









-
Annonaceae is one of the most species-rich families of Magnoliales[1], with approximately 107 genera and 2,400 species, growing in tropical and subtropical lowland forests[2−4]. Annonaceae fruits are usually rich in carbohydrates and sugars, important vitamins and minerals. The genus Annona belongs to the tribe Annoneae of subfamily Annonoideae with approximately 162 species mainly distributed in the neotropics and some species are native to Africa[1]. Annona lineages are estimated to have originated 52.5 Mya in the New World and their diversification is supposed to have started during the late early Miocene (25.6–21.8 Mya ± 3.8)[5,6]. Nowadays, several cultivated species of Annona produce edible fruits, including A. montana (mountain soursop), A. squamosa (sugar apple), A. muricata (soursop), and A. cherimola (cherimoya).
Annona montana, is popular as guanabana or false graviola due to its similarity with graviola, A. muricata. Mainly distributed in western South America, it has been cultured for its fruit in China and India[7]. A. montana also grows widely in Trinidad, and its leaves are used to treat influenza and insomnia[8]. Fruit quality is strongly related to sugar accumulation, ripening, and fruit scent. However, the molecular mechanisms of sugar accumulation, ripening, and fruit scent in A. montana are still unclear. In the present study, we report a high-quality genome of A. montana obtained using the Pacific Biosciences sequencing platform and high-resolution chromosome conformation capture (Hi-C) technology. Analysis of the A. montana genome will clarify the evolution of Annonaceae and magnoliids, thereby revealing the development and flavor formation in soursop fruit.
-
Fresh plant materials were collected from an adult A. montana growing in the South China Agriculture and Forestry University for genome sequencing. We used the modified cetyltrimethylammonium bromide (CTAB) protocol to extract total genomic DNA. The paired-end libraries (500 bp) were constructed using an Illumina protocol.
The heterozygosity and size of A. montana genome were estimated with GenomeScope[9], using the abundances of 17-nucleotide k-mers. Additionally, the PacBio 20 kb protocol (
www.pacb.com ) was used to construct Single Molecule Real-Time Sequencing (SMRT) libraries, which were subsequently sequenced on the PacBio platform. We sequenced seven cells and obtained 110.3 Gb of raw data. The fruits were collected at three developmental stages for transcriptome sequencing based on the Illumina platform.Karyotype analysis
-
Roots with active meristems were obtained by culturing sample plants. After the induction of mitosis by nitrous oxide, a large number of metaphase cells were obtained and chromosome samples were prepared. Then, the dispersed metaphase chromosome cells were obtained and the chromosome number was determined according to the karyotype analysis process. After DAPI staining, clear and intuitive chromosomes were obtained with high-resolution fluorescence microscope and CCD imaging equipment. Fluorescence probes based on telomere conserved repeat sequences, 5SrDNA and 18SrDNA probes were used for fluorescence in situ hybridization (Fish) to determine the karyotype characteristics of the species.
Genome assessment
-
The 110.3 Gb raw data after quality control was assembled in pure third-generation by Falcon[10]. Then, the BWA MEM default parameter was used to compare the second-generation data to the three generations of the arrow corrected genome[11], and the Pilon iterative correction was used three consecutive times to obtain the result. Finally, we compared the three generations of arrow corrected genome, and used Pilon v1.22[12] iterative correction three times, and obtained the genome size of A. montana. BUSCO v3[13] database (
https://anaconda.org/bioconda/busco ) was used to assess the completeness of the genome assembly.Repetitive sequence identification
-
We used the RepBase v21.12 database[14] (
www.girinst.org/repbase ) to align the homologous sequences. We used RepeatProteinMask v4.0.7 (www.repeatmasker.org ) to identify the known repeating sequences and similar sequences. Next, we used three de novo prediction softwares, including RepeatModeler[15] (www.repeatmasker.org/RepeatModeler ) and LTR_FINDER v1.06[15] (http://tlife.fudan.edu.cn/ltr_finder/ ), to identify transposable elements (TEs) in the A. montana genome. Tandem Repeats Finder v4.09[16] (http://tandem.bu.edu/trf/trf.html ) was used to determine tandem repeats across the A. montana genome. Finally, we grouped the repeat sequences with ≥ 50% identities into the same clades.Gene prediction and annotation
-
Homology-based and de novo-based predictions were used to predict protein-coding genes. Homologous proteins from five known whole-genome sequences of Antinidia chinensis, Arabidopsis thaliana, Liriodendron chinense, Populus trichocarpa, and Eucalyptus grandis were aligned to the A. montana genome sequence using Exonerate v2.2.0[17] (
www.ebi.ac.uk/Tools/psa/genewise/ ) for homolog-based prediction. Two ab initio prediction softwares, Augustus[18] (http://bioinf.uni-greifswald.de/augustus/ ) and SNAP[19] (http://homepage.mac.com/iankorf ) were employed for de novo gene prediction MAKER[20]. An online facility (http://weatherby.genetics.utah.edu/MAKER ) was used to merge the homology-based and ab initio-based gene structures following a non-redundant gene model. The annotated results of Maker were further filtered, and the following genes were selected: 1) homologous protein support for exon region < 50% and protein length < 50 amino acids; 2) TE and coding DNA sequence (CDS) of coding region overlap length > 80%.For gene annotation, seven protein databases: TrEMBL (
www.uniprot.org/ )[21], SwissProt (www.uniprot.org )[22], KEGG (www.genome.jp/kegg )[23], InterPro[24] (www.ebi.ac.uk/interpro ), NR (NCBI's non-redundant protein database), KOG[25], and GO[26], were searched and the results were aligned with Blast v2.2.31[26]. We used tRNAscan-SE 1.3.1[27] to predict tRNAs. We aligned the rRNA template sequences from the Rfam database against the genome using the BLASTN algorithm to identify rRNAs[28]. We used INFERNAL[29] (http://infernal.janelia.org/ ) in Rfam to predict the miRNAs and snRNAs, and used it against the Rfam database to predict the other ncRNAs.Genome evolution analysis
-
The gene families of 20 genomes were identified through OrthoMCL v1.4[30] (
http://orthomcl.org/orthomcl/ ). The abnormal gene families were filtered out based on OrthoMCL clustering, and CAFÉ 4.2[31] (http://sourceforge.net/projects/cafehahnlab/ ) was used to measure the expansion and contraction of orthologous gene families. We used 33 single-copy gene families of peptide sequences to establish phylogenetic relationships and estimate divergence times. The amino acid sequences of the single-copy orthologous groups were aligned using MUSCLE[32] (www.drive5.com/muscle/ ). Phylogenetic tree with 500 bootstrap replicates was constructed on RAxML. We used the Bayesian relaxed molecular clock approach to estimate the species divergence times with MCMCTREE program (http://abacus.gene.ucl.ac.uk/software/paml.html ) of the PAML package v4.7[33]. The published genomic data of A. thaliana–P. trichocarpa (100–120 Mya), Magoliineae (112.6 Mya), A. thaliana–C. arabica (111–131 Mya), A. thaliana–A. trichopoda (173–199 Mya), and A. thaliana–P. abies (289–330 Mya) were used to calibrate divergence times[34].Genome synteny
-
Genes are conserved in sequence and function during the course of evolution in collinear segments. The default parameters of JCVI v0.9.14 (
https://pypi.org/project/jcvi/ ) were employed to analyse the protein sequences of A. montana, L. chinense, and C. kanehirae, and the gene pairs were obtained in a collinear series.The Ks (substitutions per synonymous site) distribution analysis was performed to estimate WGD events in A. montana, L. chinense, and C. kanehirae genomes. We used DIAMOND to self-align the protein sequences of A. montana, L. chinense, and C. kanehira, and to extract the mutual optimal alignment in the alignment results. Finally, the Codeml in the PAML package was executed to calculate the Ks value[35,36].
