








-
Annual bluegrass (P. annua L.) is a cool-season turfgrass species present on golf courses around the world. Past US Open championships have been played on courses where putting greens are primarily composed of P. annua, indicating its ability to perform as a quality turf[1]. Mild climates are most favorable to cultivating P. annua, such as that of the Pacific Northwest, where it can be grown as a desirable turfgrass[2]; however, its low tolerance to abiotic stress, such as heat stress, diminishes P. annua performance in transitional zones where temperatures in the summer exceed 30 °C. Due to these limitations, P. annua has historically been considered a weed in transitional zones. Its high genetic diversity and prolific seed head production allow P. annua to effectively colonize courses and become the dominant stand if left unimpeded[3]. In the past, efforts have been heavily focused on controlling P. annua. Recently a shift has begun in transitional zones where golf course superintendents are moving away from eradicating P. annua and are instead trying to cultivate it[4]. In such cases, it is common to see mixed greens consisting of P. annua and creeping bentgrass (Agrostis stolonifera), another commonly cultivated cool-season turf grass species. The dynamic between these two species depends on factors such as age and the season. As A. stolonifera greens age, P. annua infestations tend to increase. Fluctuations in dominance occur annually with P. annua being the dominant competitor during spring and fall, and A. stolonifera becoming more dominant in the summer[5].
Rising temperatures in summer months make maintaining putting greens with A. stolonifera and P. annua more challenging due to heat stress, one of the major factors limiting the growth of cool-season turfgrass species. Different species of cool-season turfgrass vary in their tolerance and response to heat stress[6,7]. This is seen in mixed putting greens where heat induced summer decline often occurs earlier in populations of P. annua than those of A. stolonifera. The uneven decline in mixed stands results in an unattractive putting green. Heat stress is detrimental to cool-season turfgrass growth, causing oxidative stress, reduction in cell water content, damage to photosynthetic machinery, and decline of carbohydrate availability[8−10]. To defend against heat stress, damaged plants will undergo adjustments in molecular and physiological processes to regain homeostasis. This includes the regulation of key metabolites, such as amino acids, organic acids, and sugars that could function directly as protectants or indirectly as precursors for stress defense[11−13]. Heat tolerance of creeping bentgrass has been associated with changes in various physiological and metabolic processes, including active carbohydrate and antioxidant metabolism[14−16]; however, little is known of the mechanism governing the response of P. annua to heat stress.
The objectives of this study were to compare physiological responses between P. annua and A. stolonifera and identify differential metabolites regulated by heat stress for A. stolonifera and P. annua to unravel the metabolic mechanisms associated with heat tolerance in these two cool-season grass species. As climate change threatens to increase the intensity and frequency of heat waves, the challenges associated with maintaining cool-season grasses will increase. Understanding the mechanisms for the differential responses of P. annua and A. stolonifera is of great importance for developing strategies to improve turfgrass performance of cool-season grass species in areas with chronic heat stress and anticipated global warming.
-
Mature turf sod was collected from Rutgers Horticulture Research Farm in North Brunswick, NJ, USA for two species: creeping bentgrass (Agrostis stolonifera L.) 'Declaration' and annual bluegrass (Poa annua). A. stolonifera was taken from a well-established field, and P. annua was taken from fields that were established using mixed biotypes originally collected from Rutgers University Golf Course (Piscataway, NJ, USA) and Plainfield Country Club (Edison, NJ, USA) for over four years. Sods with roots had the thatch layer removed and were planted in plastic containers (20 cm in width, 30 cm in length, and 20 cm in depth) filled with fritted clay (Profile Products, Buffalo Grove, IL, USA). Six sod pieces (three A. stolonifera and three P. annua) were placed randomly within each container and replicated within 16 containers. Plants were maintained for 30 d in a greenhouse to allow sods to establish and subsequently relocated to two controlled growth chambers (Environmental Growth Chambers, Chagrin Falls, OH, USA) with eight containers in one chamber and eight containers in the second. Growth chambers were set to the following optimal growing conditions, controlled at 22/18 °C (day/night) average temperatures, and 750 μmol·m−2·s−1 photosynthetically active radiation at the canopy level with a 14-h photoperiod. Plants were maintained in this growth chamber for 7 d before imposing temperature treatments.
Experimental design and temperature treatments
-
After 7 d of acclimation in the growth chambers, plants were exposed to the ambient temperature (22/18 °C, day/night) as non-stress control or 35/30 °C (day/night) for heat stress. Plants in each temperature treatment were randomly placed in four growth chambers with four replicated containers in each chamber for each species. Every container had three subsamples for each grass species (container). Plants were subjected to temperature treatments for 42 d. Plants were kept well-irrigated, watering twice daily to maintain soil saturation, indicated by drainage from the bottom of containers. Fertilization using half-strength Hoagland's solution was applied weekly.
The experiment design was a split-plot design with two temperature treatments as the main plots and two grass species as sub-plots. Each treatment had four replicates and three subsamples for each species.
Physiological analysis
-
Turf quality and physiological traits were evaluated every 7 d for both control and heat stress groups throughout the 42-d temperature treatment period. Visual turf quality (TQ) was rated on a 1−9 scale, considering uniformity, color, and density, where a rating of nine indicates healthy turf, six is the minimum quality accepted, and one indicates nearly dead turf[17]. Images were taken on a Galaxy Note9 smartphone HDR dual aperture camera (Samsung Electronics Co., South Korea) and analyzed using SigmaScan Pro 5 software (Systat Software Inc., San Jose, CA, USA) to quantify turf canopy cover (percent green canopy cover) based on Karcher & Richardson[18,19].
Cell membrane stability of leaves was analyzed through the measurement of leaf electrolyte leakage (EL). Approximately 0.2 g of fresh leaf tissue was harvested from each plant and cut into 1 cm long pieces. The tissue was then submerged in 30 mL of deionized water and left on a shaker for 8−12 h at room temperature until turgid, and electrolyte content was measured using a conductance meter (YSI Incorporated, Yellow Springs, OH, USA) to get an initial reading (Ci). Tubes were autoclaved to kill plant tissue and kept on the shaker to for an additional 8 h, and the solution was measured for conductance again to determine the maximum conductance (Cmax). Percent EL was calculated with the following formula by Blum & Ebercon: EL (%) = Ci/Cmax × 100%[20].
