









-
Alfalfa (Medicago sativa L.), a polyploid legume forage, is an important crop with strong resistance to stress, and both its yield and quality are excellent[1]. In addition, alfalfa has biological nitrogen-fixing capacity, which can improve soil structure and fertility, making it an excellent crop for sustainable agriculture[2]. Previously, researchers have made several remarkable achievements in alfalfa gene expression atlas[3] and genome assembly[4−6]. Studies about alfalfa long non-coding RNAs (lncRNAs) have been rarely reported. Although Chao et al.[7] and Wan et al.[8] predicted alfalfa lncRNAs using bioinformatic methods based on long read or short read data, respectively, these lncRNAs are merely associated with leaf development and drought response. Systematic identification and characterization of alfalfa lncRNA have not been reported.
lncRNAs are a class of RNAs that are greater than 200 nt in length and have no, or low, protein coding potential[9]. Several studies reported that they expressed differently among different tissues[10−12], and they present low conservation on nucleic acid sequences among species[13]. lncRNA plays important roles in important life processes such as gene expression regulation, chromatin remodeling, and epigenetics[14−16]. lncRNA could act as a competitive endogenous RNA to regulate gene function at post-translation level. For example, transcription of an antisense lncRNA suppresses PHO84 mRNA transcription[17]. lncRNA can also act as a miRNA sponge or target mimics, binding a large number of miRNAs based on complementary base pairing, thereby positively regulating the expression of target genes[18]. Additionally, lncRNAs encode short peptides with biological functions have been found in animals and plants, such as Toddler[19], Myoregulin (MLN)[20], DWORF[21] and ENOD40[22]. And all these lncRNAs encode small peptides with 11 to 58 amino acids[19−22]. The discovery of these small peptides indicates that there is the possibility of small open read frames (sORFs) encoding short peptides in ncRNA, and the short peptides encoded by them may play some important roles in the growth and development of organisms. Therefore, it is important to systematically study the distribution and coding potential of sORFs in lncRNA. At the current stage, the research on the coding region in lncRNA is still in its infancy, more sORFs translated from lncRNA have yet to be discovered.
Full-length transcriptome sequencing is a newly developed nucleic acid sequencing technology. Compared to the second-generation sequencing technology, the full-length transcriptome sequencing obtains longer reads, and the full-length transcript can be directly obtained. Previously, we predicted alfalfa lncRNAs associated with drought response based on short read data[8]. In order to identify the lncRNA of alfalfa systematically and comprehensively, we analyzed the third-generation full-length transcriptome sequencing data generated by PacBio sequencing technology, and predicted a large number of lncRNAs using our prediction pipeline. Nucleic acid sequence conservation aomong different species was analyzed and some conserved lncRNAs were found. We also detected the expression of some sequence conserved lncRNAs and explored the potential interaction between lncRNA and miRNA. In addition, a large number of sORFs were predicted from the lncRNAs. Overall, our research obtained comprehensive omics information of alfalfa lncRNAs for the first time, and these data sets for lncRNA, miRNA and sORFs provided abundant sources to develop the research field of alfalfa lncRNA function.
-
Alfalfa (M. sativa L. 'Aohan') plants were grown in plastic pots (20 cm × 20 cm × 30 cm), cultured under natural light condition for 3 months. The alfalfa plants were watered with MS solution (pH 7.0) every 3 d. Root, node, stem, leaf and shoot apex tissue were collected respectively and frozen with liquid nitrogen immediately. Each of these tissues was collected up to 3 g for total RNA isolation.
Total RNA isolation and PacBio library construction
-
Total RNA of each sample was isolated with RNA purification reagent (Invitrogen, Carlsbad, California) according to the manufacturer's instructions. The concentration and purity of total RNA was detected with Nanodrop2000, the integrity of total RNA was checked by agarose gel electrophoresis, and RIN was quantified by Agilent2100. Then, using Clontech-SMARTer™ PCR cDNA Synthesis Kit, the total RNA was reverse transcribed into cDNA. Finally, the library was constructed with evrogen-Trimmer-2 Kit and SMRTbell Template Prep Kit 1.0.
Analysis of PacBio sequencing data
-
Analysis of PacBio sequencing data was performed by the transcriptome analysis software of Pacific Biosciences[23]. Sequences from raw data were combined into circular consensus sequence (CCS), then 5' forward primer, 3' reverse primer and polyA sequence were checked for each read. After filtering out short reads and chimeric reads, full-length non-chimeric reads (FLNCs) and non-full-length reads (NFLs) were obtained respectively. Further, in order to get unigenes, FLNCs and NFLs were clustered using cdhit software[24]. The raw data was already uploaded to the National Genomics Data Center (
www.cncb.ac.cn/ ) and the accession number is CRA009238.The pipeline to identify lncRNA from transcriptome data
-
The transcripts of alfalfa assembled from PacBio long read sequencing was used for lncRNA identification. The lncRNA identification process was described as followed: (1) all transcripts less than 200 nt were removed; (2) blast the FLNCs in NR (
www.ncbi.nlm.nih.gov/protein ), Pfam (http://pfam.xfam.org/ ), Swiss-Prot (www.ebi.ac.uk/uniprot ), KEGG (www.genome.jp/kegg ), GO (http://geneontology.org/ ) and COG (http://clovr.org/docs/clusters-of-orthologous-groups-cogs/ ) databases and removed the transcripts annotated as protein-coding sequences; and (3) screen out the putative lncRNAs by protein-coding potential using CPC2[25] and PLEK[26] software, which can be categorized as non-coding RNAs.Using the same identification pipline, we also identified lncRNAs from transcripts based on short read RNA-seq data released by AGED database (
https://plantgrn.noble.org/AGED/ )[3] . Meanwhile, we downloaded the long read sequencing lncRNA transcripts obtained from the leaves of Zhongmu 1 at different developmental stages released by Chao et al.[7] (https://static-content.springer.com/esm/art%3A10.1007%2Fs11103-018-0813-y/MediaObjects/11103_2018_813_MOESM9_ESM.fa ). In order to more comprehensively analyze the sequence characteristics of alfalfa lncRNA, we fused the two published data sets and our alfalfa (cv. Aohan) lncRNA transcripts into a new lncRNA sequence set. Further, the new lncRNA sequence set was clustered using cdhit software to prevent sequence redundancy, and the clustered sequence set was used for subsequent analysis.Sequence conservation analysis of lncRNAs
-
To reveal the sequence conservation features of alfalfa lncRNAs predicted from the long read and short read sequencing data, and the sequences of these putative lncRNAs were searched for homologs from the lncRNA sequences data sets of Arabidopsis thaliana and M. truncatula using TBtools[27] with default parameters. The homologies were screened with the cutoff of identity ≥ 90%. The lncRNA sequences of Arabidopsis thaliana were downloaded from NONCODE database[28], and the lncRNA sequences of M. truncatula was extracted from M. truncatula genome files according to the chromosome location published by Wang et al.[29].
