









-
Plant gene expression is modulated by transcription factors (TFs), regulatory proteins that recognize and bind specific DNA sequences[1]. Within this group, the WRKY gene family holds a central position, critically influencing diverse plant processes including growth and development[2,3], adaptation to stress[4,5], seed dormancy and germination control[6−8], metabolic regulation[9], senescence programs[10−12], and the production of secondary metabolites[13−15]. The WRKY superfamily, characterized by a conserved WRKY domain, stands as one of the largest plant-specific TF families known[16]. Typical WRKY protein architecture involves an N-terminal DNA-binding WRKY domain featuring the canonical WRKYGQK motif (or its variants like WRKYGKK, WRKYGRK, WRKYGEK, WRKYGNK, WRKYGHK[17], and others such as WKKY, WSKY, WKRY[18]) and a C-terminal zinc finger, predominantly C2H2 (C-X4–5-C-X22–23-HXH) or C2HC (C-X7-C-X23-HXC)[19]. Classification of WRKY TFs into four major groups (Groups I−IV) is based on the number of WRKY domains and the type of zinc finger present[20]. Group I proteins contain two WRKY domains; Group II proteins have one WRKY domain and a C2H2 finger; Group III proteins possess one WRKY domain and a C2HC finger; Group IV proteins are distinguished by an incomplete WRKY domain and the lack of a typical zinc finger motif. Extensive research indicates that WRKY TFs are key participants in signal transduction cascades and gene regulation networks activated during plant encounters with biotic and abiotic stresses[21−23]. In Malus xiaojinensis, heterologous overexpression of MxWRKY55 improved tolerance to both iron toxicity and salinity in Arabidopsis[24]. WRKY factors are also vital for pathogen defense and drought adaptation. For instance, tomato (Solanum lycopersicum) SlWRKY08 activates transcription via W-box elements, leading to heightened resistance against Pseudomonas syringae pv. Tomato (Pst) and better tolerance to salt and drought[25]. Heterologous expression of Zea mays ZmWRKY33 similarly conferred increased salt tolerance upon Arabidopsis[26]. Rice (Oryza sativa) OsWRKY54 has been implicated in salt stress responses[27]. Furthermore, WRKY TFs contribute to cold acclimation; Luffa cylindrica LcWRKY02, LcWRKY07, and LcWRKY12 respond to low temperatures[28], while Vitis amurensis VaWRKY12 has been shown to boost cold tolerance[29]. Additionally, numerous studies indicate that WRKY transcription factors are major players in ABA signal transduction[30,31]. In model plants like Arabidopsis, WRKY genes have been extensively studied for their roles in stress responses, particularly in the regulation of plant immunity and abiotic stress tolerance[32]. For instance, in Arabidopsis, the WRKY family has been shown to be integral in defense responses to pathogens and abiotic stressors, with specific WRKY genes such as AtWRKY33 being involved in regulating resistance to drought and salt stress[33].
Seashore paspalum (Paspalum Vaginatum O. Swartz, 2n = 2x = 20) is a perennial grass renowned for its exceptional tolerance to saline environments, making it an important species for turf management and forage production, particularly in coastal regions[34]. Recognized as one of the key salt-tolerant grasses, seashore paspalum has gained prominence in landscaping, golf courses, and livestock feed, contributing to agricultural sustainability and ecological restoration efforts worldwide[35,36]. As a halophyte, this plant is favored for its tolerance to salinity, drought, waterlogging, low soil pH, and its ability to adapt to low light intensity and partial shade conditions[37,38]. Seashore paspalum demonstrates significant salt tolerance through three primary mechanisms. Firstly, it effectively regulates sodium ion (Na+) accumulation, thereby preventing ion toxicity and maintaining cellular stability under saline conditions[39]. Secondly, the plant utilizes osmotic regulation by accumulating osmolytes, which help to maintain cellular water balance and facilitate adaptation to high salinity environments[40]. Lastly, Seashore paspalum strengthens its antioxidant systems to mitigate oxidative stress by scavenging reactive oxygen species (ROS), thus reducing cellular damage caused by salt stress[41]. These integrated mechanisms collectively enable seashore paspalum to thrive in salt-affected soils. The assembly of the seashore paspalum genome (PI 509022) has been completed[42], which, together with the Agrobacterium-mediated genetic transformation system, provides a foundation for research into the exploration and validation of salt tolerance genes in seashore paspalum[43]. Understanding the salt tolerance mechanisms of halophytes may provide new insights into enhancing plant salt tolerance. However, the role of the WRKY gene family in the salt tolerance of seashore paspalum has not yet been identified.
Despite the extensive research on WRKY genes in model plants, such as Arabidopsis and rice, the role of WRKY genes in other species, particularly halophytes like seashore paspalum, remains underexplored. A systematic characterization of the seashore paspalum PvWRKY gene family is presented herein, exploring evolutionary relationships, gene structural organization, conserved protein motifs, putative cis-regulatory elements, and potential protein-protein interactions. Additionally, the transcriptome analysis provides a clearer understanding of the expression patterns of WRKY genes under different durations of salt stress. This integrated approach establishes essential groundwork for subsequent research aimed at dissecting the molecular functions and regulatory networks associated with this gene family in seashore paspalum.
