










-
Jasmonates (JAs) are a class of lipid-derived phytohormones that play essential roles in plant growth, development, stress responses, and secondary metabolism regulation[1]. The JA signaling pathway modulates plant physiological processes through a cascade of molecular interactions, with JAZ (Jasmonate ZIM-domain) proteins acting as key repressors[2,3]. In the absence of JA signals, JAZ proteins interact with transcription factors such as MYC2, suppressing the expression of downstream genes[3]. Upon JA perception, JAZ proteins undergo ubiquitination and degradation via the 26S proteasome pathway, releasing MYC2 and other transcription factors to activate gene expression[4].
JAZ proteins belong to the larger Threonine-Isoleucine-Phenylalanine-Tyrosine (TIFY) superfamily, which also includes the TIFY, PPD, and ZML subfamilies[5,6]. JAZ proteins are characterized by two conserved domains: the ZIM domain and the Jas domain. The ZIM domain contains a conserved TIFY motif (TIF[F/Y]XG), essential for forming protein dimers and interacting with transcription factors such as MYC2[7]. The Jas domain, located at the protein's C-terminal end, has a conserved sequence (SLX2FX2KRX2RX5PY) critical for transcription factor binding[8−10]. Additionally, the TIFY domain interacts with the NINJA protein, collectively repressing JA signaling[11].
Gene family size and subfamily conservation differences among species are valuable indicators of plant evolutionary histories and environmental adaptations. Although copy number variations (CNVs) and gene presence/absence variants (PAVs) are common findings in pan-genomic studies, most genome-wide analyses of gene families remain restricted to single reference genomes[12]. Such approaches overlook population-level genetic diversity, including gene PAVs and CNVs. Recent pan-genomic studies in plants have demonstrated that these variations are widespread, categorizing genes into core (strictly conserved across all individuals) and variable (non-essential, absent in some individuals), classifications considered biologically meaningful. Nevertheless, comprehensive pan-genomic analyses of specific gene families, such as JAZ, remain rare.
Tea, sourced from Camellia sinensis, is one of the most widely consumed non-alcoholic beverages worldwide, conferring significant economic, health, and cultural value[13]. The distinctive quality and bioactive properties of tea are largely attributed to specialized metabolites such as polyphenols and caffeine. Recent studies have shown that jasmonic acid signaling, mediated by the JAZ gene family, plays a central role in regulating the biosynthesis of these key compounds, with jasmonic acid treatments markedly enhancing tea polyphenol and caffeine content by inducing the expression of specific genes[14,15]. Furthermore, members of the CsJAZ family display dynamic expression patterns in response to various hormonal signals (e.g., jasmonic acid, abscisic acid, gibberellin) and environmental stresses (e.g., drought, salt), highlighting their importance in tea plant growth and adaptation[16]. Despite these advances, current understanding of JAZ gene family evolution, diversity, and function in tea plants remains limited. Notably, traditional analyses based on a single reference genome often overlook gene presence-absence variations (PAVs) present across diverse genetic backgrounds. Therefore, a systematic pan-genome analysis is essential to comprehensively characterize the JAZ gene family in tea, address existing knowledge gaps, and elucidate their roles in shaping tea quality and environmental resilience.
In this study, a pan-genome-level analysis of the JAZ gene family was conducted using 22 published high-quality tea genomes. The identified JAZs were categorized into core (present in all tea plant cultivars), near-core (present in all but one or two cultivars), dispensable (present in two to twenty cultivars), and private (present only in a single variety) groups based on their distribution. Additionally, structural variations (SVs) and their impacts on gene structure, conserved domains, and protein structure were assessed. Using multi-tissue RNA-seq data, this study further explored the effects of these variations on gene expression to elucidate their evolutionary dynamics and functional significance.
-
The 22 tea plant genomes were obtained from a study by Chen et al.[12]. Arabidopsis thaliana JAZ protein sequences from A. thaliana were downloaded from the Arabidopsis Information Resource (TAIR,
www.arabidopsis.org , accessed on 10 June 2023). The hidden Markov model (HMM) profiles of the tify domain (PF06200) and Jas_motif (PF09425) were retrieved from the Pfam database (www.ebi.ac.uk/interpro/entry/pfam , accessed on 10 June 2023). JAZ domains were searched using HMMER 3.3.2, with an E-value threshold of < 1e-5[17,18]. A BLASTP search was performed using 12 AtJAZ protein sequences against 22 tea plant genomes to identify JAZs in tea plants. The candidate genes were screened out further through BLAST and HMMER double screening to identify CsJAZ family members. The identified sequences of candidate genes were further verified for the presence of conserved domains using the NCBI Conserved Domain Database (www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi , accessed on 28 July 2023) and SMART (http://smart.embl-heidelberg.de , accessed on 28 July 2023). Finally, proteins containing the tify domain and Jas_motif t were considered JAZ members. CsJAZs was categorized into core genes (present on all tea cultivar genomes), near-core genes (> 90%), dispensable genes (5%−90%), and private genes (< 5%) following the direction of Sun et al.[19]. The predicted isoelectric points (pI) and molecular weight (MW) of the CsJAZs were predicted using the tool from the ExPasy website (https://web.expasy.org/protparam ).Phylogenetic analysis and presence/absence variation of the CsJAZ family
-
The protein sequences of the JAZs from Arabidopsis and C. sinensis were used for phylogenetic analysis. Multiple sequence alignments were performed using MAFFT v7.490[20], and the maximum likelihood method (ML) phylogenetic tree was constructed using RAxML software[21]. Finally, the ML phylogenetic tree was uploaded to the iTOL (
https://itol.embl.de ) online website for optimization[22]. Using Rscript and the R package pheatmap, heatmaps were used to describe the presence/absence of each JAZ in 22 accessions[23].Ka/Ks calculation of the CsJAZs family
-
The amino acid sequence and CDS coding sequence of CsJAZs were downloaded from (
www.tea-pangenome.cn/download )[16], and the CDS sequence of AtJAZs was downloaded from (www.arabidopsis.org ). The CDS sequence of AtJAZ was used as a reference to compare with 22 tea plant cultivars. JAZ by diamond, data processing, and calculation of synonymous substitution rates (Ks) values were performed using the WGD program, and visualization was performed using the R packages ggridges and ggplot2 to plot peak ridge maps[24]. The CsJAZs protein sequence was compared with the reference genomic protein sequence ('Mingke 1' [MK1]) in both directions by BLAST, and TBtools calculated the Ka/Ks values of CsJAZs[25]. Finally, the heat map was plotted using the R package pheatmap.Analysis of the gene structure, conserved and structural domains of TPS under the influence of SV of CsJAZs
-
Multiple sequence comparisons of CsJAZs were performed using MUSCLE to obtain the position of SVs in each species, and the results were visualized using the R package ggmsa. The conserved motif analysis of CsJAZs sequences was performed using the MEME program[26]. The parameters were set as follows: the number of occurrences of each functional domain in each sequence was variable, and the maximum number of motifs was set to ten. The gene structure and conserved motifs of CsJAZ family members were visualized using the TBtools subroutine 'Gene Structure Viewer' using the files generated in the previous step and the GFF file[25].