Prediction of microRNA target mimics
-
The target mimics mechanism of lncRNA–microRNA and their potential roles in gene expression were reported in plants[18]. To explore the possibility of putative lncRNAs as microRNA targets, all lncRNA sequences were submitted to Targetfinder[30] with default parameters. Then the alignment result was screened with the cutoff of score = 0. The interaction network between miRNAs and lncRNAs was visualized with Cytoscape (version 3.6.1)[31]. And the secondary structure of lncRNAs was analyzed with RNAfold webserver (
http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi ).Small ORFs analysis of alfalfa lncRNAs
-
Small ORFs were predicted with ORFfinder[32] and MiPepid[33], respectively. The parameter of S was set as '0' when using ORFfinder to predicted sORFs. It should be noted that the sequences of transcripts containing N were removed before we predicted sORFs using MiPepid with default parameters, since the software could not recognize those sequences.
-
After cutting off low quality reads, we obtained 1,089,299 circular consensus sequences (CCS) by PacBio sequencing. 5' prime reads, 3' prime reads and polyA reads were counted and the results are listed in Table 1. In this research, we obtained 391,677 full length non-chimeric reads (FLNCs) and 687,477 non-full length reads (NFLs), which were 35.96% and 63.11% of CCS, respectively. Average FLNCs length is 2,300.8 nt. Length distribution of CCS, FLNC and NFL is shown in Fig. 1. All the FLNCs and NFLs were clustered using CD-Hit software, then 539,260 unigenes were obtained.
Table 1. Summary of reads from PacBio full-length sequencing.
Terms Number Reads of insert 1,089,299 5' prime reads 533,904 3' prime reads 569,127 Poly-A reads 549,977 Filtered short reads 665 Non-full-length reads 687,477 Full-length reads 401,157 Full-length non-chimeric reads 391,677 Average length of full-length non-chimeric reads 2,300.8 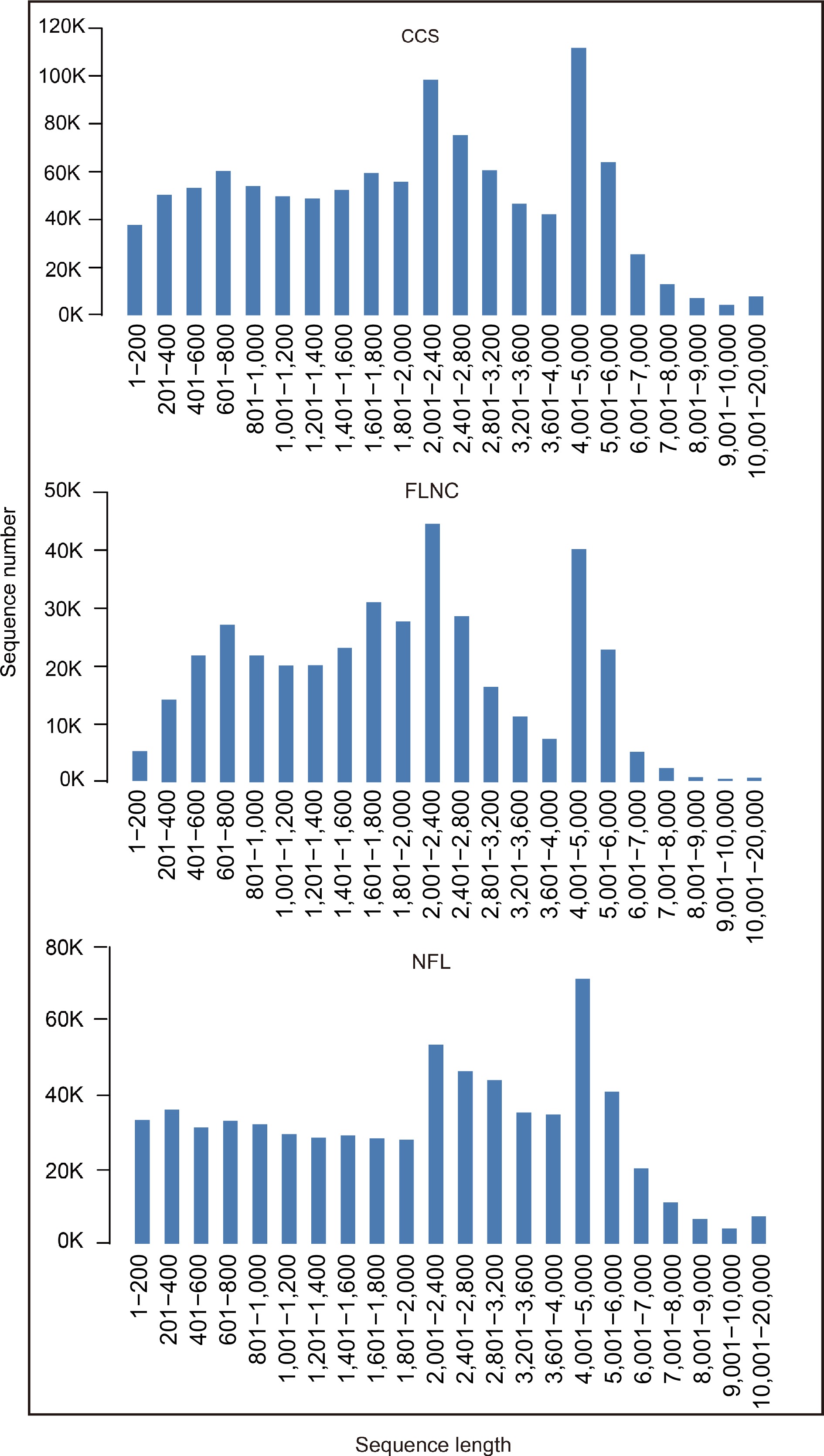
Figure 1.