-
Paspalum vaginatum genome sequences and annotations were sourced from Phytozome (
https://phytozome-next.jgi.doe.gov/ ). The WRKY domain HMM profile (PF03106) was retrieved from Pfam[44] (www.ebi.ac.uk/interpro/entry/pfam/PF03106 ). Putative PvWRKY proteins were identified by searching the predicted proteome using this HMM profile with HMMER via TBtools software[45]. Initial hits were filtered, and redundancies were manually removed. Domain confirmation of candidate sequences was performed using the NCBI Conserved Domain Database (CDD,www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml ). The CDD search parameters included an E-value < 0.01 against the CDD-62456 PSSM database, activation of composition-corrected scoring, and a maximum hit limit of 500. According to these criteria, only candidates possessing a confirmed WRKY domain were designated as members of the PvWRKY gene family.Prediction of physicochemical properties and subcellular localization of PvWRKY proteins
-
Computational prediction of key physicochemical characteristics for the identified PvWRKY protein sequences was performed using resources available on the ExPASy server (
www.expasy.org ). Specifically, theoretical parameters including molecular weight (MW, Da), isoelectric point (pI), grand average of hydropathy (GRAVY), and instability index were calculated. Concurrently, the putative subcellular localization for each PvWRKY protein was predicted using the WoLF PSORT web tool (https://wolfpsort.hgc.jp/ ).Phylogenetic analysis of PvWRKY proteins
-
Extract the WRKY protein sequences from seashore paspalum and align them with the previously classified AtWRKY proteins using the Align Protein tool included in MEGA 11[46]. Construct a phylogenetic tree using the Neighbor-Joining (NJ) method with the following parameters: the poisson model for sequence evolution, pairwise deletion for handling gaps in the alignment, bootstrap support calculated with 1,000 replicates to assess the robustness of the tree nodes, and set the Number of Threads to 3 for parallel processing and faster computation. This methodological approach will help elucidate the evolutionary relationships among the WRKY proteins and provide insights into their potential functional divergence or conservation within seashore paspalum.
Characterization of PvWRKY motifs and gene architecture
-
Conserved domains and motifs within the PvWRKY protein set were investigated. Sequences were submitted to the NCBI Conserved Domain Database (CDD,
www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml ) for domain annotation and to the MEME suite[47] (https://meme-suite.org/meme/ ) for de novo motif discovery. Parameters for MEME included: discovery of a maximum of 15 motifs, motif width constrained to 6−50 residues, and use of a zero-order background model. TBtools software was employed to visualize the resulting domain information and motif distributions relative to the gene structures.Prediction of cis-regulatory elements in PvWRKY promoters
-
Potential cis-acting regulatory elements in the promoter regions of PvWRKY genes were identified to infer regulatory potential. Utilizing the seashore paspalum genome annotation, sequences 2,000 bp upstream of the putative transcriptional start site for each PvWRKY gene were extracted using TBtools. These extracted promoter sequences were subsequently scanned for known plant regulatory motifs using the PlantCARE database resource (
https://bioinformatics.psb.ugent.be/webtools/plantcare/html/ ). The locations and types of predicted cis-elements were then imported into TBtools for visualization.Analysis of collinearity and PvWRKY evolution
-
To investigate potential gene duplication events within the PvWRKY family, synteny analysis was performed across the seashore paspalum genome. The identified 126 PvWRKY gene sequences, along with the genome annotation, were processed using the One Step McscanX utility integrated within TBtools. The underlying sequence comparisons (BLAST) were configured to retain a maximum of five hits per query and employed an E-value cutoff of 1e-10. Detected collinear blocks and syntenic gene pairs were subsequently visualized using the Advanced Circos visualization tool in TBtools.
Analysis of functional annotation for PvWRKY proteins
-
Gene Ontology (GO) functional annotation of PvWRKY proteins was conducted through the eggNOG-mapper online platform (
http://eggnog-mapper.embl.de/ )[48]. The annotation was performed using the default parameters, with the tool automatically selecting the taxonomic scope based on the input sequences, which focused on plant species. The eggNOG database (version 2.2.11) was used for orthology assignment, with a default E-value threshold of 1e-5 to select significant hits. The DIAMOND tool was employed for sequence alignment, ensuring sufficient alignment coverage for meaningful matches. Following the annotation, GO term enrichment analysis was carried out using TBtools (v2.127) for statistical visualization.Construction of the co-expression network
-
Potential protein-protein interactions (PPIs) involving selected PvWRKY proteins were predicted using the STRING database (
https://string-db.org/ ). Arabidopsis thaliana served as the reference organism for inferring homologous interactions. The analysis utilized the full STRING network data, applying a medium confidence interaction score threshold of 0.400 and a False Discovery Rate (FDR) stringency of 5%. The resulting network data, representing predicted functional associations, was imported into Cytoscape software (version 3.10.2) for visualization. Within the Cytoscape environment, node attributes (protein representation) were customized: node size and color intensity were scaled proportionally to the degree (number of connections) of each protein within the predicted network.Plant cultivation and treatment procedures
-
Seashore paspalum plants were established from individual stem nodes propagated in 30 cm × 30 cm hydroponic boxes. Plants were cultivated for 12 weeks in Hoagland's nutrient solution within a controlled greenhouse environment, characterized by a 30/25 °C day/night temperature cycle and relative humidity levels of 50% (day) and 70% (night). Under these conditions, the plants exhibited vigorous growth prior to treatment initiation.
For the experiment, control plants continued to receive standard Hoagland's solution, representing the 0 h time point. Salt stress was imposed on treatment groups by supplementing the nutrient solution with 0.2 M NaCl. Leaf and root tissues were subsequently harvested rapidly at 6, 48, and 120 h following the commencement of salt treatment. Phenotypic responses to salinity were monitored; initial leaf softening was noted at 6 h, progressing to visible dehydration and leaf curling by 48 h. These symptoms intensified at 120 h, concurrent with the onset of senescence (yellowing and withering) in basal leaves. Sampling targeted the second and third fully expanded leaves, counting from the apex, along with corresponding root tissues. Collected samples were immediately snap-frozen in liquid nitrogen and stored at −80 °C until RNA extraction.