Gene family identification
-
MADS-box genes, sugar metabolic genes and sugar transporter genes of Arabidopsis were downloaded from The Arabidopsis Information Resource (TAIR), and run them as queries in BLASTP searches against the A. montana protein sequences to identify homologous genes. The redundant sequences were discarded and the conserved protein domains were checked through CDD database (
www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi ) in automatic mode (threshold = 0.01, maximum hits = 500). The KEGG and KOG annotations of sugar metabolic and sugar transporter homologous genes in A. montana were checked, and only those genes that annotated were retained. We used the MEGA5 to align the homologous sequences of MADS-box genes[37], sugar metabolic genes and sugar transporter genes, and used the CIPRES website to construct the phylogenetic tree (www.phylo.org/portal2/ ). -
We performed a detailed characterization of A. montana chromosomes (2n = 2x = 14) (Supplemental Fig. S1) by a combination of in situ hybridization techniques, fluorochrome banding and karyomorphological analysis. Survey analysis showed that the A. montana genome had a low level of heterozygosity, corresponding to a genome size of 1.09 Gb (Supplemental Fig. S2). We obtained 110.3 Gb raw data from the de novo whole-genome sequencing of A. montana using the Pacific Biosciences RS II sequencing platform (Supplemental Table S1). We assembled 974.35 Mb of the genome with a contig N50 value of 7.89 Mb (Supplemental Table S2). The completeness of the assembled genome was 90.80% based on the analysis of Benchmarking Universal Single-Copy Orthologs (BUSCO) (Supplemental Table S3). A 92.12 Gb clean data was obtained by a sequencing library of genome-wide chromosome conformation capture (Hi-C) and used it for further scaffolding (Supplemental Table S1). We anchored a total of 973.54 Mb (99.92%) of the genome that was mapped to seven pseudochromosomes, the lengths of which ranged from 90.58 Mb to 188.31 Mb (Supplemental Tables S4, S5). The heat map of the interaction between the pseudochromosomes indicates that the Hi-C assembly of the A. montana genome is of very high quality (Supplemental Fig. S3).
Gene prediction and annotation
-
A total of 26,399 protein-coding genes were annotated in the A. montana genome, of which 25,933 predicted protein-coding genes were functionally annotated (Supplemental Table S6). The 14,280 (54.09%) genes were annotated in KEGG Orthologue (KO) terms, and 23,577 (89.31%) genes were annotated in Gene Ontology (GO) terms (Supplemental Table S7). The average length of a protein-coding gene in A. montana was 5,630.77 bp, the average length of a coding DNA sequence (CDS) was 1,201.92 bp, the average number of exons per gene was 4.95, the average length of an exon was 242.87 bp, and the average length of an intron was 890.22 bp (Supplemental Fig. S4 & Supplemental Table S6). The A. montana genome contained 51 miRNAs, 625 tRNAs, 2,832 rRNAs, and 82 snRNAs (Supplemental Table S8). In addition, we performed CEGMA (Core Eukaryotic Genes Mapping Approach) and BUSCO assessments and found the completeness of the annotated genome to be 91.13% and 96.59%, respectively (Supplemental Table S9).
Through the combination of homology-based searching and de novo prediction, we found that 61.58% of the A. montana genome consists of repetitive sequences (Supplemental Fig. S5 & Supplemental Tables S10, S11), which is comparable to Liriodendron chinense (61.6%)[38], but smaller than that of Phoebe bournei (~68.51%)[39], and larger than that of Cinnamomum kanehirae (~47.84%)[40], Litsea cubeba (~55.47%)[41], and A. muricata (~54.87%)[42]. Long terminal repeats (LTRs) accounted for 49.52% of the repetitive sequences and 3.42% of the total DNA in A. montana (41.28% in A. muricata, followed by DNA repeats 7.29%).
Evolution of gene families
-
The expansion and contraction analysis showed that 73 gene families were expanded, resulting in Magnoliales, and 769 families were contracted in the lineage (Fig. 1a). In A. montana, 479 gene families were expanded, and 1,226 gene families were contracted (Fig. 1a). Enrichment analysis found that the significantly expanded gene families in A. montana were especially enriched in GO terms of 'cellular protein metabolic process', 'organomercury catabolic process' and 'alkylmercury lyase activity', and in the KO terms of 'endocytosis' (Supplemental Tables S12 & S13), it may relate to Mercury metal ion resistance. The significantly contracted gene families in A. montana were especially enriched in the GO terms of 'active transmembrane transporter activity' and 'cation-transporting ATPase activity', and in the KEGG pathway of 'purine metabolism' (Supplemental Tables S12 & S13). In addition, a total of 337 unique genes in A. montana gene families were found to be specifically enriched in the GO terms of 'aminoglycan catabolic process' and 'O-methyltransferase activity', and in the KEGG pathway of 'fatty acid degradation' and 'biosynthesis of secondary metabolites' (Supplemental Tables S12 & S13).
