









-
To ensure timely floral transition, plants have acquired an intricate network of genetic pathways driven by various environmental and developmental cues[1]. Flower development takes place over three main phases, including (i) signal integration, (ii) meristem determination, and (iii) organ determination[2]. Key regulators directing each of these phases have been extensively studied in Arabidopsis, including the identification of the floral signal integrator SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (SOC1)[3].
SOC1 has been shown to act as an activator of floral transition in many plant species[4−8]. In Arabidopsis, SOC1 is directly regulated by an upstream floral repressor complex, involving FLOWERING LOCUS C (FLC) and SHORT VEGETATIVE PHASE (SVP), which acts in response to flowering signals received from the vernalization and autonomous pathways[9]. Contrarily, SOC1 is activated through the photoperiodic pathway using the central floral regulator CONSTANS (CO), via the activity of FLOWERING LOCUS T (FT)[10]. In addition, SOC1 has been shown to interact with AGL24, another floral signal integrator, to induce floral meristem identity through the activation of LEAFY (LFY) which, in turn, drives part of the floral organ determination phase[11,12]. During the early stages of floral initiation and meristem determination, SOC1 acts redundantly with SVP and AGL24 to prevent the precocious differentiation of floral organs by directly repressing SEPALLATA3 (SEP3)[2,13].
In Arabidopsis, floral organ determination is driven by organ identity genes which are categorised as either A-function (APETALA1 [AP1] and AP2), B-function (AP3 and PISTILLATA [PI]), C-function (AGAMOUS [AG]) or E-function (SEP1-4), according to the ABCE flower model[14,15]. Each of these genes, with one exception (AP2), encode for MADS-box proteins which interact with each other to form different combinations of multimeric complexes that drive the development of each floral organ type, namely sepals (A and E), petals (A, B, and E), stamens (B, C, and E), and carpels (C and E)[15].
Flower development differs greatly between Arabidopsis and grapevine (Vitis vinifera) with regards to flower anatomy, floral inducive signals, and lifecycle (reviewed in Carmona et al.[16]), and, as such, variations in their flowering gene networks are to be expected. Nevertheless, it has recently been reported that the expression patterns of Arabidopsis and grapevine floral organ identity gene homologs are well conserved[17]. In addition, previous research characterizing grapevine homologs of the FT and SVP genes indicated similar flowering functions to those held in Arabidopsis[18,19], suggesting that key regulators of the general flowering pathway may be partly conserved between these two plant species.
VviSOC1a, a SOC1 homolog of grapevine, has previously been shown to reduce flowering time in Arabidopsis, thereby identifying as a possible floral activator involved in signal integration[18]. Currently, however, no research efforts have been made in identifying VviSOC1a downstream targets or characterizing its gene regulatory network. In addition, its potential function in floral organ identity has not yet been explored. To better understand the role of VviSOC1a in flowering regulation, this study conducted phenotypic and gene expression analyses in overexpressing (OE) tomato (Solanum lycopersicum) lines. The association gene network and expression profile of VviSOC1a were also investigated.
-
Protein sequence alignments were performed with CLUSTALW[20] on the MEGAX software[21]. A phylogenetic tree was subsequently constructed using the Neighbor-Joining method[22] and p-distance model[23], with bootstrap values calculated for 100 replicates[24]. The list of VviSOC1a associated genes, computed using the Vitis OneGenE method[25], was retrieved by querying the
http://ibdm.disi.unitn.it/onegene/vv/onegene-vv.php andhttp://vitis.onegenexp.eu websites with the V1 gene ID (VIT_15s0048g01250). Genes were selected according to relative frequency values (FreI > 0.5) and used for Gene Ontology (GO) enrichment analysis on the ShinyGO 0.77 database[26], under default settings and false discovery rates (FDR) < 0.05. Developmental and tissue-specific expression data is based on the grapevine expression atlas[27]. Hierarchical clustering of expression data was created using the Expression Atlases application within the Vitis Visualization (Vitviz) platform (www.vitviz.tomsbiolab.com ).Plant transformation and growth conditions
-
The coding sequence of VviSOC1a was PCR-isolated from cultivar 'Pinot Noir' complementary DNA (cDNA) and cloned into pCXSN, driven by the constitutive CaMV 35S promoter[28]. Primer sequences used for gene isolation are listed in Supplemental Table S1. Transgenic tomato lines were generated through Agrobacterium-mediated transformation of cultivar 'Ailsa Craig' cotyledons as previously described[29], with the exception of using zeatin (2 μg/mL) and indole-3-acetic acid (IAA) (0.01 μg/mL) as plant hormones for shoot regeneration. Growth chamber conditions for plant regeneration were set at ± 23 °C and a 16-h photoperiod. Transgenic plants were transplanted in potting soil and grown in a greenhouse equipped with supplemental lighting (200−250 μmol/m2/s), for further use in gene expression and phenotypic analysis. As reference plants, ripening inhibitor (rin) mutants were grown up concurrently under the same conditions described above[30].