RNA extraction and illumina sequencing procedures
-
Total RNA was isolated from leaf, and root materials harvested at 0, 6, 48, and 120 h post-treatment. The isolation utilized the Plant Total RNA Extraction Kit (TIANGEN Biotech, Beijing, China), strictly adhering to the provided protocol. Subsequently, cDNA was generated employing the PrimeScript RT reagent kit, which included a gDNA elimination step (Takara, Dalian, China). Rigorous quality control was performed to verify RNA sample purity and integrity; assessments included NanoDrop 2000 spectrophotometry (Thermo Fisher Scientific, MA, USA), agarose gel electrophoresis, and evaluation with an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). Samples advancing to library construction met stringent criteria: a minimum of 1 μg total RNA, an OD260/280 absorbance ratio of 1.8 or higher, and an RNA Integrity Number (RIN) value of 6.5 or greater. Illumina-compatible sequencing libraries were prepared using the NEBNext Ultra™ RNA Library Prep Kit (NEB, Ipswich, MA, USA). In total, eight libraries were created, comprising four leaf (L-0, L-6, L-48, L-120) and four root (R-0, R-6, R-48, R-120) replicates across the time course. Library sequencing was conducted by Biomarker Technologies (Beijing, China) via the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA).
Transcript abundance data (FPKM values) obtained from the RNA-seq analysis served as the basis for calculating expression fold changes (FC). These calculations compared each salt treatment time point (6, 48, and 120 h) against the 0 h control condition. For comparative analysis and visualization, the resulting FC values were normalized via log2 transformation. Heatmaps illustrating the log2(FC) differential expression patterns across the experimental time points were subsequently generated using Tbtools software.
RNA isolation and quantitative RT-PCR analysis
-
Total RNA, extracted using the MiniBEST Plant RNA Extraction Kit (Takara), was reverse-transcribed into cDNA with the PrimeScript™ RT-PCR Kit (Takara). Expression levels of target genes were quantified via qRT-PCR. Gene-specific primers (detailed in Supplementary Table S1) were designed using Primer3Plus, targeting Tm values of 55−65 °C, lengths of 19−22 bp, and amplicon sizes of 100−300 bp. qRT-PCR was performed on a Bio-Rad CFX96 Touch system (Bio-Rad). Individual 20 μL reactions contained 10 μL 2× SYBR® Green I premix, 5 μL cDNA template, 1 μL forward primer, 1 μL reverse primer, and 3 μL ddH2O. The cycling conditions were: 50 °C for 2 min, 95 °C for 2 min, then 40 cycles of 95 °C for 15 s and 60 °C for 34 s. Product specificity was validated by melt curve analysis. Relative gene expression was calculated using the 2−ΔΔCᴛ method[49], normalized against the internal reference gene PvActin1[50] (chosen for its expression stability under salt stress). All quantifications were performed using three biological replicates for each sample condition.
Statistical analysis
-
Experimental reliability was ensured through the use of three independent biological replicates. Hypothesis testing was performed using SPSS software (version 17.0). Differences among group means were initially assessed with one-way ANOVA. Specific pairwise comparisons were then conducted using Duncan's multiple range test, where appropriate. Statistical significance for all tests was established at p < 0.05. Data throughout this report are presented as mean values ± standard error (SE) based on these replicates.
-
In this study, a combination of HMM prediction and conserved domain analysis was used to jointly identify the WRKY family genes in seashore paspalum. A total of 126 PvWRKY genes were identified at the whole-genome level (Supplementary Table S2). Compared to the 103 PbWRKY family members in Pyrus bretschneideri[51], 160 TaWRKY members in Triticum aestivum L.[52], 76 WRKY family members in Lactuca sativa[53], 121 WRKY gene family members in Phyllostachys edulis[54], and 68 WRKY gene family members in Betula platyphylla Suk.[55], the number of PvWRKY genes in seashore paspalum was relatively higher. According to the analysis of the physicochemical properties of PvWRKY proteins (Supplementary Table S3), amino acid lengths of these proteins ranged from 134 (PvWRKY09) to 1,403 (PvWRKY17), with an average of 371 residues, highlighting significant variation among the proteins. Molecular weights varied from 14,441.93 to 157,368.97 Da, with a mean of 39,827 Da. Predicted isoelectric points (pI) range from 4.81 to 10.49. Instability indices ranged between 39.34 and 70.69, classifying most PvWRKY proteins as unstable, except for PvWRKY37, which had an index of 39.34 and was considered stable. All 126 PvWRKY proteins were predicted to have negative hydrophilicity indices, indicating that the members of this gene family are hydrophilic. Subcellular localization predictions suggest that while a few PvWRKY proteins are localized to chloroplasts (9.52%), cytoplasm (0.80%), mitochondria (0.80%), and plasma membranes (2.38%), the majority are primarily found in the nucleus (84.92%).
Classification and phylogenetic analysis of PvWRKYs
-
To explore the evolutionary relationships among the 126 members of the PvWRKY gene family in seashore paspalum, a phylogenetic tree was built using multiple sequence alignment, including WRKY sequences from Arabidopsis thaliana (Fig. 1)[56]. The phylogenetic tree included 75 AtWRKY and 126 PvWRKY proteins, which were classified into three groups (I, II, and III) based on conserved domain characteristics. Group I contained 22 members, with subgroup I N having 10 members and subgroup I C having 12 members, all of which possess two conserved WRKY domains. Group II, comprising 58 members, was further divided into five subgroups (II.a, II.b, II.c, II.d, and II.e). Subgroup II.a included four members, II.b had 11 members, II.c contained 22 members, II.d consisted of 10 members, and II.e had 11 members. Most PvWRKY proteins in this group contained a complete WRKYGQK domain. Group III consisted of 46 members, typically characterized by a single WRKY domain and a zinc finger domain (PF10533).