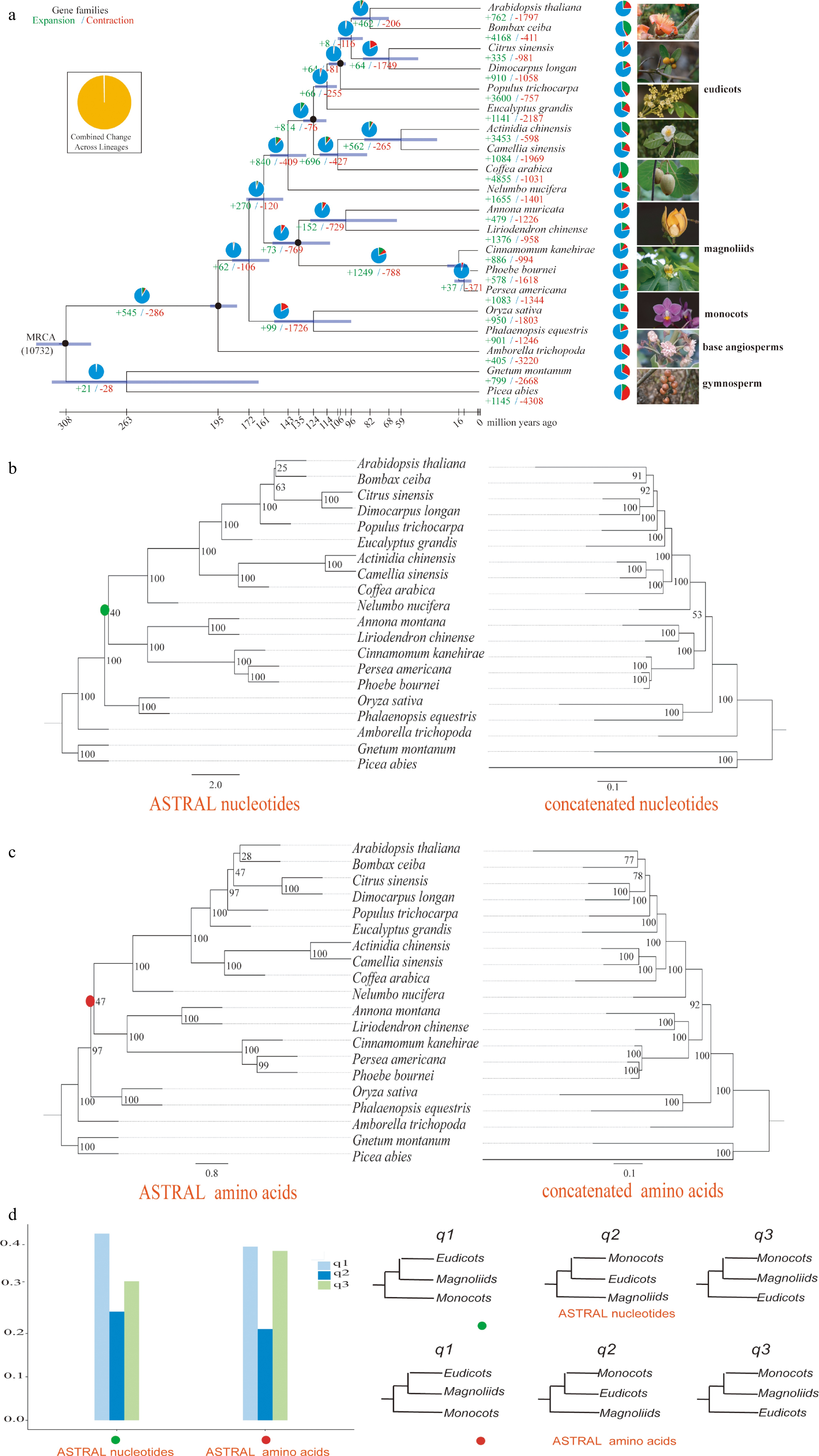
Figure 1.
Phylogenetic tree, concatenated and ASTRAL trees of A. montana. (a) Phylogenetic tree based on the Bayesian method. (b) ASTRAL (left) and concatenated (right) trees constructed based on nucleotide sequences. (c) ASTRAL (left) and concatenated (right) trees constructed based on amino acid sequences. (d) Comparison of q-values of ASTRAL trees based on nucleotide and amino acids sequences.
The phylogenetic relationships among magnoliids, eudicots, and monocots
-
The evolutionary position of magnoliids remains uncertain[38−44]. We extracted single-copy families from 20 different plant genomes for phylogenetic tree construction, including basal angiosperms, five magnoliids, ten eudicots, two monocots, and two gymnosperms (Supplemental Fig. S6 & Supplemental Table S14). The Bayesian tree indicated that A. montana and L. chinense formed a subclade (Magnoliales), which was sister to the subclade formed by C. kanehirae, P. bournei, and P. americana (Laurales) (Fig. 1a). Magnoliales and eudicots diverged approximately 159.55 Mya, Magnoliales and Laurales diverged approximately 139.59 Mya, and Magnoliaceae (L. chinense) and Annonaceae (A. montana) diverged approximately 98.76 Mya (Fig. 1a).
Incomplete lineage sorting (ILS) in early angiosperms may confuse the resolution of early diverging branches in angiosperms, such as the divergence of monocots, eudicots, and magnoliids. Therefore, we used nucleotide and protein sequences to construct ASTRAL and concatenated trees. The results show that magnoliids are sisters to eudicots after their common ancestor diverged from monocots with lower approval ratios (Fig. 1b, c). The q-value in ASTRAL was used to display the percentage of gene trees in support of different topologies. The results show that magnolias and eudicots are sister groups as the main topology (q1), magnolias and monocots-eudicots are sister groups as the first topology (q2), and magnolias and monocots are sister groups as the second topology (q3) (Fig. 1d). Furthermore, these support rates did not exceed 50%, and q1 was significantly higher than q2 and q3 (Fig. 1d). Thus, the Bayesian tree support that magnoliids are sisters to eudicots. Because of ILS during the rapid divergence of early diverging branches in angiosperms, ASTRAL trees showed that magnoliids might also be sisters to monocots−eudicots or monocots.
Whole genome duplication (WGD) event and divergence in C. kanehirae, L. chinense, and A. montana
-
The distribution of Ks values in A. montana and L. chinense genomes showed one clear peak, which was greater than that of A. montana−L. chinense (Fig. 2a). This indicates that after the common ancestors of A. montana and L. chinense shared a WGD, A. montana and L. chinense differentiated.

Figure 2.
Whole genome duplication (WGD) analysis. (a) Ks distribution in A. montana and L. chinense. (b) Ks distribution in L. chinense and C. kanehirae. (c) Collinear relationship between L. chinense and C. kanehirae. From (c), we found several collinearity regions that satisfy L. chinense: C. kanehirae = 2:4, which indicates that L. chinense underwent WGD once and C. kanehirae underwent WGD twice after diverging.
To determine whether this WGD event was shared by the common ancestor of C. kanehirae and L. chinense, we analysed the distribution of Ks values in C. kanehirae and the Ks differentiation peak of C. kanehirae−L. chinense (Fig. 2b). The C. kanehirae genome showed two Ks peaks, Ks1 ≈ 0.5–0.6 and Ks2 ≈ 0.85–0.95, while the L. chinense genome showed one Ks peak, Ks ≈ 0.7. The Ks differentiation peak of C. kanehirae−L. chinense was between the peak of Ks1 and the peak of Ks2 in the genomes of C. kanehirae. This indicates that after the common ancestor of C. kanehirae and L. chinense experienced an ancient WGD event (Ks2 ≈ 0.85–0.95), C. kanehirae and L. chinense differentiated, and then C. kanehirae alone experienced a recent WGD event (Ks1 ≈ 0.5–0.6). However, the Ks differentiation peak of the L. chinense genome (Ks ≈ 0.7) was smaller than that of C. kanehirae−L. chinense (Ks ≈ 0.825) (Fig. 2b), indicating that a WGD event occurred after the differentiation of C. kanehirae and L. chinense, and L. chinense experienced a WGD event.
To determine whether the WGD event of L. chinense is unique to L. chinense, we determined the differentiation of C. kanehirae and L. chinense by constructing a gene tree and carrying out collinearity analysis. The results from the gene tree and collinearity relations both showed that after C. kanehirae and L. chinense differentiated, L. chinense experienced one WGD event and C. kanehirae experienced two WGD events (Fig. 2c & Supplemental Fig. S7).
Based on the above results, our WGD analysis indicated that after the common ancestor of A. montana−L. chinense, and C. kanehirae differentiated, the common ancestor of A. montana−L. chinense experienced one WGD event, and C. kanehirae experienced two WGD events. Neither A. montana nor L. chinense had its own WGD event.
