









-
As a major staple crop, today maize accounts for approximately 40% of total worldwide cereal production (
http://faostat.fao.org/ ). Since its domestication ~9,000 years ago from a subgroup of teosinte (Zea mays ssp. parviglumis) in the tropical lowlands of southwest Mexico[1], its cultivating area has greatly expanded, covering most of the world[2]. Human's breeding and utilization of maize have gone through several stages, from landraces, open-pollinated varieties (OPVs), double-cross hybrids (1930s-1950s) and since the middle 1950s, single-cross hybrids. Nowadays, global maize production is mostly provided by single-cross hybrids, which exhibit higher-yielding and better stress tolerance than OPVs and double-cross hybrids[3].Besides its agronomic importance, maize has also been used as a model plant species for genetic studies due to its out-crossing habit, large quantities of seeds produced and the availability of diverse germplasm. The abundant mutants of maize facilitated the development of the first genetic and cytogenetic maps of plants, and made it an ideal plant species to identify regulators of developmental processes[4−6]. Although initially lagging behind other model plant species (such as Arabidopsis and rice) in multi-omics research, the recent rapid development in sequencing and transformation technologies, and various new tools (such as CRISPR technologies, double haploids etc.) are repositioning maize research at the frontiers of plant research, and surely, it will continue to reveal fundamental insights into plant biology, as well as to accelerate molecular breeding for this vitally important crop[7, 8].
-
During domestication from teosinte to maize, a number of distinguishing morphological and physiological changes occurred, including increased apical dominance, reduced glumes, suppression of ear prolificacy, increase in kernel row number, loss of seed shattering, nutritional changes etc.[9] (Fig. 1). At the genomic level, genome-wide genetic diversity was reduced due to a population bottleneck effect, accompanied by directional selection at specific genomic regions underlying agronomically important traits. Over a century ago, Beadle initially proposed that four or five genes or blocks of genes might be responsible for much of the phenotypic changes between maize and teosinte[10,11]. Later studies by Doebley et al. used teosinte–maize F2 populations to dissect several quantitative trait loci (QTL) to the responsible genes (such as tb1 and tga1)[12,13]. On the other hand, based on analysis of single-nucleotide polymorphisms (SNPs) in 774 genes, Wright et al.[14] estimated that 2%−4% of maize genes (~800−1,700 genes genome-wide) were selected during maize domestication and subsequent improvement. Taking advantage of the next-generation sequencing (NGS) technologies, Hufford et al.[15] conducted resequencing analysis of a set of wild relatives, landraces and improved maize varieties, and identified ~500 selective genomic regions during maize domestication. In a recent study, Xu et al.[16] conducted a genome-wide survey of 982 maize inbred lines and 190 teosinte accession. They identified 394 domestication sweeps and 360 adaptation sweeps. Collectively, these studies suggest that maize domestication likely involved hundreds of genomic regions. Nevertheless, much fewer domestication genes have been functionally studied so far.
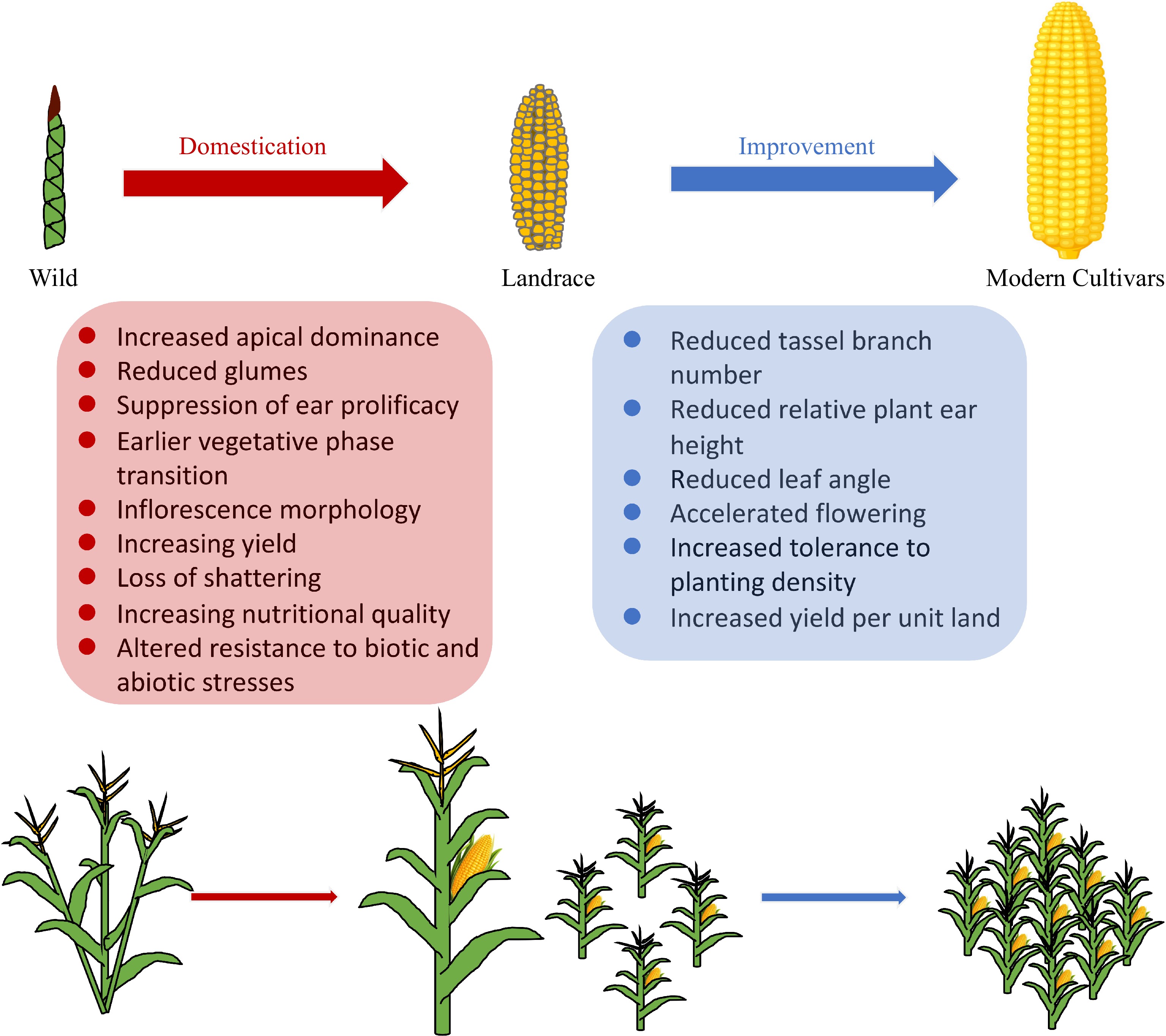
Figure 1.