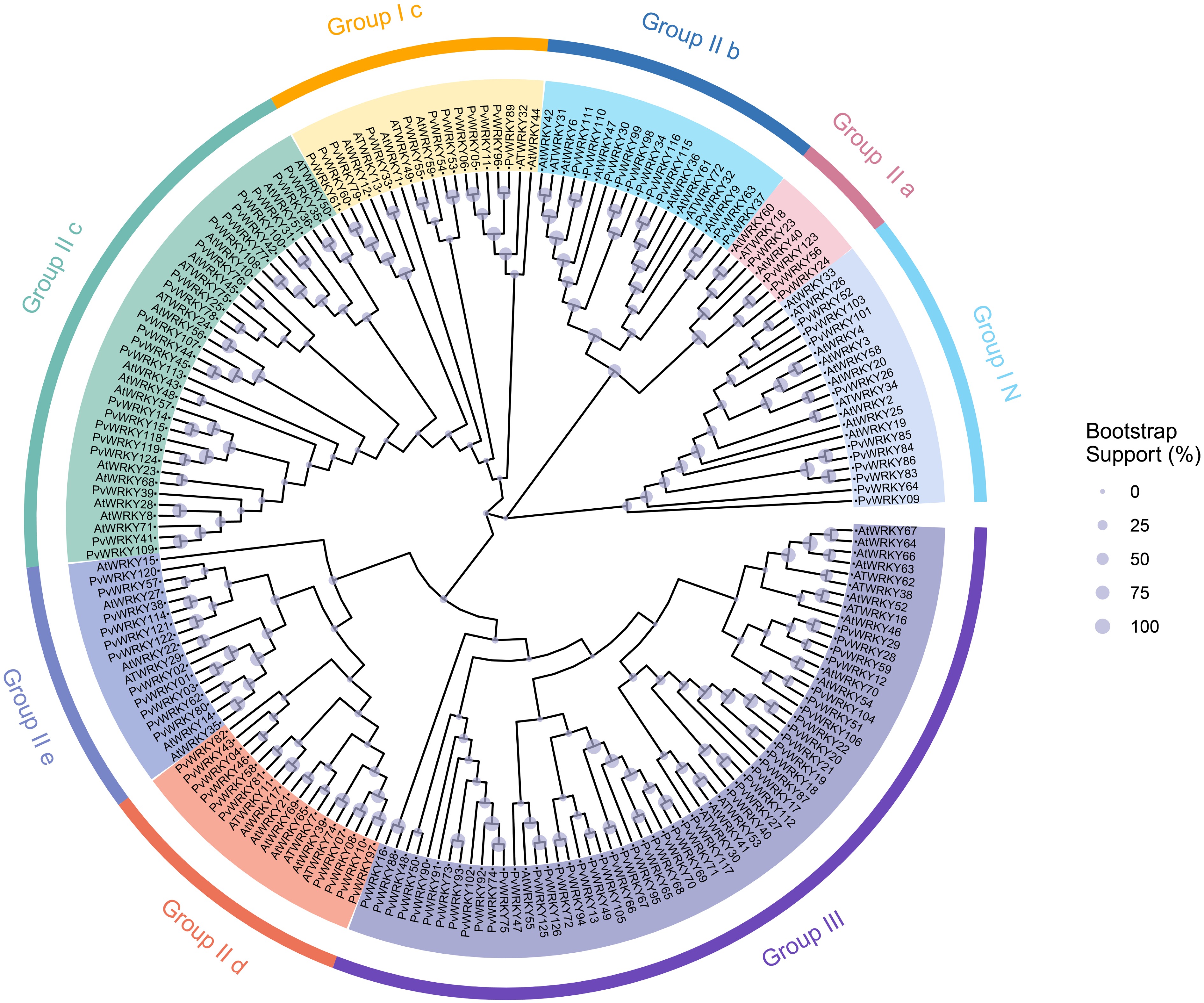
Figure 1.
Phylogenetic clustering analysis of the 126 identified WRKY proteins from seashore paspalum. Different colors represent distinct subfamilies. The purple sections on the branches indicate bootstrap values, with larger bootstrap values corresponding to larger purple sections.
Analysis of motifs and gene structure in PvWRKYs
-
In addition to the phylogenetic analysis, a motif analysis was conducted on the PvWRKY proteins using the MEME Suite to investigate their conserved domains and functional roles further. A total of 10 distinct motifs were identified, ranging in length from 11 to 50 amino acids, with highly significant E-values between 2.7 × 10−1504 and 5.4 × 10−91, reflecting strong conservation across the PvWRKY family (Fig. 2; Supplementary Table S4). Notably, motif 1 and motif 4 contained the highly conserved WRKYGQK core sequence, which is critical for the DNA-binding function of WRKY proteins. MEME analysis also revealed that motifs 1 and 2 were the most conserved, widely present across various PvWRKY proteins, while motifs 9 and 10, though longer, appeared only in specific branches of the phylogenetic tree. A combined analysis of the phylogenetic tree and gene structure revealed that closely related PvWRKY genes often share similar motif patterns. Additionally, motif 2 was enriched in basic amino acids (R and K), suggesting a potential role in nuclear targeting[57], while motif 6 contains numerous potential phosphorylation sites (T, Y, S), which are typically associated with protein interactions, indicating their possible involvement in nuclear localization or protein-protein interactions. This aligns with the subcellular localization predictions discussed in this study. As observed from the phylogenetic analysis, almost all PvWRKY genes, except for PvWRKY84 and PvWRKY85, contain motif 1 and motif 3, exhibiting high conservation, particularly in groups I and II. However, motifs 5 and 9 are conserved only in certain subclades of group I, suggesting that these genes may have specialized biological functions. Furthermore, the gene structure analysis showed significant differences in UTR regions and exon-intron structures among different PvWRKY genes, particularly in group I, where some genes possess more exons, hinting at more complex regulatory functions.

Figure 2.