Gene expression analysis
-
RNA was extracted using the Spectrum™ Plant Total RNA Kit (Sigma-Aldrich, Burlington, MA, USA) and On-Column DNase I Digestion Set (Sigma-Aldrich, Burlington, MA, USA), according to the manual. First-strand cDNA was synthesized with an UltraScript 2.0 cDNA Synthesis Kit (PCR Biosystems, London, England). Real-time qPCR analysis was performed using gene-specific primers and qPCRBIO SyGreen Mix (PCR Biosystems, London, England) on a ViiA 7 instrument (Applied Biosystems, CA, USA) under default parameters. For transgene quantification, three plants were analyzed per independently transformed line, while for downstream target genes, one plant (each) was used for three independently transformed lines. Two technical replicates were performed per sample. Relative expression levels were calculated using the LinRegPCR program for amplification data analysis[31] and the delta-delta Ct method[32]. The qPCR data was normalized against Expressed (Exp) gene expression[33]. Primers used for the qPCR experiments are listed in Supplemental Table S1.
Chlorophyll and carotenoid quantification
-
At the anthesis stage, flowers were sampled and pooled from three plants per independently transformed line. Photosynthetic pigments were extracted from 50 mg of fresh petal tissue with 600 μL of pre-chilled acetone : water 80:20 (v/v), according to a previously described protocol[34]. Absorbance (A) was measured at 470, 647, 663, and 750 nm, using the Synergy™ 2 Plate Reader (BioTek, Winooski, VT, USA). Total chlorophyll a/b and carotenoids were calculated as suggested[34]:
$ \mathrm{Chlorophyll}\; \mathit{a} \;(\mathrm{C} \mathit{a} )\;({\text μ}{\rm g/mL})=12.25 \mathit{A} _{ \mathrm{663}} -{2.79} \mathit{A} _{ \mathrm{647}} $ $ \mathrm{Chlorophyll}\; \mathit{b} \;(\mathrm{C} \mathit{b})\; ({\text μ}{\rm g/mL})=21.50 \mathit{A} _{ \mathrm{647}}- \mathrm{5.10} \mathit{A} _{ \mathrm{663}} $ $ \mathrm{Carotenoids\;({\text μ}{\rm g/mL})=(1,000} \mathit{A} _{ \mathrm{470}} -\mathrm{1.82C} \mathit{a}- \mathrm{85.02C} \mathit{b} \mathrm{)/198} $ Statistical analysis
-
A two-tailed t-test was performed to compare means, where p ≤ 0.1, p ≤ 0.05, and p ≤ 0.01, were considered low, moderate, and high significant differences, respectively.
-
Molecular phylogenetic analysis grouped the VviSOC1a protein with previously characterized SOC1 clade members of the Type II MIKCc MADS-box family (Fig. 1). VviSOC1a shared highest amino acid similarity with SlTM3 (63%), PsSOC1 (58%), GhSOC1 (55%), and FaSOC1 (53%), which have primarily been shown to regulate flowering time, in addition to having roles in meristem branching control[6,7,35,36].

Figure 1.
Phylogenetic analysis of VviMADS8/SOC1a with functionally characterised proteins of the Type II MIKCc MADS-box family SOC1 clade, including SlSISTER OF TOMATO MADS-box gene 3 (SlSTM3, Solyc01g092950), SlTM3 (XP_025887600), PsSOC1 (AHJ60268), GhSOC1 (NP_001314583), FaSOC1 (AFR90178), SaMADSA (U25696), AtSOC1 (NP_182090), GmSOC1-like (NP_001236377), MtSOC1a (XP_003623808), ZmSOC1 (NP_001105152), OsMADS50 (NP_001388955), TaSOC1 (AB281427), OsMADS56 (NP_001390992), DnAGL19 (AMO66151), and DoSOC1 (AGK07583), as well as the closely related VviSOC1b (VIT_16s0022g02400) and VviSOC1c (VIT_02s0025g04650). AtAP1 (NP_177074) was selected as the outgroup. The scale bar represents the number of amino acid differences per site. The tree was generated using the Neighbor-Joining method and p-distance model[22,23]. Numbers at each node indicate bootstrap values calculated from 100 replicates[24]. At, Arabidopsis thaliana; Dn, Dendrobium nobile; Do, Dendrobium orchid; Fa, Fragaria × ananassa; Gh, Gossypium hirsutum; Gm, Glycine max; Mt, Medicago truncatula; Os, Oryza sativa; Ps, Paeonia suffruticosa; Sa, Sinapis alba; Sl, Solanum lycopersicum; Vvi, Vitis vinifera; Ta, Triticum aestivum; Zm, Zea mays.