Length distribution of CCS, FLNC and NFL. Circular consensus sequences (CCS); Full length non-chimeric reads (FLNC); Non-full length reads (NFL).
Identification of lncRNAs from alfalfa long and short read RNA-seq data
-
The assembled transcripts of alfalfa based on PacBio sequencing were used for lncRNA identification. This file contains 539,260 reliably expressed transcripts. We dumped transcripts with length < 200 nt. Then CPC2 and PLEK software were introduced to screen out transcripts with low or without coding potential, and 174,345 transcripts were filtered out. Further, we filtered out the transcripts with known protein-coding genes by mapping transcripts to pfam, Nr, Swissprot, KEGG, GO and COG database, and 45,116 transcripts as expressed putative lncRNAs were left.
The length distribution of lncRNAs is shown in Fig. 2, and three obvious peaks were consistent with the fraction size of CCS and FLNC. The length distribution analysis showed that more than 38.87% of the lncRNAs were in the range of 200 to 2,000 bp, and about 61.14% of the lncRNAs were in the range of 2,001 to 4,000 bp.
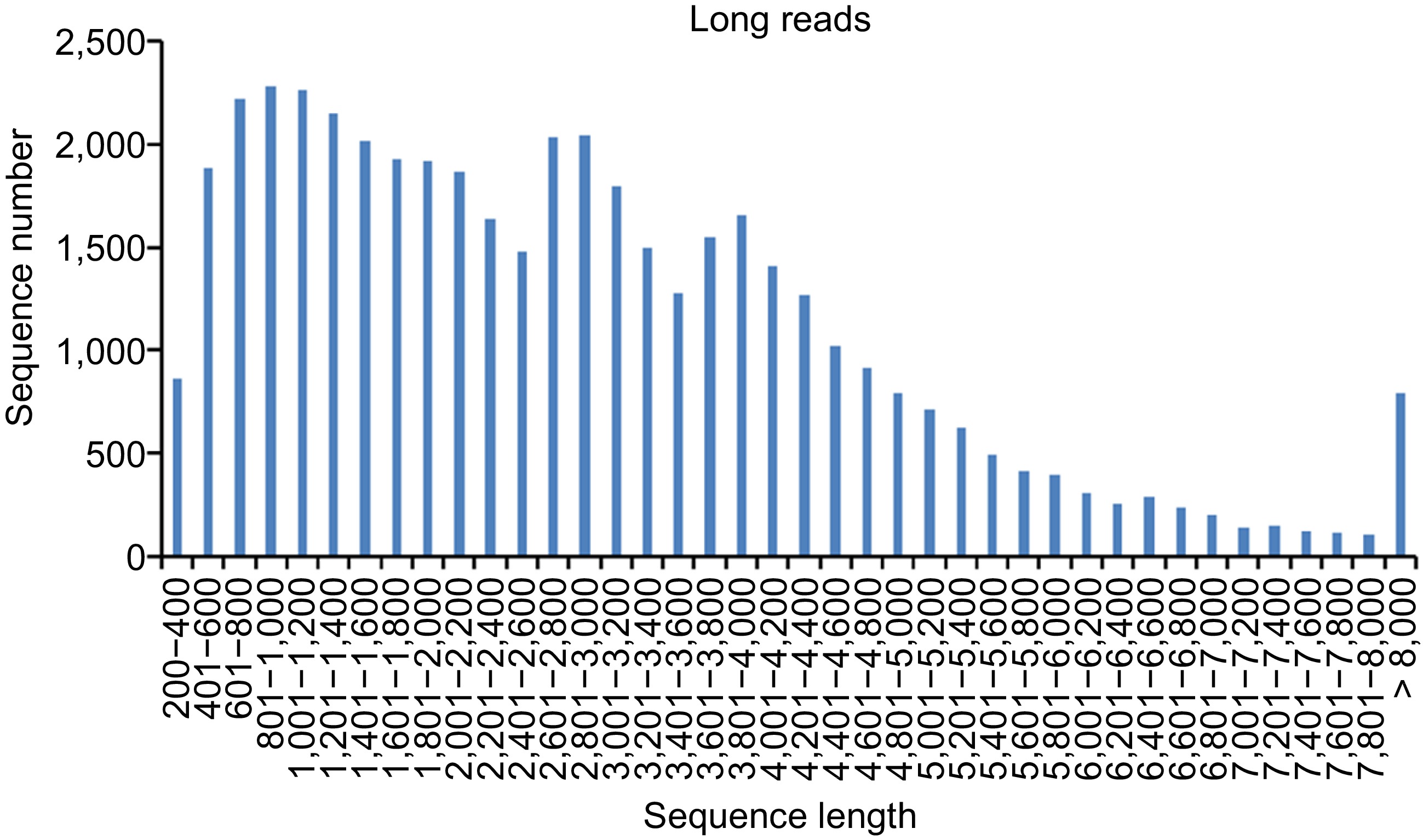
Figure 2.
Length distribution of lncRNAs identified from PacBio sequencing.