Analysis of protein tertiary structure of JAZs under the influence of SV
-
The SV-containing CsJAZs protein sequences were uploaded to the Interpro database (
www.ebi.ac.uk/interpro/search/sequence ) to obtain structural domain information. To further analyze the three-dimensional structural changes under the influence of SV, via the AlphaFold2 online toolhttps://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb ) to analyze the 3D structure of CsJAZs[27]. The sequences with pLDDT scores greater than 90 were considered to be approximations of the true structure, and the sequences with pLDDT > 90 were extracted using an in-house Perl script and finally visualized by PyMOL.RNA-seq data analysis
-
RNA-seq data for tea plants were obtained from Chen et al.[12]. After quality control of the raw data using fastp software, all clean reads were mapped to the C. sinensis genome using hisat2 software with default parameters. Gene expression levels were quantified by calculating transcripts per million (TPM) values using featureCount. The CsJAZs expression in different tissues was visualized using the R package pheatmap with log10 (TPM + 1).
-
A pan-genome-wide analysis was performed to identify JAZ family members across 22 tea plant genomes using BLASTP searches and conserved domain validations (Pfam, CDD, and SMART). A total of 21 CsJAZs were identified and classified as two core genes (present in all 22 genomes), three near-core genes (present in 20–21 genomes), ten dispensable genes (present in 2–19 genomes), and six private genes (unique to single genomes) (Supplementary Table S1). The predicted CsJAZs ranged from 137 to 451 amino acids in length (mean: 292 residues), with molecular weights varying from 15.6 to 48.2 kDa (mean: 31.3 kDa) and isoelectric points (pI) between 4.66 and 10.57 (mean: 8.56) (Supplementary Table S2). Based on comparisons with JAZs protein structure in A. thaliana (AtJAZs) classification[5], CsJAZs were assigned to five groups: group I and III contained a single member each, group II had two members, group IV comprised four members, and group V was the largest, with thirteen members (Fig. 1a). Notably, six private genes (CsJAZ16–CsJAZ21) were uniquely detected in single tea plant cultivars (C. sinensis var. assamica (CSA) 'Guihong 3' [GH3], 'Yinghong 9' [YH9], C. sinensis var. sinensis (CSS) 'Jiaming 1" [JM1], 'Longjing 43' [LJ43], and 'Zhuyeqi' [ZYQ]), suggesting potential contributions to cultivar-specific traits (Fig. 1b).
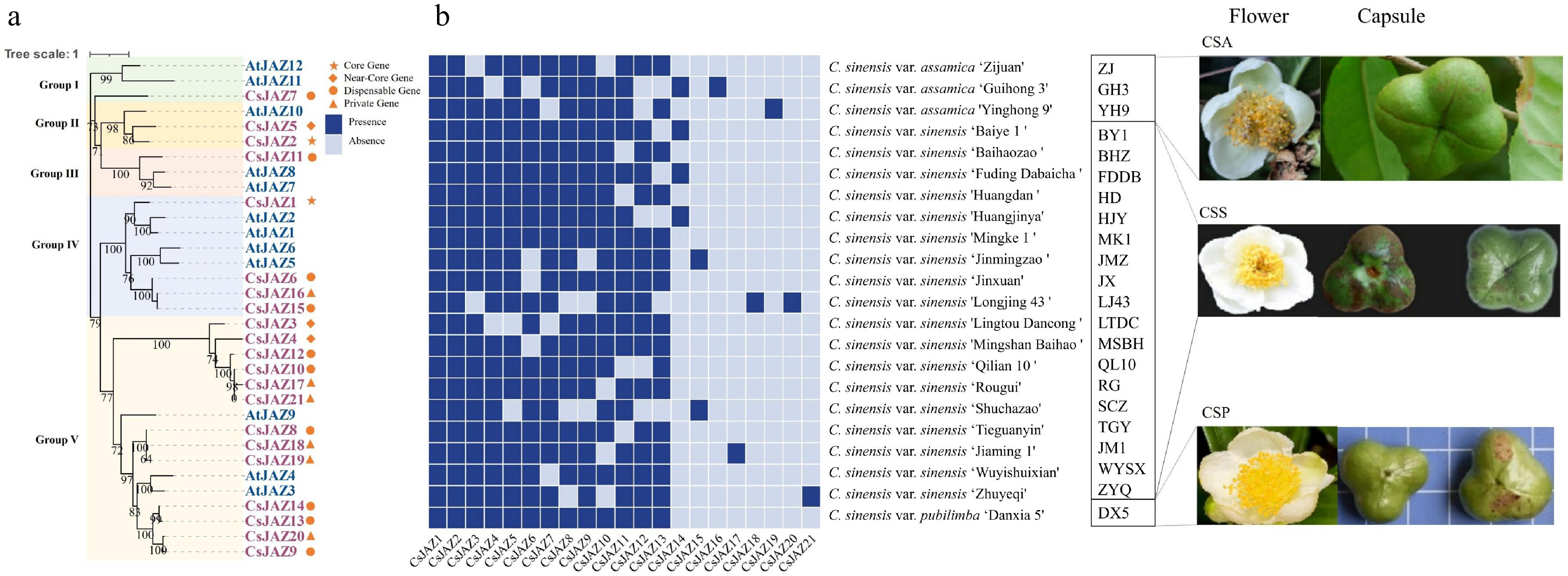
Figure 1.