Analysis of motifs and gene structure in PvWRKYs. (a) Motif analysis of WRKYs in seashore paspalum. (b) WRKY gene structure in seashore paspalum.
Analysis of Cis-elements in the PvWRKY Gene Family
-
To explore the regulatory potential of PvWRKY genes in hormone and stress signaling pathways, sequences 2,000 bp upstream of the predicted transcriptional start site for each gene were computationally extracted. These promoter regions were subsequently scanned for putative cis-acting regulatory elements using the PlantCARE database resource. The analysis, summarized in Fig. 3 (and detailed in Supplementary Table S5), identified 11 distinct categories of motifs predicted to be involved in hormone or stress responsiveness within the PvWRKY promoters. Prominent among these were elements associated with abscisic acid (ABRE), gibberellin, and auxin signaling, as well as motifs implicated in responses to methyl jasmonate (MeJA), drought (e.g., MYB binding sites), wounding, and defense/stress generally.

Figure 3.
Analysis of cis-elements in PvWRKY TFs. Cis-elements related to various environmental stress responses or hormonal interactions are displayed in different colors.
As depicted in Fig. 3, the cis-acting regulatory element associated with MeJA responsiveness was the most prevalent element, appearing 640 times, with 115 PvWRKY genes containing this element. Notably, PvWRKY117 had 20 occurrences, PvWRKY103 had 16, and PvWRKY58 had 14 occurrences. ABRE (ABA-responsive elements) appeared 562 times in total across 116 PvWRKY genes, with 13 occurrences each in the promoter regions of PvWRKY38, PvWRKY46, and PvWRKY31. Furthermore, the gibberellin-responsive element was identified 86 times in 66 PvWRKY genes, with four occurrences in PvWRKY70 and three occurrences each in PvWRKY04 and PvWRKY34.
The presence of ABRE motifs in 116 PvWRKY genes suggests a significant role for ABA signaling in modulating PvWRKY gene expression under stress conditions, particularly salt stress. ABRE motifs are known to mediate ABA-dependent transcriptional regulation, which is crucial for plant adaptation to salinity. The identification of ABRE motifs in multiple PvWRKY genes, including those with high occurrences like PvWRKY38, PvWRKY46, and PvWRKY31, implies that these genes may be involved in ABA-mediated signaling pathways under salt stress. Recent studies have shown that ABA-WRKY interactions play a key role in regulating the plant's response to salinity by activating or repressing specific genes involved in stress tolerance mechanisms[58]. This suggests that the ABRE-containing PvWRKY genes could be integral to the salt stress response, facilitating the plant’s adaptive strategies by regulating downstream stress-related genes.
These results suggest that the expression of PvWRKY genes is strongly regulated by hormonal signals, particularly MeJA and ABA, and also involved in the plant's response to various abiotic stresses. The high frequency of MeJA- and ABA-responsive elements indicates that these phytohormones may play a crucial role in mediating the expression of PvWRKY genes under stress conditions.
Collinearity and evolution analysis of PvWRKYs
-
A systematic chromosomal localization and cross-species collinearity analysis of the target gene family was conducted in this study, unveiling its distribution characteristics and evolutionary relationships within the genome. Initially, all members of the gene family were precisely mapped to the chromosomes of the target species. The results demonstrate that these genes are distributed across 10 chromosomes, exhibiting specific patterns of clustering and dispersion (Fig. 4a). Notably, significant clustering of gene family members was observed on chromosomes 2, 3, 5, and 9, suggesting the occurrence of gene expansion events such as tandem duplications, in these regions.
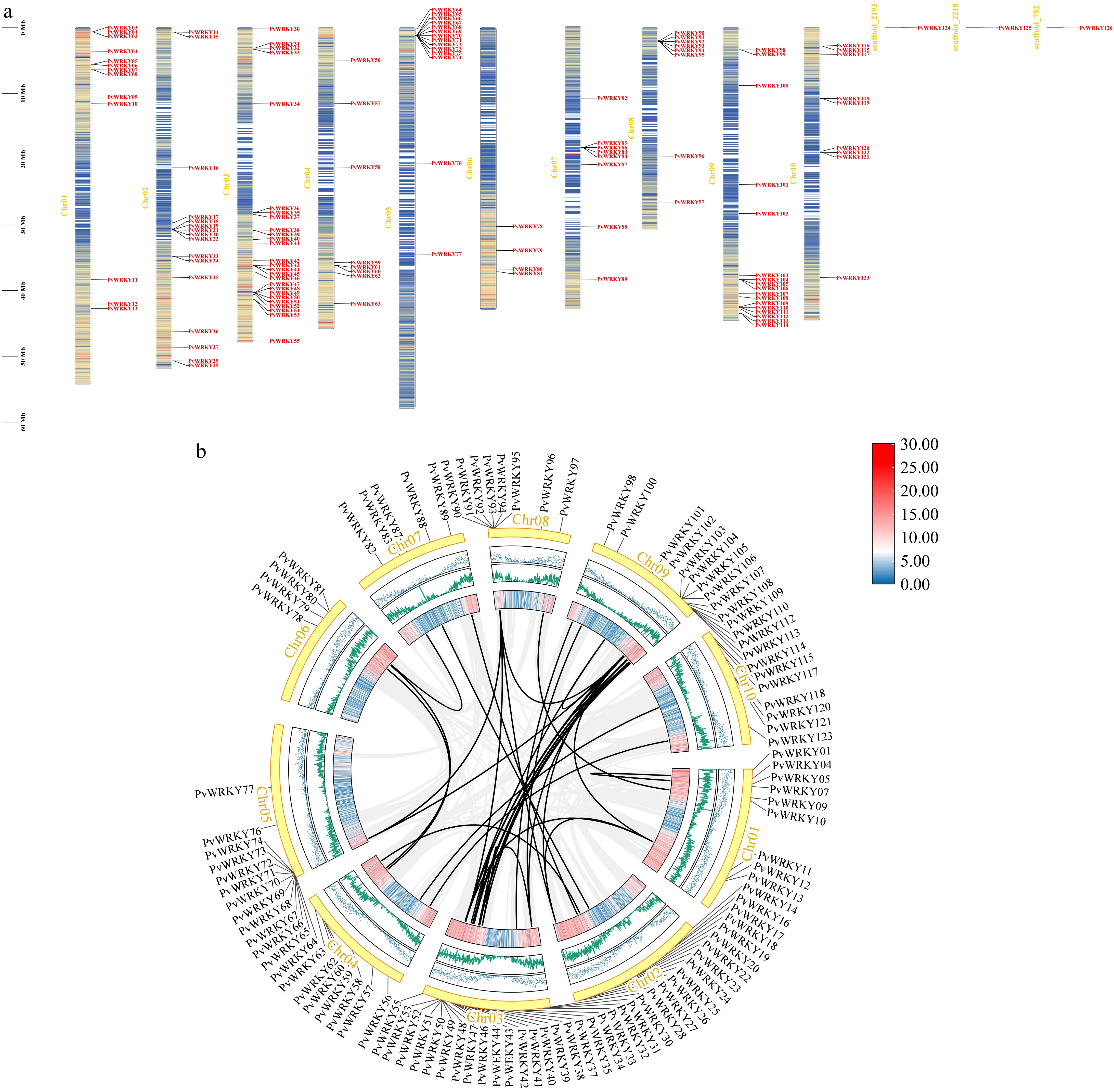
Figure 4.