We also identified lncRNAs from transcripts based on short read RNA-seq data released by AGED database[3] using the same identification pipline. And we got 37,733 lncRNA transcripts from the short reads RNA-seq data. The details of these short read based lncRNAs are listed in Supplemental Table S1. The IDs and gene expression data in Supplemental Table S1 were retrived from the AGED database.
Length distribution of lncRNAs identified from short read RNA-seq was statisticed (Fig. 3). The result showed that more than 50% of the identified lncRNAs were in the range of 200 to 2,000 bp, and about 30% of the lncRNAs were more than 2,000 bp in length.
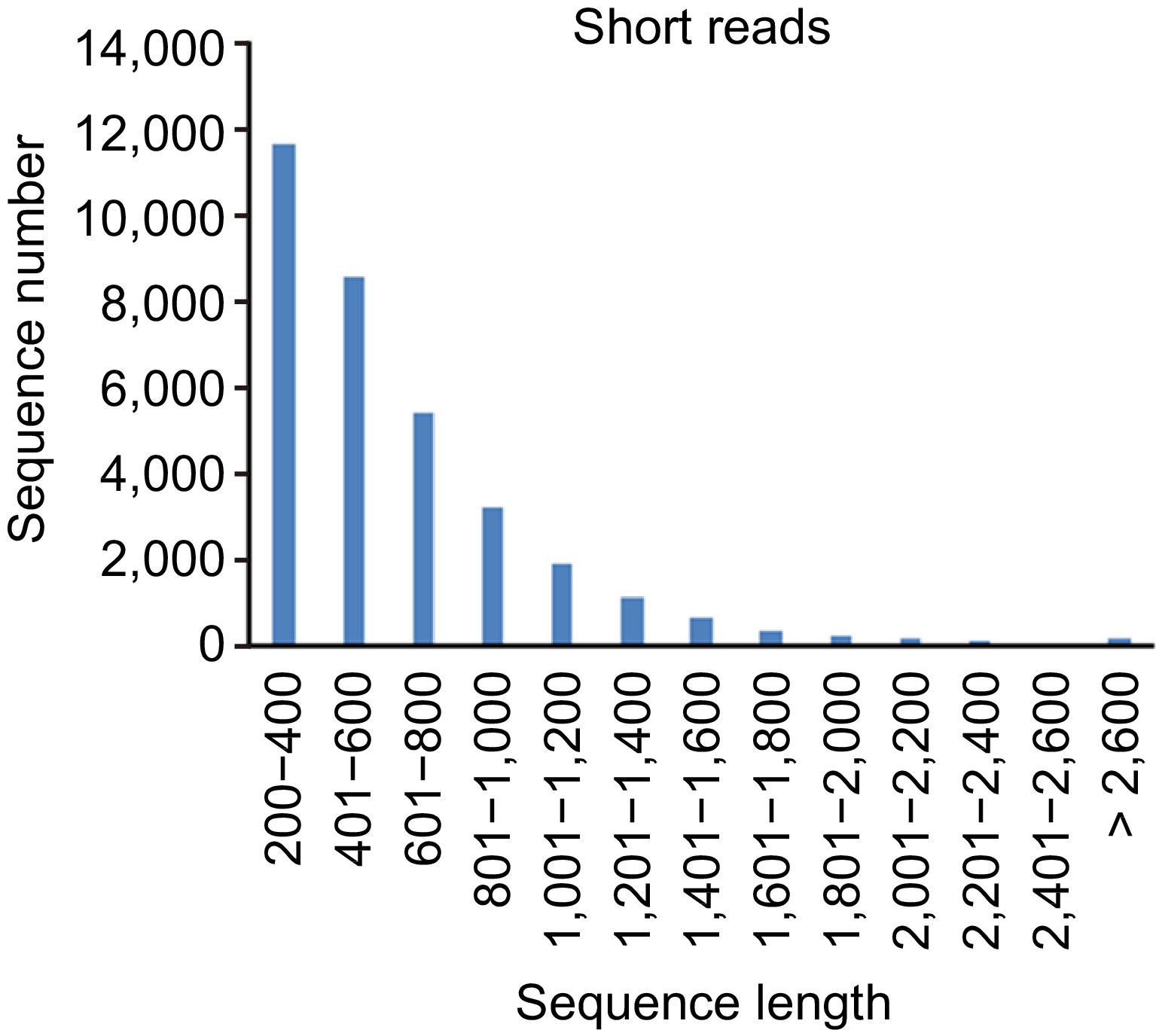
Figure 3.
Length distribution of lncRNAs identified from short read RNA-seq data.
Sequence conservation of lncRNAs between species
-
We tried to find out lncRNAs highly conserved among species by aligning the alfalfa lncRNA sequences with M. truncatula[29] and Arabidopsis thaliana[28] lncRNAs sequences, respectively. The alignment result showed that only five lncRNAs were homologous with M. truncatula and A. thaliana lncRNAs (Supplemental Table S2). And we noticed that two of the five lncRNAs, fl11.68878518.31_2627_CCS and fl8.47251612.31_2377_CCS, aligned their targets with high identity and great hit length. Then we blast their sequences in NCBI using blastn, and found that the two lncRNAs may be derived from alfalfa chloroplast or mitochondrial genomes, since fl11.68878518.31_2627_CCS contains a fraction of 18S ribosomal RNA in alfalfa chloroplast genome, and fl8.47251612.31_2377_CCS is a part of large subunit ribosomal RNA in alfalfa mitochondrial genome. The alignment results of the novel lncRNA (fl11.68878518.31_2627_CCS) of alfalfa with its homologies in A. thaliana and M. truncatula are shown in Supplemental Fig. S1.
Identification and characterizition of plastid lncRNAs
-
Considering the above results and the fact that the plastid genome is more conservative than the nuclear genome, we hypothesized that there may be highly conserved lncRNAs in the plastid genome among species. In order to more comprehensively analyze the sequence characteristics of alfalfa lncRNA, we fused three sets of alfalfa lncRNA transcripts, including the long readsequencing lncRNA transcripts obtained from the leaves of Zhongmu 1 at different developmental stages[7], the short reads sequencing lncRNA transcripts from different tissues[3], and the long reads sequencing lncRNA transcripts from different tissues of alfalfa (cv. Aohan) obtained in this study. A sequence set containing 88704 lncRNA transcripts for subsequent analysis was obtained by transcript clustering.