(a) Phylogenetic tree of JAZ gene family in tea plant and Arabidopsis. (b) Heatmap of the presence and absence of 21 JAZs in 22 tea plant cultivars.
Selection pressures acting on CsJAZs
-
To understand the selective pressures influencing CsJAZs evolution, the synonymous substitution rate (Ks) and the ratio of non-synonymous to synonymous substitutions (Ka/Ks) were analyzed across 22 tea plant cultivars. Ks distribution revealed two distinct peaks across all cultivars, indicating two recent gene duplication events specific to the JAZ family (Fig. 2a). Cultivars 'Baihaozao' (BHZ) and 'Huangdan' (HD) exhibited smaller Ks values for the most recent duplication event, suggesting these duplications occurred more recently. Additionally, 15 cultivars—C. sinensis var. sinensis 'Baiye 1' (BY1), 'BHZ', 'HD', 'Lingtou Dancong' (LTDC), 'Huangjinya' (HJY), 'MK1', 'Fuding Dabaicha' (FDDB), 'Jinxuan' (JX), 'Longjing 43' (LJ43), 'Mingshan Baihao' (MSBH), 'Rougui' (RG), 'Zhuyeqi' (ZYQ), 'Shuchazao' (SCZ), 'Tieguanyin' (TGY), and 'JM1'—had notably shorter intervals between the two duplication peaks compared to the remaining seven cultivars. Further, most CsJAZs exhibited Ka/Ks ratios below 1.0, indicating that they are likely subject to purifying selection, which maintains sequence conservation by removing deleterious mutations. In contrast, CsJAZ1 (in 'FDDB' and 'MSBH'), CsJAZ8 (in 'YH9', C. sinensis var. pubilimba 'Danxia 5' [DX5], 'BY1', 'JM1', 'LJ43', and C. sinensis var. sinensis 'Qilan 10' [QL10]), and CsJAZ9 (in 'JM1' and 'LJ43') — displayed Ka/Ks values greater than 1.0, which may suggest either positive selection or relaxed selective constraints during tea domestication. To further assess these signals, PAML branch-site model analyses were performed on CsJAZ1, CsJAZ8, and CsJAZ9. However, these analyses did not yield significant likelihood ratio test statistics nor identify any codons under positive selection using BEB analysis. Thus, robust evidence for adaptive evolution in these genes was not obtained from the branch-site model analysis (Fig. 2b; Supplementary Table S3).
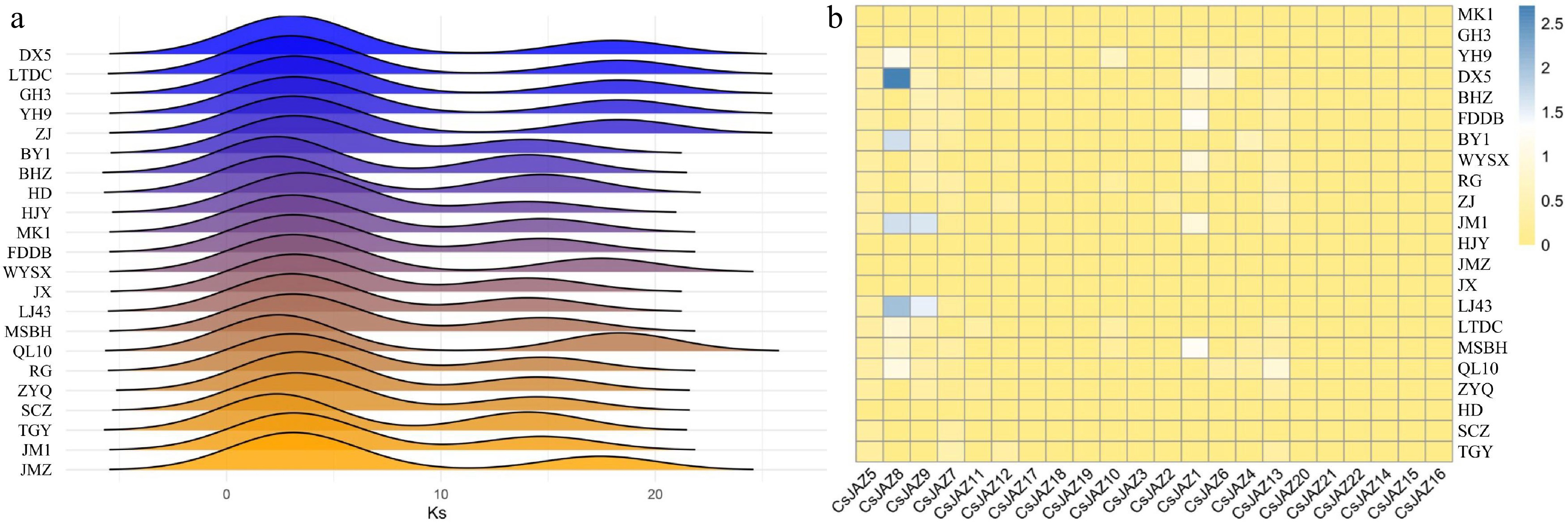
Figure 2.