Chromosomal mapping and syntenic relationships of PvWRKY genes. (a) The physical distribution of the 126 identified PvWRKY genes across the seashore paspalum chromosomes is shown, with locus IDs labeled according to their genomic positions. (b) Synteny networks are displayed, where homologous gene pairs connected by black lines indicate segmental duplication events contributing to WRKY family expansion. Gray background linkages outline conserved syntenic blocks, while black-highlighted pairs represent duplicated WRKY genes.
Tandem duplications likely contributed to the expansion of the WRKY gene family in Paspalum vaginatum, facilitating the diversification of WRKY genes and their functional adaptation. To further elucidate their evolutionary trajectories, a collinearity analysis was performed. The analysis revealed varying degrees of collinearity among all chromosomes, with 19 pairs of genes between chromosomes 3 and 9 exhibiting significant collinearity (Fig. 4b). These conserved collinear blocks suggest that the genomic structures in these regions have remained relatively stable throughout species diversification, potentially preserving the functions of gene family members.
Moreover, based on collinearity analysis, potential whole-genome duplication (WGD) and chromosomal rearrangement events were detected. Further analysis of the collinear blocks identified several gene family members within duplicated genomic regions. These duplications, likely stemming from historical whole-genome duplication events, have facilitated the expansion and functional diversification of the gene family. The phylogenetic analysis (Fig. 1) also revealed distinct clades within the WRKY gene family, which corresponded to different groups, such as Group I, II, and III. The clustering of tandemly duplicated genes in certain groups further supports the hypothesis of functional divergence. Gene pairs such as PvWRKY83, PvWRKY84, PvWRKY85, and PvWRKY86, particularly exhibit high sequence similarity, and hint at recent duplication events. These neogenes may have undergone functional divergence, such as neofunctionalization or subfunctionalization, playing roles in the adaptive evolutionary processes of the species.
Functional annotation analysis of PvWRKY proteins
-
To elucidate the functional roles associated with PvWRKY proteins, Gene Ontology (GO) annotation followed by enrichment analysis was conducted. The analysis focused on the three principal GO domains: biological process (BP), molecular function (MF), and cellular component (CC). When analyzing differentially expressed genes (DEGs) belonging to the PvWRKY family, a predominant enrichment was observed for terms within the BP category. This finding suggests substantial involvement of PvWRKY proteins in pathways governing transcriptional control and plant development (Fig. 5; Supplementary Table S6).