Main traits of maize involved in domestication and improvement.
-
During maize domestication, a most profound morphological change is an increase in apical dominance, transforming a multi-branched plant architecture in teosinte to a single stalked plant (terminated by a tassel) in maize. The tillers and long branches of teosinte are terminated by tassels and bear many small ears. Similarly, the single maize stalk bears few ears and is terminated by a tassel[9,12,17]. A series of landmark studies by Doebley et al. elegantly demonstrated that tb1, which encodes a TCP transcription factor, is responsible for this transformation[18, 19]. Later studies showed that insertion of a Hopscotch transposon located ~60 kb upstream of tb1 enhances the expression of tb1 in maize, thereby repressing branch outgrowth[20, 21]. Through ChIP-seq and RNA-seq analyses, Dong et al.[22] demonstrated that tb1 acts to regulate multiple phytohormone signaling pathways (gibberellins, abscisic acid and jasmonic acid) and sugar sensing. Moreover, several other domestication loci, including teosinte glume architecture1 (tga1), prol1.1/grassy tillers1, were identified as its putative targets. Elucidating the precise regulatory mechanisms of these loci and signaling pathways will be an interesting and rewarding area of future research. Also worth noting, studies showed that tb1 and its homologous genes in Arabidopsis (Branched1 or BRC1) and rice (FINE CULM1 or FC1) play a conserved role in repressing the outgrowth of axillary branches in both dicotyledon and monocotyledon plants[23, 24].
Teosinte glume architecture 1 (tga1) and reduced glumes in maize
-
Teosinte ears possess two ranks of fruitcase-enclosed kernels, while maize produces hundreds of naked kernels on the ear[13]. tga1, which encodes a squamosa-promoter binding protein (SBP) transcription factor, underlies this transformation[25]. It has been shown that a de novo mutation occurred during maize domestication, causing a single amino acid substitution (Lys to Asn) in the TGA1 protein, altering its binding activity to its target genes, including a group of MADS-box genes that regulate glume identity[26].
Grassy tillers1 (gt1) and suppression of prolificacy in maize
-
Prolificacy, the number of ears per plants, is also a domestication trait. It has been shown that grassy tillers 1 (gt1), which encodes an HD-ZIP I transcription factor, suppresses prolificacy by promoting lateral bud dormancy and suppressing elongation of the later ear branches[27]. The expression of gt1 is induced by shading and requires the activity of tb1, suggesting that gt1 acts downstream of tb1 to mediate the suppressed branching activity in response to shade. Later studies mapped a large effect QTL for prolificacy (prol1.1) to a 2.7 kb 'causative region' upstream of the gt1gene[28]. In addition, a recent study identified a new QTL, qEN7 (for ear number on chromosome 7). Zm00001d020683, which encodes a putative INDETERMINATE DOMAIN (IDD) transcription factor, was identified as the likely candidate gene based on its expression pattern and signature of selection during maize improvement[29]. However, its functionality and regulatory relationship with tb1 and gt1 remain to be elucidated.
UPA2 and leaf angle
-
Smaller leaf angle and thus more compact plant architecture is a desired trait for modern maize varieties. Tian et al.[30] used a maize-teosinte BC2S3 population and cloned two QTLs (Upright Plant Architecture1 and 2 [UPA1 and UPA2]) that regulate leaf angle. Interestingly, the authors showed that the functional variant of UPA2 is a 2-bp InDel located 9.5 kb upstream of ZmRAVL1, which encodes a B3 domain transcription factor. The 2-bp Indel flanks the binding site of the transcription factor Drooping Leaf1 (DRL1)[31], which represses ZmRAVL1 expression through interacting with Liguleless1 (LG1), a SBP-box transcription factor essential for leaf ligule and auricle development[32]. UPA1 encodes brassinosteroid C-6 oxidase1 (brd1), a key enzyme for biosynthesis of active brassinolide (BR). The teosinte-derived allele of UPA2 binds DRL1 more strongly, leading to lower expression of ZmRAVL1 and thus, lower expression of brd1 and BR levels, and ultimately smaller leaf angle. Notably, the authors demonstrated that the teosinte-derived allele of UPA2 confers enhanced yields under high planting densities when introgressed into modern maize varieties[30, 33].
GLOSSY15 (Gl15) and vegetative phase change
-
Maize plants exhibit salient vegetative phase change, which marks the vegetative transition from the juvenile stage to the adult stage, characterized by several changes in maize leaves produced before and after the transition, such as production of leaf epicuticular wax and epidermal hairs. Previous studies reported that Glossy15 (Gl15), which encodes an AP2-like transcription factor, promotes juvenile leaf identity and suppressing adult leaf identity. Ectopic overexpression of Gl15 causes delayed vegetative phase change and flowering, while loss-of-function gl15 mutant displayed earlier vegetative phase change[34]. In another study, Gl15 was identified as a major QTL (qVT9-1) controlling the difference in the vegetative transition between maize and teosinte. Further, it was shown that a pre-existing low-frequency standing variation, SNP2154-G, was selected during domestication and likely represents the causal variation underlying differential expression of Gl15, and thus the difference in the vegetative transition between maize and teosinte[35].