Ka/Ks values of CsJAZ. (a) Distribution of Ks values of CsJAZ in 22 tea plant cultivars. (b) Heatmap of the frequency of occurrence of different tea plant cultivars at each JAZ.
SVs influence different CsJAZ gene structures and expression
-
Three SV-affected genes were examined: CsJAZ4, CsJAZ9, and CsJAZ12, comparing their sequences across representative tea plant varieties (CSS, CSA, var. pubilimba) (Figs 3, 4; Supplementary Figs S1−S3). CsJAZ4 exhibited a 77-bp insertion in cultivars 'Zijuan' (ZJ), 'DX5', and 'MK1' relative to the reference cultivar 'FDDB', resulting in significant expression changes in 'ZJ' (Fig. 3b). CsJAZ9 had a 315-bp deletion in 'ZJ', 'JX', and 'MK1' relative to 'DX5', correlating with elevated expression in 'DX5' (Fig. 3c). CsJAZ12 showed deletions of 86 and 58 bp in 'LTDC', 'DX5', and 'MK1' relative to 'ZJ', leading to higher expression in 'ZJ' compared to deletion cultivars (Fig. 3d). These findings underscore that SVs significantly impact gene expression patterns of CsJAZ9 and CsJAZ12.
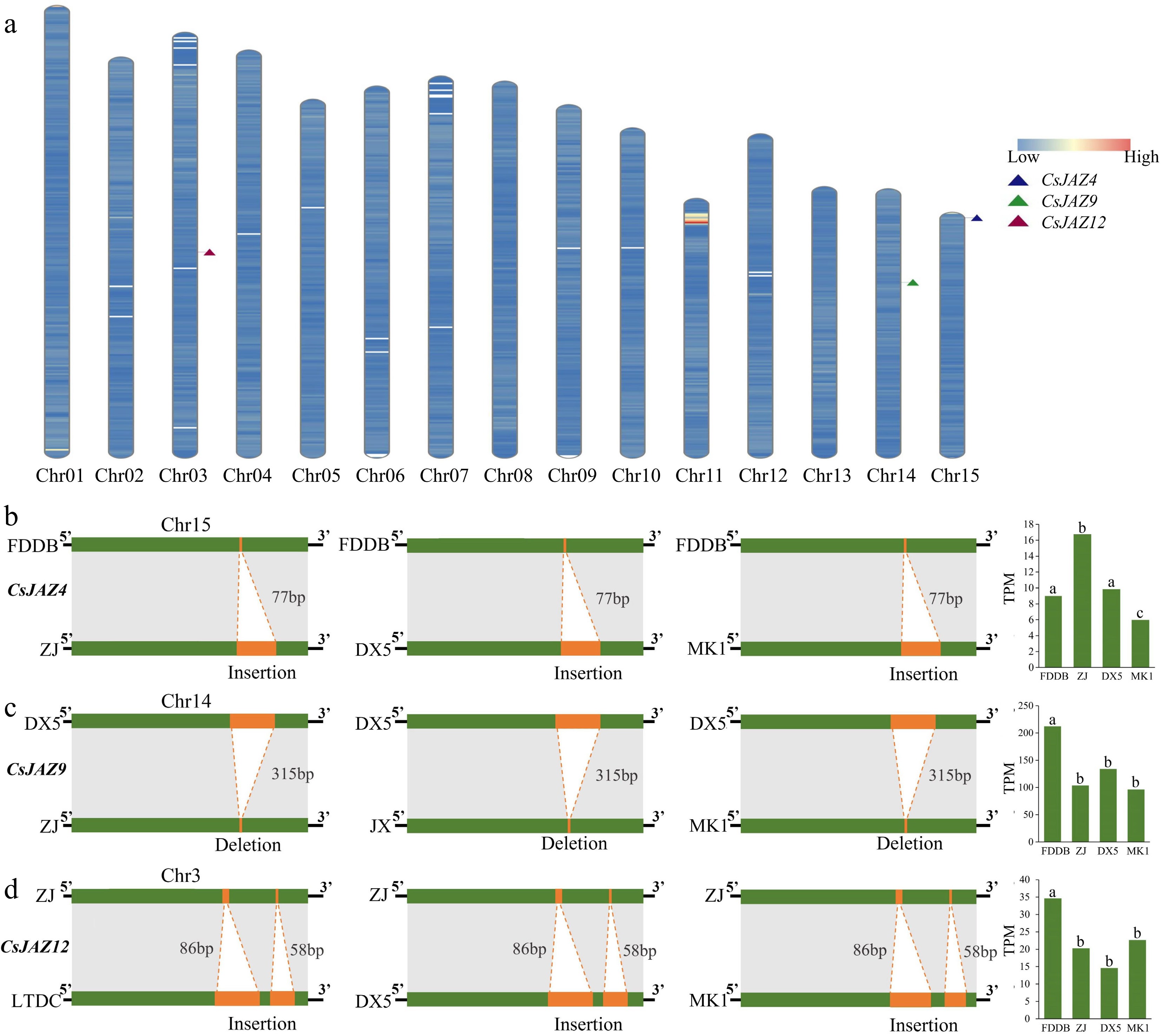
Figure 3.
Effect of SVs on genes. (a) Location of genes on chromosomes, using the 'MK1' genome as a template. (b) Effect of SV insertion and deletion on CsJAZ4 and significant effect of SVs on the expression of JAZ4 in different tea plant cultivars. (c) Effect of SV insertion and deletion on CsJAZ9 and significant effect of SVs on the expression of JAZ9 in different tea plant cultivars. (d) Effect of SV insertion and deletion on CsJAZ12 and significant effect of SVs on the expression of JAZ12 in different tea plant cultivars. The bar graph shows the TPM expression matrix of tea plant leaves.