Metabolic analysis
-
Leaf tissue was collected from both species in the control and heat stress groups 42 d after heat stress was initiated. Leaf samples were freeze-dried and ground with a mortar and pestle. Then 20.0 mg of ground tissue was pooled from the three sub-plants of each container to get one metabolite sample for each species per container. Samples were stored at −80 °C until ready for metabolite analysis. Metabolite content was analyzed by LC-MS using the methods described in Errickson & Huang[21]. The 20 mg samples were resuspended in 1 mL 40:40:20 methanol:acetonitrile:water solution and left at ambient temperatures for a 10-min period. To neutralize sample mixtures, 50 μL of 15% (m/v) NH4HCO3 was added before diluting them four times in the methanol:acetonitrile:water solution. The diluted samples were centrifuged at 4 °C at 16,000 g for 10 min, and supernatant was subsequently moved into a fresh tube.
Metabolite identification and concentration were completed using a Vanquish Horizon UHPLC system (Thermo Fisher Scientific, USA) with an XBridge Amide column (150 mm × 2.1 mm, 2.5 μm particle size, Waters), using a gradient of solvent A (95%:5% H2O:acetonitrile with 20 mM acetic acid, 40 mM ammonium hydroxide, pH 9.4) and solvent B (20%:80% H2O:acetonitrile with 20 mM acetic acid, 40 mM ammonium hydroxide, pH 9.4). The gradient was 0 min, 100% B; 3 min, 100% B; 3.2 min, 90% B; 6.2 min, 90% B; 6.5 min, 80% B; 10.5 min, 80% B; 10.7 min, 70% B; 13.5 min, 70% B; 13.7 min, 45% B; 16 min, 45% B; 16.5 min, 100% B; and 22 min, 100% B. Separation was carried out at the following settings: flow rate at 300 μL·min−1, column temperature at 25 °C, autosampler temperature at 4 °C, and an injection volume of 5 μL. Mass spectrometry scans were obtained in both negative and positive ion modes with a revolution of 70,000 at m/z 200, and there was an automatic gain control target of 3 × 106 and m/z scan range of 72−1,000. Metabolite data was obtained using the MAVEN software package (mass accuracy windows: 5 ppm)[22].
Statistical analysis
-
Treatment differences for TQ, physiological traits, and metabolite content between the heat stress and control groups were compared for each species by the analysis of variance test (SAS v9.2). Significant differences between treatments were tested using Fisher's protected LSD at α = 0.05. Metabolite content from LC-MS was analyzed using a t-test to compare the heat stress of each species to the corresponding control at 42 d, where the fold-change (FC) threshold = 1.0 and p-value = 0.05. From this, the relative changes seen within each species were used to identify any differential responses between A. stolonifera and P. annua. Volcano plots, partial least squares discriminant analysis, heat maps, and metabolic pathway enrichment analysis were generated using the MetaboAnalyst 5.0 website with R statistical programs (
www.metaboanalyst.ca/MetaboAnalyst/ModuleView.xhtml ). -
Turf quality (TQ) of heat-stressed plants relative to that of control plants for each species (% of control) was compared between the two species to better elucidate their differential responses to heat stress due to the intrinsic and genetic variations in TQ for A. stolonifera and P. annua under normal or non-stress conditions (Fig. 1). Heat-stressed A. stolonifera had significantly higher TQ relative to its control plants, compared to that of P. annua during 21–42 d of heat stress. The decline in TQ during heat stress was more severe for P. annua than for A. stolonifera.
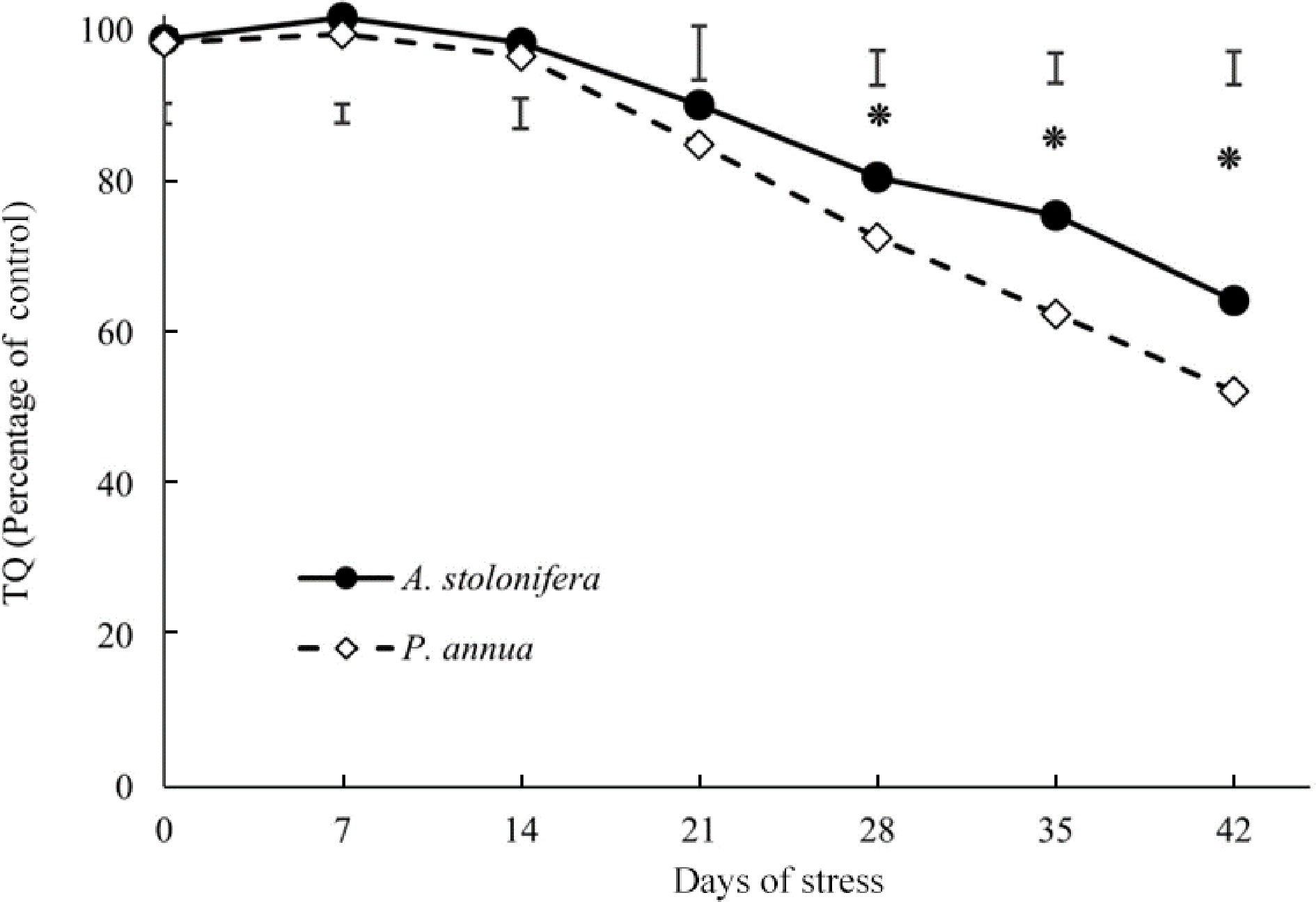
Figure 1.