To further explore the predicted function of VviSOC1a in floral transition, GO enrichment analysis of the VviSOC1a association gene network, computed using the Vitis OneGenE method[25] (Supplemental Table S2), was performed. From this analysis it was found that genes interacting with VviSOC1a were significantly enriched in biological processes related to flower development and vernalization response (Supplemental Fig. S1). Of particular interest were 14 grapevine genes that were identified as putative homologs of regulators involved in the general flowering pathway of Arabidopsis[2,37], including those related to floral signal integration (VviSOC1b, VviSOC1c, and VviSVP2-7) and floral meristem determination (VviFRUITFULL1 [VviFUL1]), as well as genes predicted to regulate floral organ identity in grapevine (VviAG1, VviAG2, VviAP3b, VviFUL1, VviSEP1, and VviSEP3)[17] (Fig. 2). Another important gene is the TERMINAL FLOWER1 (TFL1) homolog, VviTFL1, which was previously characterized as a repressor of floral development[38].
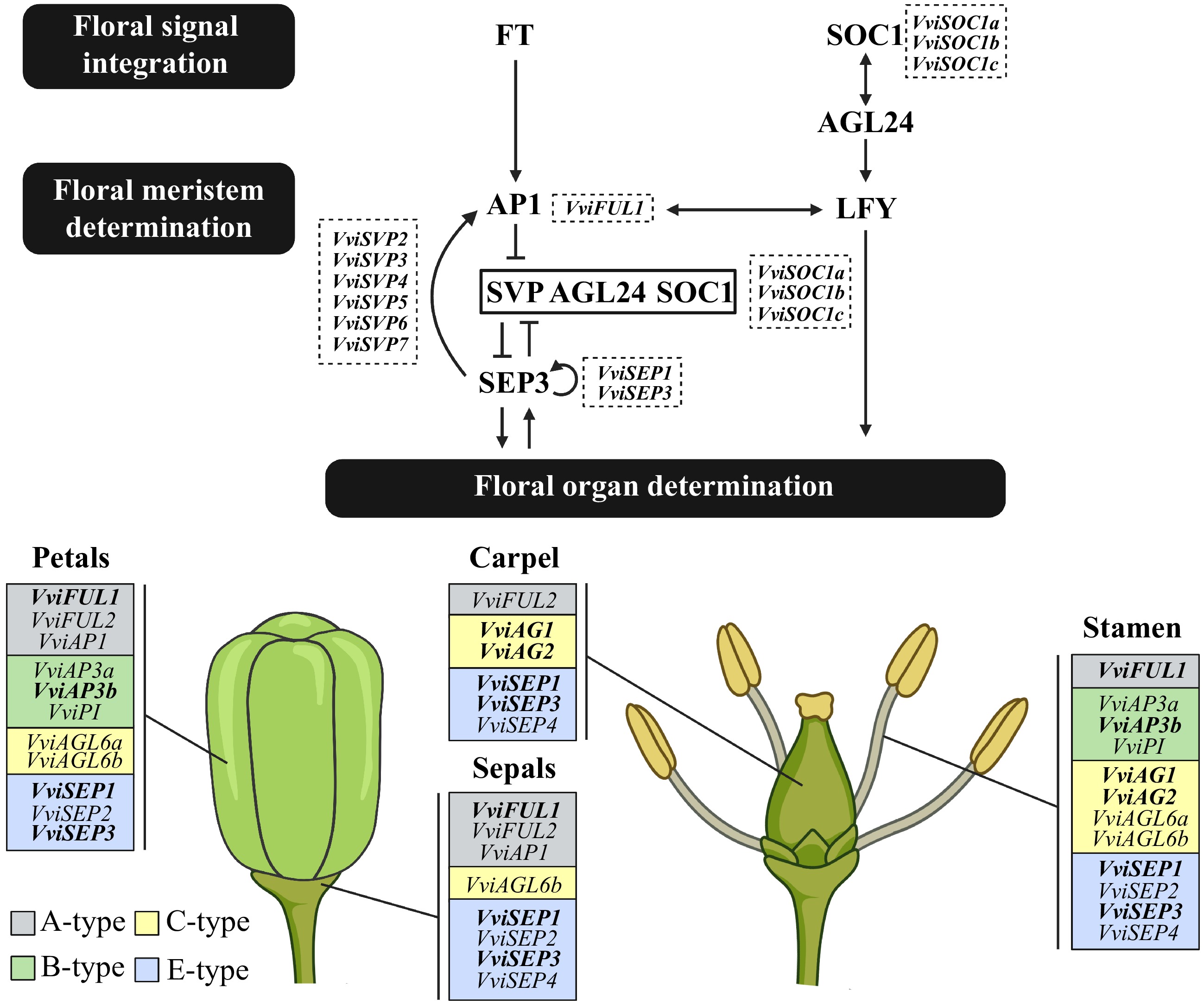
Figure 2.