Compared to the reference cultivar 'FDDB', CsJAZ4 contained a 77-bp insertion in cultivars 'ZJ', 'DX5', and 'MK1', coinciding with significant differences in gene expression levels in 'ZJ' and 'MK1' relative to 'FDDB' (Fig. 4a). Similarly, CsJAZ9 exhibited a 315-bp deletion in 'ZJ', 'JX', and 'MK1' compared to the reference 'DX5', potentially explaining its higher expression in 'DX5' (Fig. 4b). For CsJAZ12, deletions of 86 and 58 bp occurred in 'LTDC', 'DX5', and 'MK1' compared to the reference 'ZJ', correlating with elevated expression observed in 'ZJ' (Fig. 4c). Together, these results indicate that structural variations substantially shape expression patterns of these CsJAZs.
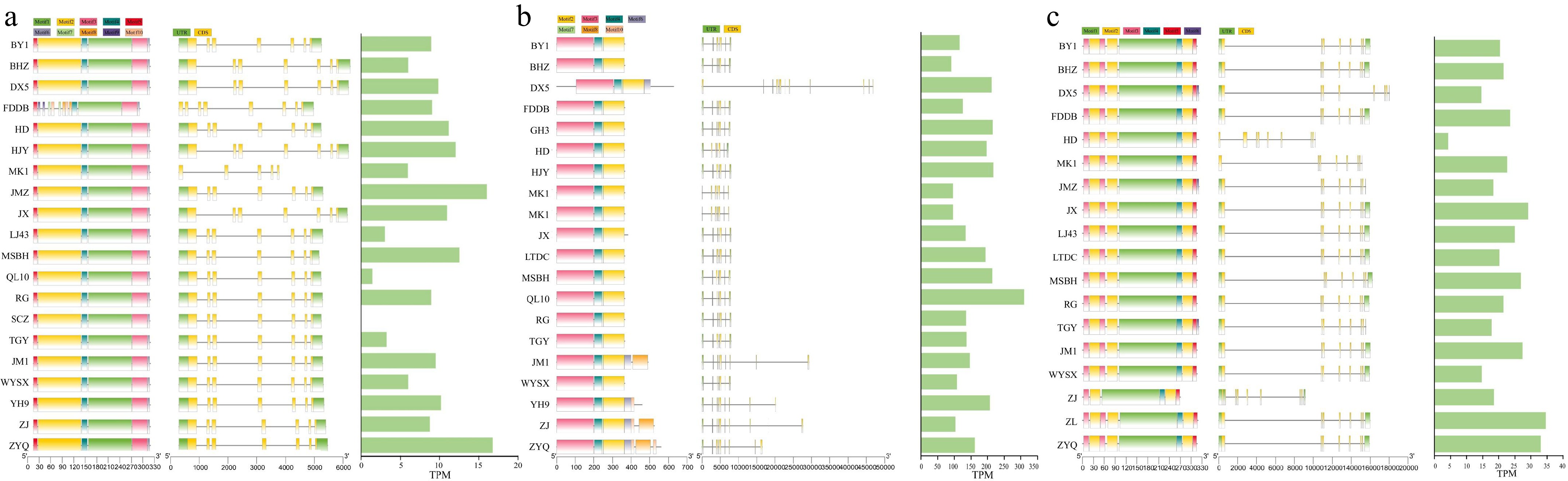
Figure 4.
Gene structure and TPM values in 22 tea plant genomes. (a) Gene structure and TPM values of CsJAZ4 in 22 tea plant genomes. (b) Gene structure and TPM values of CsJAZ9 in 22 tea plant genomes. (c) Gene structure and TPM values of CsJAZ12 in 22 tea plant genomes. TPM values represent transcript abundance in leaf tissue, which was used as a representative condition for assessing expression variation across accessions, where 'QL10' and 'SCZ' are mixed tissues (containing leaves).
Effects of SVs on CsJAZ protein structure
-
To assess how SVs influence protein conformation, the three-dimensional structures of CsJAZ4, CsJAZ9, and CsJAZ12 were predicted (using cultivar 'MK1' as reference) with AlphaFold2. This method assigns each amino acid residue a confidence score (pLDDT: 0–100), with values above 90 indicating highly reliable structural predictions. Regions scoring above 90 overlapped substantially with conserved functional domains (Fig. 5). Specifically, CsJAZ4 and CsJAZ12 each contained the conserved CCT, ZnF-GATA, and Tify domains, whereas CsJAZ9 included the conserved Tify and Jas-motif domains. The presence of these intact core domains suggests that, despite SV-induced sequence alterations, the key structural features of these CsJAZs are likely maintained, though their functional integrity requires further experimental validation.