Relative changes (% of control) in turf quality (TQ) of heat-stressed plants compared to the respective control plants in A. stolonifera and P. annua during 42 d of heat stress. Vertical bars indicate least significant difference (LSD) values demonstrating significant differences (*) between the two grass species (p < 0.05) at a given day of heat stress.
Percent green canopy cover of heat-stressed plants declined significantly below the non-stress control level for both P. annua and A. stolonifera during 28−42 d of heat stress. The decline in percent green canopy cover was more pronounced in P. annua than A. stolonifera during heat stress (Fig. 2).
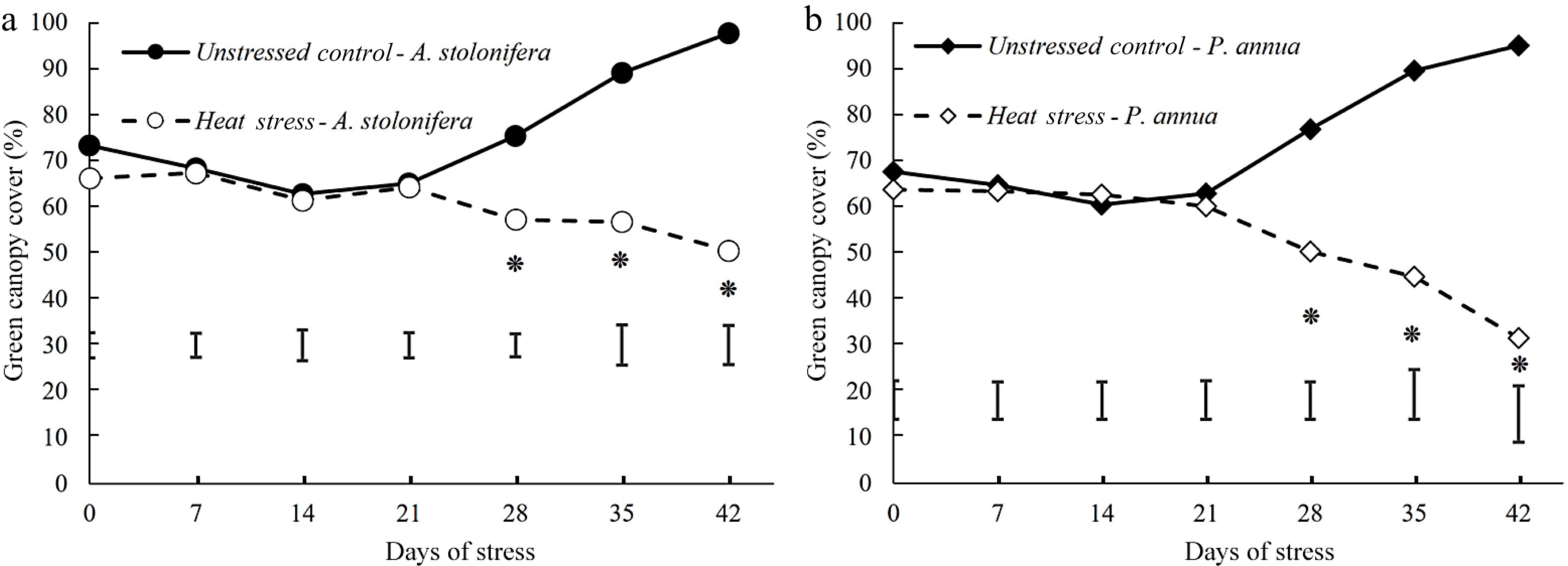
Figure 2.
Changes (%) of green canopy cover during 42 d of heat stress relative to their respective control plants for (a) A. stolonifera and (b) P. annua. Vertical bars indicate least significant difference (LSD) values demonstrating significant differences (*) between the two grass species (p < 0.05) at a given day of heat stress.
Leaf electrolyte leakage (EL) increased to a significantly higher level in heat-stressed plants from 21 to 42 d in P. annua. The increases in EL induced by heat stress in P. annua were more severe than those of A. stolonifera, as the significantly higher EL in heat-stressed plants relative to the control plants did not occur until 42 d in A. stolonifera. The present increases in EL due to heat stress were greater in P. annua than those in A. stolonifera (Fig. 3).
