









-
Sugars as the main soluble solid component are important nutrients and a key factor influencing the flavor quality of fruits[1]. They also play a crucial role in regulating the expression of fruit-related genes, as well as plant growth and development[2], stress responses[3], and other developmental processes. In leaves, sugars are produced as important photoassimilates and are loaded into the phloem system through the symplasm pathway. They are then unloaded in sink organs, such as fruits and flower, through the apoplast pathway[4]. Sugar transporters that play an indispensable role in phloem loading, nectar secretion, and reproductive tissue development. Many studies have explored the mechanism of sucrose transport from source cells to sink cells, which involves the synergistic effects of multiple transporters[4−8]. At present, sugar transporters in plants are classified into two types: Sugars Will Eventually be Exported Transporters (SWEETs) and major facilitator superfamily (MFS) transporters. Sugars Will Eventually be Exported Transporters (SWEETs) have been newly identified in plants in recent years[9]. Sugar transporters of MFS are further classified into MSTs and SUTs[10], which primarily facilitate sugar influx into the cytosol. However, some MSTs, namely the tonoplast sugar transporter (TST) and the vacuolar glucose transporter (VGT) involved in transporting sugars from the cytosol to vacuoles and act as H+/sugar antiporters[11]. Both MSTs and SUTs consist of 12 transmembrane α-helices and mediate membrane transport of different sugars[12,13].
SWEET transporters differ from the classic 12 transmembrane structural domains of the major facilitator superfamily. The typical characteristics of SWEET genes include seven TM domains, including two MtN3_saliva domains, which are connected to a low conserved single TM, forming a 3-1-3 symmetric structure[14]. Phylogenetic analysis shows that members of SWEET can be divided into four clades. Clades I, Clades II and Clades IV are mainly hexose transporters, while Clade III is mainly a sucrose transporter[15]. There is substantial evidence suggesting that SWEET protein in Clade I and II may transport glucose, most of the Clade III SWEET proteins are sucrose transporters, and Clade IV evolved from other SWEET clades to primarily act as vacuolar transporters, regulating fructose transport[14]. Members of the SWEET family are widely distributed, and SWEET genes are found not only in plants but also in prokaryotes and animals[16]. With the development of plant genome research, the identification and functional study of SWEET family genes have been carried out in many plant species, and the number of SWEET family members varies significantly among different plants. For example, the SWEET family consists of 17 members in Arabidopsis, 22 in Saccharum spontaneum, 20 in strawberry, 15 in pomegranate, 19 in jujube, 29 in tomato, and 33 in apple[17−23]. Previous studies have reported that members of the SWEET gene family are involved in many important physiological processes of plant growth and development, including nectar production, seed, and pollen development, and the regulation of phloem loading, phloem transport, phloem unloading, abiotic stress, and pathogen interaction by regulating carbohydrate compounds[18, 24−26]. SWEET proteins are also associated with flower, fruit, and seed development. For example, AtSWEET8 mainly affected fertility during early inflorescence development, and AtSWEET13 mainly affected fertility during late inflorescence development[27]. The SWEET gene family was also involved in plant interactions with pathogens, such as VvSWEET4, which shows strong up-regulation of expression with infection of Botrytis cinerea[28, 29].
Furthermore, SWEET proteins are key factor in regulating the distribution of soluble sugars, which is closely related to plant stress resistance[26]. Overexpression of two homologues AtSWET16 and AtSWEET17 in Arabidopsis thaliana can improve the cold resistance of transgenic plants[26, 30]. Overexpression of AtSWEET15 show more sensitivity to salt stress, and loss of function mutations in AtSWEET15 show higher salt tolerance[31]. Drought stress affects the redistribution of carbohydrates in plants[32]. Under water deficiency conditions, the expression of AtSWEET11 and AtSWEET12 in leaves and roots increased, accompanying an increase in the transportation capacity of sucrose from leaves to roots, indicating that plants regulate the redistribution of carbohydrates by regulating the expression of SWEETs under water deficiency conditions[33].
Actinidia polygama (Sieb. & Zucc.) Maxim. is a perennial vine plant, its fruit contains many unique and interesting flavors and many nutrients, including organic acids, amino acids, flavonoids, dietary fiber, and vitamins C and E. In addition, the leaves, fruits, stems, and roots also were exploited as nutraceuticals or medicine. Therefore, A. polygama as a valuable wild resource has very high nutritional and medicinal value. However, the current domestication of A. polygama is not complete, particularly in terms of improving fruit quality. During the ripening process of A. polygama fruit, sugar accumulation is closely related to fruit quality and edible ability. In this study, we identified the SWEET gene family members of A. polygama, and analyzed the physical and chemical properties, phylogenetic relationship, gene structure, and promoter elements of SWEET family proteins. We also studied the expression of SWEET gene family members in different organs of A. polygama, which laid a foundation for further study on the structure and function of the SWEET gene family.
-
By searching the SWEET gene on the website (ID: PRJDB13926), a total of 23 SWEET gene family members of A. polygama were screened out, and nine SWEET subfamily members were eliminated because of lack of the conserved structures. According to the physicochemical property table (Table 1), the amino acid quantity of the SWEET gene family ranged from 233 to 304. The molecular weight ranges from 25,895.58 to 33,192.12. The isoelectric point is between 6.96 and 9.71, and the stability coefficient of ApSWEET4, ApSWEET5 and ApSWEET14 proteins was greater than the remaining 11 proteins. The aliphatic index was 94.76~118.24, which was a stable protein. Furthermore, most of the SWEET genes were found to be present on the cell membrane, rarely on the chloroplast and Golgi apparatus.
Table 1. Physicochemical properties of SWEET gene family proteins.
Gene ID Number of amino acids Molecular weight pI Asp + Glu Arg + Lys Instability index Predicted location ApSWEET1 275 31,093 8.17 17 19 37.35 (stable) Cell membrane ApSWEET2 244 27,049.86 6.86 16 16 35.79 (stable) Cell membrane ApSWEET3 304 33,192.12 9.49 19 30 36.79 (stable) Chloroplast ApSWEET4 254 29,132.9 7.61 19 20 46.56 (unstable) Cell membrane ApSWEET5 259 28,602.2 9.71 13 24 45.53 (unstable) Cell membrane ApSWEET6 234 25,895.58 8.48 14 16 35.70 (stable) Cell membrane ApSWEET7 238 26,891.1 9.18 13 19 36.22 (stable) Cell membrane ApSWEET8 237 26,653.71 8.87 13 17 34.25 (stable) Cell membrane ApSWEET9 253 27,530.9 9.5 12 23 29.02 (stable) Cell membrane
Golgi apparatusApSWEET10 252 27,529.8 9.49 12 20 25.31 (stable) Cell membrane ApSWEET11 236 25,918.83 9.26 8 14 36.71 (stable) Cell membrane ApSWEET12 234 25,977.87 9.03 9 14 36.26 (stable) Cell membrane ApSWEET13 238 26,391.35 9.01 9 14 38.49 (stable) Cell membrane ApSWEET14 233 26,641.9 9.36 15 24 43.15 (unstable) Cell membrane Phylogenetic and structural analysis of SWEET gene family members in A. polygama
-
To study the phylogenetic relationships among SWEET genes in A. polygama and other plant species, a neighbor-joining phylogenetic tree was constructed by aligning 14 ApSWEET sequences, 17 AtSWEET sequences, and 14 VvSWEET sequences (Supplemental Tables S1–S3). Apparently, 45 proteins were clustered into four different groups (Fig. 1). In detail, six ApSWEETs (ApSWEET9, 10, 11, 12, 13, 14) showed high homology with three AtSWEETs (AtSWEET1–3) and three VvSWEETs (VvSWEET2a, 2b, 3) in group I. In group II, three ApSWEETs (ApSWEET5, 7, 8) were clustered with five AtSWEETs (AtSWEET4–8) and three VvSWEETs (VvSWEET5a, 5b, 7). ApSWEET1/4 were homologous to seven AtSWEETs (9–15) and five VvSWEETs (VvSWEET9, 10, 11, 12, 15) in group III. Three ApSWEET (ApSWEET2, 3, 6), two AtSWEETs (AtSWEET16, 17) and three VvSWEETs (VvSWEET17a, 17b, 17d) were included in group IV. The exon-intron structural evolution showed that most of the ApSWEET contained six exons, except for ApSWEET5 and ApSWEET14 which contained five exons (Fig. 2). ApSWEET2 and ApSWEET4 had the shortest and longest sequence, respectively. ApSWEET11, ApSWEET12, and ApSWEET13 had similar exon-intron structures. In addition, ApSWEET1 and ApSWEET4, ApSWEET3, and ApSWEET6 also showed similar structures, these genes belong to the same group. These results suggested that ApSWEETs in the same group shared similar exon-intron organizations.