-
A number of studies documented evidence that tassels replace upper ears1 (tru1) is a key regulator of the conversion of the male terminal lateral inflorescence (tassel) in teosinte to a female terminal inflorescence (ear) in maize. tru1 encodes a BTB/POZ ankyrin repeat domain protein, and it is directly targeted by tb1, suggesting their close regulatory relationship[36]. In addition, a number of regulators of maize inflorescence morphology, were also shown as selective targets during maize domestication, including ramosa1 (ra1)[37, 38], which encodes a putative transcription factor repressing inflorescence (the ear and tassel) branching, Zea Agamous-like1 (zagl1)[39], which encodes a MADS-box transcription factor regulating flowering time and ear size, Zea floricaula leafy2 (zfl2, homologue of Arabidopsis Leafy)[40, 41], which likely regulates ear rank number, and barren inflorescence2 (bif2, ortholog of the Arabidopsis serine/threonine kinase PINOID)[42, 43], which regulates the formation of spikelet pair meristems and branch meristems on the tassel. The detailed regulatory networks of these key regulators of maize inflorescence still remain to be further elucidated.
-
Kernel row number (KRN) and kernel weight are two important determinants of maize yield. A number of domestication genes modulating KRN and kernel weight have been identified and cloned, including KRN1, KRN2, KRN4 and qHKW1. KRN4 was mapped to a 3-kb regulatory region located ~60 kb downstream of Unbranched3 (UB3), which encodes a SBP transcription factor and negatively regulates KRN through imparting on multiple hormone signaling pathways (cytokinin, auxin and CLV-WUS)[44, 45]. Studies have also shown that a harbinger TE in the intergenic region and a SNP (S35) in the third exon of UB3 act in an additive fashion to regulate the expression level of UB3 and thus KRN[46].
KRN1 encodes an AP2 transcription factor that pleiotropically affects plant height, spike density and grain size of maize[47], and is allelic to ids1/Ts6 (indeterminate spikelet 1/Tassel seed 6)[48]. Noteworthy, KRN1 is homologous to the wheat domestication gene Q, a major regulator of spike/spikelet morphology and grain threshability in wheat[49].
KRN2 encodes a WD40 domain protein and it negatively regulates kernel row number[50]. Selection in a ~700-bp upstream region (containing the 5’UTR) of KRN2 during domestication resulted in reduced expression and thus increased kernel row number. Interestingly, its orthologous gene in rice, OsKRN2, was shown also a selected gene during rice domestication to negatively regulate secondary panicle branches and thus grain number. These observations suggest convergent selection of yield-related genes occurred during parallel domestication of cereal crops.
qHKW1 is a major QTL for hundred-kernel weight (HKW)[51]. It encodes a CLAVATA1 (CLV1)/BARELY ANY MERISTEM (BAM)-related receptor kinase-like protein positively regulating HKW. A 8.9 Kb insertion in its promoter region was find to enhance its expression, leading to enhanced HKW[52]. In addition, Chen et al.[53] reported cloning of a major QTL for kernel morphology, qKM4.08, which encodes ZmVPS29, a retromer complex component. Sequencing and association analysis revealed that ZmVPS29 was a selective target during maize domestication. They authors also identified two significant polymorphic sites in its promoter region significantly associated with the kernel morphology. Moreover, a strong selective signature was detected in ZmSWEET4c during maize domestication. ZmSWEET4c encodes a hexose transporter protein functioning in sugar transport across the basal endosperm transfer cell layer (BETL) during seed filling[54]. The favorable alleles of these genes could serve as valuable targets for genetic improvement of maize yield.
In a recent effort to more systematically analyze teosinte alleles that could contribute to yield potential of maize, Wang et al.[55] constructed four backcrossed maize-teosinte recombinant inbred line (RIL) populations and conducted detailed phenotyping of 26 agronomic traits under five environmental conditions. They identified 71 QTL associated with 24 plant architecture and yield related traits through inclusive composite interval mapping. Interestingly, they identified Zm00001eb352570 and Zm00001eb352580, both encode ethylene-responsive transcription factors, as two key candidate genes regulating ear height and the ratio of ear to plant height. Chen et al.[56] constructed a teosinte nested association mapping (TeoNAM) population, and performed joint-linkage mapping and GWAS analyses of 22 domestication and agronomic traits. They identified the maize homologue of PROSTRATE GROWTH1, a rice domestication gene controlling the switch from prostrate to erect growth, is also a QTL associated with tillering in teosinte and maize. Additionally, they also detected multiple QTL for days-to-anthesis (such as ZCN8 and ZmMADS69) and other traits (such as tassel branch number and tillering) that could be exploited for maize improvement. These lines of work highlight again the value of mining the vast amounts of superior alleles hidden in teosinte for future maize genetic improvement.
ZmSh1 and seed shattering
-
Loss of seed shattering was also a key trait of maize domestication, like in other cereals. shattering1 (sh1), which encodes a zinc finger and YABBY domain protein regulating seed shattering. Interesting, sh1 was demonstrated to undergo parallel domestication in several cereals, including rice, maize, sorghum, and foxtail millet[57]. Later studies showed that the foxtail millet sh1 gene represses lignin biosynthesis in the abscission layer, and that an 855-bp Harbinger transposable element insertion in sh1 causes loss of seed shattering in foxtail millet[58].