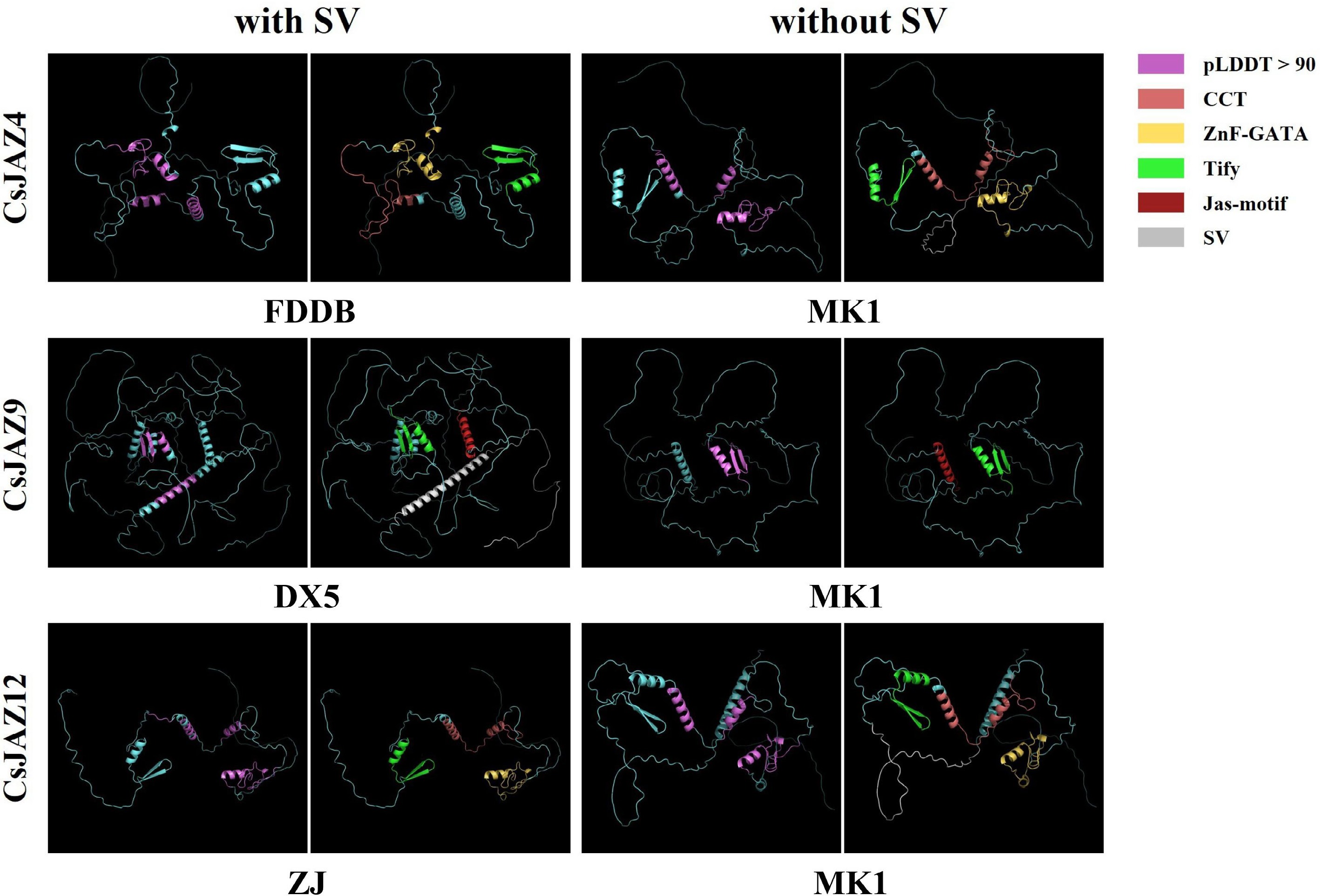
Figure 5.
Protein tertiary structure of JAZs under the influence of SV. Purple represents pLDDT > 90 indicating that the residue has a very high model confidence; pink represents the CCT motif, yellow represents the ZnF-GATA, green represents the Tify domain, red represents the Jas-motif domain, grey represents SV, and blue represents the amino acid sequence.
Transcriptome analyses and evolutionary patterns of CsJAZs
-
Transcriptome sequencing data were analyzed across four tissues (flower, root, stem, leaf) in 22 tea plant cultivars to characterize gene expression. Expression analysis confirmed the transcription of over half of the identified CsJAZs in at least one tissue (Fig. 6a), supporting the accuracy of our pan-genome predictions. Clustering analyses grouped genes according to their expression profiles: genes highly expressed in all tissues, genes uniformly low or absent across tissues, and genes lacking expression, specifically in flowers, formed distinct clusters. Among the most consistently expressed genes were CsJAZ1, CsJAZ2, CsJAZ6, CsJAZ9, CsJAZ13, and CsJAZ14, highlighting their potential roles as core regulatory genes across diverse tissues.
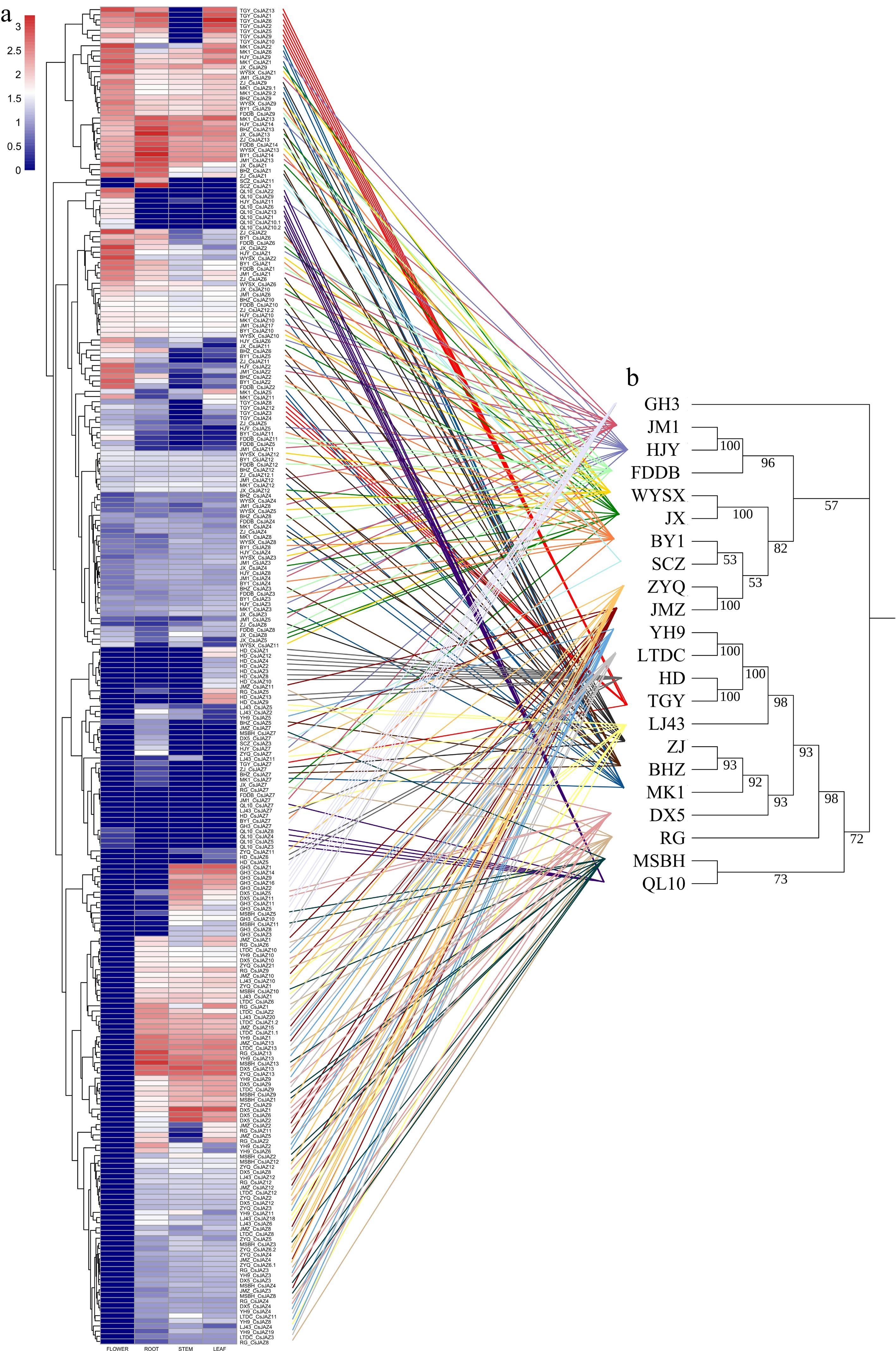
Figure 6.
(a) The heatmap of expression profile clustering in the pangenome of CsJAZs from 22 tea plant cultivars in four tissues (flower, root, stem, leaf). (b) Species phylogeny tree. Gene expression values were normalized using log10(TPM + 1) transformation. Comparison of gene expression clustered branching plots (left tree) with species phylogenies (right tree) via connecting lines (different species highlighted in different colors).
A comparative analysis of gene expression patterns against phylogenetic relationships among the 22 tea plant cultivars revealed clear patterns of transcriptomic divergence. Cultivars clustered into three distinct phylogenetic branches: 'GH3' formed a distinct lineage; 'JM1', 'HJY', 'FDDB', 'WYSX', 'JX', 'BY1', 'SCZ', 'ZYQ', and 'JMZ' grouped closely; 'YH9', 'LTDC', 'HD', 'TGY', 'LJ43', 'ZJ', 'BHZ', 'MK1', 'DX5', 'RG', 'MSBH', and 'QL10' comprised a separate lineage (Fig. 6b). By aligning transcriptomic data clusters with these phylogenetic relationships, evolutionary divergence was visualized in expression patterns, demonstrating how pan-genomic and transcriptomic data integration elucidates the evolutionary dynamics of gene families.
-
Integrating pan-genomic and transcriptomic approaches offers a robust framework for unraveling gene family evolution and functional diversity beyond traditional single-genome analyses[28,29]. This approach enhances our understanding of gene function, evolutionary dynamics, and cultivar-specific traits, providing a model for future genomic investigations in tea and other plant species.