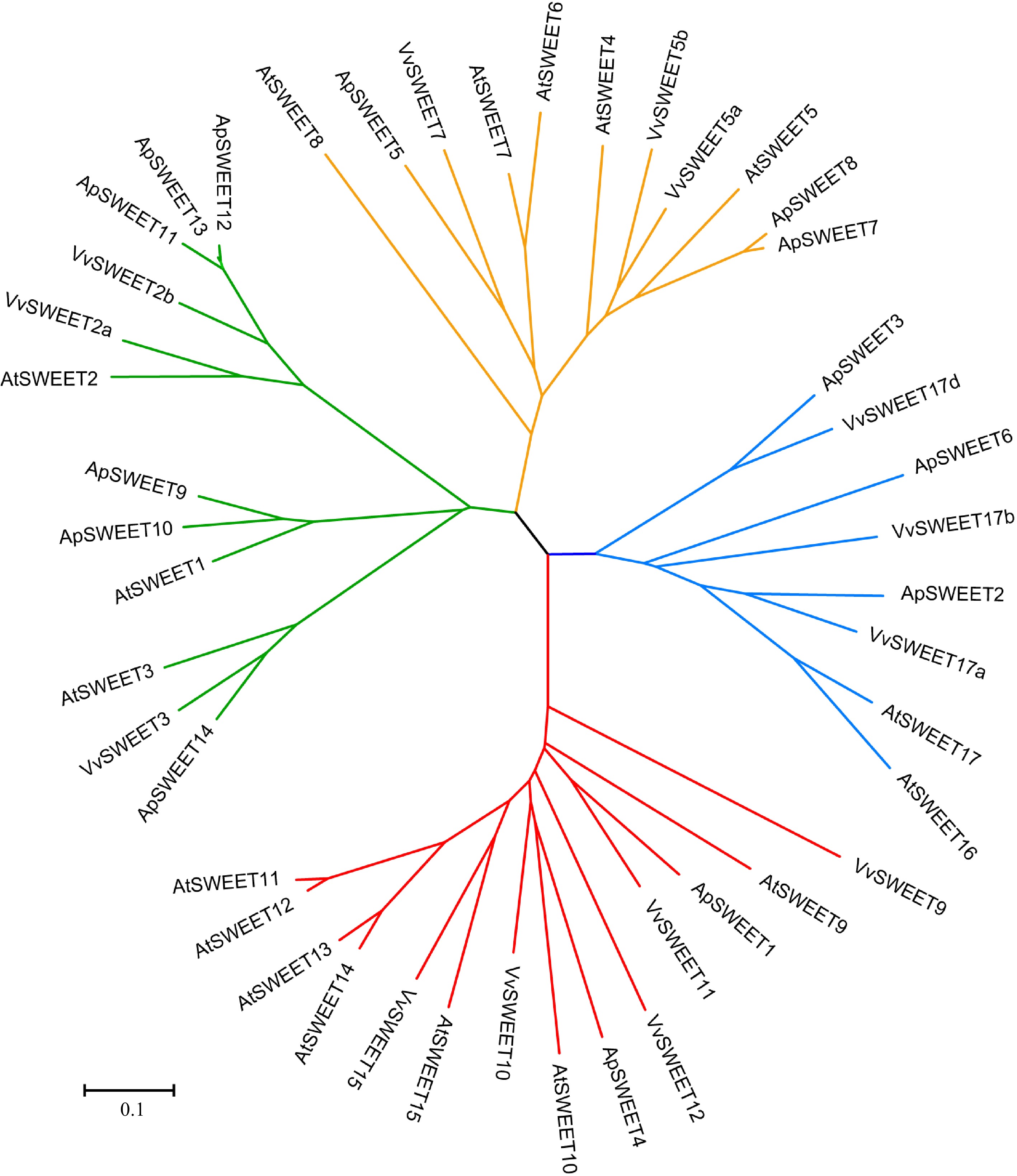
Figure 1.
Phylogenetic analysis of the ApSWEETs from A. polygama, Arabidopsis thaliana, and Vitis vinifera. The Neighbor-joining tree was drawn using MEGA7.0 with 1,000 bootstraps. The roman numbers (I–IV) labeled with various colors indicate different clades: green – Clade I, orange – Clade II, red – Clade III, blue – Clade IV.
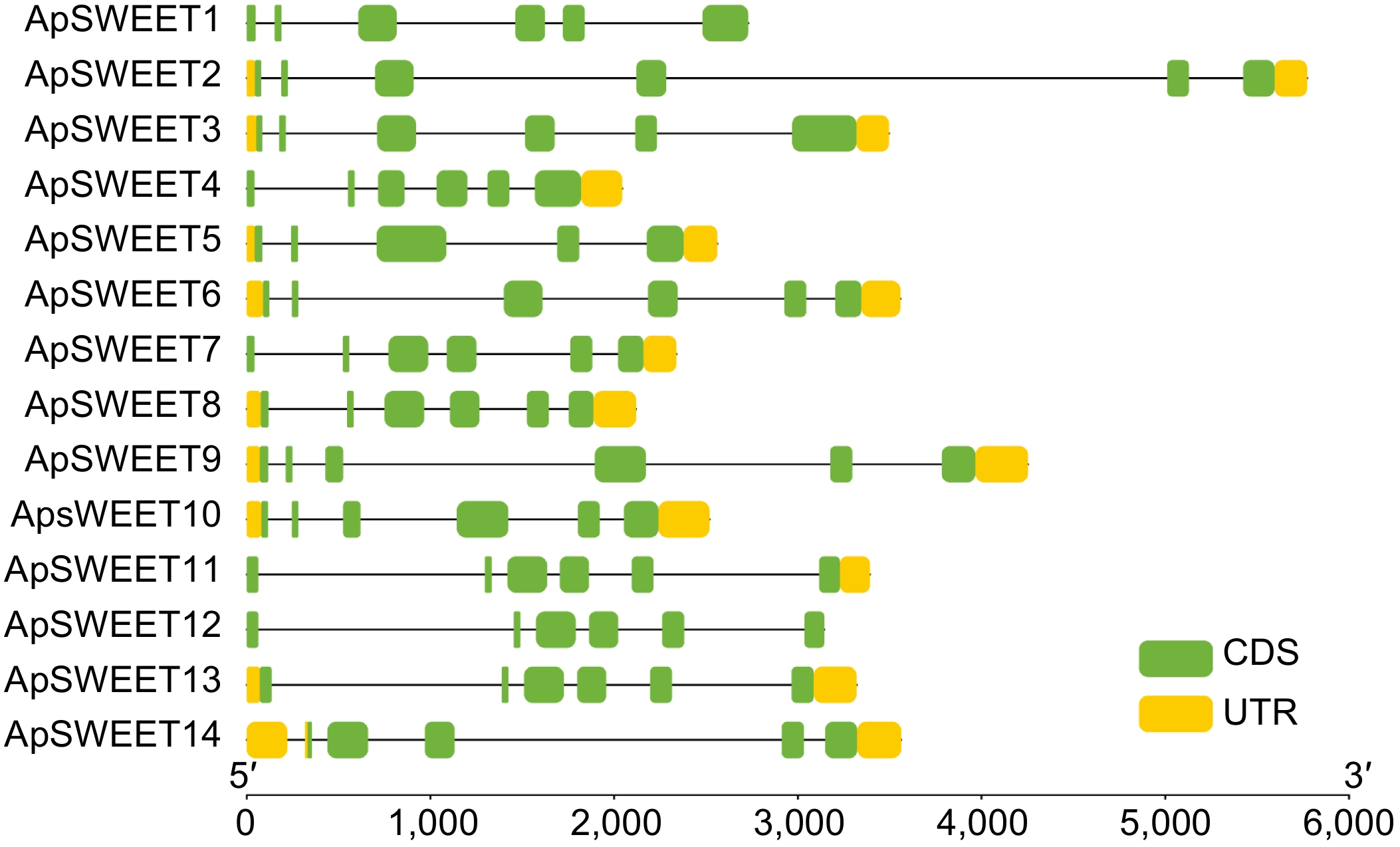
Figure 2.
ApSWEET gene structure of A. polygama.
Analysis of conserved motif of ApSWEET gene family
-
The Motif1~Motif10 conserved motifs were found in the SWEET gene of A. polygama. (Fig. 3), whereas motifs 1, 2, 4, and 5 were observed in all ApSWEET members. In addition, Motif3 was observed in 13 ApSWEET members except ApSWEET4. Six genes lacked Motif6, and five genes lacked Motif7. Only ApSWEET13, ApSWEET12 and ApSWEET2 genes contained Motif8, ApSWEET10, ApSWEET5 and ApSWEET4 contained Motif9, ApSWEET1 and ApSWEET14 contained Motif10. Most of the conserved motifs had a relatively consistent relationship with the evolutionary tree with the same order of number, suggesting that these genes had strong conserved structures and similar gene functions. We confirmed that the ApSWEET proteins also contained P-loop, MtN3-slv, and transmembrane domain (Fig. 4). The typical structure of plant SWEET proteins consists of seven predicted transmembrane (7-TM) helices forming two MtN3_slv domains (triple-helix bundles, THB) connected by a linker transmembrane helix (TM4). All ApSWEET genes comprise two sugar transporter domains for intercellular exchange.