Nutritional quality
-
In addition to morphological traits, a number of physiological and nutritional related traits have also been selected during maize domestication. Based on survey of the nucleotide diversity, Whitt et al.[59] reported that six genes involved in starch metabolism (ae1, bt2, sh1, sh2, su1 and wx1) are selective targets during maize domestication. Palaisa et al.[60] reported selection of the Y1 gene (encoding a phytoene synthase) for increased nutritional value. Karn et al.[61] identified two, three, and six QTLs for starch, protein and oil respectively and showed that teosinte alleles can be exploited for the improvement of kernel composition traits in modern maize germplasm. Fan et at.[62] reported a strong selection imposed on waxy (wx) in the Chinese waxy maize population. Moreover, a recent exciting study reported the identification of a teosinte-derived allele of teosinte high protein 9 (Thp9) conferring increased protein level and nitrogen utilization efficiency (NUE). It was further shown that Thp9 encodes an asparagine synthetase 4 and that incorrect splicing of Thp9-B73 transcripts in temperate maize varieties is responsible for its diminished expression, and thus reduced NUE and protein content[63].
-
Teosintes is known to confer superior disease resistance and adaptation to extreme environments (such as low phosphorus and high salinity). de Lange et al. and Lennon et al.[64−66] reported the identification of teosinte-derived QTLs for resistance to gray leaf spot and southern leaf blight in maize. Mano & Omori reported that teosinte-derived QTLs could confer flooding tolerance[67]. Feng et al.[68] identified four teosinte-derived QTL that could improve resistance to Fusarium ear rot (FER) caused by Fusarium verticillioides. Recently, Wang et al.[69] reported a MYB transcription repressor of teosinte origin (ZmMM1) that confers resistance to northern leaf blight (NLB), southern corn rust (SCR) and gray leaf spot (GLS) in maize, while Zhang et al.[70] reported the identification of an elite allele of SNP947-G ZmHKT1 (encoding a sodium transporter) derived from teosinte can effectively improve salt tolerance via exporting Na+ from the above-ground plant parts. Gao et al.[71] reported that ZmSRO1d-R can regulate the balance between crop yield and drought resistance by increasing the guard cells' ROS level, and it underwent selection during maize domestication and breeding. These studies argue for the need of putting more efforts to tapping into the genetic resources hidden in the maize’s wild relatives. The so far cloned genes involved in maize domestication are summarized in Table 1. Notably, the enrichment of transcription factors in the cloned domestication genes highlights a crucial role of transcriptional re-wiring in maize domestication.
Table 1. Key domestication genes cloned in maize.
Gene Phenotype Functional annotation Selection type Causative change References tb1 Plant architecture TCP transcription factor Increased expression ~60 kb upstream of tb1 enhancing expression [18−22] tga1 Hardened fruitcase SBP-domain transcription factor Protein function A SNP in exon (K-N) [25, 26] gt1 Plant architecture Homeodomain leucine zipper Increased expression prol1.1 in 2.7 kb upstream of the promoter region increasing expression [27, 28] Zm00001d020683 Plant architecture INDETERMINATE DOMAIN transcription factor Protein function Unknown [29] UPA1 Leaf angle Brassinosteroid C-6 oxidase1 Protein function Unknown [30] UPA2 Leaf angle B3 domain transcription factor Increased expression A 2 bp indel in 9.5 kb upstream of ZmRALV1 [30] Gl15 Vegetative phase change AP2-like transcription factor Altered expression SNP2154: a stop codon (G-A) [34, 35] tru1 Plant architecture BTB/POZ ankyrin repeat protein Increased expression Unknown [36] ra1 Inflorescence architecture Transcription factor Altered expression Unknown [37, 38] zfl Plant architecture Transcription factor Altered expression Unknown [40, 41] UB3 Kernel row number SBP-box transcription factor Altered expression A TE in the intergenic region; [44−46] SNP (S35): third exon of UB3
(A-G) increasing expression of UB3 and KRNKRN1/ids1/Ts6 Kernel row number AP2 Transcription factor Increased expression Unknown [47, 48] KRN2 Kernel row number WD40 domain Decreased expression Unknown [50] qHKW1 Kernel row weight CLV1/BAM-related receptor kinase-like protein Increased expression 8.9 kb insertion upstream of HKW [51, 52] ZmVPS29 Kernel morphology A retromer complex component Protein function Two SNPs (S-1830 and S-1558) in the promoter of ZmVPS29 [53] ZmSWEET4c Seed filling Hexose transporter Protein function Unknown [54] ZmSh1 Shattering A zinc finger and YABBY transcription factor Protein function Unknown [57, 58] Thp9 Nutrition quality Asparagine synthetase 4 enzyme Protein function A deletion in 10th intron of Thp9 reducing NUE and protein content [63] ZmMM1 Biotic stress MYB Transcription repressor Protein function Unknown [69] ZmHKT1 Abiotic stress A sodium transporter Protein function SNP947-G: a nonsynonymous variation increasing salt tolerance [70] ZmSRO1d-R Drought resistance and production PolyADP-ribose polymerase and C-terminal RST domain Protein function Three non-synonymous variants: SNP131 (A44G), SNP134 (V45A) and InDel433 [71] -
After its domestication from its wild progenitor teosinte in southwestern Mexico in the tropics, maize has now become the mostly cultivated crop worldwide owing to its extensive range expansion and adaptation to diverse environmental conditions (such as temperature and day length). A key prerequisite for the spread of maize from tropical to temperate regions is reduced photoperiod sensitivity[72]. It was recently shown that CENTRORADIALIS 8 (ZCN8), an Flowering Locus T (FT) homologue, underlies a major quantitative trait locus (qDTA8) for flowering time[73]. Interestingly, it has been shown that step-wise cis-regulatory changes occurred in ZCN8 during maize domestication and post-domestication expansion. SNP-1245 is a target of selection during early maize domestication for latitudinal adaptation, and after its fixation, selection of InDel-2339 (most likely introgressed from Zea mays ssp. Mexicana) likely contributed to the spread of maize from tropical to temperate regions[74].