JAZ proteins play crucial roles in plant development, flowering, stress responses, and plant-pathogen interactions[5,30]. Studies in A. thaliana revealed that AtJAZ1 positively regulates flowering time and seed traits, whereas AtJAZ4 influences hypocotyl and petiole elongation by repressing auxin- and cytokinin-associated regulators[31,32]. Additionally, overexpression of AtJAZ8, AtJAZ9, or AtJAZ13 reduces jasmonate sensitivity, thus affecting root growth[33]. While JAZ family members have been extensively characterized in model plants, investigations based solely on a single reference genome often fail to capture a species' full genetic diversity and complexity[34,35].
Here, a pan-genomic approach was leveraged, analyzing 22 high-quality tea plant genomes to understand the JAZ family[12] comprehensively. 21 CsJAZs were identified, categorizing them into core (present in all cultivars), near-core, dispensable, and private (unique to specific cultivars) groups. Core genes likely fulfill essential biological roles, while private genes (CsJAZ16–CsJAZ21), identified exclusively in cultivars such as 'JM1', 'LJ43', 'YH9', and 'ZYQ', potentially underpin cultivar-specific traits. Notably, CsJAZ3, CsJAZ4, CsJAZ10, CsJAZ12, CsJAZ17, and CsJAZ21 cluster closely in the phylogeny and display mutually exclusive presence-absence patterns across cultivars (Fig. 1b), suggesting functional redundancy. The mutually exclusive distribution may ensure the maintenance of critical JAZ-mediated functions despite gene loss in individual genomes[29].
Evolutionary analysis based on Ks and Ka/Ks ratios indicated distinct selective pressures across CsJAZ family members. Two recent duplication events were evident from the Ks distributions, with particularly recent duplications in cultivars 'BHZ' and 'HD'. Most CsJAZs exhibited Ka/Ks ratios below 1.0, indicating pervasive purifying selection and evolutionary constraints that likely preserve essential gene functions. In contrast, CsJAZ1 (in 'FDDB' and 'MSBH'), CsJAZ8 (in 'YH9', 'DX5', 'BY1', 'JM1', 'LJ43', and 'QL10'), and CsJAZ9 (in 'JM1' and 'LJ43') showed Ka/Ks ratios greater than 1.0, which may reflect either episodes of positive selection or relaxed functional constraints. However, further branch-site model analyses using PAML did not yield significant likelihood ratio tests or identify any codons under positive selection, suggesting that the elevated Ka/Ks values may not robustly support adaptive evolution in these gene lineages. These findings highlight the complexity and potential limitations of inferring positive selection from Ka/Ks ratios alone, especially in gene families subject to recent duplication and structural variation.