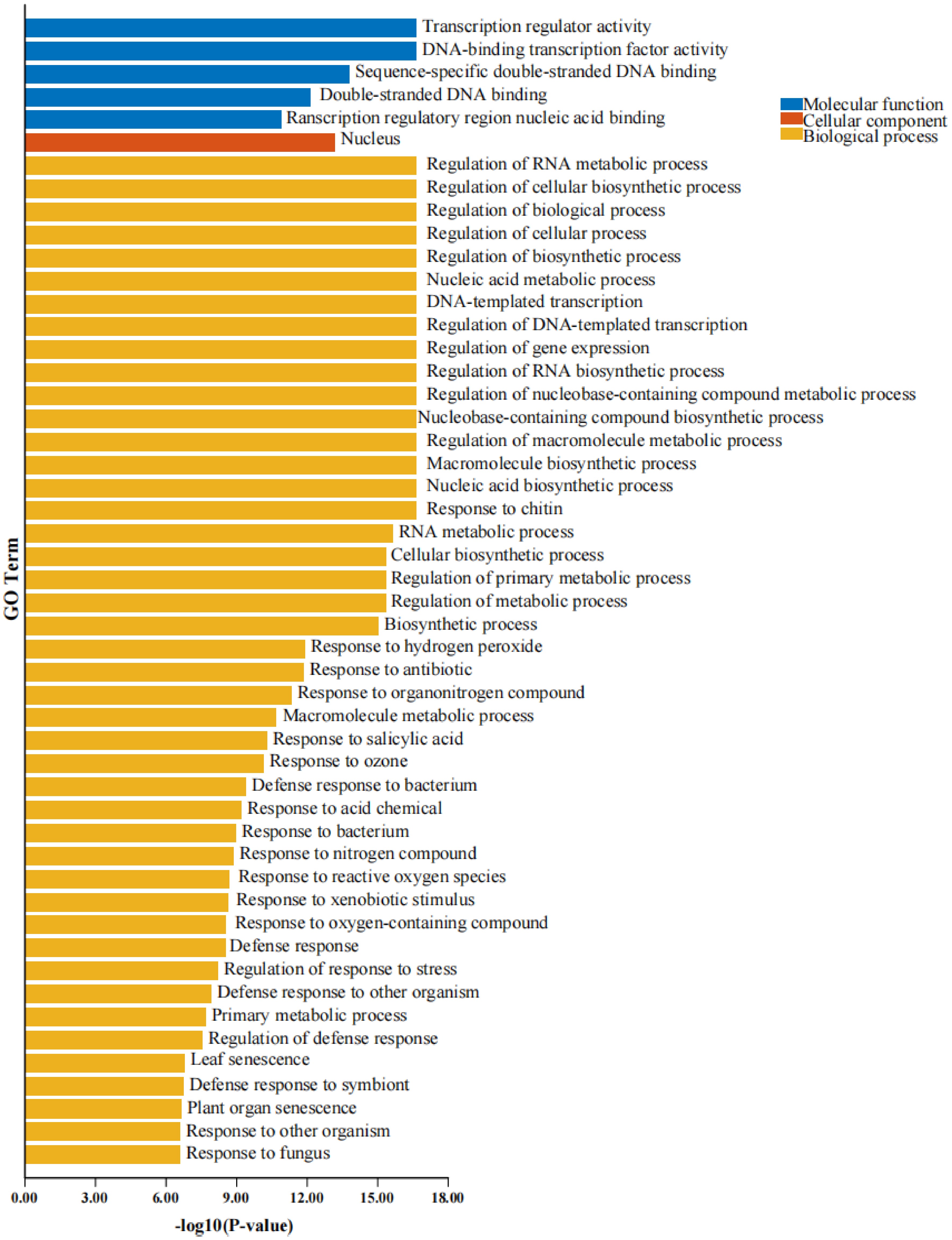
Figure 5.
GO enrichment analysis of WRKY proteins. Blue indicates molecular function (MF), orange represents biological process (BP), and yellow corresponds to cellular component (CC). The x-axis displays −log10(p-value), which reflects the statistical significance of the enriched GO terms for WRKY proteins. A higher value indicates greater enrichment significance.
Within the Biological Process (BP) category, GO enrichment highlighted the central involvement of PvWRKY proteins in governing cellular metabolic processes, regulating transcription (GO:0045893), and mediating responses to diverse stimuli (biotic and abiotic). Further detailed enrichment pointed to specific roles in RNA biosynthesis, overall gene expression control, and responses to oxidative stress, ozone, salicylic acid, and general environmental factors. This reinforces the critical contribution of PvWRKYs to plant growth, including organogenesis (e.g., leaf senescence, GO:0010150; root development, GO:0048364), and stress adaptation mechanisms. The Molecular Function (MF) analysis strongly corroborated the identity of PvWRKYs as transcription factors. Significant enrichment was observed for terms such as transcription regulator activity (GO:0030528), DNA-binding transcription factor activity (GO:0003700), and sequence-specific DNA binding (GO:0043565). Their role in interacting with regulatory DNA regions was further underscored by enrichment for transcription cis-regulatory region binding (GO:0001067), positioning them as key players in complex regulatory networks. Finally, Cellular Component (CC) annotations indicated that PvWRKY proteins are predominantly localized within the nucleus (GO:0005634) and, to a lesser extent, associated with membrane-bound organelles (GO:0043231). This localization pattern aligns perfectly with their established functions as transcriptional regulators operating primarily within the nucleus.
Expression profiles of PvWRKY genes under salt stress
-
Transcriptomic analysis using RNA-seq data elucidated the expression behavior of the PvWRKY gene family in seashore paspalum roots and leaves under varying salt stress durations. Conversion to FPKM values allowed for the observation of substantial expression level changes for multiple PvWRKY genes as salt stress progressed (Fig. 6). A prominent finding was the differential expression of individual genes between root and leaf tissues, suggesting divergent functional roles in these organs during salt adaptation. Moreover, a common pattern observed was the similar expression behavior among WRKY genes belonging to the same phylogenetic cluster, potentially indicating functional correlation or synergistic action within these groups.