MADS-box gene
-
The MADS-box gene family plays an important role in several plant processes, such as floral development, flowering time control, and fruit ripening regulation[45]. In the present study, 46 MADS-box genes were identified in the A. montana genome, which were classified into type I and type II genes based on phylogenetic analysis (Supplemental Fig. S8 & Supplemental Table S15). We subdivided 13 type I MADS-box genes into three subfamilies (Mα, Mγ, and Mβ) (Supplemental Fig. S8 & Table 1) with four and two members in Mβ and Mγ, respectively, and seven members in Mα (the orthologues had duplicated). Type I genes have been reported to be associated with the development of embryo, female gametophyte[46], central cell, and endosperm[47,48] but its specific role in A. montana is yet to be studied.
Table 1. MADS-box genes in A. montana, A. thaliana, P. bournei.
Category A. thaliana P. bournei A. montana Type II (Total) 46 34 33 MIKC 40 28 28 MIKC* 6 6 5 Mδ 0 0 0 Type I (Total) 62 30 13 Ma 25 23 7 Mβ 21 4 7 Mγ 16 3 2 Total 108 64 46 In the type II gene, there were 28 and five members in the MIKCC-type and MIKC*-type of genes, respectively (Supplemental Fig. S8 & Table 1). MIKC* regulation plays an important role in pollen gene expression[49,50]. There was only one gene from A-class and two genes from AGL6-class. In a previous study, the ANR1 and AGL12 genes have been reported to play an important role in root development[51]. A. montana contains five members in the ANR1 and AGL12 clades (Supplemental Fig. S8 & Table 1). The growth of A. montana requires strong roots, which may be the reason for more genes related to root development in A. montana. Nevertheless, there were no FLC subfamily genes in A. montana, indicating that this family might have been lost (Supplemental Fig. S8 & Table 1). This could be because A. montana does not require vernalisation for flowering, similar to rice[52].
Fruit development and ripening
-
Fruit development is a complex process that involves many changes in colour, size, texture, nutritional components, and sugar content[53]. To comprehensively characterise the genes related to the development and quality of A. montana fruit, RNA-Seq was performed at three crucial stages (small fruit (SF), medium fruit (MF), and big fruit (BF) stages) of fruit development in A. montana (Supplemental Fig. S9). We identified 6537 differentially expressed genes related to the fruit development of A. montana (Supplemental Fig. S10). The MADS-box genes MADS-RIN and AGL1 are involved in the expansion and ripening of fruits such as tomato, banana, and watermelon[53−55]. In the A. montana MADS-box gene family, we identified six MADS transcription factors in the AGL1 and RIN clades (Supplemental Table S15). Of which, five genes (Amo010931, Amo001372, and Amo024502 in the RIN clade, and Amo005810 and Amo014903 in the AGL1 clade) were highly expressed in the small, medium and big fruit stages of A. montana fruit (Supplemental Fig. S11). We also identified that one B-PI gene (Amo014395) was highly expressed in the small, medium and big fruit stages; and one AGL11 gene (Amo014526) was highly expressed in the small and medium fruit stages. These eight genes were highly expressed throughout fruit development, indicating that they could have evolved to participate in other functions, in addition to ripening and fruit expansion.
Sugar metabolism and accumulation during A. montana fruit development genes encoding key enzymes involved in sugar metabolism
-
Sugar can be converted to fructose and glucose by vacuolar acid invertase (vAINV), cell wall invertase (CWINV), and neutral invertase (NINV)[56]. We identified ten genes encoding invertase, including three CWINVs (Amo021330, Amo007261, and Amo006983), six NINVs (Amo016363, Amo012168, Amo015907, Amo020421, Amo023294, and Amo010001) and one vAINV (Amo007259) (Supplemental Table S16). Amo021330 and Amo007261 were clustered together and were sisters to AtCWINV6 (Fig. 3a), which is a fructan exohydrolase (FEH) that can degrade both inulin-type and levan-type fructans[57]. Three AmoNINVs (Amo016363, Amo012168, and Amo015907) were clustered in the α clade and another three AmoNINVs (Amo020421, Amo023294, and Amo010001) were clustered in the β clade (Fig. 3a). Transcriptome analysis showed that all CWINV orthologous genes from A. montana had low expression in small, medium and big stages of A. montana (Fig. 3b). AmovAINV (Amo007259) was highly expressed in the small fruit stage and decreased during the later stages of development of A. montana fruit (Fig. 3b). Two NINV genes (Amo016363 and Amo010001) were highly expressed in the small, medium and big stages, and one NINV gene (Amo012168) was highly expressed in the small fruit stage (Fig. 3b). These results indicate that AINV and NINV genes may play an important role in controlling sucrose concentration in the cytosol of A. montana fruit, especially in the small fruit stage.
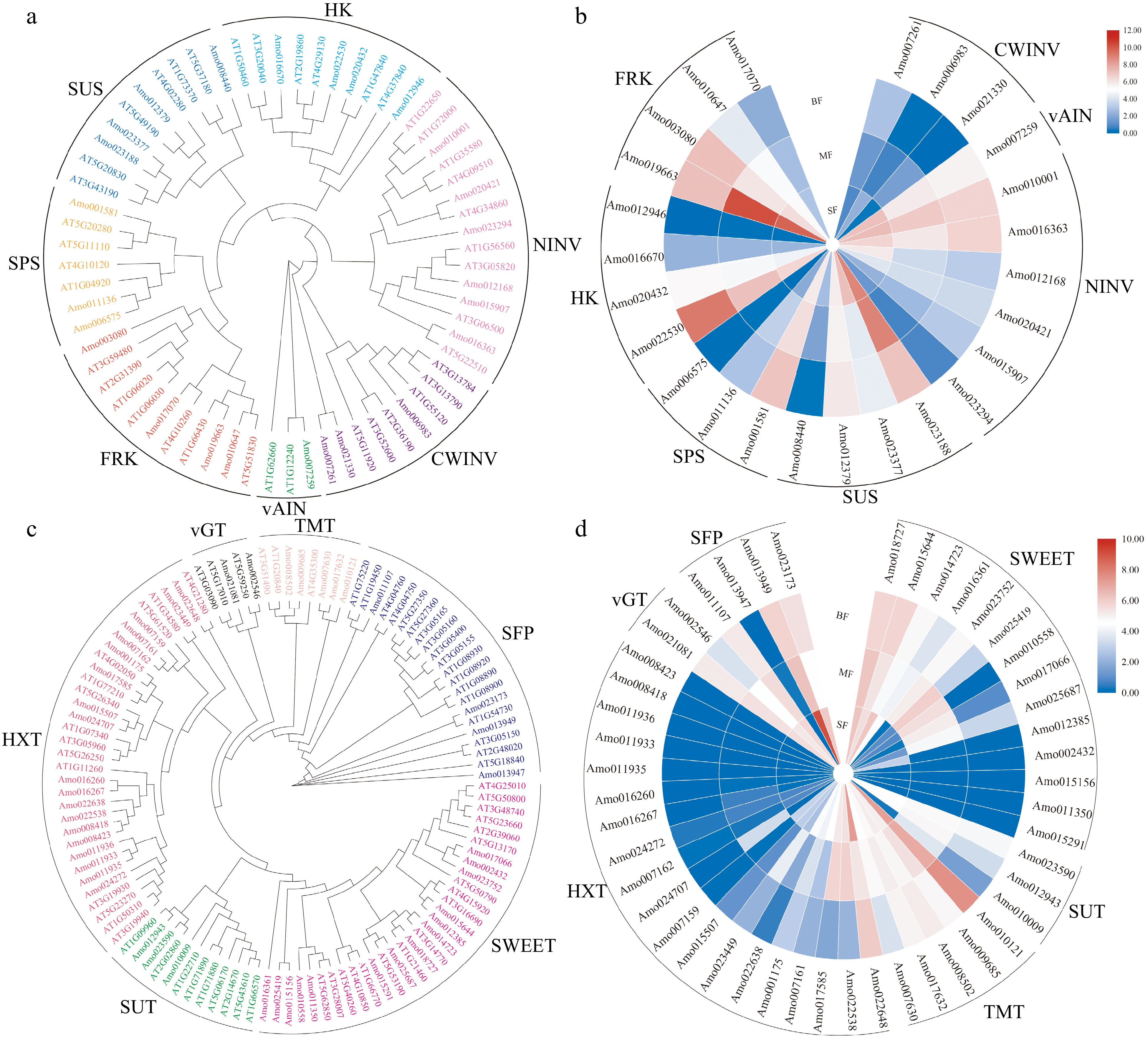
Figure 3.