ZCN8 interacts with the basic leucine zipper transcription factor DLF1 (Delayed flowering 1) to form the florigen activation complex (FAC) in maize. Interestingly, DFL1 was found to underlie qLB7-1, a flowering time QTL identified in a BC2S3 population of maize-teosinte. Moreover, it was shown that DLF1 directly activates ZmMADS4 and ZmMADS67 in the shoot apex to promote floral transition[75]. In addition, ZmMADS69 underlies the flowering time QTL qDTA3-2 and encodes a MADS-box transcription factor. It acts to inhibit the expression of ZmRap2.7, thereby relieving its repression on ZCN8 expression and causing earlier flowering. Population genetic analyses showed that DLF1, ZmMADS67 and ZmMADS69 are all targets of artificial selection and likely contributed to the spread of maize from the tropics to temperate zones[75, 76].
In addition, a few genes regulating the photoperiod pathway and contributing to the acclimation of maize to higher latitudes in North America have been cloned, including Vgt1, ZmCCT (also named ZmCCT10), ZmCCT9 and ZmELF3.1. Vgt1 was shown to act as a cis-regulatory element of ZmRap2.7, and a MITE TE located ~70 kb upstream of Vgt1 was found to be significantly associated with flowering time and was a major target for selection during the expansion of maize to the temperate and high-latitude regions[77−79]. ZmCCT is another major flowering-time QTL and it encodes a CCT-domain protein homologous to rice Ghd7[80]. Its causal variation is a 5122-bp CACTA-like TE inserted ~2.5 kb upstream of ZmCCT10[72, 81]. ZmCCT9 was identified a QTL for days to anthesis (qDTA9). A Harbinger-like TE located ~57 kb upstream of ZmCCT9 showed the most significant association with DTA and thus believed to be the causal variation[82]. Notably, the CATCA-like TE of ZmCCT10 and the Harbinger-like TE of ZmCCT9 are not observed in surveyed teosinte accessions, hinting that they are de novo mutations occurred after the initial domestication of maize[72, 82]. ZmELF3.1 was shown to underlie the flowering time QTL qFT3_218. It was demonstrated that ZmELF3.1 and its homolog ZmELF3.2 can form the maize Evening Complex (EC) through physically interacting with ZmELF4.1/ZmELF4.2, and ZmLUX1/ZmLUX2. Knockout mutants of Zmelf3.1 and Zmelf3.1/3.2 double mutant presented delayed flowering under both long-day and short-day conditions. It was further shown that the maize EC promote flowering through repressing the expression of several known flowering suppressor genes (e.g., ZmCCT9, ZmCCT10, ZmCOL3, ZmPRR37a and ZmPRR73), and consequently alleviating their inhibition on several maize florigen genes (ZCN8, ZCN7 and ZCN12). Insertion of two closely linked retrotransposon elements upstream of the ZmELF3.1 coding region increases the expression of ZmELF3.1, thus promoting flowering[83]. The increase frequencies of the causal TEs in Vgt1, ZmCCT10, ZmCCT9 and ZmELF3.1 in temperate maize compared to tropical maize highlight a critical role of these genes during the spread and adaptation of maize to higher latitudinal temperate regions through promoting flowering under long-day conditions[72,81−83].
In addition, Barnes et al.[84] recently showed that the High Phosphatidyl Choline 1 (HPC1) gene, which encodes a phospholipase A1 enzyme, contributed to the spread of the initially domesticated maize from the warm Mexican southwest to the highlands of Mexico and South America by modulating phosphatidylcholine levels. The Mexicana-derived allele harbors a polymorphism and impaired protein function, leading to accelerated flowering and better fitness in highlands.
Besides the above characterized QTLs and genes, additional genetic elements likely also contributed to the pre-Columbia spreading of maize. Hufford et al.[85] proposed that incorporation of mexicana alleles into maize may helped the expansion of maize to the highlands of central Mexico based on detection of bi-directional gene flow between maize and Mexicana. This proposal was supported by a recent study showing evidence of introgression for over 10% of the maize genome from the mexicana genome[86]. Consistently, Calfee et al.[87] found that sequences of mexicana ancestry increases in high-elevation maize populations, supporting the notion that introgression from mexicana facilitating adaptation of maize to the highland environment. Moreover, a recent study examined the genome-wide genetic diversity of the Zea genus and showed that dozens of flowering-related genes (such as GI, BAS1 and PRR7) are associated with high-latitude adaptation[88]. These studies together demonstrate unequivocally that introgression of genes from Mexicana and selection of genes in the photoperiod pathway contributed to the spread of maize to the temperate regions.
The so far cloned genes involved in pre-Columbia spread of maize are summarized in Fig. 2 and Table 2.