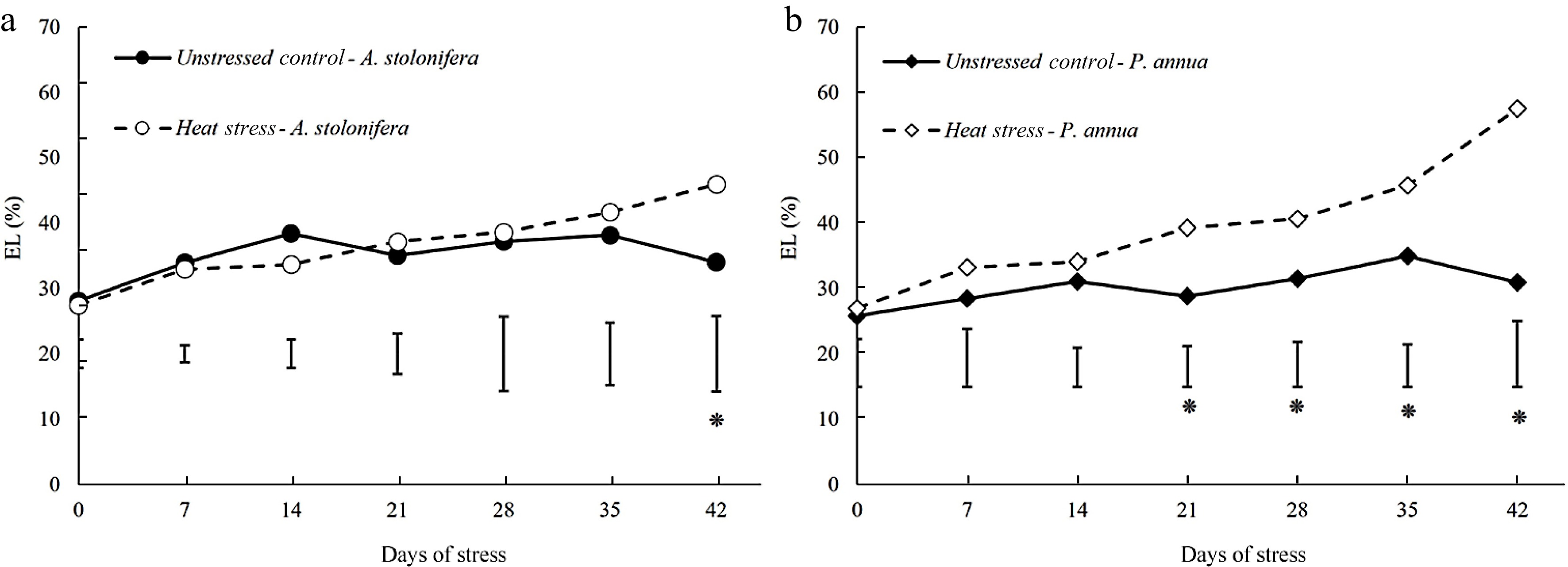
Figure 3.
Changes in electrolyte leakage (EL) during 42 d of heat stress relative to their respective control plants for (a) A. stolonifera and (b) P. annua. Vertical bars indicate least significant difference (LSD) values demonstrating significant differences (*) between the two grass species (p < 0.05) at a given day of heat stress.
Differential metabolites regulated by heat stress in A. stolonifera and P. annua
-
A total of 55 metabolites relevant to plant processes were identified using LC-MS (Table 1).
Table 1. Mass spectrometry properties of differentially regulated metabolites by heat stress (42 d) in A. stolonifera and P. annua.
No. RT (min) Metabolite Mz No. RT (min) Metabolite Mz 1 4.93 γ-Acetamidobutyrate 144.07 29 4.42 Lactate 89.02 2 7.47 GABA 102.06 30 4.03 Leucine 130.09 3 10.79 Aconitate 173.01 31 11.04 Lysine 145.10 4 2.60 Adenine 134.05 32 9.24 Malate 133.01 5 2.72 Adenosine 266.09 33 8.82 Malonic acid 103.00 6 6.63 Alanine 88.04 34 4.66 Methionine 148.04 7 8.32 Alpha-Ketoglutarate 145.01 35 3.43 Mevalonate 147.07 8 4.75 Arabitol 151.06 36 9.88 Dinucleotide 662.10 9 7.81 Asparagine 131.05 37 3.69 Phenylalanine 164.07 10 9.05 Aspartate 132.03 38 12.14 Phosphocholine 242.08 11 11.33 Citrate 191.02 39 5.77 Proline 114.06 12 4.04 Cytidine 242.08 40 2.82 Pyruvate 87.01 13 5.15 Fructose 179.06 41 9.62 Raffinose 503.16 14 9.23 Fumarate 115.00 42 2.66 Riboflavin 375.13 15 5.88 Glucose 179.06 43 7.70 Serine 104.04 16 11.78 Glucose 6-phosphate 259.02 44 5.83 Sorbitol 181.07 17 10.81 Glucose 1-phosphate 259.02 45 11.94 Stachyose 665.21 18 8.80 Glucuronic acid 193.04 46 8.81 Succinic acid 117.02 19 8.48 Glutamate 146.05 47 7.36 Sucrose 341.11 20 8.90 Glutamine 145.06 48 5.76 Thiamine 263.10 21 6.03 Glycerate 105.02 49 6.94 Threonine 118.05 22 7.11 Glycine 74.02 50 2.06 Thymidine 241.08 23 3.44 Guanine 150.04 51 3.47 Tryptophan 203.08 24 4.28 Guanosine 282.08 52 5.17 Tyrosine 180.07 25 9.17 Histidine 154.06 53 10.56 UDP-Glucose 565.05 26 8.41 Inositol 179.06 54 2.81 Uridine 243.06 27 9.95 Isocitrate 191.02 55 5.47 Valine 116.07 28 4.45 Isoleucine 130.09 The partial least-squares discriminant analysis (PLS-DA) detected a difference in metabolic composition between the control and heat stress groups in both A. stolonifera and P. annua leaf tissue samples collected at 42 d of heat stress (Fig. 4).