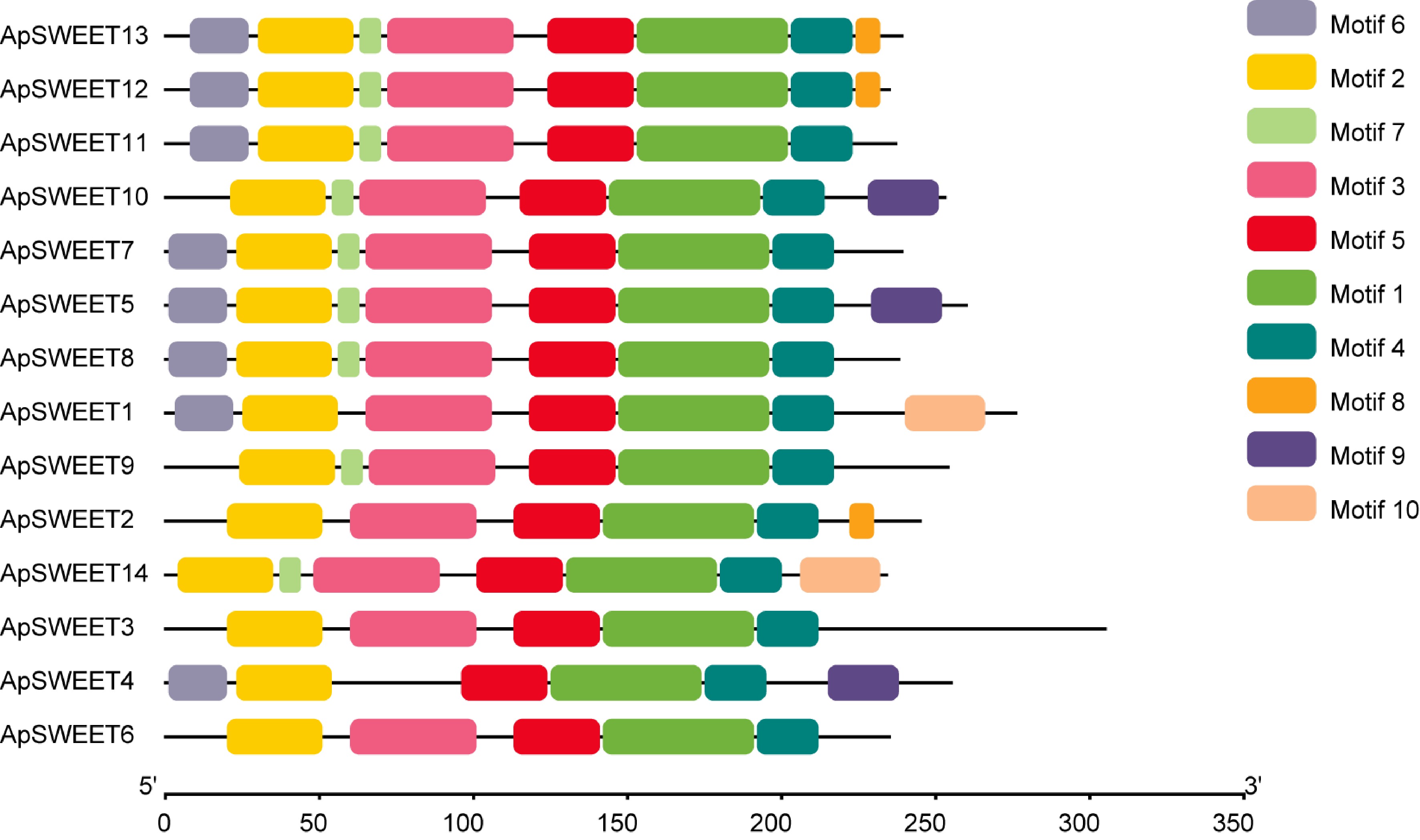
Figure 3.
The conserved motif analyses of ApSWEETs proteins.
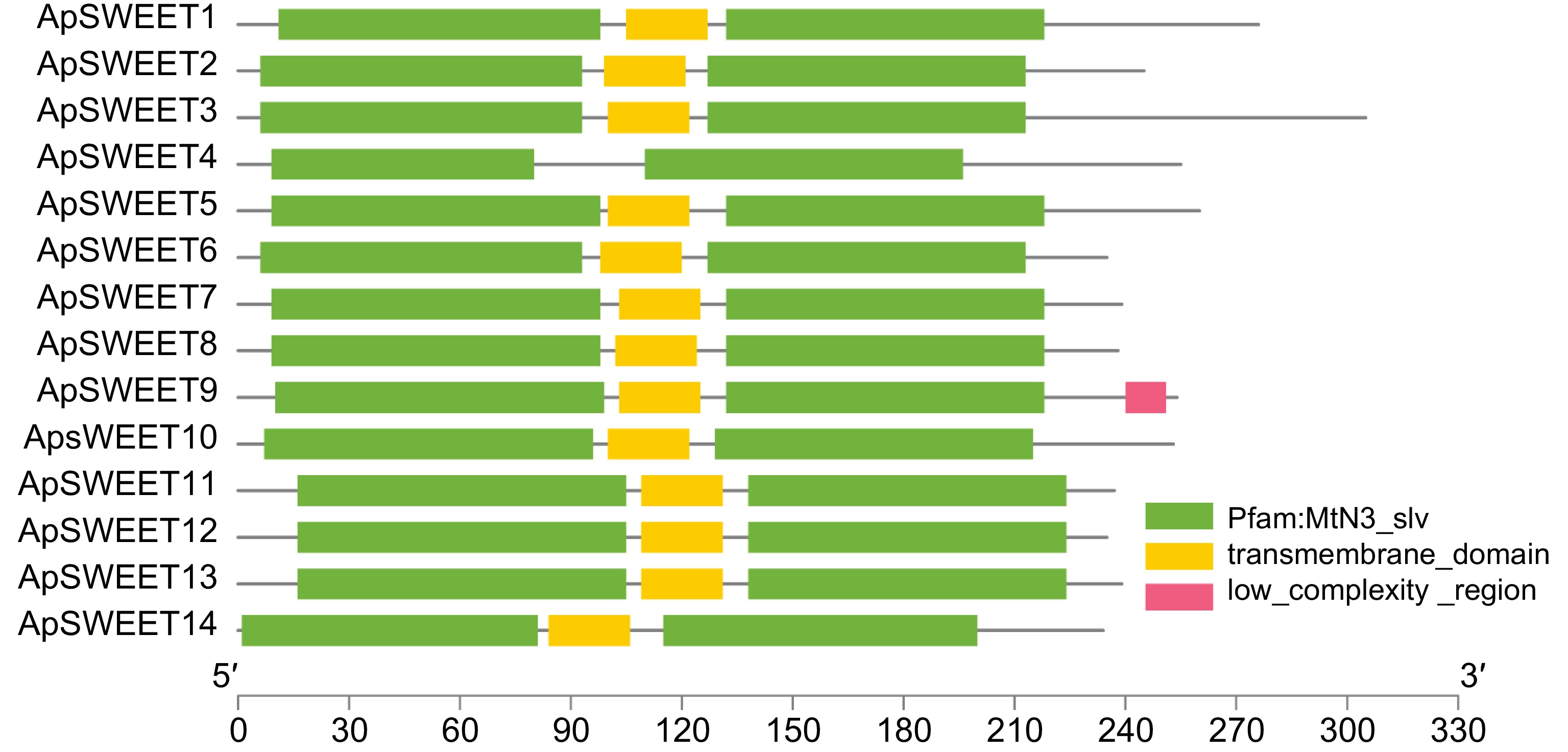
Figure 4.
Conserved structural domains of ApSWEETs.
Secondary and tertiary structure analysis of ApSWEET gene family members
-
SOPMA was used to analyze the secondary structure of the SWEET gene family in A. polygama, including Alpha helix, chain extension structure, Beta turn, and Random coil (Table 2). These results showed that Alpha helix and Random coil were significantly higher than the chain extension structure and Beta turn. In addition, the Alpha helix proportion of ApSWEET6 was the highest, and ApSWEET9 was the lowest. Meanwhile, the proportion of chain extension structure in ApSWEET9 was the highest, and ApSWEET3 was the lowest. The beta turn of ApSWEET8 was the highest, and ApSWEET4 was the lowest. A random coil of ApSWEET4 was the highest, but ApSWEET6 was the lowest.