SVs, including insertions and deletions, significantly impacted CsJAZ structures and expression patterns. For instance, SVs within CsJAZ4, CsJAZ9, and CsJAZ12 altered their expression levels across cultivars. Such SV-induced modifications can also result in loss or alteration of functional domains, complicating traditional bioinformatics identification. Nevertheless, genes with these altered sequences may retain critical biological functions. Similar phenomena have been documented in other tea genes: structural variants in GluRS disrupt domains involved in chlorophyll synthesis pathways, affecting leaf coloration[12], and insertion of LTR/Gypsy elements upstream of CsMYB114 correlates strongly with anthocyanin-related leaf pigmentation[12,36,37]. Thus, SVs are crucial in shaping cultivar-specific phenotypic diversity.
Transcriptome analyses across multiple tissues revealed stable expression of core CsJAZs (CsJAZ1 and CsJAZ2), underscoring their fundamental roles. Several CsJAZs showed tissue-specific or cultivar-specific expression patterns, likely reflecting adaptive specialization, such as stress responses. Previous studies indicated that CsJAZ3, CsJAZ5, CsJAZ6, CsJAZ8, and CsJAZ12 positively regulate the ICE-CBF/DREB1 pathway in response to cold stress[34]. Our comparative analyses between phylogenetic relationships and gene expression patterns further illustrated that closely related tea plant cultivars share similar transcriptional profiles, highlighting conserved expression patterns linked to evolutionary history.
-
This study conducted a comprehensive pan-genome analysis of the JAZ family across 22 high-quality tea plant genomes, identifying 21 CsJAZs categorized as two core, three near-core, ten dispensable, and six private genes. These genes are clustered into five groups based on comparisons with the Arabidopsis JAZ classification. Evolutionary analyses revealed that CsJAZ1, CsJAZ8, and CsJAZ9 underwent positive selection during tea domestication, as indicated by elevated Ka/Ks values (> 1). SVs significantly influenced gene structure, conserved protein domains, and expression patterns, particularly for CsJAZ4, CsJAZ9, and CsJAZ12. Transcriptome analyses across multiple tissues demonstrated consistently high expression for CsJAZ1, CsJAZ2, CsJAZ6, CsJAZ9, CsJAZ13, and CsJAZ14, highlighting their functional importance across tea plant cultivars. Collectively, these findings enhance our understanding of the evolutionary dynamics and functional diversity of the JAZ family, providing valuable genomic resources and insights for future genetic improvement and stress adaptation studies in tea plants.
This work was supported by the National Guidance of Local Science and Technology Development Fund of China (Grant No. [2023]009), the National Natural Science Foundation of China (Grant No. 32260086), and the Cultivation Project of Guizhou University (Grant No. Gzu.2020No.65).
-
The authors confirm their contributions to the paper as follows: study conception and design, and manuscript editing and revision: Niu S, Xiong B; data analysis: Xiong B, Yang Y, Li Q; figures and manuscript constructing: Yang Y; experiments performing: Li Q; materials providing and facilities testing: Niu S; manuscript writing: Xiong B. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 Classification of JAZs.
- Supplementary Table S2 Molecular characteristics of CsJAZs.
- Supplementary Table S3 Ka/Ks value of the frequency of occurrence of different tea plants cultivars at each JAZ.
- Supplementary Fig. S1 CDS sequence comparison of CsJAZ4.
- Supplementary Fig. S2 CDS sequence comparison of CsJAZ9.
- Supplementary Fig. S3 CDS sequence comparison of CsJAZ12.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Xiong B, Yang Y, Li Q, Niu S. 2025. Evolutionary dynamics and functional characterization of Jasmonate ZIM-domain (JAZ) genes across Camellia sinensis pan-genome. Beverage Plant Research 5: e036 doi: 10.48130/bpr-0025-0027 

Evolutionary dynamics and functional characterization of Jasmonate ZIM-domain (JAZ) genes across Camellia sinensis pan-genome
- Received: 15 April 2025
- Revised: 26 June 2025
- Accepted: 01 July 2025
- Published online: 17 November 2025
Abstract: Jasmonate ZIM-domain (JAZ) proteins regulate critical processes in plants, including growth, development, secondary metabolism, and responses to biotic and abiotic stresses. Previous studies primarily focused on single reference genomes, neglecting gene presence-absence variations (PAV) across populations. Investigating the JAZ gene family at the pan-genomic scale is thus essential to fully understand its evolutionary and functional dynamics in tea plants. Here, 22 high-quality Camellia sinensis genomes were analyzed, and 21 JAZs exhibiting substantial presence-absence variability were identified. These included two core genes (present in all 22 genomes), three near-core genes (present in 20–21 genomes), ten dispensable genes (present in 2–19 genomes), and six private genes (unique to single genomes). Phylogenetic analysis categorized these JAZs into five distinct groups, aligning with the AtJAZ family. Selection pressure analysis revealed positive selection (Ka/Ks > 1) acting on CsJAZ1, CsJAZ8, and CsJAZ9, suggesting adaptive roles during tea domestication. Structural variants (SVs) significantly impacted gene expression and structural integrity; notably, CsJAZ4, CsJAZ9, and CsJAZ12 exhibited differential expression when affected by SVs. RNA-seq analysis across four tissues from the 22 tea plant cultivars showed consistently high expression of CsJAZ1, CsJAZ2, CsJAZ6, CsJAZ9, CsJAZ13, and CsJAZ14, highlighting their potential fundamental roles. This study elucidates the JAZ gene family's evolutionary complexity and functional versatility within the tea plant pan-genome. These findings provide valuable insights for future research into CsJAZ functions and serve as a model for pan-genomic analyses of gene families in other plant species.
-
Key words:
- JAZ family /
- Pan-genome /
- Structural variation /
- Expression pattern