Then we carried out the alignments between the fused lncRNA sequence set and chloroplast/mitochondrial genomes of different species, and found some lncRNAs had homologies in the chloroplast/mitochondrial genome. Firstly, we found that 62, 21, 25, 47 and 79 lncRNAs had homologies in the chloroplast genomes of Arabidopsis (AtC), rice (OsC), oats (AsC), M. truncatula (MtC) and alfalfa (MsC), respectively (Supplemental Table S3, Fig. 4). This result implies that some of the identified lncRNAs may be plastid lncRNAs. interestingly, it was found that sequences of conting86830 and conting82120 lncRNAs were highly conserved in the chloroplast genomes of the above five species, which implies these lncRNAs may play important roles in plant growth and development.
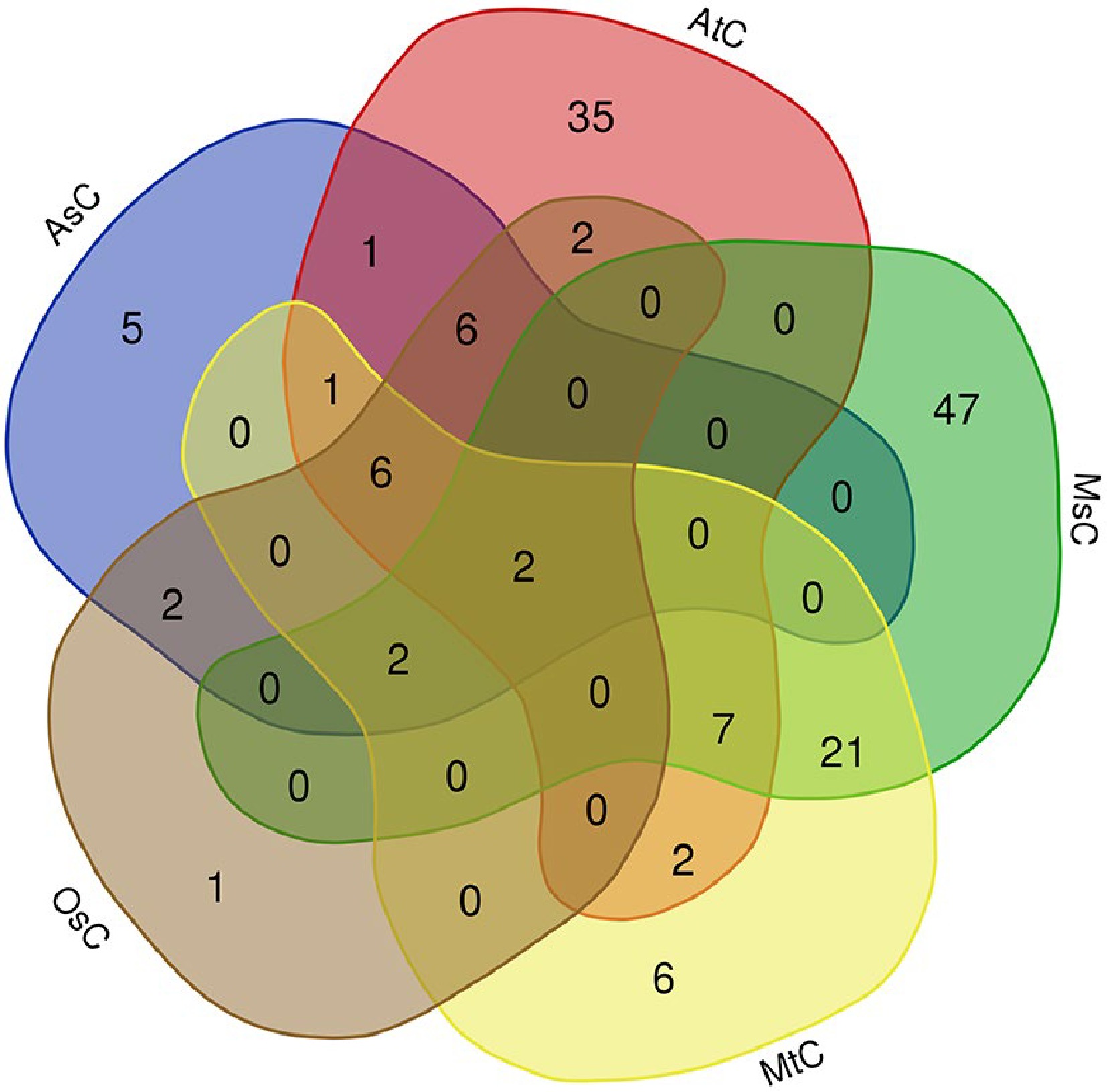
Figure 4.
Venn diagram of alfalfa lncRNA homologies in chloroplast genomes of Arabidopsis (AtC), rice (OsC), oats (AsC), M. truncatula (MtC) and alfalfa (MsC), respectively.
Secondly, we found that 109 lncRNAs had homologies in the mitochondrial genomes of Arabidopsis (AtM), rice (OsM), M. truncatula (MtM) (Supplemental File S1, Supplemental Table S4). And the venn diagram showed that it had the most homologies in the M. truncatula mitochondrial genome as expected, and 28 lncRNAs own homologies in all the three mitochondrial genomes (Supplemental Fig. S2).
lncRNAs associated miRNAs
-
In order to explore the lncRNAs associated with miRNAs, all lncRNA sequences were submitted to TargetFinder and mapped to miRNAs of M. truncatula. Then we found that 85 miRNAs could be mapped to 34 lncRNAs without mismatch, which implies these lncRNAs may be precursors or targets of the 85 miRNAs (Supplemental Table S5). The relationship between these lncRNAs and miRNAs was shown in Fig. 5. For convenience of presentation, the miRNAs belonging to the same family were collapsed into one node. The details of this network were list in Supplemental Table S5. To further investigate relationship between these lncRNAs and miRNAs, we submitted the sequences of 34 lncRNAs into RNAfold and analyzed secondary structure of the lncRNAs. We found sequences of 16 miRNAs were located at hairpin area in secondary structures of 19 lncRNAs (Supplemental Table S5).