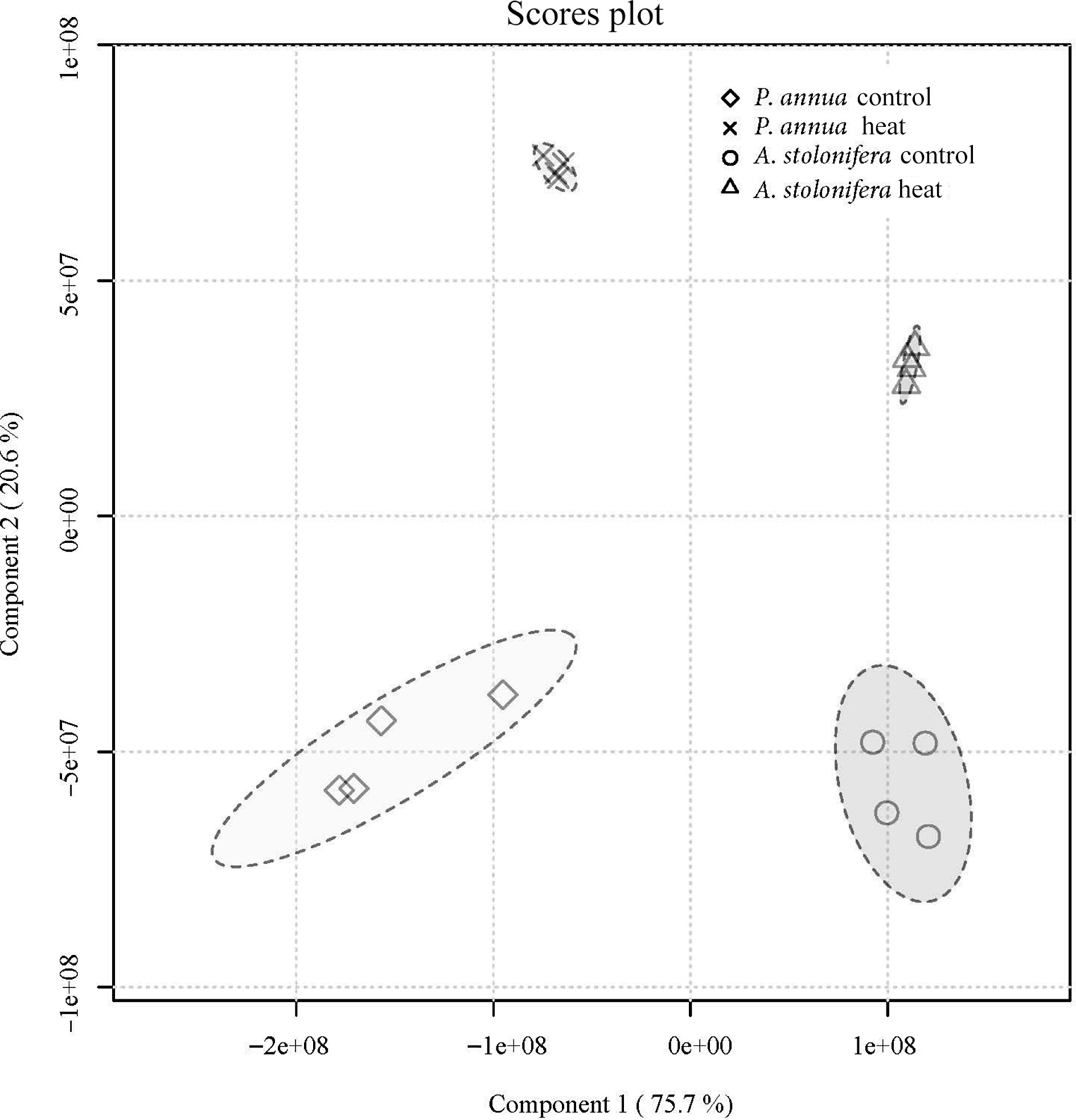
Figure 4.
Partial least-squares discriminant analysis of metabolic composition for A. stolonifera under heat stress compared to control conditions at 42 d of heat stress for P. annua.
A total of 17 metabolites were upregulated and 22 were downregulated in A. stolonifera, while P. annua had 21 significantly upregulated and 26 significantly downregulated metabolites due to heat stress (Fig. 5a & b). Many metabolites were regulated differentially between P. annua and A. stolonifera under heat stress relative to their respective control plants, including organic acids, amino acids, nucleic acids, and carbohydrates. In both A. stolonifera and P. annua, 12 of the same metabolites were upregulated, with five unique metabolites in A. stolonifera and nine unique metabolites in P. annua. There were 17 of the same metabolites downregulated in both species, with five uniquely downregulated in A. stolonifera and nine uniquely downregulated in P. annua.
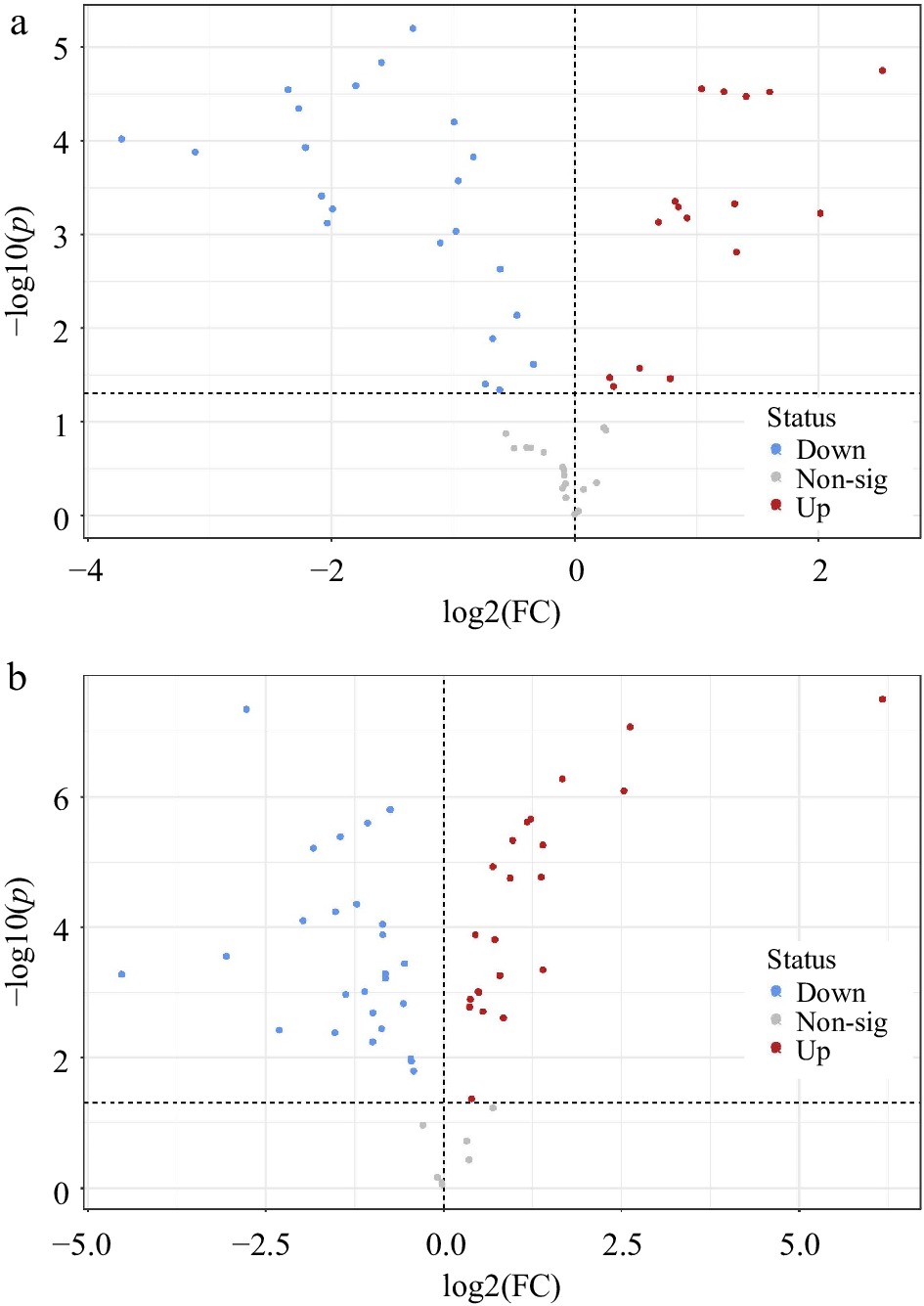
Figure 5.