Schematic diagram highlighting the flowering-related genes (featured in bold) present in the VviSOC1a association network, computed using the Vitis OneGenE method. This general flowering pathway, from floral signal integration to floral organ determination, is adapted from Wagner[37], Lee & Lee[2], and Palumbo et al.[17]. Grapevine homologs of floral signal integration and meristem determination genes are placed in the dotted boxes, while those associated with floral organ determination are categorised as either A-type (grey boxes), B-type (green boxes), C-type (yellow boxes) or E-type (blue boxes), based on the predicted ABCE flower model[15]. Created using
BioRender.com .Analysis of genome-wide transcriptomic data across 54 grapevine tissues and developmental stages[27] revealed that VviSOC1a is predominantly expressed during vegetative growth, except the seedling stage (Supplemental Fig. S2). In particular, VviSOC1a expression tends to be highest in younger tissue types, for example in younger leaves and stems, as well as in the latent buds and during budburst. VviSOC1a is also strongly expressed during early flowering, and especially in pollen grains, but has little to no expression during berry development or ripening (Supplemental Fig. S2). When comparing expression profiles, VviSOC1a shares similar trends to associated putative grapevine homologs involved in floral signal integration and meristem determination (Fig. 2), where expression mainly occurs in the vegetative and flowering tissues, and is mostly absent at fruit stages (Supplemental Fig. S3). Contrarily, putative grapevine homologs involved in floral organ determination display opposite patterns, in that gene expression occurs almost exclusively in the reproductive tissues, such as during later flowering stages and throughout berry development (Supplemental Fig. S3).
Overexpressing VviSOC1a leads to floral reversion and sepal elongation
-
Transgenic tomato lines showed significant changes to flower morphology, where the most striking phenotype observed was the formation of leaf-like sepals (Figs 3 & 4). In severe cases, impacts on normal floral development, such as absent, dwarfed, and loosely packed reproductive organs, resulted in plant sterility (Fig. 3a−h). Three VviSOC1a-OE tomato lines showing comparable, albeit weaker, floral reversion phenotypes were further assessed (Fig. 3i−k & Supplemental Fig. S4). For the two higher expressing lines (Fig. 3i, j), a significant elongation in curly sepal growth was observed compared to the wild type (Fig. 3l & Supplemental Fig. S5), but not in the case of the lowest expresser (Fig. 3k). This was particularly evident when comparing isolated floral organs (Fig. 4a−c). In addition, the extensive sepal growth found in the highest expresser (Fig. 4a) resembled the rin mutant flower (Fig. 4d). Apart from alterations to the sepal size, there was a notable development of greener sepaloid petal tissue in the transgenic flowers (Fig. 4a−c). To further assess petal color, photosynthetic pigment quantification of all three independently transformed lines (Fig. 4e), as well as the two highest expressing lines (Fig. 4f), confirmed that a significant increase in total petal chlorophyll (a + b) content was observed for the higher expressers (Fig. 4e). Post-anthesis, transgenic lines showed further sepal elongation, however, did not display noticeable changes in length between different mature fruit stages (Fig. 5a, b). As observed with the flowers, the red-ripe fruit sepal phenotype of the highest expresser (line 3) was comparable to the rin mutant (Fig. 5b).