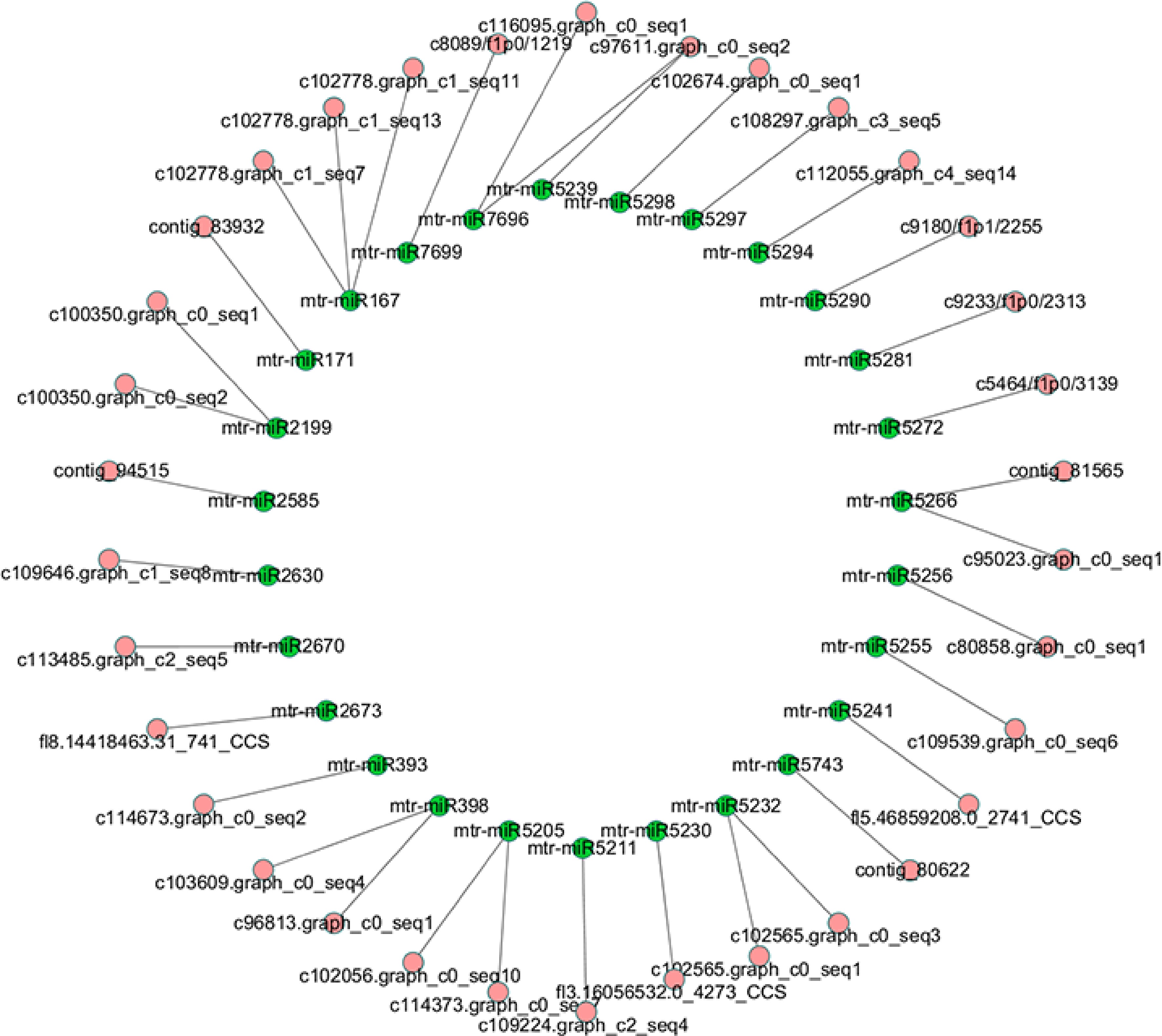
Figure 5.
Network of miRNAs and their target lncRNAs.
Small ORFs analysis of alfalfa lncRNAs
-
Small ORFs were predicted with ORFfinder and MiPepid. A total of 2,334,873 sORFs was predicted by ORFfinder from 88,558 lncRNAs and 2,617,979 sORFs were predicted by MiPepid from 85,710 lncRNAs (Supplemental Fig. S3). Sequences of sORFs predicted by the two methods were list in Supplemental Files S2 & S3. Further, we investigated relationships between sequence length of lncRNA and number of sORF which it contains. Figure 6 shows that there is a positive correlation between the length of the transcripts and the number of sORFs predicted in the transcripts, that is, the longer the transcript, the more candidate sORFs it contains.