Table 2. Secondary structure analysis of ApSWEET family members.
Alpha
helixExtended
strandBeta turn Random
coilApSWEET1 45.09% 16.73% 2.18% 36.00% ApSWEET2 41.80% 22.54% 2.87% 32.79% ApSWEET3 49.01% 15.46% 5.26% 30.26% ApSWEET4 37.80% 18.11% 0.79% 43.31% ApSWEET5 35.91% 21.24% 4.63% 38.22% ApSWEET6 50.00% 21.79% 3.85% 24.36% ApSWEET7 40.34% 21.85% 4.20% 33.61% ApSWEET8 40.51% 21.94% 5.91% 31.65% ApSWEET9 33.99% 25.30% 4.35% 36.36% ApSWEET10 46.83% 17.06% 3.97% 32.14% ApSWEET11 42.37% 19.49% 5.08% 33.05% ApSWEET12 41.88% 20.94% 4.27% 32.91% ApSWEET13 46.22% 19.33% 2.52% 31.93% ApSWEET14 36.48% 21.89% 3.86% 37.77% SWISS-MODEL was used for homology modeling analysis (Fig. 5), and it was found that the three-dimensional structure of ApSWEET proteins could be roughly divided into two categories. ApSWEET1, ApSWEET2, ApSWEET4, ApSWEET5, ApSWEET6, ApSWEET7 and ApSWEET8 was clustered into one group, and ApSWEET3, ApSWEET9, ApSWEET10, ApSWEET11, ApSWEET12, ApSWEET13 and ApSWEET14 was clustered into one group.
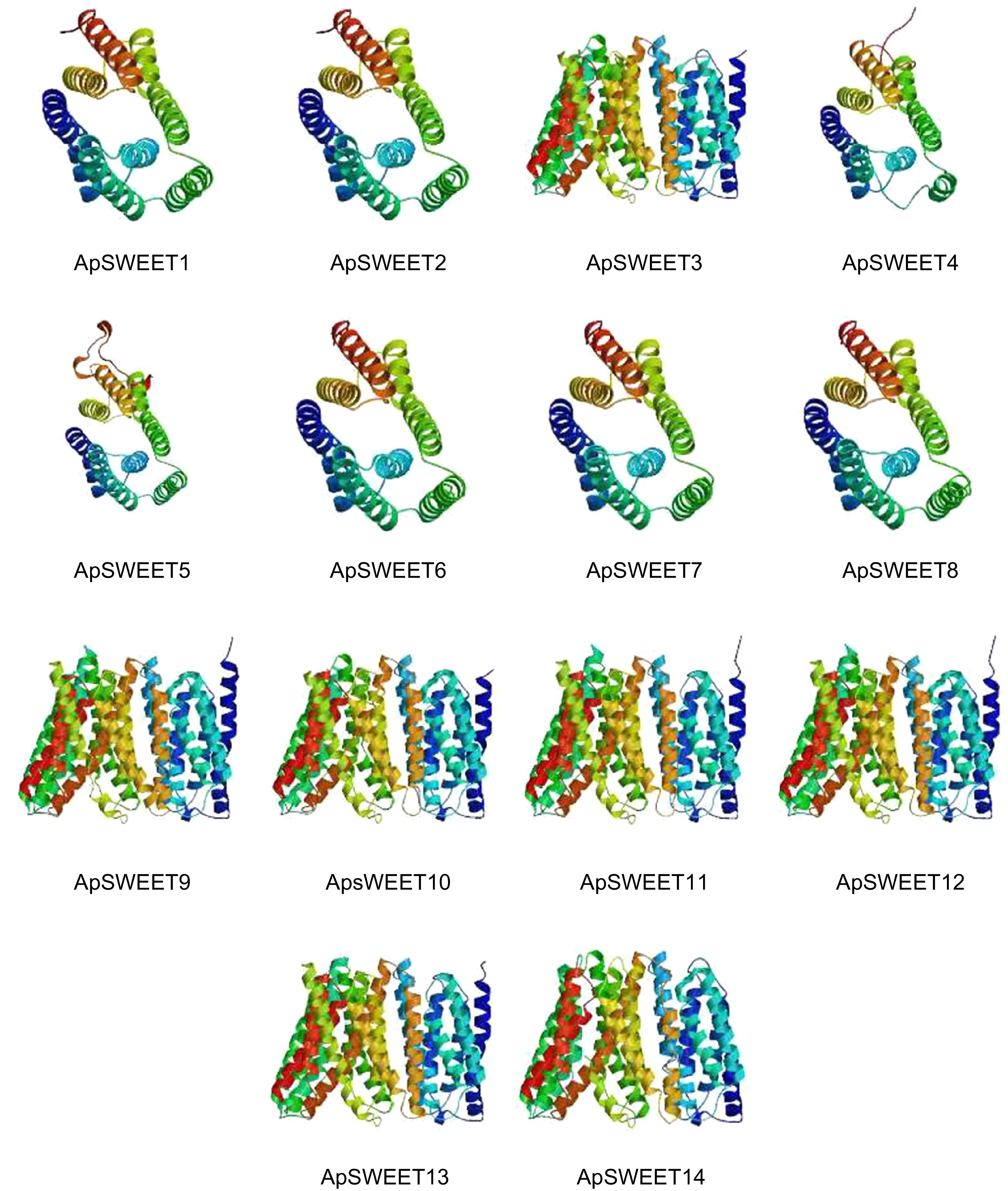
Figure 5.
Single protein structure of ApSWEETs.
Chromosomal localization and collinear analysis of the ApSWEET gene
-
Chromosome analysis of genes (Fig. 6) showed that except for chromosome ApChr19, other genes were evenly distributed on 11 chromosomes. Three ApSWEETs genes were distributed in clusters on chromosomes ApChr19, ApSWEET12, and ApSWEET13 may be due to gene replication. According to the collinearity analysis diagram (Fig. 7), ApSWEET2 and ApSWEET6, ApSWEET4 and ApSWEET7, ApSWEET11 and ApSWEET3/12/13 exist collinearity, which may be obtained by chromosome fragment replication.
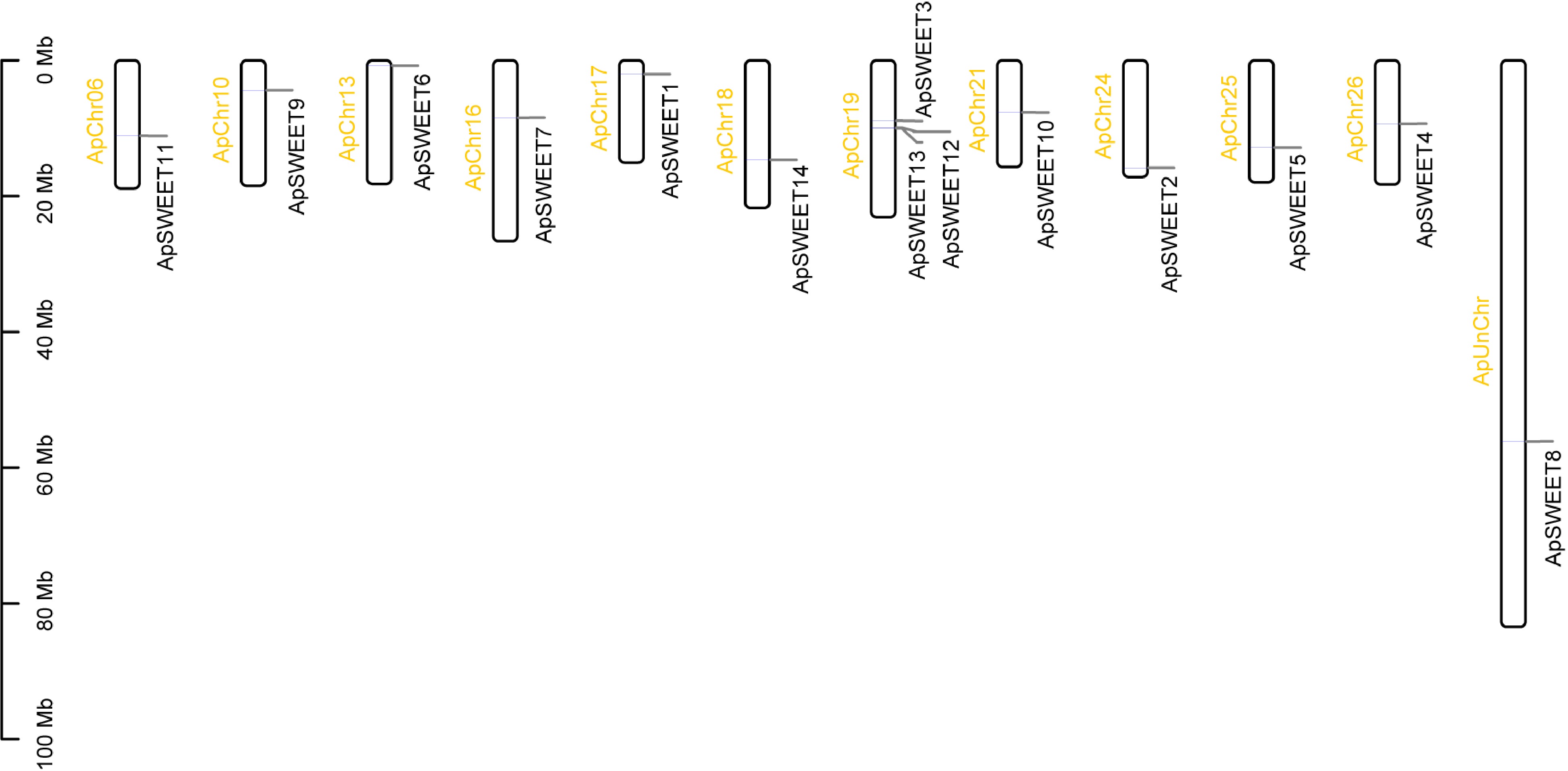
Figure 6.