Figure 3.
Whole-flower phenotypes of tomato lines overexpressing VviSOC1a. (a)−(h) Transgenic lines developed leaf-like sepals and displayed severe floral dwarfing phenotypes resulting in plant sterility. Transgenic lines displayed moderate floral phenotypes of (i), (j) sepal extension and curling compared to (k) line 6 and (l) wild type plants.
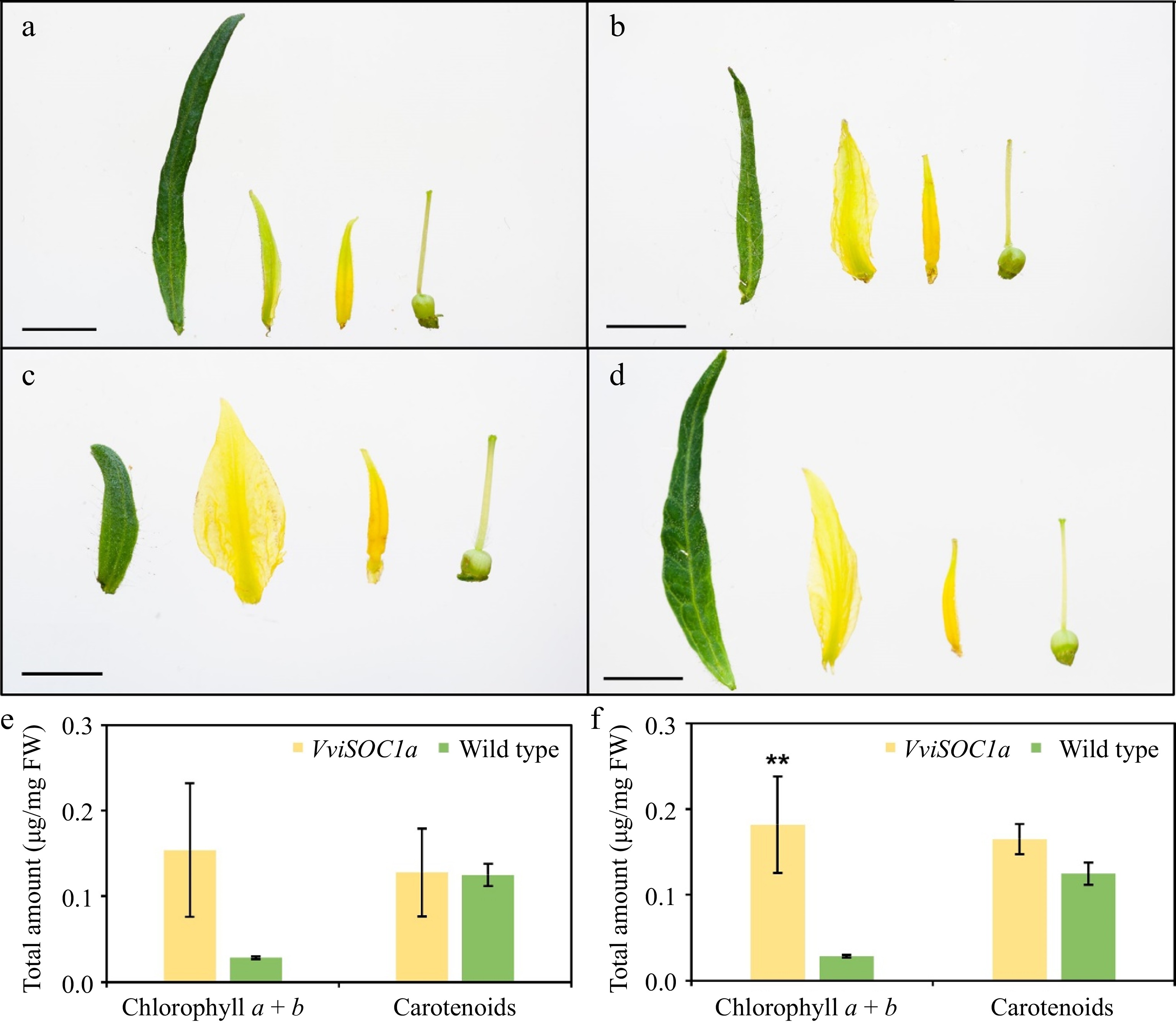
Figure 4.
Floral organ phenotypes of tomato lines overexpressing VviSOC1a. Dissected floral organs (left to right: sepal, petal, stamen, and carpel) of (a) line 3 and (b) line 2 developed greener petals than (c) wild type, as well as (d) longer sepals that resemble the flowers of the rin mutant[30]. Scale bars represent 0.5 cm. Total chlorophyll (a + b) and carotenoid petal content of (e) three independently transformed lines and (f) two high expressing lines. Bars represent the mean and standard error of three biological replicates (n = 3), where (**) indicates moderate (p ≤ 0.05) significant differences compared to wild type.

Figure 5.
Fruit sepal phenotypes of tomato lines overexpressing VviSOC1a. Sepal development is displayed at two different fruit developmental stages, including (a) mature green and (b) red ripe. (a) Elongated sepals observed for lines 2 and 3 compared to line 6 and wild type. (b) Comparable sepal extension observed for line 3 and the rin mutant[30] in relation to wild type.
VviSOC1a represses flowering and ripening-related genes
-
Gene expression analysis in young leaf (Fig. 6a) and floral sepal (Fig. 6b) tissues showed that VviSOC1a expression repressed the downstream tomato AP1/FUL (SlFUL2 and SlMACROCALYX [SlMC]), SVP (SlJOINTLESS [SlJ]), and SEP3 (SlCMB1, SlRIN, and SlMBP21) homologs, which are well-characterized flowering genes in Arabidopsis[2], in addition to the SOC1 homolog, SlSOC1-like. However, the down-regulation of these targets was tissue-dependent. It was further observed that the overexpression of VviSOC1a down-regulated genes involved in ripening (Fig. 6a, b), including SlCHALCONE SYNTHASE1 (SlCHS1), SlMYB12, and SlRIN[39−41], as well as SlFUL2, which regulates both flowering and ripening[42,43]. Since each of these ripening-related genes has been shown to regulate and/or impact cuticle deposition, the transcript levels of important cuticle regulators and biosynthetic enzymes were also analyzed in the leaf tissue (Fig. 6c). It was found that cuticle regulators, SlMIXTA, and SlSHINE3 (SlSHN3)[44], were significantly repressed in VviSOC1a-OE lines, along with the downstream target gene SlCYP77. Contrarily, VviSOC1a was observed to induce the expression of other cuticle-related genes involved in the biosynthetic pathway, namely SlLACS2 and SlGDSL1 (Fig. 6c). Significantly down-regulated genes that are associated with sepal size regulation in tomato[45−47] are highlighted in Fig. 6d. Importantly, these genes are homologous to flowering pathway members that form part of a positive feedback loop that is repressed in the presence of SOC1[2,37].