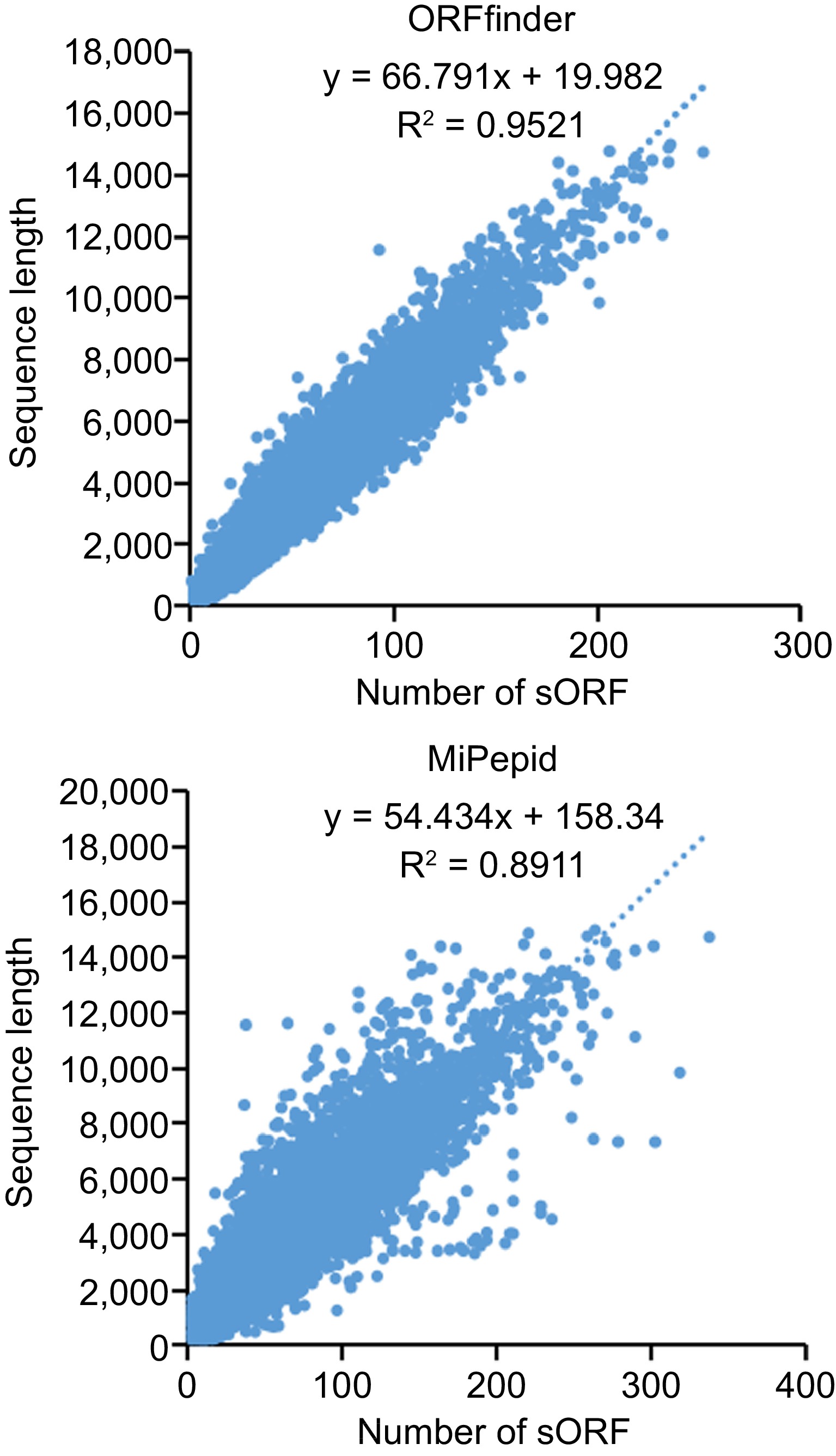
Figure 6.
The correlation between the length of lncRNA and the quantity of small open read frames (sORFs).
-
In this research, we sequenced RNA samples isolated from four different alfalfa tissues with PacBio sequencing technology and finally got 391,677 FLNCs from the sequencing data. The number of FLNC is about 2.6 times that of the previous report[7], which prefigures more sequence information containing in our dataset. Based on our long read sequencing data and the other two published alfalfa transcripts, the genome-wide lncRNAs were predicted by using the pepline developed by ourselves, the sequence conservation and small peptide coding of these lncRNAs were analyzed. It is found that quantity of lncRNAs predicted from transcripts of different experimental materials varies greatly. We identified 45,116 lncRNAs from our full-length RNA-seq data derived from four different alfalfa tissues. However, Chao et al. identified 20,915 lncRNAs from alfalfa leaf[7]. The difference between the two studies may be caused by prediction methods, or more likely caused by factors such as genotype, physiological state, development stage, tissue type, since expression of lncRNA is tissue specific and stage specific[10−12]. Actually, we detected expression level of some lncRNAs using quantitative real-time PCR. Supplemental Fig. S4 showed that the expression level of some lncRNAs is tissue specific indeed, such as fl5.23462008.3573_24_CCS and fl12.43843667.4342_55_CCS. The expression level of fl12.43843667.4342_55_CCS was higher in leaf than other tissues, which indicates it may be associated with leaf development or photosynthesis.
The sequence conservation analysis revealed that homology of lncRNAs was extremely low between alfalfa and other species. The result supported the current point that lncRNAs present low conservation on nucleic acid sequences among species[10,13]. Although most of the lncRNAs present low sequence conservation, we still found some lncRNAs annotated as chloroplast genomic sequence showed high sequence homology among species. We blast the predicted lncRNA sequences with the chloroplast genomes of several species to systematically identify sequence conserved plastid lncRNAs. The results showed that the number of lncRNAs aligned to the chloroplast genome of alfalfa was the largest, followed by M. truncatula and Arabidopsis, and finally oats and rice. We also blasted the lncRNA sequences with three mitochondrial genomes, the results also showed the same characteristics. The number of lncRNA aligned to the mitochondrial genome of M. truncatula was the largest, followed by Arabidopsis and rice. The alignment results showed that the sequence conservation of some lncRNAs from mitochondria or chloroplasts is very high, such as fl11.68878518.31_2627_CCS (Fig. 4). LncRNAs in mitochondrial DNA have been found previously in animals[34], and also present high sequence conservation among species, which is in agreement with our result.
In addition, we also found that some interesting lncRNAs of which only dozens of bp nucleotide showed high homology with the mitochondrial or chloroplast genomic sequences. These homologous sequences may be some conserved motifs or the result of the exchange between nuclear genome and plastid genome. These short sequences are likely to have some biological functions, since these kinds of lncRNAs may play roles of miRNA sponges or transcription suppressors.