Volcano plots indicating significantly different metabolites in leaf tissue at 42 d of heat stress compared to unstressed control plants of (a) A. stolonifera and (b) P. annua (p < 0.05).
Metabolites with the most notable differences in their responses to 42 d of heat stress between the two species were organic acids (Table 2). Most organic acids exhibited decreases in their content or were downregulated in both grass species, except for two organic acids. Various organic acids and intermediates involved in respiration in glycolysis and the tricarboxylic acid (TCA) cycle were downregulated only in P. annua, including glucose-1-phosphate, glucose-6-phosphate, pyruvate, lactate, fumarate, and malate, but these remained unchanged in A. stolonifera in response to heat stress. Glycerate was downregulated to a greater degree in P. annua (−1.83) than in A. stolonifera (−1.11). Glucuronic acid was upregulated in both species but to a greater extent in A. stolonifera.
Table 2. Relative content of notable metabolites in heat-stressed plants (42 d) compared to control levels expressed as log2(fold-change (FC)) in A. stolonifera and P. annua (p < 0.05).
Metabolite class Significant metabolites A. stolonifera P. annua log2(FC) p-value log2(FC) p-value Sugar Sucrose 2.0116 0.000594 0.49699 0.00099707 Stachyose 1.5947 3E-05 1.3962 0.00045046 Raffinose 1.037 2.78E-05 0.84152 0.0024677 Glucose 0.8465 0.000509 0.55068 0.0019506 Fructose 0.31411 0.04186 0.37504 0.0012791 UDP-Glucose −0.61714 0.002339 −0.56454 0.0014869 Organic acid/carbon intermediates Glycerate −1.1096 0.00123 −1.8291 6.1183E-06 Glucose 1-phosphate NS NS −0.42047 0.015974 Glucose 6-phosphate NS NS −0.85721 0.000090569 Lactate NS NS −1.5261 0.0041586 Pyruvate NS NS −0.87007 0.0036087 Fumarate NS NS −0.81981 0.00051454 Malate NS NS −0.81785 0.000603 Aconitate NS NS −2.317 0.003775 Citrate −0.73972 0.039597 −0.45058 0.011317 Succinic acid −0.83789 0.000149 −1.1086 0.00096848 Glucuronic acid 0.81941 0.000441 0.44683 0.00013123 Malonic acid 0.68336 0.000738 −1.5208 0.000057636 NAD 1.3216 0.001537 NS NS Amino acids Phenylalanine −2.0384 0.000755 1.175 2.4601E-06 Tryptophan 0.9163 0.000664 1.3705 0.000017093 Tyrosine −0.47894 0.007285 0.48675 0.00097056 Serine −1.8034 2.56E-05 −1.2194 0.000044349 Threonine −2.0837 0.000386 −1.9735 0.000079449 Asparagine −3.7262 9.53E-05 −4.5243 0.00052899 Glutamine −1.9935 0.000535 −0.9921 0.0020488 Aspartate −3.1222 0.000131 −3.0542 0.00027844 Glutamate −2.2705 4.5E-05 −1.4502 4.1223E-06 Lysine 0.28221 0.03375 0.36616 0.0016675 Histidine NS NS 0.39498 0.042833 Alanine −0.96231 0.000266 −0.85354 0.00013055 Valine −0.34439 0.024368 0.69188 0.000011814 Isoleucine NS NS 1.2272 0.000002184 Leucine NS NS 0.97381 4.6598E-06 Glycine −0.98193 0.000923 −0.99736 0.0056936 Proline 0.78009 0.034569 2.6224 8.5101E-08 Nucleic acids Thymidine 2.5231 1.77E-05 6.1698 3.1863E-08 Guanine 1.2196 2.98E-05 0.71981 0.00015414 Adenosine −0.62274 0.045692 0.93481 0.000017701 Guanosine −1.3337 6.29E-06 −1.3762 0.0010679 Uridine −1.5916 1.45E-05 −1.0659 2.5287E-06 Adenine −2.3584 2.82E-05 −2.7694 4.5431E-08 Cytidine −0.99572 6.27E-05 −0.54768 0.00036086 'NS' denotes metabolites that had no significant change in concentration between heat-stressed and non-stressed control plants. Most carbohydrates or sugars, including sucrose, stachyose, raffinose, and glucose exhibited increases in their content in both species exposed to 42 d of heat stress but were elevated to a greater extent in A. stolonifera than in P. annua, with the exception of fructose, which was upregulated to a similar extent in both grass species (Table 2). The UDP-glucose was downregulated in both grass species.
Most amino acids exhibited a decline in their content or were downregulated under heat stress in both grass species, including serine, threonine, asparagine, glutamine, aspartate, glutamate, alanine, and glycine, which were downregulated to a similar extent in the two species (Table 2). Tryptophan, lysine, and proline were upregulated in both species with a greater fold-change (FC) in P. annua. Phenylalanine, tyrosine, and valine were downregulated in A. stolonifera but upregulated in P. annua. Histidine, isoleucine, and leucine were all exclusively upregulated in P. annua in response to heat stress.