Analysis of genes related to sugar metabolism and accumulation in A. montana fruit development. (a) Phylogenetic analysis of genes related to sugar metabolism. (b) Expression pattern of key enzyme genes involved in sugar metabolism. (c) Phylogenetic analysis of genes related to sugar transporters. (d) Expression pattern of sugar transporters. 'SF' represents the small fruit stage, 'MF' represents medium fruit stage, 'BF' represents big fruit stage.
Sucrose synthase (SUS) is one of the most important enzymes involved in sucrose synthesis and hydrolysis[58]. A substantial activity of this enzyme has been related to the rapid accumulation of hexoses in some fruits. Arabidopsis thaliana SUS genes have been divided into three subfamilies, including SUSA, SUS1, and SUS2[58]. We identified four SUS gene family members from A. montana fruit (Amo012379, Amo023188, Amo023377, and Amo008440), and classified them into three subfamilies based on phylogenetic analysis (Fig. 3a & Supplemental Table S16). The number of SUS genes in A. montana is less than those in A. thaliana (six SUS genes)[58], peach (five SUS genes)[59], and apple (five SUS genes)[56]. Transcriptome analysis shows that Amo012379 and Amo023188 were highly expressed in the small, medium and big fruit stages, while Amo023377 was highly expressed in the small fruit stage (Fig. 3b). These results suggest that Amo012379, Amo023377 and Amo023188 may be largely responsible for the total SUS activities in A. montana fruit, and this makes rapid metabolism of the imported sucrose at the early stage of A. montana fruit development.
Sucrose phosphate synthase (SPS), which is one of the key enzymes in sucrose synthesis, uses fructose 6-phosphate (F6P) and uridine diphosphate (UDP)-glucose as substrates[60]. Moreover, SPS is a key enzyme that controls carbon flux towards sucrose and has been divided into three subfamilies in A. thaliana: subfamilies A, B, and C[60]. We identified three SPS genes (Amo011136, Amo001581, and Amo006575) in A. montana and divided them into three subfamilies based on phylogenetic information (Fig. 3a & Supplemental Table S17). The number of SPS genes in A. montana is less than those in A. thaliana (four SPS genes)[60], wheat (five SPS genes)[61], and apple (five SPS genes)[56]. AtSPSC had little effect on sucrose accumulation in A. thaliana and pear fruit[62]. However, no SPSC gene was observed in A. montana (Fig. 3a). Amo001581 was highly expressed in the small, medium and big fruit stages, with the highest expression at the big fruit stage. Amo011136 showed low expression in medium and big fruit stages. These results suggest that the SPS genes play an important role in sucrose synthesis at the late stage of A. montana fruit (Fig. 3b).
Hexokinase (HK) can catalyse glucose phosphorylation and participates in plant sugar induction and sugar signal transduction[63]. We found four orthologs of HK in the A. montana genome (Fig. 3a & Supplemental Table S16). Amo016670 was an ortholog gene of AtHKL1 and AtHKL2 that belong to HK 'group 3'[63]. Amo020432 had high homology with AtHK3 that belongs to the HK 'group 4'. Amo012946 was homologous to AtHKL3 that belongs to HK 'group 5'. Amo022530 had high homology and shared the same clade as AtHK1 and AtHK2, that is, 'group 6'. Amo022530 was highly expressed in the small, medium and big fruit stages; Amo020432 showed medium expression in the small, medium and big fruit stages; and the expression of these two genes was maximum at the big fruit stage (Fig. 3a). The higher expression levels of HK genes in the late stage of A. montana fruit suggest that they may be related to fast utilization of the glucose released from starch breakdown.
Fructokinase (FRK) can phosphorylate fructose to glucose 6-phosphate (G6P) and fructose 6-phosphate[64]. We identified four FRK genes in the A. montana genome (Fig. 3a & Supplemental Table S16). All FRK genes in A. montana were expressed in small, medium and big fruit stages of A. montana. Moreover, Amo019663 was highly expressed in the small and medium fruit stages of A. montana (Fig. 3b). The higher expression of Amo019663 in the small and medium fruit stages of suggests that it may play an important role in efficient utilization of fructose in young fruit, and fructose accumulation during fruit cell expansion.
In conclusion, we found that most genes encoding key enzymes involved in sugar metabolism were highly expressed during fruit development. At the small and medium fruit stages of fruit development, the genes vAIN, SUS, and FRK were highly expressed (Fig. 3b), enabling the fruit to rapidly metabolize the imported sugars to satisfy the requirements of energy and intermediates for cell division and growth during early development. Eventually, with the decrease in energy and carbon skeleton requirements in fruit development, the expression of these three enzymes decreased (Fig. 3b). The expression of HK and SPS genes increased during the development of fruit, and the peak expression was observed at the big fruit stage of fruit development (Fig. 3b). The decreased expression of FRK (Fig. 3b) shows that less fructose is metabolised and more is available for accumulation during fruit development. Thus, the breakdown of starch at the late stage of fruit development, and up-regulation of sucrose synthesis by SPS contributes significantly to the continuous sugar accumulation in the vacuoles and make the total soluble sugar reach its maximum level at maturity.
Genes encoding key sugar transporters
-
The SWEET family of sugar transporters can be classified into four clades: the first and second clades mainly transport glucose, the third clade mainly transports sucrose, and the fourth clade mainly transports fructose[65]. We identified 14 SWEET genes in A. montana and divided them into four subfamilies based on phylogenetic information (Fig. 3c & Supplemental Table S16). Of these, four genes (Amo015156, Amo011350, Amo002432, and Amo015291) were not expressed in A. montana fruit (Fig. 3d). Moreover, Amo018727 and Amo015644 were highly expressed in small, medium and big fruit stages of A. montana fruit; Amo014723 was highly expressed in small fruit stage; Amo023752, Amo025419, Amo010558, and Amo017066 were highly expressed in medium fruit stage. Amo018727 was clustered with AtSWEET1 (Fig. 3c), which mainly transported glucose[65]. Amo014723 formed a clade with AtSWEET2 (Fig. 3c), which mainly transported 2-deoxyglucose[66]. Amo015644 and Amo012385 formed a clade, and were sisters to AtSWEET16 and AtSWEET17 (Fig. 3c), which mainly transport fructose[67,68]. The decreases of expression level in SWEET genes with fruit development suggest that these genes might not be involved in sucrose accumulation in A. montana fruit towards maturity.