To discover lncRNAs function as miRNA sponge, we mapped the lncRNAs to miRNAs of M. truncatula, to try to figure out how the lncRNAs work with miRNAs. As expected, it was found that 34 lncRNAs could bind with one or more miRNAs including miR167, miR171, miR393 and miR398, through complete complementary base pairing. Considering that miRNA play vital roles in the processes of plant growth, development and stress response. According to the result from secondary structure analysis, 16 miRNAs were located at hairpin area in secondary structures of 19 lncRNAs, which implies these lncRNAs maybe pri-miRNAs of the 16 alfalfa miRNAs, such as mtr-miR167b-5p, mtr-miR171g, mtr-miR393a and mtr-miR398b. In previous reports, mtr-miR167b-5p is responsive to arbuscular mycorrhizal fungi (AMF) colonization[35], and mtr-miR171h modulates AMF colonization of Medicago truncatula by targeting NSP2[36]. As targets of miR167 and miR171, contig_83932, c102778.graph_c1_seq7, c102778.graph_c1_seq11 and c102778.graph_c1_seq13 may also associated with AMF colonization. MiR393 targeting c114673.graph_c0_seq2 regulates the homeostasis of auxin signaling[37], and miR398 targeting c103609.graph_c0_seq4 and c96813.graph_c0_seq1 regulates plant responses to salt stress, water deficit, high sucrose, copper and phosphate deficiency and bacterial infection[38]. Therefore, these lncRNAs may be linked to the plant hormone and stress regulatory networks.
More and more studies proved that sORF-encoded micro peptides play important roles in regulating various biological activities[19−22]. Using bioinformatic methods, we found more than 96% lncRNAs identified in this study could encode small peptides, which suggests that lncRNA has great potential to regulate some life processes through the synthesis of small peptides, although the existence of these small peptides needs further experimental verification. To date, there isn't a bioinformatic way developed to annotated biological function of sORFs yet, and functional characterization of sORFs for plants is far behind that of other species. So, except for experimental methods, bioinformatic methods for investigating sORF function should be developed as soon as possible.
-
In this study, we identified alfalfa lncRNAs from combined long and short read sequencing data, resulting in a tremendous number of putative lncRNAs. We also reported a set of plastid lncRNAs in plant and predicted sORFs of alfalfa lncRNAs for the first time. Our research not only provides abundant sequence information of alfalfa lncRNA, but also offers a fresh perspective to study them.
-
The authors confirm contribution to the paper as follows: study conception and design: Li Y, Sun Y; data collection: Li Y; analysis and interpretation of results: Li Y, Wang C, Cui H, Jia F, Kang J; draft manuscript preparation: Li Y, Zhu K, Ma C. All authors reviewed the results and approved the final version of the manuscript.
-
The raw data of PacBio sequencing was uploaded to the National Genomics Data Center (
www.cncb.ac.cn ) and the accession number is CRA009238. The supplemental files are available in Figshare (DOI:10.6084/m9.figshare24564784 ). This study was financially supported by the National Natural Science Foundation of China (No. 32271763).
-
The authors declare that they have no conflict of interest.
- Supplemental Table S1 Details of the lncRNAs predicted form short reads RNA-seq data.
- Supplemental Table S2 Blast result of the five lncRNAs which were homologous with M. truncatula and Arabidopsis thaliana lncRNAs.
- Supplemental Table S3 Homologies in the chloroplast genomes of Arabidopsis (AtC), rice (OsC), oats (AsC), M. truncatula (MtC) and alfalfa (MsC), respectively.
- Supplemental Table S4 Homologies in the mitochondrial genomes of Arabidopsis (AtM), rice (OsM), M. truncatula (MtM).
- Supplemental Table S5 Details of the miRNA-lncRNA interaction network.
- Supplemental File S1 The source of data in Figure 4.
- Supplemental File S2 Sequences of sORF candidates predicted by ORFfinder.
- Supplemental File S3 Sequences of sORF candidates predicted by MiPepid.
- Supplemental Fig. S1 Alignment of the nucleotide sequences of fl11.68878518.31_2627_CCS and its two homologies in in A. thaliana (NONATHT003850.1) and M. truncatula (chr4_490072_490302). Red and white backgrounds indicate conserved and non-conserved residues, respectively.
- Supplemental Fig. S2 Venn diagram of alfalfa lncRNA homologies in mitochondrial genomes of Arabidopsis (AtM), rice (OsM), M. truncatula (MtM).
- Supplemental Fig. S3 Venn diagram of sORF numbers predicted by ORFfinder and MiPepid, respectively.
- Supplemental Fig. S4 Relative expression level of 6 lncRNAs in different tissues, such as shoot apex (A), leaf (L), node (N) and root (R).
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Li Y, Wang C, Cui H, Zhu K, Jia F, et al. 2023. Identification of alfalfa lncRNAs based on PacBio sequencing. Grass Research 3:26 doi: 10.48130/GR-2023-0026 

Identification of alfalfa lncRNAs based on PacBio sequencing
- Received: 17 September 2023
- Accepted: 27 November 2023
- Published online: 18 December 2023
Abstract: Alfalfa is an important forage crop worldwide. lncRNAs are considered to be a class of functional biomacromolecules, while little is known about lncRNAs in alfalfa. In this study, RNAs from different tissues of alfalfa were sequenced and analyzed with full-length transcriptome sequencing technology. Based on our full-length sequencing and public RNA-seq data, we identified 88,563 lncRNAs, approximately 96.5% of total lncRNAs may encode small ORFs. The results of sequence conservation analysis showed most alfalfa lncRNAs shared low sequence conservation with those in other plant species. Some lncRNAs originating from plastid genome were revealed. And we found that 34 lncRNAs could be precursors or targets of 85 miRNAs. Our research generated the most comprehensive sequence set of alfalfa lncRNAs so far, and revealed some plastid originated lncRNAs with high sequence conservation.
-
Key words:
- lncRNA /
- Alfalfa /
- Full-length transcriptome sequencing /
- Sequence conservation /
- Small ORF