Chromosome location of ApSWEET gene family.
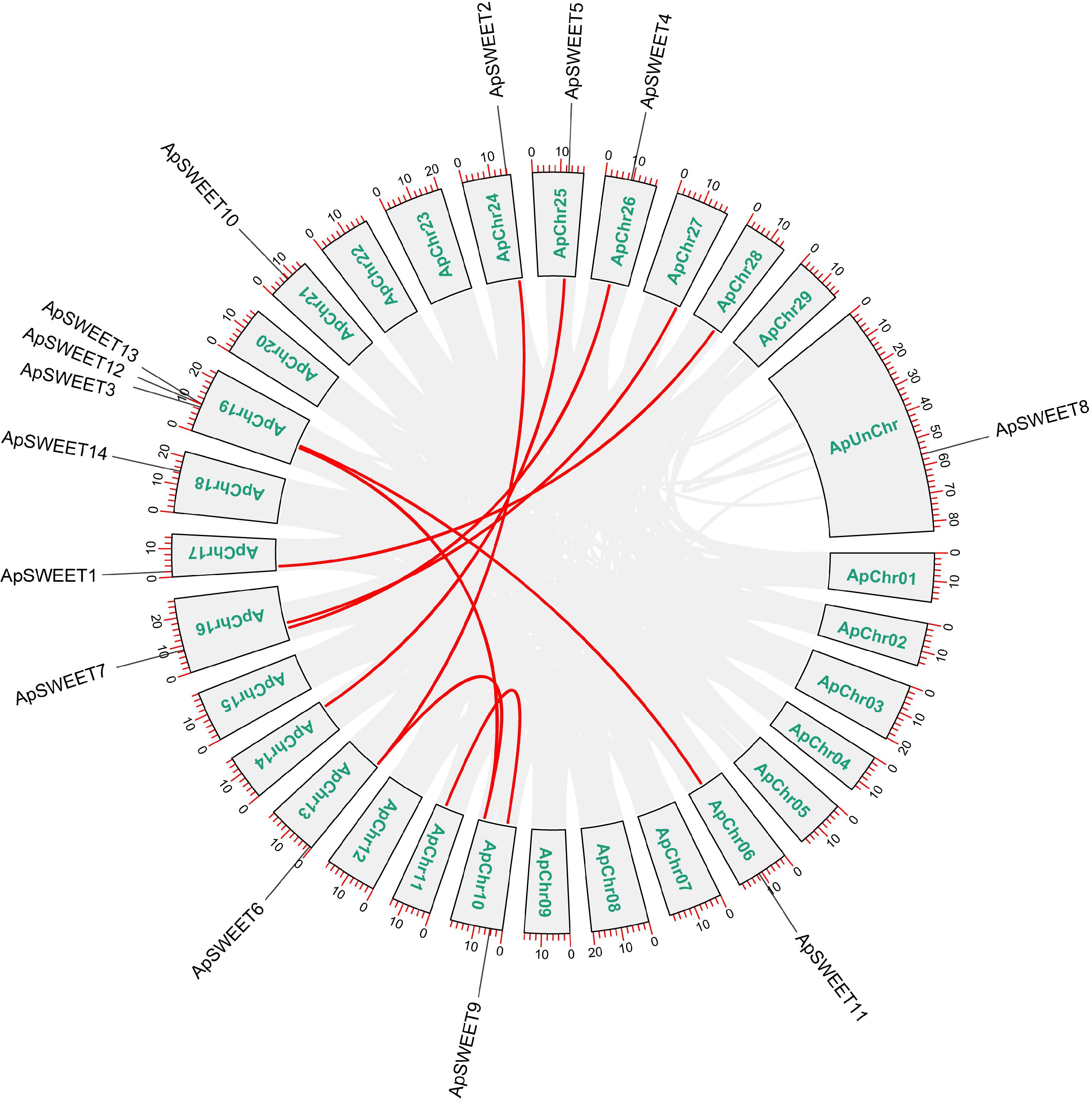
Figure 7.
Collinearity analysis of the ApSWEET gene family.
Promoter analysis of the ApSWEET gene family
-
In order to study the potential regulatory factors of the ApSWEET gene, the 2,000 bp promoter region of this family was analyzed (Fig. 8), and 88 elements in promoter regions of all ApSWEETs genes were predicted. The results showed that response elements such as low temperature, light, and hormone appeared in most gene promoter regions, indicating that genes may be affected by low temperature, light, and hormone levels. They were classified into three groups based on their functional associations: stresses (ARE, DRE, STRE, LTR, MBS, and MYC), hormones (ABRE, TATC-box, CGTCA motif/TGACG motif, HD-Zip1, P-Box, GARE-motif, GA-motif, ERE, and TCA-element) and light (GT1-motif, TCCC-motif, TCT-motif, G-Box, Gap-box, LAMP-element). Among these elements, six elements were responsive to stress, ten elements were responsive to hormones, and six elements were responsive to light. Four development-related elements are responsive to meristem expression (CAT-box), cis-regulatory element involved in endosperm expression (GCN4), involved in endosperm-specific negative expression (AACA), and seed-specific regulation (RY). These findings indicated that ApSWEETs may respond to hormones or be involved in plant growth and stress resistance.
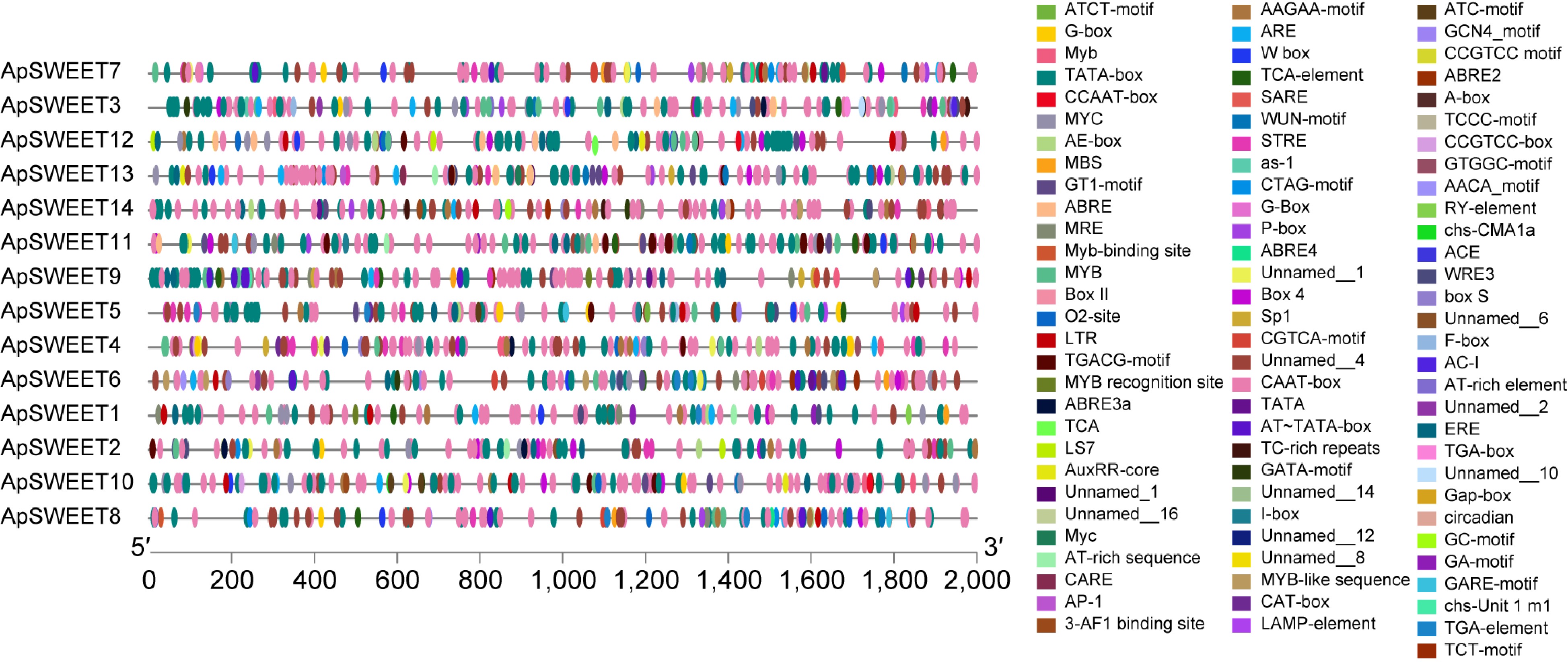
Figure 8.