The sucrose transporter (SUT) is mainly responsible for transmembrane transport and distribution of sucrose[56]. We identified three orthologous genes of SUT in the A. montana genome (Fig. 3c & Supplemental Table S16). Amo023590 had high homology with AtSUT3, which belonged to SUC 'group 3' according to Braun & Slewinski[69]. Amo012943 was in an independent clade with AtSUT4 in 'group 4'. Amo010009 formed a clade with other SUT genes of A. thaliana. Both AmoSUTs were expressed in small, medium and big fruit stages, and the expression level was decreased with the development of fruit (Fig. 3d). This result suggests that the SUTs transport sucrose into cytosol from apoplast or vacuole primarily in the early stage of A. montana fruit.
Tonoplast sugar transporters (TMTs) play an essential role in sugar partitioning, immobilisation, and accumulation during fruit development and ripening[70]. We identified five orthologous genes of TMT in A. montana (Fig. 3c & Supplemental Table S16). Amo017632, Amo007630, and Amo010121 shared high similarities with the amino acid sequence of AtTMT1; Amoq009685 and Amo008502 showed high homology with AtTMT2 and AtTMT3 (Fig. 3c). Transcriptome analysis showed that the expression level of Amo007630 decreased with fruit development, whereas those of Amo010121, Amo009685, Amo017632 and Amo008502 were consistent during fruit development. These results indicate that TMT genes may play an important role in the accumulation of fructose and sucrose with the fruit development of A. montana.
The hexose transporter (STP/HXT) is a monosaccharide transporter that can transport hexoses such as glucose, fructose, and mannose across the membrane[71]. We identified 19 orthologs of HXT in A. montana and divided them into seven clades based on phylogenetic information (Fig. 3c & Supplemental Table S16). Of the 19 genes, 16 genes exhibited low or no expression in A. montana fruit, while Amo022648 was highly expressed in small, medium and big fruit stages, and Amo022538 and Amo017585 were highly expressed in the small and medium fruit stages (Fig. 3d). This suggests that the HXT genes may transport the hexoses into vacuole in the early stage of A. montana fruit development.
Vacuolar glucose transporter (vGT) is a hexose transporter of the vacuolar membrane, which plays a vital role in all aspects of plant development[72]. In the present study, we demonstrate two of the AtvGT homologs, and divide them into two clades based on phylogenetic information (Fig. 3c & Supplemental Table S16). Amo002546 is a sister to AtvGT3, and Amo021081 forms a clade with AtvGT1 and AtvGT2 (Fig. 3c). Amo002546 was highly expressed at the small fruit stage, and Amo021081 was highly expressed at the small, medium and big fruit stages. These results indicate that the vGTs mainly translocate the glucose into vacuole in the early stage of A. montana fruit development (Fig. 3d).
The sugar-porter family protein (SFP) is a monosaccharide transporter subfamily[73]. We identified four SFP orthologous genes of A. montana and divided them into four clades based on phylogenetic information (Fig. 3c & Supplemental Table S16). Amo013949 was highly expressed at the small fruit stage, and moderately expressed at the medium and big fruit stages. Amo011107 was moderately expressed at the small, medium and big fruit stages. Amo023173 exhibited a moderate expression in the medium and big fruit stages. Amo013949 was highly expressed in the small fruit stage, whereas Amo013947 showed negligible expression in the small, medium and big fruit stages. Thus, the monosaccharides were mainly accumulated at the early stage of A. montana fruit development (Fig. 3d).
Most sugar transporter genes were initially expressed at high levels, but most of them showed low expression during the later stages of fruit development, indicating that sugar was rapidly introduced into fruits to meet the needs of energy and intermediate products for cell division and growth at the early stage of fruit development.
Fruit softening related metabolism
-
Fruits lose firmness during ripening, and the ripening of fleshy fruit is related to starch degradation or cell wall metabolism[74,75]. The genes involved in these two pathways were investigated in this study. We identified 32 members of eight gene families involved in the starch degradation pathway in the genome of A. montana (Supplemental Table S17). A. montana has more genes related to starch degradation than Eucalyptus grandis (15 genes), Punica granatum (28 genes), and A. thaliana (23 genes), but less than Psidium guajava (44 genes)[74]. The major degradation enzymes in A. montana are β-amylase (BAM) and glucan phosphorylase (PHS), which account for 56.29% of the starch degrading enzymes in A. montana. More members of PHS and BAM were detected in A. montana than in any other plant that we surveyed (Supplemental Table S17). We also identified α-glucosidase (AGL), 4-α-glucanotransferase (DPE), phosphoglucan (PWD), and isoamylase (ISA) in A. montana (Supplemental Table S17), which are other key enzymes in the starch degradation pathway that are not present in eucalyptus, pomegranate, or guava[74]. The expression of most genes related to starch degradation increased with fruit development in A. montana (Supplemental Fig. S12), suggesting that starch degradation plays an important role in fruit softening.
A total of 193 genes encoding ten key enzymes involved in cell wall degradation were identified (Supplemental Table S17). The main cell wall degrading enzymes in A. montana included polygalacturonase (PG), xyloglucan endotransglucosylase (XET), beta-glucosidase (BG), and pectin methylesterase (PME), which account for 72.02% of the starch degrading enzymes in A. montana. Our results showed that several enzymes involved in cell wall degradation were highly expressed in fruits during ripening, although not consistently (Supplemental Fig. S13). Some genes encoding β-galactosidase, pectin methylesterase, endoglucanase, beta-glucosidase, xyloglucan endotransglucosylase, and pectate lyase showed increasingly higher expression during fruit ripening (Supplemental Fig. S13), indicating that cell wall degradation may also be useful in fruit softening in A. montana.
Together, these results indicate that the joint action of cell wall degrading and starch degrading enzymes may result in A. montana fruit softening. Glucose is the major product of starch degradation, and the energy required for producing volatile compounds is generally provided by glucose during fruit ripening[75]. Starch degradation is known to play an important part in the softening of fruits such as banana, persimmon, and guava[74]. Thus, the reference genome of A. montana may contribute to studies on both ripening and softening mechanisms, and the shelf life enhancement of A. montana.
Aroma volatiles in A. montana fruit
-
Aroma significantly contributes to flavour, which directly affects the commercial quality of fruit[76]. To date, several volatile chemicals have been detected in fresh fruits. The lipoxygenase pathway is one of the main pathways for the synthesis of volatile chemicals, such as esters, alcohols, and ketones[77]. We analysed the four key enzymes in the lipoxygenase pathway, namely, alcohol dehydrogenase (ADH), lipoxygenase (LOX), alcohol acyltransferase (AAT), and hydroperoxide lyase (HPL) (Supplemental Table S18).