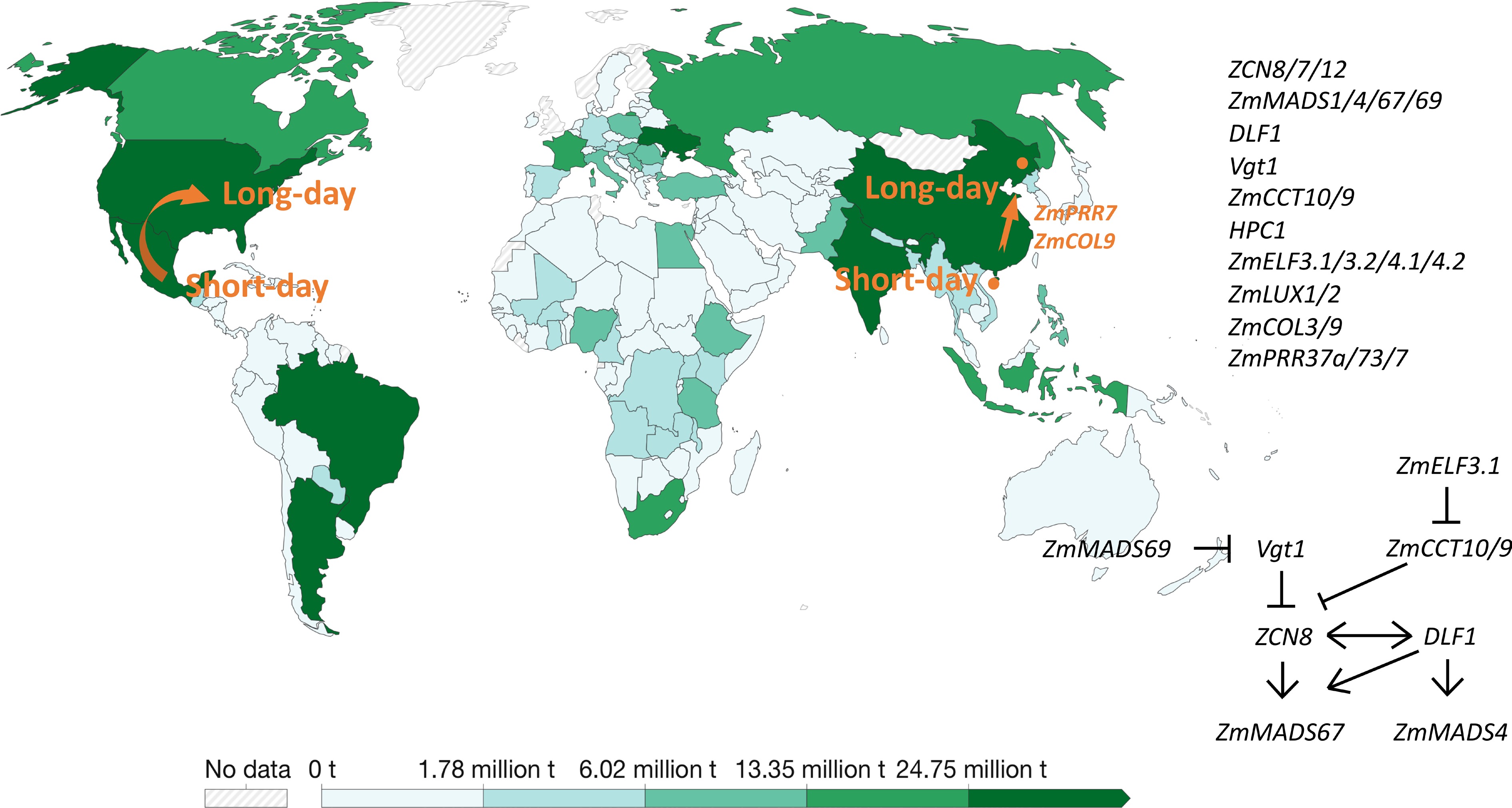
Figure 2.
Genes involved in Pre-Columbia spread of maize to higher latitudes and the temperate regions. The production of world maize in 2020 is presented by the green bar in the map from Ritchie et al. (2023). Ritchie H, Rosado P, and Roser M. 2023. "Agricultural Production". Published online at OurWorldInData.org. Retrieved from: 'https:ourowrldindata.org/agricultural-production' [online Resource].
Table 2. Flowering time related genes contributing to Pre-Columbia spread of maize.
Gene Functional annotation Causative change References ZCN8 Florigen protein SNP-1245 and Indel-2339 in promoter [73, 74] DLF1 Basic leucine zipper transcription factor Unknown [75] ZmMADS69 MADS-box transcription factor Unknown [76] ZmRap2.7 AP2-like transcription factor MITE TE inserted ~70 kb upstream [77−79] ZmCCT CCT-domain protein 5122-bp CACTA-like TE inserted ~2.5 kb upstream [72,81] ZmCCT9 CCT transcription factor A harbinger-like element at 57 kb upstream [82] ZmELF3.1 Unknown wo retrotransposons in the promote [84] HPC1 Phospholipase A1 enzym Unknown [83] ZmPRR7 Unknown Unknown [88] ZmCOL9 CO-like-transcription factor Unknown [88] -
Subsequent to domestication ~9,000 years ago, maize has been continuously subject to human selection during the post-domestication breeding process. Through re-sequencing analysis of 35 improved maize lines, 23 traditional landraces and 17 wild relatives, Hufford et al.[15] identified 484 and 695 selective sweeps during maize domestication and improvement, respectively. Moreover, they found that about a quarter (23%) of domestication sweeps (107) were also selected during improvement, indicating that a substantial portion of the domestication loci underwent continuous selection during post-domestication breeding.
Genetic improvement of maize culminated in the development of high planting density tolerant hybrid maize to increase grain yield per unit land area[89, 90]. To investigate the key morphological traits that have been selected during modern maize breeding, we recently conducted sequencing and phenotypic analyses of 350 elite maize inbred lines widely used in the US and China over the past few decades. We identified four convergently improved morphological traits related to adapting to increased planting density, i.e., reduced leaf angle, reduced tassel branch number (TBN), reduced relative plant height (EH/PH) and accelerated flowering. Genome-wide Association Study (GWAS) identified a total of 166 loci associated with the four selected traits, and found evidence of convergent increases in allele frequency at putatively favorable alleles for the identified loci. Moreover, genome scan using the cross-population composite likelihood ratio approach (XP-CLR) identified a total of 1,888 selective sweeps during modern maize breeding in the US and China. Gene ontology analysis of the 5,356 genes encompassed in the selective sweeps revealed enrichment of genes related to biosynthesis or signaling processes of auxin and other phytohormones, and in responses to light, biotic and abiotic stresses. This study provides a valuable resource for mining genes regulating morphological and physiological traits underlying adaptation to high-density planting[91].
In another study, Li et al.[92] identified ZmPGP1 (ABCB1 or Br2) as a selected target gene during maize domestication and genetic improvement. ZmPGP1 is involved in auxin polar transport, and has been shown to have a pleiotropic effect on plant height, stalk diameter, leaf length, leaf angle, root development and yield. Sequence and phenotypic analyses of ZmPGP1 identified SNP1473 as the most significant variant for kernel length and ear grain weight and that the SNP1473T allele is selected during both the domestication and improvement processes. Moreover, the authors identified a rare allele of ZmPGP1 carrying a 241-bp deletion in the last exon, which results in significantly reduced plant height and ear height and increased stalk diameter and erected leaves, yet no negative effect on yield[93], highlighting a potential utility in breeding high-density tolerant maize cultivars.