Nucleic acid changes were similar between species. Guanosine, uridine, adenine, and cytidine were downregulated by heat stress to a similar extent in both grass species (Table 2). Thymidine and guanine were upregulated in both species, but to a greater level for thymidine in P. annua. Adenosine exhibited upregulation in P. annua but downregulation in A. stolonifera at 42 d of heat stress.
-
The superior heat tolerance of A. stolonifera relative to P. annua was characterized by the lesser extent of declines in TQ and green canopy cover and lower EL in heat-stressed plants compared to the respective control for each species. Metabolomic analysis in this study identified some metabolites and their relevant metabolic pathways that could explain some of the differences in heat tolerance between the two grass species, as presented above. To better elucidate metabolic pathways affected by heat stress in the two grass species that could be associated with the differences in heat tolerance, only the metabolites that exhibited differential responses to heat stress in each species relative to their respective non-stress controls are discussed in detail below (Fig. 6).
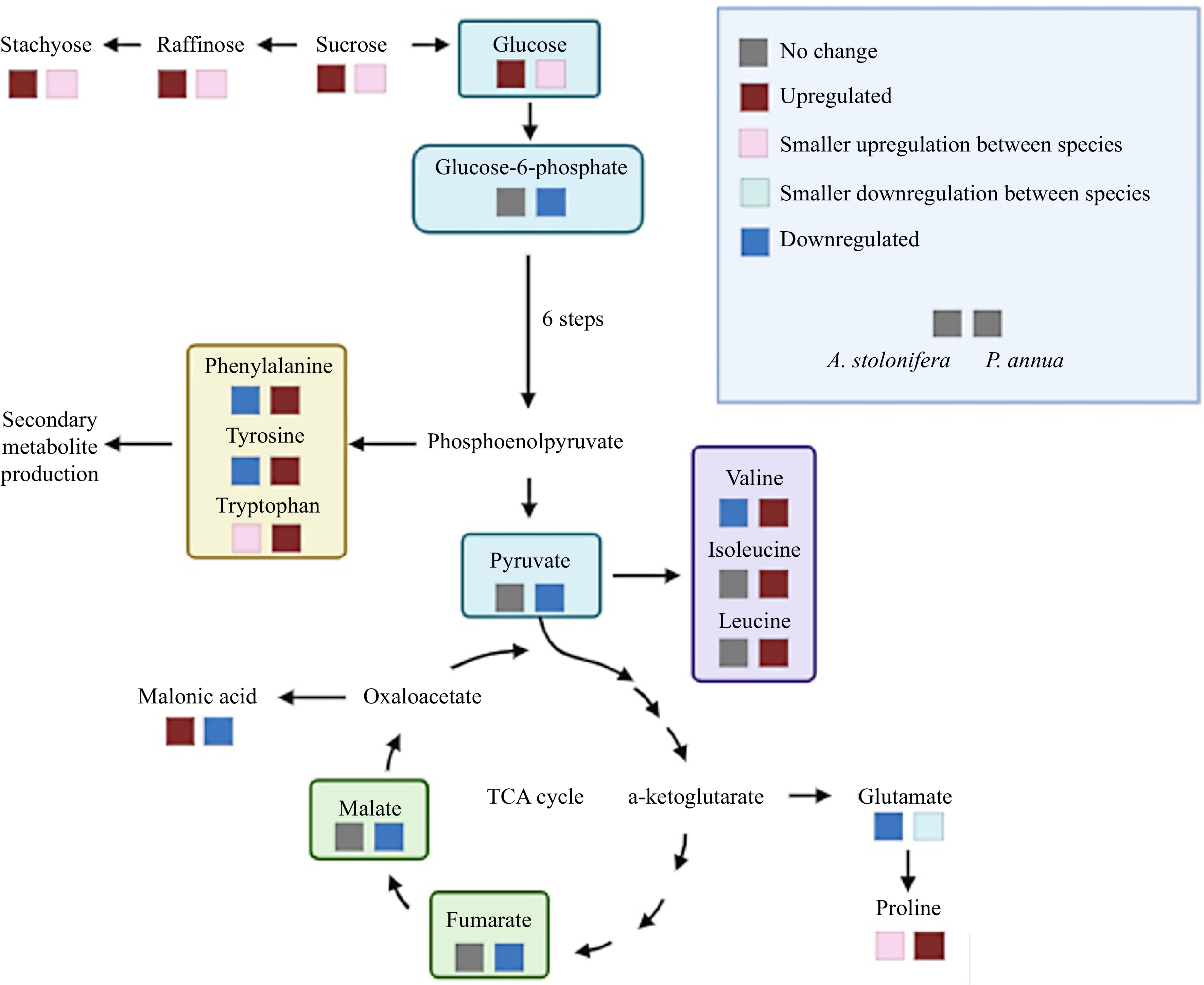
Figure 6.
Metabolic pathways illustrating metabolites differentially up-regulated (increase in content) or down-regulated (decrease in content) by heat stress in P. annua and A. stolonifera.
Carbon metabolism, in the processes of photosynthesis and respiration, plays essential roles in regulating plant tolerance to abiotic stress, including heat stress[23]. In this study, several carbohydrates or sugars, including sucrose, stachyose, raffinose, and glucose exhibited a greater extent of increases in their content in A. stolonifera than those in P. annua in response to heat stress compared to their respective non-stress controls; however, in response to heat stress, the significant downregulation of organic acids, including malate, fumarate, pyruvate, aconitate, malonic acid, lactate, glucose-1-phosphate, and glucose-6-phosphate, was observed only in P. annua or was more severe in P. annua than in A. stolonifera. Those organic acids and carbon intermediates are key metabolites in glycolysis and the tricarboxylic acid (TCA) cycle of respiration and support energy production, affecting plant responses to high temperatures[24,25]. It is particularly interesting to find that glucuronic acid and malonic acid, which are not directly involved in energy production, exhibited different response patterns to heat stress from those organic acids involved in respiration. Glucuronic acid was upregulated in response to heat stress, but to a greater extent in A. stolonifera than in P. annua. It has been found to play roles in stress protection by acting as an inducer for the synthesis of heat shock proteins (HSP) in mammalian cells[26]. Kim et al. reported that glucuronic acid induced the synthesis of HSP70 and that exogenous treatment of cells significantly enhanced tolerance to heat shock. Malonic acid was upregulated in response to heat stress in A. stolonifera, while it was downregulated in P. annua[26]. The increases in malonic acid content during drought stress have been associated with enhancement of drought tolerance through modulation of osmotic potential[27]; however, the roles of either glucuronic acid or malonic acid in plant tolerance to heat stress are unclear and deserve further investigation. The differential metabolic changes to heat stress between the two grass species suggested the maintenance of active carbohydrate and organic acid metabolism for energy production in respiration, and increased metabolites for stress protection could contribute to the superior heat tolerance of A. stolonifera relative to P. annua.
