant 'Meyer' under cold acclimated and non-cold acclimated conditions. The differentially expressed genes showed different patterns in the two cultivars in response to cold acclimation. Integrating with previous studies, several genes were identified across the transcriptome, proteome, and QTL mapping to be associated with cold acclimation and freezing tolerance, such as LEA, CIPK, POD, HSF, HSP, MPK, PSII as well as multiple transcription factors. These findings strengthen the connection of these genes with resistance to cold and freeze stress, and upon further study, may be utilized to facilitate future breeding efforts to increase freeze tolerance in Z. japonica.
-
The authors confirm contribution to the paper as follows: funding acquisition: Milla-Lewis SR; study conception and design: Yu X, Milla-Lewis SR; data collection: Brown JM, Holloway HMP; resource and methodology: Tuong TD, Patton AJ, DaCosta M, Livingston DP; analysis and interpretation of results: Brown JM, Weldt CE, Yu X; draft manuscript preparation: Brown JM, Yu X; review and editing: Yu X, Milla-Lewis SR. All authors reviewed the results and approved the final version of the manuscript.
-
All the raw sequencing data were deposited into the database of sequence read archive (SRA) in National Center for Biotechnology Information (NCBI) under BioProject ID: PRJNA1040203. The other datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
This research was supported in part by funding provided by the NC State University Plant Breeding Consortium, the NC State University Center for Turfgrass Environmental Research and Education, and the United States Golf Association. The authors are appreciative to Dr. Rui Shi from North Carolina State University for manuscript reviewing.
-
The authors declare that they have no conflict of interest. Susana R. Milla-Lewis is the Editorial Board member of Grass Research who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and the research groups.
- Supplemental File 1 List of differentially expressed genes (DEGs) in different comparisons.
- Supplemental File 2 Gene Ontology (GO) classification of the DEGs identified in different comparisons.
- Supplemental File 3 KEGG classification of the DEGs identified in different comparisons.
- Supplemental File 4 List of candidate acclimation response genes that be identified both in QTL regions and differentially expressed genes. QTL regions were previously reported in Brown et al.[15].
- Supplemental File 5 List of candidate acclimation response genes that were also differentially expressed in protein abundance in a previous study[16].
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Brown JM, Weldt CE, Holloway HMP, Tuong TD, Patton AJ, et al. 2023. Transcriptomic analysis of zoysiagrass (Zoysia japonica) provides novel insights into the molecular basis of cold acclimation. Grass Research 3:25 doi: 10.48130/GR-2023-0025 

Transcriptomic analysis of zoysiagrass (Zoysia japonica) provides novel insights into the molecular basis of cold acclimation
- Received: 27 September 2023
- Accepted: 17 November 2023
- Published online: 05 December 2023
Abstract: Zoysia japonica (Z. japonica) is a warm-season perennial turfgrass commonly grown in the southeastern United States for its relatively low input requirements and general tolerance to drought, shade, and salinity. Improving freezing tolerance is critical for Z. japonica, as it could extend the northern boundaries that the species is able to be grown in. To deepen our understanding of the molecular basis of freezing tolerance in Z. japonica, a transcriptomic approach was utilized to identify genes involved in cold acclimation. 'Meyer', a freeze tolerant cultivar, and 'Victoria', a freeze susceptible cultivar were subjected to cold acclimation and non-cold acclimation treatments to determine the number of differentially expressed genes (DEGs). In response to cold acclimation, a total of 4,609 DEGs were up-regulated and 3,605 DEGs were down-regulated in 'Meyer', while 3,758 DEGs were up-regulated and 3,516 DEGs were down-regulated in 'Victoria'. GO and KEGG enrichment analysis showed several diverse pathways and biological processes, including photosynthesis, transmembrane transport, and plant hormone signal transduction. Integrating these information with previous studies on proteomics and QTL mapping, several candidate genes were identified to be associated with cold acclimation and freezing tolerance across different studies, such as LEA, CIPK, POD, HSF, HSP, MPK, PSII and multiple transcription factors. The candidate genes identified in this study showed great value in potentially being a target for future selection efforts for freeze-tolerant Z. japonica.
-
Key words:
- Z. japonica /
- Cold acclimation /
- Freeze tolerance /
- Transcriptome /
- Differentially expressed genes