-
Shade avoidance syndrome (SAS) is a set of adaptive responses triggered when plants sense a reduction in the red to far-red light (R:FR) ratio under high planting density conditions, commonly manifested by increased plant height (and thus more prone to lodging), suppressed branching, accelerated flowering and reduced resistance to pathogens and pests[94, 95]. High-density planting could also cause extended anthesis-silking interval (ASI), reduced tassel size and smaller ear, and even barrenness[96, 97]. Thus, breeding of maize cultivars of attenuated SAS is a priority for adaptation to increased planting density.
Extensive studies have been performed in Arabidopsis to dissect the regulatory mechanism of SAS and this topic has been recently extensively reviewed[98]. We recently showed that a major signaling mechanism regulating SAS in Arabidopsis is the phytochrome-PIFs module regulates the miR156-SPL module-mediated aging pathway[99]. We proposed that in maize there might be a similar phytochrome-PIFs-miR156-SPL regulatory pathway regulating SAS and that the maize SPL genes could be exploited as valuable targets for genetic improvement of plant architecture tailored for high-density planting[100].
In support of this, it has been shown that the ZmphyBs (ZmphyB1 and ZmphyB2), ZmphyCs (ZmphyC1 and ZmphyC2) and ZmPIFs are involved in regulating SAS in maize[101−103]. In addition, earlier studies have shown that as direct targets of miR156s, three homologous SPL transcription factors, UB2, UB3 and TSH4, regulate multiple agronomic traits including vegetative tillering, plant height, tassel branch number and kernel row number[44, 104]. Moreover, it has been shown that ZmphyBs[101, 105] and ZmPIF3.1[91], ZmPIF4.1[102] and TSH4[91] are selective targets during modern maize breeding (Table 3).
Table 3. Selective genes underpinning genetic improvement during modern maize breeding.
Gene Phenotype Functional annotation Selection type Causative change References ZmPIF3.1 Plant height Basic helix-loop-helix transcription factor Increased expression Unknown [91] TSH4 Tassel branch number Transcription factor Altered expression Unknown [91] ZmPGP1 Plant architecture ATP binding cassette transporter Altered expression A 241 bp deletion in the last exon of ZmPGP1 [92, 93] PhyB2 Light signal Phytochrome B Altered expression A 10 bp deletion in the translation start site [101] ZmPIF4.1 Light signal Basic helix-loop-helix transcription factor Altered expression Unknown [102] ZmKOB1 Grain yield Glycotransferase-like protein Protein function Unknown [121] In a recent study to dissect the signaling process regulating inflorescence development in response to the shade signal, Kong et al.[106] compared the gene expression changes along the male and female inflorescence development under simulated shade treatments and normal light conditions, and identified a large set of genes that are co-regulated by developmental progression and simulated shade treatments. They found that these co-regulated genes are enriched in plant hormone signaling pathways and transcription factors. By network analyses, they found that UB2, UB3 and TSH4 act as a central regulatory node controlling maize inflorescence development in response to shade signal, and their loss-of-function mutants exhibit reduced sensitivity to simulated shade treatments. This study provides a valuable genetic source for mining and manipulating key shading-responsive genes for improved tassel and ear traits under high density planting conditions.
-
Nowadays, global maize production is mostly provided by hybrid maize, which exhibits heterosis (or hybrid vigor) in yields and stress tolerance over open-pollinated varieties[3]. Hybrid maize breeding has gone through several stages, from the 'inbred-hybrid method' stage by Shull[107] and East[108] in the early twentieth century, to the 'double-cross hybrids' stage (1930s−1950s) by Jones[109], and then the 'single-cross hybrids' stage since the 1960s. Since its development, single-cross hybrid was quickly adopted globally due to its superior heterosis and easiness of production[3].
Single-cross maize hybrids are produced from crossing two unrelated parental inbred lines (female × male) belonging to genetically distinct pools of germplasm, called heterotic groups. Heterotic groups allow better exploitation of heterosis, since inter-group hybrids display a higher level of heterosis than intra-group hybrids. A specific pair of female and male heterotic groups expressing pronounced heterosis is termed as a heterotic pattern[110, 111]. Initially, the parental lines were derived from a limited number of key founder inbred lines and empirically classified into different heterotic groups (such as SSS and NSS)[112]. Over time, they have expanded dramatically, accompanied by formation of new 'heterotic groups' (such as Iodent, PA and PB). Nowadays, Stiff Stalk Synthetics (SSS) and PA are generally used as FHGs (female heterotic groups), while Non Stiff Stalk (NSS), PB and Sipingtou (SPT) are generally used as the MHGs (male heterotic groups) in temperate hybrid maize breeding[113].
With the development of molecular biology, various molecular markers, ranging from RFLPs, SSRs, and more recently high-density genome-wide SNP data have been utilized to assign newly developed inbred lines into various heterotic groups, and to guide crosses between heterotic pools to produce the most productive hybrids[114−116]. Multiple studies with molecular markers have suggested that heterotic groups have diverged genetically over time for better heterosis[117−120]. However, there has been a lack of a systematic assessment of the effect and contribution of breeding selection on phenotypic improvement and the underlying genomic changes of FHGs and MHGs for different heterotic patterns on a population scale during modern hybrid maize breeding.