The cis-elements in the promoter sequences of ApSWEETs gene in A. polygama.
Expression analysis of ApSWEET gene
-
Real-time fluorescence PCR was used to detect the expression of ApSWEET members in leaves, stems, flowers, roots, mature fruits (Fig. 9) and fruits of different developmental stages (Fig. 10). These results showed that the expression level of ApSWEET2, ApSWEET3, ApSWEET10, ApSWEET11 and ApSWEET13 was higher in leaves, the expression level of ApSWEET2, ApSWEET3, ApSWEET10, ApSWEET11, ApSWEET9, and ApSWEET13 was higher in stem, the expression level of ApSWEET5 and ApSWEET11 was higher in flower, the expression level of ApSWEET3, ApSWEET10, and ApSWEET11 was higher in the root, the expression level of ApSWEET1, ApSWEET10 and ApSWEET11 was higher in fruit. Most of the SWEET genes were found to be ubiquitously expressed in all tissues except for ApSWEET1 and ApSWEET5, the two genes were specifically expressed in the fruit and flower.
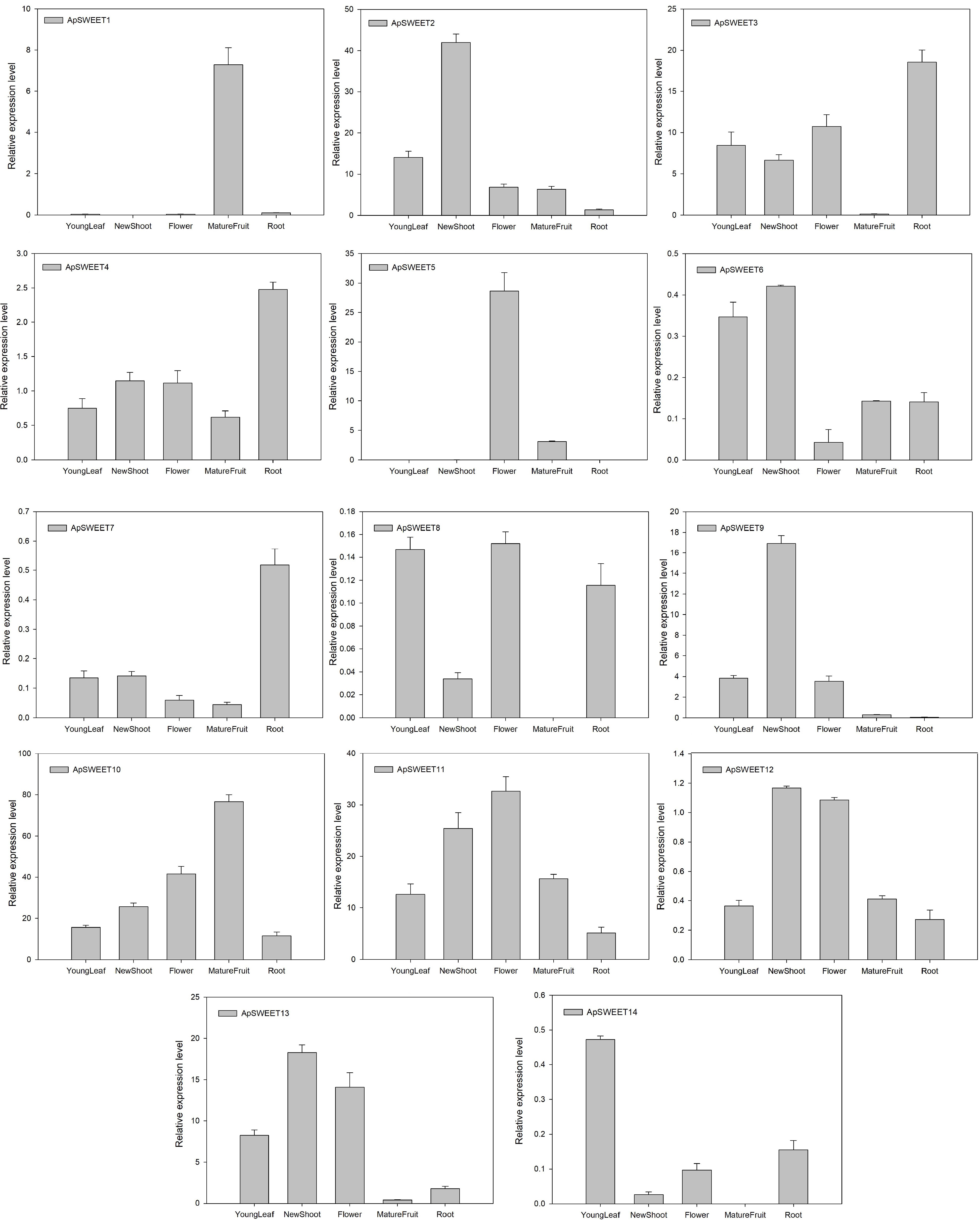
Figure 9.
ApSWEET expression of different tissues.
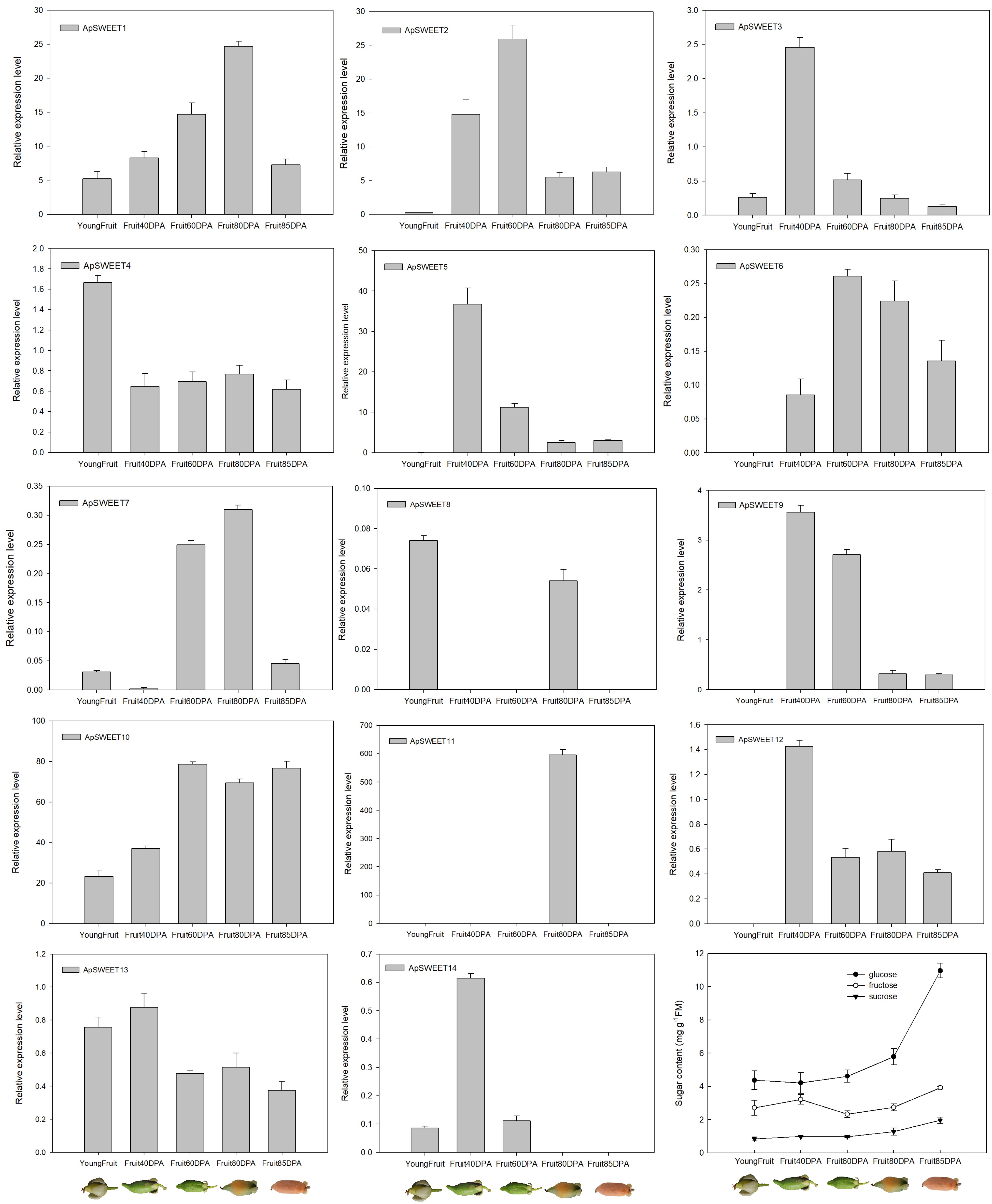
Figure 10.