The difference in heat tolerance for P. annua and A. stolonifera was also associated with differential changes in amino acid metabolism, which plays critical roles in plant tolerance to heat stress[28]. Proline, tryptophan, and lysine were upregulated to a greater extent in P. annua than in A. stolonifera in response to heat stress, while phenylalanine, tyrosine, valine, isoleucine, and leucine were upregulated only in heat-stressed P. annua, although most amino acids exhibited a decline in their content in both grass species exposed to heat stress. Proline accumulation has been widely reported in various plant species exposed to abiotic stresses and is considered a metabolic stress indicator[12,29]. Tryptophan, phenylalanine, and tyrosine belong to the aromatic amino acid family (AAA) and act as precursors or intermediates in the synthesis pathway of secondary metabolites and signaling molecules in response to abiotic stresses[29,30]. The upregulation of AAA in P. annua could reflect heat injury and/or the inefficient use of AAA to produce secondary metabolites involved in stress defense; however, the roles of secondary metabolites in the differential heat response between P. annua and A. stolonifera are yet to be investigated. The degradation of lysine and some branched-chain amino acids (BCAAs), including valine, isoleucine, and leucine has been positively associated with dehydration tolerance in arabidopsis (Arabidopsis thaliana)[31], although in this study, the accumulation of lysine, valine, isoleucine, and leucine observed in P. annua following prolonged periods of heat stress could be the result of metabolic impairment or inefficient use of those amino acids in the downstream metabolism in P. annua.
In summary, physiological analysis demonstrated the superior heat tolerance of A. stolonifera relative to P. annua. The comparative metabolic profiling analysis identified some metabolites that were upregulated uniquely or to a greater extent in heat-tolerant A. stolonifera (sucrose, stachyose, raffinose, glucose, glucuronic acid, and malonic acid) and some that were up-regulated uniquely or to a greater extent (proline, tryptophan, histidine, phenylalanine, tyrosine, valine, isoleucine, and leucine) or downregulated (malate, fumarate, pyruvate, aconitic acid, malonic acid, lactate, glucose-1-phosphate, and glucose-6-phosphate) in heat-sensitive P. annua in response to heat stress. Those differential metabolites are mainly in carbohydrate and organic acid metabolism, as well as amino acid metabolism, and are involved in photosynthesis, respiration, secondary metabolism, and stress protection. Those distinct metabolites present in the two grass species were associated with the difference in heat tolerance for A. stolonifera vs P. annua and could be used as biomarkers or incorporated into biofertilizers to improve the heat tolerance of P. annua. Furthermore, future research to identify molecular factors involved in the synthesis of the differential heat-responsive metabolites between the two grass species differing in heat tolerance may provide further insight into mechanisms for improving heat tolerance in P. annua and other cool-season turfgrasses.
-
The authors confirm contribution to the paper as follows: data collection: McBride S, Rossi S; data analysis and draft manuscript preparation: McBride S; developed research ideas, experimental design, acquired funding, and revised manuscript: Huang B. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article.
Thanks go to the Tri-State Turfgrass Research Foundation and New Jersey Agricultural Experiment Station for funding support.
-
The authors declare no conflict of interest. Bingru Huang is the Editorial Board member of Grass Research who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and the research groups.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
McBride S, Rossi S, Huang B. 2024. Differential metabolic responses to heat stress associated with interspecific variations in stress tolerance for annual bluegrass and creeping bentgrass. Grass Research 4: e013 doi: 10.48130/grares-0024-0011 

Differential metabolic responses to heat stress associated with interspecific variations in stress tolerance for annual bluegrass and creeping bentgrass
- Received: 12 March 2024
- Revised: 10 April 2024
- Accepted: 10 April 2024
- Published online: 03 June 2024
Abstract: Annual bluegrass (Poa annua) typically exhibits early or more severe turf quality decline than creeping bentgrass (Agrostis stolonifera) during summer months when they are co-present on golf course putting greens. The mechanisms underlying the difference in heat tolerance between the two species are largely unknown. This study was conducted to compare physiological responses between P. annua and A. stolonifera and to identify metabolites and associated metabolic pathways underlying the differential heat tolerance. Plants were grown in controlled growth chambers at 22/17 °C (day/night, non-stress control) and 35/30 °C (heat stress) for 42 d. Visual turf quality, percent green canopy cover, and leaf electrolyte leakage were measured weekly. Metabolomic analysis was performed with leaf tissues collected at 42 d of heat stress. Relative to control plants, heat-stressed P. annua exhibited more severe declines in health across physiological parameters than A. stolonifera. Comparative metabolomic analysis of heat-stressed plants relative to the respective control identified metabolites that were upregulated uniquely or to a greater extent in A. stolonifera (sucrose, stachyose, raffinose, and glucose, glucuronic acid, and malonic acid) or P. annua (proline, tryptophan, lysine, phenylalanine, tyrosine, valine, isoleucine, and leucine) and those uniquely downregulated (malate, fumarate, pyruvate, aconitic acid, malonic acid, lactate, and glucose-1-phosphate and glucose-6-phosphate) in P. annua. Those distinct metabolites are mainly in photosynthesis, respiration, secondary metabolism, and stress protection. They were associated with the difference in heat tolerance for A. stolonifera vs P. annua and could be used as biomarkers or incorporated into biofertilizers to improve P. annua heat tolerance.
-
Key words:
- Stresses /
- Differential /
- Metabolic /
- Responses /
- Associated /
- Interspecific