To systematically assess the phenotypic improvement and the underlying genomic changes of FHGs and MHGs during modern hybrid maize breeding, we recently conducted re-sequencing and phenotypic analyses of 21 agronomic traits for a panel of 1,604 modern elite maize lines[121]. Several interesting observations were made: (1) The MHGs experienced more intensive selection than the FMGs during the progression from era I (before the year 2000) to era II (after the year 2000). Significant changes were observed for 18 out of 21 traits in the MHGs, but only 10 of the 21 traits showed significant changes in the FHGs; (2) The MHGs and FHGs experienced both convergent and divergent selection towards different sets of agronomic traits. Both the MHGs and FHGs experienced a decrease in flowering time and an increase in yield and plant architecture related traits, but three traits potentially related to seed dehydration rate were selected in opposite direction in the MHGs and FHGs. GWAS analysis identified 4,329 genes associated with the 21 traits. Consistent with the observed convergent and divergent changes of different traits, we observed convergent increase for the frequencies of favorable alleles for the convergently selected traits in both the MHGs and FHGs, and anti-directional changes for the frequencies of favorable alleles for the oppositely selected traits. These observations highlight a critical contribution of accumulation of favorable alleles to agronomic trait improvement of the parental lines of both FHGs and MHGs during modern maize breeding.
Moreover, FST statistics showed increased genetic differentiation between the respective MHGs and FHGs of the US_SS × US_NSS and PA × SPT heterotic patterns from era I to era II. Further, we detected significant positive correlations between the number of accumulated heterozygous superior alleles of the differentiated genes with increased grain yield per plant and better parent heterosis, supporting a role of the differentiated genes in promoting maize heterosis. Further, mutational and overexpressional studies demonstrated a role of ZmKOB1, which encodes a putative glycotransferase, in promoting grain yield[121]. While this study complemented earlier studies on maize domestication and variation maps in maize, a pitfall of this study is that variation is limited to SNP polymorphisms. Further exploitation of more variants (Indels, PAVs, CNVs etc.) in the historical maize panel will greatly deepen our understanding of the impact of artificial selection on the maize genome, and identify valuable new targets for genetic improvement of maize.
-
The ever-increasing worldwide population and anticipated climate deterioration pose a great challenge to global food security and call for more effective and precise breeding methods for crops. To accommodate the projected population increase in the next 30 years, it is estimated that cereal production needs to increase at least 70% by 2050 (FAO). As a staple cereal crop, breeding of maize cultivars that are not only high-yielding and with superior quality, but also resilient to environmental stresses, is essential to meet this demand. The recent advances in genome sequencing, genotyping and phenotyping technologies, generation of multi-omics data (including genomic, phenomic, epigenomic, transcriptomic, proteomic, and metabolomic data), creation of novel superior alleles by genome editing, development of more efficient double haploid technologies, integrating with machine learning and artificial intelligence are ushering the transition of maize breeding from the Breeding 3.0 stage (biological breeding) into the Breeding 4.0 stage (intelligent breeding)[122, 123]. However, several major challenges remain to be effectively tackled before such a transition could be implemented. First, most agronomic traits of maize are controlled by numerous small-effect QTL and complex genotype-environment interactions (G × E). Thus, elucidating the contribution of the abundant genetic variation in the maize population to phenotypic plasticity remains a major challenge in the post-genomic era of maize genetics and breeding. Secondly, most maize cultivars cultivated nowadays are hybrids that exhibit superior heterosis than their parental lines. Hybrid maize breeding involves the development of elite inbred lines with high general combining ability (GCA) and specific combining ability (SCA) that allows maximal exploitation of heterosis. Despite much effort to dissect the mechanisms of maize heterosis, the molecular basis of maize heterosis is still a debated topic[124−126]. Thirdly, only limited maize germplasm is amenable to genetic manipulation (genetic transformation, genome editing etc.), which significantly hinders the efficiency of genetic improvement. Development of efficient genotype-independent transformation procedure will greatly boost maize functional genomic research and breeding. Noteworthy, the Smart Corn System recently launched by Bayer is promised to revolutionize global corn production in the coming years. At the heart of the new system is short stature hybrid corn (~30%−40% shorter than traditional hybrids), which offers several advantages: sturdier stems and exceptional lodging resistance under higher planting densities (grow 20%−30% more plants per hectare), higher and more stable yield production per unit land area, easier management and application of plant protection products, better use of solar energy, water and other natural resources, and improved greenhouse gas footprint[127]. Indeed, a new age of maize green revolution is yet to come!
This work was supported by grants from the Key Research and Development Program of Guangdong Province (2022B0202060005), National Natural Science Foundation of China (32130077) and Hainan Yazhou Bay Seed Lab (B21HJ8101). We thank Professors Hai Wang (China Agricultural University) and Jinshun Zhong (South China Agricultural University) for valuable comments and helpful discussion on the manuscript. We apologize to authors whose excellent work could not be cited due to space limitations.
-
The authors declare that they have no conflict of interest. Haiyang Wang is an Editorial Board member of Seed Biology who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and his research groups.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press on behalf of Hainan Yazhou Bay Seed Laboratory. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang M, Kong D, Wang H. 2023. Genomic landscape of maize domestication and breeding improvement. Seed Biology 2:9 doi: 10.48130/SeedBio-2023-0009 

Genomic landscape of maize domestication and breeding improvement
- Received: 20 January 2023
- Accepted: 22 May 2023
- Published online: 03 August 2023
Abstract: Maize (Zea mays ssp. mays) is the most productive crop worldwide now, and it is widely used as food, feed and raw materials for various industrial products. The continuous increase of maize yield is a testament of the success of plant breeding and modern agriculture. During domestication and historical breeding, humans has imposed strong selection on its morphological and physiological traits that benefit ecological adaptation, increase in yield and nutritional value, and harvesting. Recent advance in maize functional genomics studies has greatly deepened and expanded our understanding of the molecular and genetic bases of maize domestication and genetic improvement. In this article, we summarize the key traits and regulatory genes that underlie domestication and post-domestication genetic improvement of maize, and provide a forward outlook as to how the knowledge can be harnessed to accelerate future maize breeding.
-
Key words:
- Maize /
- Domestication /
- Improvement /
- Molecular bases /
- Genetic variants