ApSWEET expression of different fruit development stage.
During fruit development, only ApSWEET5 had higher expression at an early stage of fruit development, ApSWEET1, ApSWEET2, ApSWEET10, and ApSWEET11 had higher expression at the mid and late stages of fruit development (Fig. 10). In addition, the glucose content in A. polygama is higher than the fructose and sucrose content from the initial measurement on June 11th 2022 (Fig. 10). In the final measurement on September 27th 2022, the glucose content is 2.7 times and 5.4 times higher than the fructose content and sucrose content. Therefore, glucose content was the highest, followed by fructose, and sucrose content was the lowest during A. polygama development.
After the mature fruits are harvested, only ApSWEET1 shows strong expression at different storage periods. ApSWEET10 showed strong expression only on the first day after harvest, but the expression levels of ApSWEET11 gradually decrease with prolonged fruit storage time (Fig. 11).
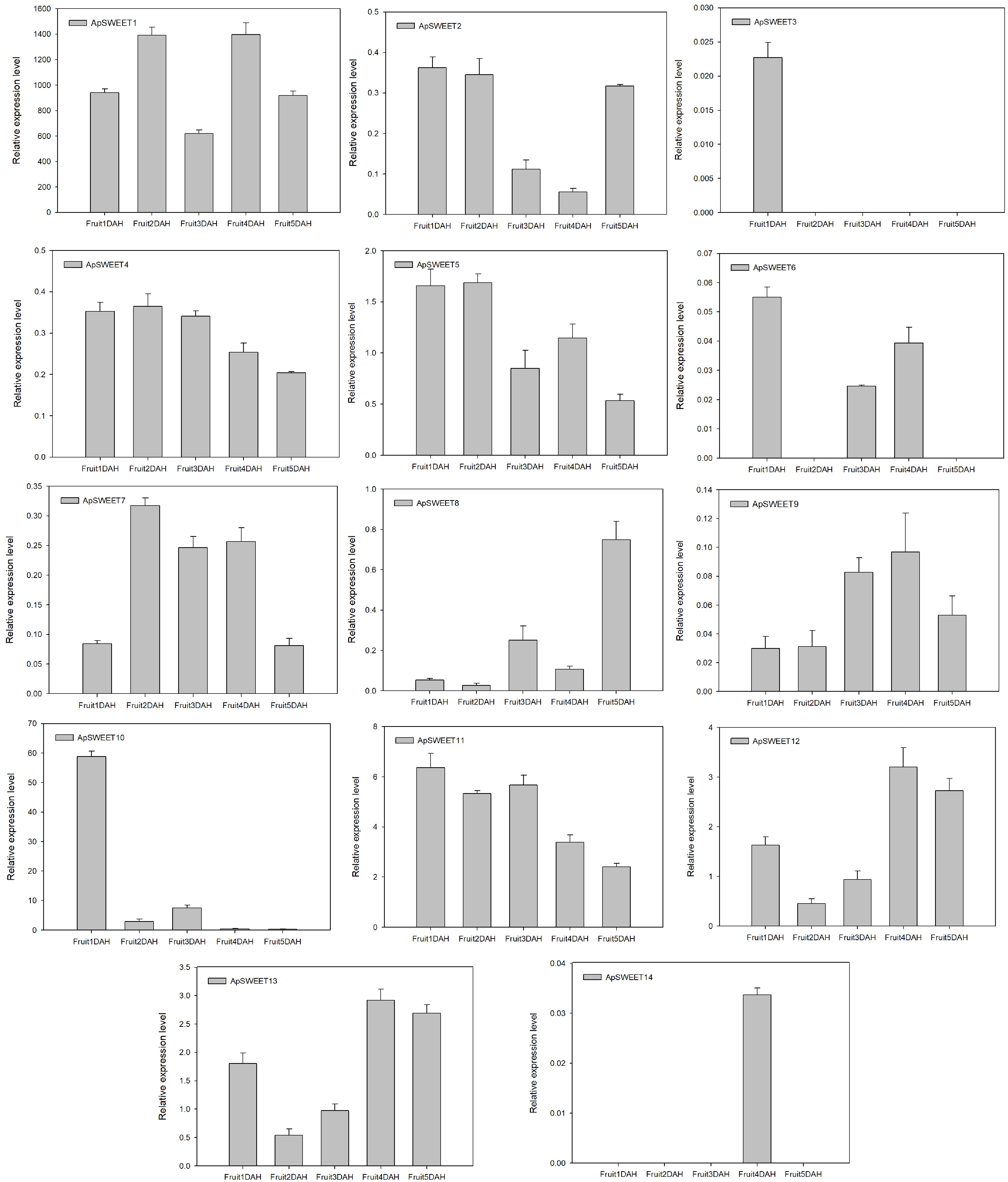
Figure 11.
ApSWEET expression of different storage periods.
-
Although SWEET genes have been extensively studied in various species, such as Arabidopsis thaliana[17], Hemerocallis fulva[34], Chinese jujube[14], Malus[35] and Litchi chinensis[36], the presence of SWEET gene family in A. polygama has not been reported. In this study, we identified 14 members of the SWEET gene family in A. polygama. The number is less than three in Arabidopsis and seven in rice[17, 18]. Gene duplication has been shown to contribute to the expansion of SWEET genes in soybean and potato, enabling them to adapt to environmental changes. This could explain why these species have more SWEET proteins than others. Moreover, different predictions of their physical and chemical properties suggest that SWEET genes may have diverse functions in plants[17]. Eleven of the identified genes were located on different chromosomes, consistent with findings from Arabidopsis. It is speculated that different members of the gene family may perform specific biological functions in different plant tissues. However, further research is needed to elucidate their exact roles in plant growth and development.
The structure and number of conserved motifs among members of the SWEET gene family were similar, and there was also a relative relationship between the evolution tree and gene structure. For example, ApSWEET11, ApSWEET12 and ApSWEET13 exhibited similar gene structures and belonged to the same clade. However, some gene structures displayed noticeable differences, suggesting the occurrence of expansion, reduction, or mutation during the evolution of the SWEET gene family. These variations may be associated with the diverse functions of the genes. The SWEET family members generally consist of five to six exons in their structures. It is speculated that the diversity of gene function may arise from the loss or addition of exons during the evolutionary process of these family genes. Additionally, all the members of SWEET gene family in A. polygama contained four highly conserved motifs: Motif1, Motif2, Motif4 and Motif5. These conserved motifs may play a key role in the biological function of the SWEET protein of A. polygama.
The expression patterns in different organs are closely correlated with gene function and serve as a predictor of biological functions. Numerous studies have reported the involvement of SWEET genes in various physiological processes, which usually were associated with specific tissue expression patterns. Our results also revealed that some SWEET genes were ubiquitously expressed in the flower, fruit tissues, root, leaf and stem. For example, ApSWEET5 and ApSWEET11 were highly expressed in flower. Some studies have reported that SWEET genes may play an important role in reproductive development. For example, AtSWEET13 and AtSWEET14 were found to be expressed in the anther wall, responsible for facilitating sucrose efflux into locules to support pollen development and maturation[37], and mutations in AtSWEET9 had been shown to impair nectar secretion[38,39]. These genes were specifically expressed in pollen. Similarly, OsSWEET11 and SbSWEET9-3 were highly expressed in the panicle[40, 41], suggesting that these genes may be essential for reproduction. Additionally, ApSWEET1 and ApSWEET5 exhibited specific expression in fruit. Similarly, high expression of VvSWEET transporters in flowers and berries highlighted a putative important role in sugar partitioning during flower and fruit development[4]. Furthermore, ApSWEET3 and ApSWEET10 displayed high expression in the roots (Fig. 5), while ApSWEET2, ApSWEET9, and ApSWEET10 were relatively highly expressed in the leaves and stem, similar to the function of AtSWEET17 as fructose transporter[42], these genes were proposed to participate in the phloem loading of photoassimilate in leaves of A. polygama. Our results suggested these genes may be involved in flower development, as well as the short and long-distance transportation and distribution of sugars. However, the regulatory mechanisms underlying the expression of these genes require further clarification.