LOX is a type of non-heme iron-containing dioxygenase, which is ubiquitous in plants and animals, and contributes to fruit aroma[78]. We identified 11 LOX genes in A. montana (Fig. 4). The number of LOX genes in A. montana is greater than those in A. thaliana (six LOX genes)[79], and less than grape (18 LOX genes)[80] and pear (23 LOX genes)[78]. Of the 11 LOX genes, two (Amo016127 and Amo013980) in A. montana were highly expressed during fruit development, and two (Amo015486 and Amo018849) were highly expressed in the medium stage, suggesting that Amo016127 and Amo013980 were involved in the production of C9/C13 volatiles.
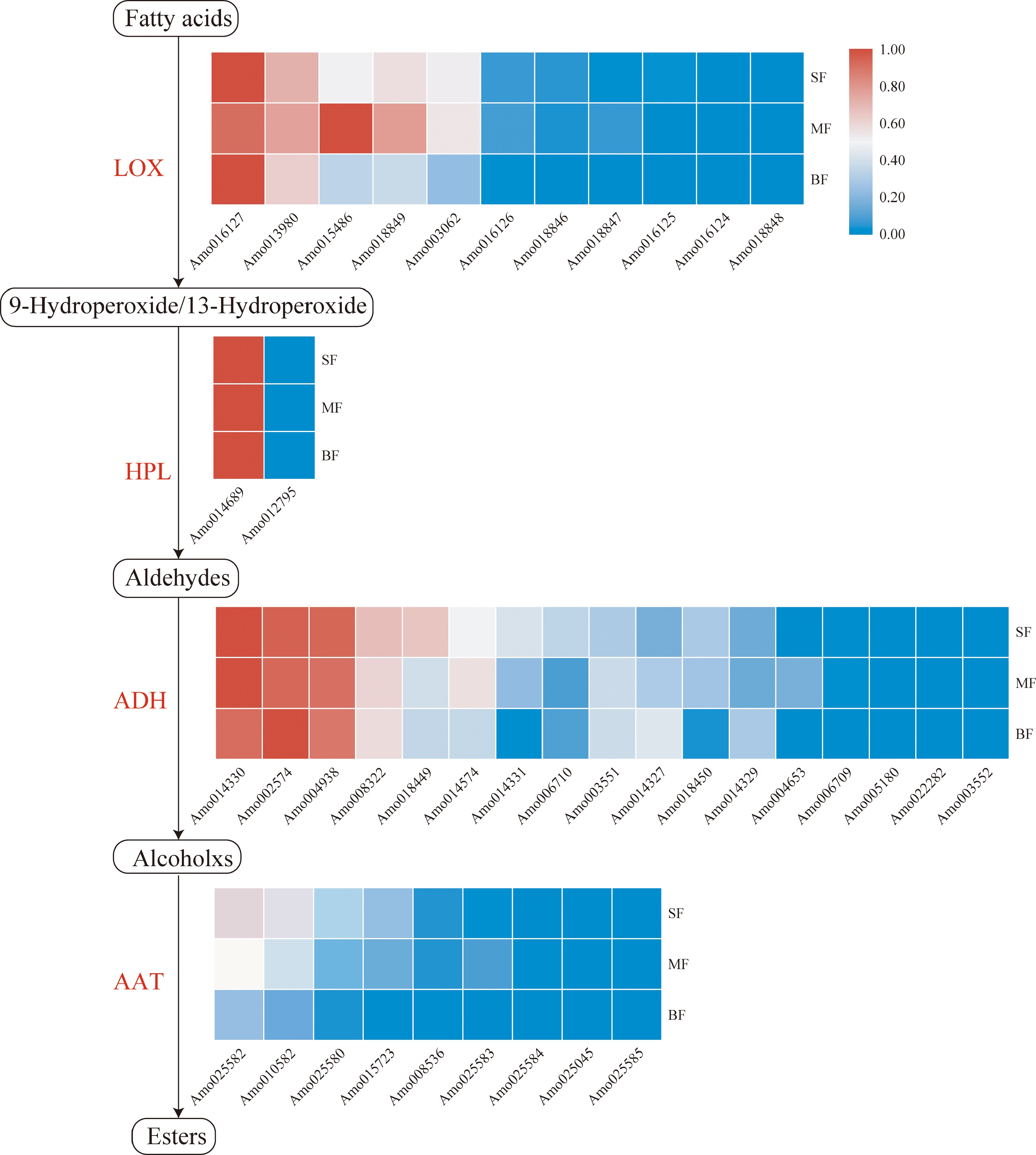
Figure 4.
Fatty acid pathway in A. montana fruit. Tissue-specific relative expression profiles (red-blue scale) of genes implicated in fatty acid pathway (heat map). Intermediates are shown in black, and the enzymes (Supplemental Table S18) involved at each step are shown in red. 'SF' represents the small fruit stage, 'MF' represents medium fruit stage, 'BF' represents big fruit stage.
HPL is an enzyme downstream of the lipoxygenase pathway, and its catalytic product is the main component of volatiles in fruits[81]. We identified two HPL genes (Amo012795 and Amo014689) in A. montana (Fig. 4). Amo014689 was the main gene involved in the production of aldehydes and was highly expressed during fruit ripening.
ADH is a member of the dehydrogenase enzyme superfamily in plants and plays an important role in fruit ripening and aroma production[82]. Seventeen ADH genes were identified in A. montana, which were more than those in melon (13 ADH genes)[81], Glycine max (six ADH genes)[83], Pinus banksiana (seven ADH genes)[84], and rice (two ADH genes)[85]. Of the 17 ADH genes, four genes (Amo014330, Amo002574, Amo004938, and Amo008322) were moderately or strongly expressed in all the fruit developmental stages (Fig. 4), while Amo018449 and Amo014574 showed moderate expression in the small and medium fruit stages, respectively, and seven other genes showed low expression during fruit ripening.
AAT is a key enzyme that controls the biosynthesis of esters, such as ethyl benzoate[86,87]. We identified nine AAT genes in A. montana, which were less than those in Actinidia chinensis (30 AAT genes)[88]. The expression of AAT genes in A. montana fruit decreased during fruit ripening (Fig. 4), suggesting that the volatile esters were not the major aroma components in A. montana.
In the lipoxygenase pathway, the first three enzymes were highly expressed at all developmental stages of A. montana, while the expression of the last enzyme decreased during fruit ripening, which indicates that the volatile alcohols were the major aroma components in A. montana.
-
Annonaceae is one of the important families in magnoliids, presenting morphological diversity with high economic and ornamental value, and distributed widely in tropical and subtropical lowland forests[2−4]. A. montana is popular as guanabana or false graviola due to its similarity with graviola[7]. Genomic data can reveal the evolutionary position of Magnoliales and Annonaceae and facilitate the molecular study of its significant features. Here, we assembled the first high-quality and chromosome-level genome of A. montana in A. montana using PacBio sequencing with Hi-C technology. This genome assembly will be a useful genetic resource for Annonaceae.