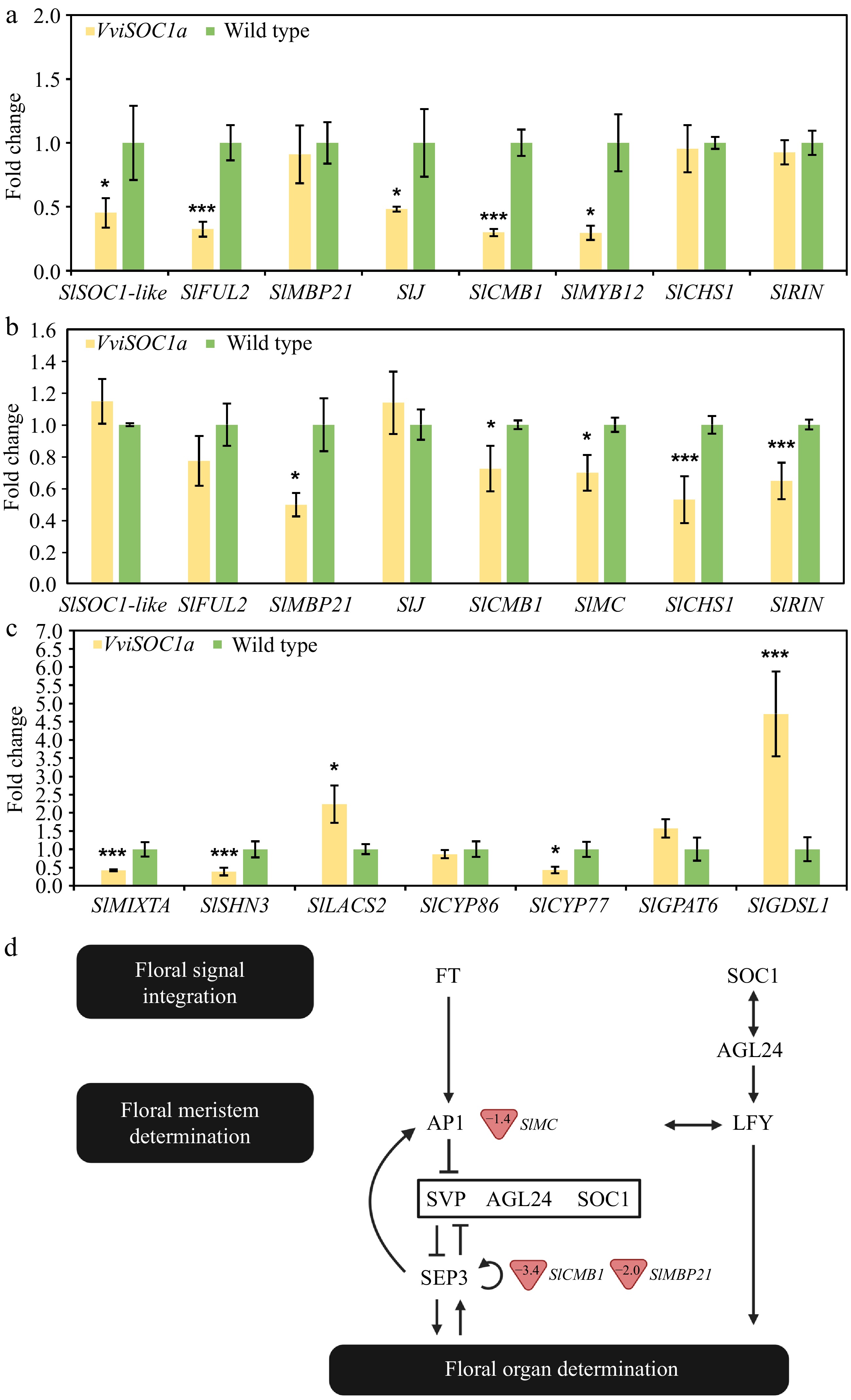
Figure 6.
Expression analysis of putative VviSOC1a downstream target genes in acclimated overexpressing tomato lines using real-time qPCR analysis. Flowering and ripening-related gene expression in (a) young leaves and (b) open-flower sepals. (c) Cuticle-related gene expression in young leaf tissues. Bars represent the mean and standard error of three independently transformed biological replicates (n = 3), where (*) and (***) indicate low (p ≤ 0.1) and high (p ≤ 0.01) significant differences compared to wild type, respectively. (d) Schematic diagram of the general flowering pathway[2,37] showing tomato homologs involved in sepal size regulation with altered expression in the VviSOC1a-OE lines. Values in red triangles represent fold change.
-
The MADS-box SOC1 protein plays a vital function in the integration of external stimuli, such as those derived from temperature, photoperiod, hormone, and age-related pathways, to drive timely floral transition[2]. SOC1 gene homologs, in a multitude of plant species, have been shown to hold conserved functions in positively regulating the flowering time, causing hastened or delayed flowering phenotypes in constitutively expressed or knockout lines, respectively[4−8,35,36,48−52]. These studies include the SOC1 homologs of tree peony (PsSOC1), cotton (GhSOC1), and strawberry (FaSOC1), which were found to share the highest protein homology (53%−58%) with VviSOC1a. While VviSOC1a has previously been reported to cause early bolting in Arabidopsis[18], the current study found no obvious changes to flowering time in VviSOC1a-OE tomato lines. However, SOC1 may hold divergent functions in tomato, since two previously characterized tomato SOC1 gene homologs, SlSTM3 and SlTM3, are involved in the promotion of floral meristem branching[36], as opposed to the repressive role held by SOC1 in Arabidopsis[53]. Further, only SlSTM3 displayed regulatory functions promoting floral transition, with SlTM3 expression (sharing higher sequence similarity with VviSOC1a, Fig. 1) having no effect on flowering time[36]. Nevertheless, VviSOC1a is expected to hold a function in floral signal integration in grapevine due to its increased expression in vegetative tissues, particularly young leaves and stems where floral inducive stimuli are perceived (Supplemental Fig. S2). VviSOC1a expression also persists into early flowering stages, although at a much lower level. This expression profile agrees with those described for SOC1 in Arabidopsis[54], and other crops such as kiwifruit (Actinidia spp.)[55] and cotton[7].