In fruit, sugar (such as glucose, sucrose and fructose) is an important index that determines the quality of fruit. Many studies have reported functions of SWEET genes in both sink and source organs, particularly in fruits like tomato, grape, apple and Chinese jujube. The different expression patterns of the SWEET gene during fruit development are closely related to their function and can be used to predict biological functions. For instance, in apples, the expression of SWEET genes at young (MdSWEET1.1/2, MdSWEET2.4 and MdSWEET3.5) and ripe fruit development stages were different[23]. In grape, the expression of VvSWEET10, VvSWEET12, and VvSWEET15 was higher in young fruit, but VvSWEET15 was more abundant in mature fruit[28]. Furthermore, SlSWEET7a, SlSWEET14 and SlSWEET15 in tomato fruit are responsible the for transportation of glucose, fructose and sucrose[43], and VvSWEET4 acted as a glucose transporter in grape[28]. In our study, seven ApSWEET genes were expressed at five different fruit development stage. Among them, three ApSWEET genes exhibited high expression at the early stage of fruit development, and two ApSWEET genes showed high expression in ripe fruit. Moreover, there is a certain correlation between gene expression and homology, with some genes belonging to the same clade as ApSWEET1, VvSWEET10, VvSWEET11 and VvSWEET15[4, 23]. On the basis of sugar content during fruit development, it was also speculated that ApSWEET1 and two ApSWEET (ApSWEET2 and ApSWEET3) may be involved in the transportation and distribution of sucrose and fructose during fruit ripening, while ApSWEET10 and ApSWEET11 may play a role in transportation and distribution of glucose during fruit ripening.
-
Sampled plants were grown outdoors on the campus of Jilin Agricultural University, Jilin, China (43°48'48'' N, 125°24'15'' E). The annual precipitation is 867 mm, and the annual highest and lowest temperatures are 35 and −40 °C, respectively. Each field plot was divided into three subplots, and the seedlings were planted in each plot (3 m × 4 m). The phosphorous (4.12 ± 0.31 g/Kg), nitrogen (32.17 ± 1.98 g/Kg) and potassium (3.51 ± 0.19 g/Kg) concentrations were sufficient. Seedlings in all experiments were of uniform size. The expression of ApSWEET genes was detected in A. polygama has different tissues and fruits at different development stages. Leaf, flower, root, stem, and fruit were collected. Fruit collected at 10, 40, 60, 80, and 85 d after full bloom respectively. After the mature fruits are harvested, fruit were stored for 1, 2, 3, 4, and 5 d. All fresh plant samples were collected with three independent replicates and immediately frozen in liquid nitrogen, then stored at −80 °C.
Identification of gene family members
-
Actinidia polygama SWEET gene family was identified by protein Blast of the 17 Arabidopsis SWEET proteins against the Actinidia polygama genome database (
https://figshare.com/s/f46aea0009a54a6a0528 ).The NCBI CDD (
www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi ) and PFAM (http://pfam.sanger.ac.uk/ ) website were used to search for the conserved domains of the candidate members.Physicochemical properties and structural analysis
-
For the protein sequences encoded by the gene family members obtained above, Expasy (
http://web.expasy.org/ ) was used to predict their molecular weight, isoelectric point, stability and other physicochemical properties, respectively. WoLFPSORT (www.genscript.com/wolf-psort.html ) was used to predict protein subcellular localization, and TMHMM (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 ) used to predict transmembrane structure. The gene structure of SWEET gene family members of Actinidia polygama was analyzed according to the location information of introns and exons on chromosomes. MEME was used to predict the motif of the protein-conserved domain. The analysis results were visualized using TBtools and modified by AI. The online software SOPMA (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html ) was used to predict the secondary structure of SWEET protein, including random curling, chain extension structure, alpha helix and beta-turn. Using the SWISS-MODEL to analyze homology modeling (www.swissmodel.expasy.org ).Construction of molecular evolutionary tree
-
The MUSCLE was used for sequence comparison of the gene family members and was beautified with GENEDOC. The comparison results were clipped with trim Al and analyzed with IQ-TREE evolutionary tree. Finally, the exhibited (
http://tree.bio.ed.ac.uk/software/figtree/ ) for the beautification of the illustration.Chromosome localization and collinearity analysis of gene family
-
Based on the whole genome and location information downloaded from the Actinidia polygama genome database (
https://figshare.com/s/f46aea0009a54a6a0528 ), TBtools was used to conduct chromosome localization, collinearity, and gene tandem repeat event analysis for all SWEET gene family members of Actinidia polygama, and the results were visualized. MCScan was used to compare the whole genome sequence of Actinidia polygama, and the collinearity relationship was obtained. The homologous gene map was drawn with TB tools.Gene family promoter analysis
-
The 2 kb nucleotide sequences upstream of the transcription starting points of 14 genes of the gene family were predicted using PlantCARE, and TBtools software was used for visualization.
RNA extraction and real-time PCR
-
To validate the reliability of RNA-Seq, qRT-PCR for transcripts was carried out as described by Yu et al.[44]. Total RNA was extracted from leaves, flowers, root, stem, and fruit sampled simultaneously. For each experiment, 1 µg of clean RNA was converted to cDNA using the PrimeScript™ RT reagent Kit (TaKaRa Bio., Dalian, China) according to the manufacturer's protocol. Gene-specific primers were designed using Primer5.0 software (Premier Biosoft). Gene expression was performed using the SYBR Green Real-time PCR kit (TaKaRa Bio). ACTIN was used as a housekeeping gene after examining its constitutive expression pattern from the RNA-seq results. Relative gene expression levels were calculated with the 2−ΔΔCᴛ method[45]. The sequences of the primers used for qRT-PCR are listed in Supplemental Table S4.
-
The authors confirm contribution to the paper as follows: study conception and design: Wang ZX, Wang YP; performing the research: Chen L, Song HF, Liu JX, Jiang XX, Ai J. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
This study was supported by the Natural Science Foundation of China (to ZXW, GLS and JA, 31870673) and Jilin Province Development and Reform Commission, Grant/Award (Number: 2022C037-1); Department of Science and Technology of Jilin Province, Grant/Award (Numbers: 20210204083YY, 202101013697JC). This work complies with Chinese law. We thank Professor Ya-dong Li from the Jilin Agricultural University, Changchun, for providing recommendations for data collection.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Li Chen, Hui-Fang Song, Jia-Xin Liu
- Supplemental Table S1 The protein sequences of SWEET genes from A. polygama.
- Supplemental Table S2 The protein sequences of AtSWEETs.
- Supplemental Table S3 The protein sequences of VvSWEETs.
- Supplemental Table S4 The primer sequences of ApSWEET genes for qRT-PCR.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Chen L, Song HF, Liu JX, Jiang XX, Ai J, et al. 2024. Genome-wide identification and expression profiling of the SWEET family in Actinidia polygama (Sieb. & Zucc.) Maxim.. Fruit Research 4: e017 doi: 10.48130/frures-0024-0010 

Genome-wide identification and expression profiling of the SWEET family in Actinidia polygama (Sieb. & Zucc.) Maxim.
- Received: 11 December 2023
- Revised: 10 February 2024
- Accepted: 17 February 2024
- Published online: 06 May 2024
Abstract: Sugar was transported from photosynthetic source cells to sink cells, sugar efflux transporter protein (sugars will eventually be exported to transporters, SWEETs) play an important role in the process. Although SWEET family members had been identified in many plants, transcriptome or genomics analysis of Actinidia polygama SWEET genes remains uncharacterized. In this study, 14 SWEET genes of Actinidia polygama were identified by protein Blast. The structural characteristics of SWEET genes showed that the number of amino acids encoded by the gene family was between 233 and 304, the relative molecular weight was between 25,918.83 and 33,192.12, the isoelectric point was within the range of 6.96 to 9.71, 14 ApSWEET from Actinidia polygama and the known grape and Arabidopsis SWEETs were divided into four clades (I, II, III, and IV) according to the phylogenetic relationships. The gene structure analysis showed that most of ApSWEET genes have six exons and five introns except ApSWEET5 and ApSWEET14. All ApSWEET proteins also contained P-loop, MtN3-slv, and transmembrane domain. Expression patterns of 14 ApSWEET in different organs and at different fruit developmental stages were analyzed. ApSWEET1 and ApSWEET5 exhibited tissue-specific expression, whereas other genes were more ubiquitously expressed. ApSWEET1, ApSWEET10, and ApSWEET11 exhibited higher expression in fruit. The results of this study provide insights into the characteristics of the SWEET genes in Actinidia polygama and may serve as a basis for further functional studies of such genes.
-
Key words:
- Actinidia polygama /
- SWEET gene family /
- Sugar transport /
- Gene expression analysis