The previous studies on Liriodendron chinense[38], Cinnamomum kanehirae[40], Persea americana[89], Phoebe bournei[39] and Litsea cubeba[41], discussed the phylogenetic relationship between magnoliids, monocots and eudicots. The phylogenetic analysis based on single-copy nucleotide and amino acid sequences of this study indicated that A. montana and L. chinense formed a subclade (Magnoliales), which was sister to the subclade formed by C. kanehirae, P. bournei, and P. americana (Laurales). The Bayesian tree supported that magnoliids are sisters to eudicots. Because of ILS during the rapid divergence of early diverging branches in angiosperms, ASTRAL trees showed that magnoliids might also be sisters to monocots−eudicots or monocots. Therefore, this study supports that the magnoliids are sisters to eudicots.
We assembled 947.35 Mb of the A. montana genome and annotated 26,399 protein-coding genes. We identified 46 MADS-box genes, but no FLC subfamily genes were observed in A. montana. We screened out 25 members of the sugar metabolic genes and 47 members of sugar transporter genes in A. montana. The sugar metabolism and accumulation are regulated by sugar metabolic genes and sugar transporter genes during fruit development. Our analysis indicated that the metabolism of sugars in fruits is highly regulated by the developmental process. We recognised 32 genes involving starch degradation and 193 genes encoding ten key enzymes related to cell wall degradation. These genes may be related to the softening of the A. montana fruit. We also mined 39 genes encoding four key enzymes related to the lipoxygenase pathway in A. montana fruit and postulated that alcohols may be the main volatile aromatic compounds in A. montana fruit. The reference-quality A. montana genome sequence will assist in the efforts to conserve genome-wide genetic diversity in the genus Annona and provide a new insight into the fruit development, softening, aroma, and sugar accumulation in A. montana.
Data availability statement
-
Genome sequences were submitted to the National Genomics Data Center (NGDC). Whole genome assemblies were deposited in BioProject/GSA under accession codes PRJNA940321.
This research was jointly funded by The Project of the Forestry Administration of Guangdong Province to Guangda Tang (Grant No. YUE CAI NONG [2019] No. 51; No YUE CAI ZI HUAN [2020] No. 130), The National Natural Science Foundation of China (No. 31870199) to Zhongjian Liu, and the Forestry Peak Discipline Construction Project of Fujian Agriculture and Forestry University (72202200205) to Zhongjian Liu and Siren Lan.
-
The authors declare that they have no conflict of interest.
-
# These authors contributed equally: Guangda Tang, Guizhen Chen, Jianhao Ke
- Supplemental Fig. S1 The karyotype analysis of A. montana.
- Supplemental Fig. S2 Genome size and heterozygosity of A. montana estimated using 17 k-mer distribution.
- Supplemental Fig. S3 High-through chromosome conformation capture (Hi-C) contact data mapped to the genome of A. montana.
- Supplemental Fig. S4 Gene structure prediction results statistics.
- Supplemental Fig. S5 The sequence divergence rate of A. montana genome.
- Supplemental Fig. S6 Comparison of the number of homologous genes between the genomes of 20 species.
- Supplemental Fig. S7 Gene tree of C. kanehirae, L. chinense, and A. montana.
- Supplemental Fig. S8 Phylogenetic tree of MADS-box gene from A. montana, P. bournei, and A. thaliana.
- Supplemental Fig. S9 Pictures of the three stages of fruit development in A. montana genome.
- Supplemental Fig. S10 The Venn diagram of differentially expressed genes related to A. montana fruit development.
- Supplemental Fig. S11 Expression patterns of MADS-box genes in the three stages of A. montana fruit development.
- Supplemental Fig. S12 Expression patterns of starch degradation pathway in A. montana fruit.
- Supplemental Fig. S13 Expression patterns of cell wall degradation in A. montana fruit.
- Supplemental Table S1 The statistics of sequencing raw data from Pacific sequencing.
- Supplemental Table S2 Summary statistics of the final genome assembly of A. montana.
- Supplemental Table S3 Benchmarking Universal Single-Copy Orthologs (BUSCO) assessment of the A. montana assembly genome.
- Supplemental Table S4 The statistics of contig clustering results.
- Supplemental Table S5 Length statistics of seven chromosomes in contig cluster.
- Supplemental Table S6 The prediction of gene structures of A. montana genome.
- Supplemental Table S7 The statistics of functional annotation results of A. montana genome.
- Supplemental Table S8 The statistics of the annotation of non-coding RNAs in the A. montana genome.
- Supplemental Table S9 Core Eukaryotic Genes Mapping Approach (CEGMA) and Benchmarking Universal Single-Copy Orthologs (BUSCO) assessment of the A. montana annotated genome.
- Supplemental Table S10 The statistics result of repeat sequences in A. montana genome.
- Supplemental Table S11 The classification of repeat sequence in A. montana genome.
- Supplemental Table S12 GO enrichment result of significant expansion, contraction, and unique genes of A. montana.
- Supplemental Table S13 KEGG enrichment result of significant expansion, contraction, and unique genes of A. montana.
- Supplemental Table S14 The statistics of the result of clustered gene families in 20 species.
- Supplemental Table S15 MADS-box genes of A. montana genome.
- Supplemental Table S16
- Supplemental Table S16 Sugar metabolic and transporter genes of A. montana genome.
- Supplemental Table S17 Number of genes related to fruit softening in the A. montana genome.
- Supplemental Table S18 Number of predicted genes encoding enzymes involved in volatile chemicals in A. montana genome.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Tang G, Chen G, Ke J, Wang J, Zhang D, et al. 2023. The Annona montana genome reveals the development and flavor formation in mountain soursop fruit. Ornamental Plant Research 3:14 doi: 10.48130/OPR-2023-0014 

The Annona montana genome reveals the development and flavor formation in mountain soursop fruit
- Received: 02 March 2023
- Accepted: 23 May 2023
- Published online: 11 July 2023
Abstract: Annona is a genus of family Annonaceae within the magnoliids and plays a crucial role in revealing the evolution of magnolias. Annona species provide important fruit resources. Here, we report a chromosome-level genome assembly of A. montana, an edible and ornamental fruit species. Integration with other genomes provides clear evidence that the magnoliids were sisters to eudicots, and the ASTRAL trees showed discordance in the phylogenetic position of magnoliids, which might be caused by incomplete lineage sorting (ILS). Whole genome duplication (WGD) analysis showed that the common ancestor of A. montana and Liriodendron chinense experienced a WGD event, and this WGD event occurred after the splitting of Magnoliales and Laurales. We identified the gene family expansions and contractions in Annonaceae. Based on the identification of MADS-box gene families, we inferred the pathway integrators of morphological regulation, the occurrence of florescence and the development of fruit in A. montana. In addition, we identified key sugar transporter genes and the key enzyme genes related to sugar accumulation in A. montana fruit. The gene function analysis indicated that starch and cell wall degradation might be the main reasons for the softening of A. montana fruit. Furthermore, aromatic alcohols were suggested be the main volatile aromatic compounds in A. montana fruit. Our results provide the genetic basis of fruit development, softening, aroma, and sugar accumulation in A. montana and the evolution and diversification of Annonaceae.
-
Key words:
- Annona montana /
- Fruit development /
- Softening /
- Aroma /
- Sugar accumulation