Floral organ specification is repressed by VviSOC1a through the down-regulation of floral organ identity genes
-
SOC1 holds another function during the early stages of floral meristem establishment in Arabidopsis, where it acts redundantly with SVP and AGL24 to negatively regulate organ identity genes[2]. This function is crucial for timely flowering and the prevention of premature organ differentiation. In relation to this, floral reversion phenotypes, including the development of enlarged leaf-like sepals and sepaloid-type petals, have commonly been reported for these three genes across multiple plant species[4,19,56−59]. Similarly, the overexpression of VviSOC1a led to sepal elongation and the formation of leaf-like sepals (in severe cases), as well as greener sepaloid petals, in tomato plants (Figs 3−5). This phenotype may be, in part, explained by the repressive activity of SOC1 on SEP3, an E-function floral gene[13]. SEP3 directly interacts with AP1, an A-function floral identity protein, to drive sepal and petal specification[56]. In tomato, as in Arabidopsis, SEP3 (SlCMB1) and AP1 (SlMC) gene homologs have been characterized as sepal size regulators since their knockdown or knockout has led to elongated sepal phenotypes[47,56,60,61]. This function is further supported by the well-known rin mutant, which is the product of a knockout mutation involving the fusion of SlRIN (an E-function SEP gene) and SlMC[30]. In this mutant, leaf-like sepals are formed that are comparable to those observed in VviSOC1a overexpression lines (Figs 4 & 5). SlCMB1 has also been shown to directly interact with another E-function protein, namely SlMBP21, which holds a similar sepal size regulatory role[47,62]. Since all four genes (SIMC, SlRIN, SlCMB1, and SlMBP21) were shown to be down-regulated in VviSOC1a-OE sepal tissue (Fig. 6), it may be assumed that their combined suppression at later stages of floral organ development is what led to the observed sepal phenotype. Further, it is expected that SlCMB1 acts as a key contributor to the development of sepal-like petals and defective plant organs which, in acute cases, lead to sterile VviSOC1a transgenic lines (Fig. 3). This is because SEP3 is known to activate, in concert with LFY, B-function, and C-function identity genes to specify the petal, stamen, and carpel organs (Fig. 2), with double mutants developing comparable floral organ defects and vegetative petals in Arabidopsis[13]. The repressive activity of VviSOC1a on the SlCMB1 gene in tomato is likely conserved in grapevine, given that VviSOC1a and VviSEP3 display opposite expression profiles across vegetative and reproductive tissues (Supplemental Fig. S3). Despite the redundant functions held by SOC1 and SVP in repressing organ identity (Fig. 2), these two genes play antagonistic roles in Arabidopsis during the early floral transition, as floral activators and repressors, respectively[9]. This may explain the significant down-regulation of the tomato SVP homolog, SlJ, observed in young leaf tissues (where floral induction takes place), but not in the sepals of the VviSOC1a-OE lines (Fig. 6).
VviSOC1a activity has a downstream impact on cuticle and ripening pathways
-
Since carpel specification precedes fruit development and ripening, it was speculated that VviSOC1a may impact the regulation of genes driving these later reproductive stages, through down-regulation of the SlCMB1 gene. In SlCMB1-silenced tomato lines, ripening-related genes, including SlRIN and SlFUL2 were significantly repressed, leading to delayed ripening phenotypes[63]. Although phenotypic ripening analyses were not conducted in the current study, these genes, in addition to those forming part of the flavonoid pathway (SlMYB12 and SlCHS), were also down-regulated in the VviSOC1a-OE tomato lines, supporting the likely impact of SlCMB1 suppression on ripening regulation (Fig. 6). However, gene expression was only analyzed in young leaves and sepals. Thus, confirmation of the relationship between VviSOC1a and ripening within a fruit background may be necessary to draw a more accurate conclusion. Previous studies have also shown that each of the ripening-related genes modulated in this study impact fruit cuticular properties linked to crop quality traits[39−42]. As such, further analyses of the transcript levels of well-known cuticle genes were also performed in young leaf tissues, revealing that VviSOC1a may also affect cuticle formation, though contrasting findings were observed. For example, key cuticle regulators (SlMIXTA and SlSHN3) were significantly down-regulated, while VviSOC1a activated the expression of genes coding for cuticle biosynthetic enzymes (SlLACS2 and SlGDSL1)[44,64,65] (Fig. 6). In conclusion, further characterization is needed to better understand the impact of VviSOC1a function on ripening and fruit cuticle formation.
VviSOC1a may hold a conserved perennial crop function in bud break regulation
-
Functional divergence has been reported for SOC1 homologs in woody perennial plants, such as grapevine, which display significantly different growth habits to Arabidopsis. For one, floral induction in grapevine is believed to occur in the latent primary buds during summer, while flower development takes place the following spring after bud burst, a phenological event requiring a vernalization period[16,66]. In the woody perennials kiwifruit and poplar (Populus tremula × alba), SOC1 homologs have been shown to promote early bud break in overexpressing lines[55,67]. Interestingly, SOC1 was also found to hold a function in determining the annual growth habit of Arabidopsis, with soc1 ful double mutants displaying perennial growth characteristics[68]. The heightened expression observed for VviSOC1a in latent buds supports its predicted function in floral induction (Supplemental Fig. S2). The fact that VviSOC1a also displays expression peaks during bud burst stages, suggests that it may hold an additional role in bud dormancy release as well, similar to other woody perennials, however, functional confirmation is required.
-
SOC1 is an important regulator of flowering that has primarily been associated with floral initiation, though, additional functions related to the prevention of precocious floral organ development and the control of bud dormancy release have also been reported. The grapevine gene homolog, VviSOC1a, has previously been shown to reduce flowering time in Arabidopsis. In the current study, a new function involving the repression of floral organ differentiation was identified through heterologous expression in tomato. This role is likely mediated through the suppression of SIMC, SlRIN, SlCMB1, and SlMBP21, gene expression. An additional function for VviSOC1a relating to the regulation of bud dormancy release, based on gene expression data, is further postulated, though, experimental confirmation is still required. Another interesting result is the repressive activity of VviSOC1a on ripening and cuticle-related genes, since these hold important functions related to crop stress tolerance and quality[69]. As such, future research may benefit from the further characterization of VviSOC1a to better understand its influence on these crop traits.
-
The authors confirm their contribution to the paper as follows: study conception and design: Jolliffe JB, Lashbrooke JG; data collection: Jolliffe JB; analysis and interpretation of results: Jolliffe JB, Lashbrooke JG, Moser C, Pilati S; draft manuscript preparation: Jolliffe JB. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
The Lashbrooke lab (Stellenbosch University) is funded by Winetech, South Africa. This work was funded by a FIRST grant from Fondazione Edmund Mach, Italy.
-
The authors declare that they have no conflict of interest.
- Supplemental Table S1 Details of primers used for gene isolation and qPCR analysis.
- Supplemental Table S2 Details of genes present in the VviSOC1a association gene network.
- Supplemental Fig. S1 GO enrichment analysis of genes associated with VviSOC1a.
- Supplemental Fig. S2 VviSOC1a gene expression profile.
- Supplemental Fig. S3 Expression profile of VviSOC1a and associated genes.
- Supplemental Fig. S4 Transgene expression analysis of VviSOC1a in tomato lines.
- Supplemental Fig. S5 Sepal length data for VviSOC1a transgenic tomato lines.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Jolliffe JB, Moser C, Pilati S, Lashbrooke JG. 2024. The grapevine SOC1 homolog, VviMADS8/SOC1a, regulates floral organ specification in tomato. Fruit Research 4: e029 doi: 10.48130/frures-0024-0023 

The grapevine SOC1 homolog, VviMADS8/SOC1a, regulates floral organ specification in tomato
- Received: 04 April 2024
- Revised: 02 June 2024
- Accepted: 24 June 2024
- Published online: 09 August 2024
Abstract: The MADS-box protein SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (SOC1) is a key floral activator that coordinates external and internal stimuli to ensure timely floral transition. During early development, SOC1 represses floral organ identity to prevent premature differentiation and, thus, is also linked to the successful development of functional flowers. In woody perennials, SOC1 has established a divergent function in the regulation of bud dormancy release. Apart from reducing flowering time in Arabidopsis, little is known about the function of VviSOC1a and its gene regulatory network. In this study, VviSOC1a was functionally characterized through overexpression in tomato, where it was found to promote the development of leaf-like sepals and petals with an increased accumulation of chlorophyll. In severe cases, overexpression of VviSOC1a led to the formation of defective floral organs resulting in plant sterility phenotypes. Gene expression analyses revealed the significant downregulation of important floral organ identity genes in tomato, such as SIMC, SlRIN, SlCMB1, and SlMBP21. Additional downstream impacts on ripening and cuticle-associated gene expression warrant further characterization of VviSOC1a within the context of these crop traits. In silico analysis of the VviSOC1a expression profile revealed patterns distinctive of genes involved in floral induction. This, in combination with an association gene network significantly enriched in flower developmental processes, supports a predicted function for VviSOC1a in floral initiation and floral organ specification.
-
Key words:
- SOC1 /
- Grapevine /
- Flower /
- Organ identity /
- Transcription factor