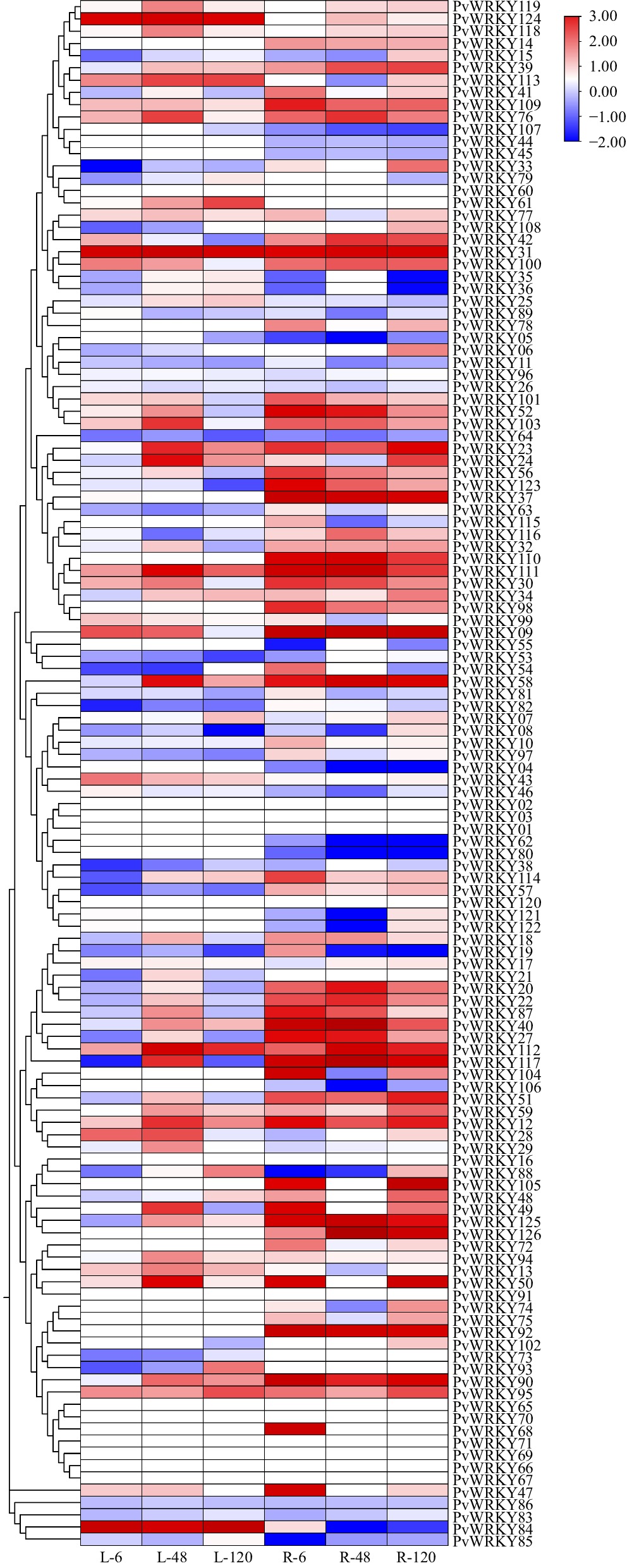
Figure 6.
Heatmap of PvWRKY gene expression under 0.2 mol/L NaCl salt stress. The heatmap illustrates the expression levels of PvWRKY genes in leaves and roots. This heatmap displays the expression levels of PvWRKY genes in leaf (L) and root (R) tissues under salt stress at different time points. The time points, labeled as L-6, L-48, and L-120 for leaves, and R-6, R-48, and R-120 for roots, represent gene expression at 6, 48, and 120 h post-salt treatment, respectively. Red shading indicates high gene expression levels, while blue shading represents low gene expression levels. The heatmap was created using TBtools software, with the intensity of red and blue providing an indication of gene expression variation across the conditions.
Building on these observations, the potential roles of PvWRKY genes in plant salt tolerance were predicted. Specifically, genes such as PvWRKY105, PvWRKY117, and PvWRKY126 showed notably high expression in roots, which increased significantly with prolonged salt stress, highlighting their essential roles in root response and salt tolerance. In contrast, PvWRKY84, PvWRKY58, and PvWRKY90 were highly expressed in leaves, suggesting their key involvement in the foliar response to salt stress. Interestingly, genes like PvWRKY12, PvWRKY31, PvWRKY111, and PvWRKY112 exhibited high expression levels in both roots and leaves, with stable or increasing expression trends across various salt stress time points. This suggests that these genes may play central roles in the plant's overall salt tolerance mechanisms (Fig. 6).
Additionally, PvWRKY09 and PvWRKY58 showed significantly higher expression in roots than in leaves, showing an upregulation trend under salt stress, which may be associated with the initial root response to salt stress. In contrast, PvWRKY84 exhibited a marked increase in expression in leaves under salt stress, while its expression in roots decreased, indicating its specific role in the foliar adaptation mechanisms to salt stress.
Validation of transcriptomic data through qRT-qPCR
-
Based on RNA-seq data, eight genes—PvWRKY10, PvWRKY22, PvWRKY34, PvWRKY52, PvWRKY56, PvWRKY76, PvWRKY87, and PvWRKY123—were selected for qRT-PCR validation. The qRT-PCR results validated the expression patterns of these PvWRKY genes in the leaves and roots of seashore paspalum under salt stress at 0, 6, 48, and 120 h.
As shown in Fig. 7, PvWRKY10 displayed an overall increase in expression in both leaves and roots under salt stress, with its expression reaching a peak in roots at 6 h, followed by a slight decline, though it remained above baseline levels. PvWRKY22 exhibited a biphasic expression pattern in leaves, initially decreasing before rising again, peaking at 48 h. In roots, its expression significantly increased at 6 and 48 h, with a slight decline at 120 h, suggesting a role in the mid-term response to salt stress. PvWRKY34 exhibited a significant decrease in leaf expression at 6 h, followed by a gradual increase at 48 and 120 h. In roots, its expression increased at 6 and 120 h but decreased at 48 h, indicating differential regulation across tissues. For PvWRKY52, little change was observed in leaf expression, but its expression significantly increased in roots at 6 and 48 h, before declining at 120 h, suggesting it may primarily function in early and mid-term salt stress responses in roots. PvWRKY56 exhibited stable expression in leaves, while its expression in roots steadily increased under salt stress, suggesting a persistent role in root salt tolerance. PvWRKY76 showed elevated expression levels in both leaves and roots at 6 and 48 h, followed by a decrease at 120 h, indicating its involvement in the early and mid-term stages of the salt stress response. PvWRKY87 reached peak expression in both leaves and roots at 48 h, with lower levels at 6 and 120 h, suggesting a key function in the mid-term response to salt stress. PvWRKY123 showed minimal variation in leaf expression, but its expression significantly increased in roots at 6 and 48 h, followed by a decrease at 120 h, indicating its potential role in the early and mid-term salt stress responses in roots. These findings indicate that the PvWRKY genes display time-dependent and tissue-specific regulatory patterns under salt stress, likely playing crucial roles at different stages of the salt stress response. Additionally, the expression trends observed in qRT-PCR closely aligned with those from the RNA-seq data, reinforcing the reliability of the RNA-seq results.
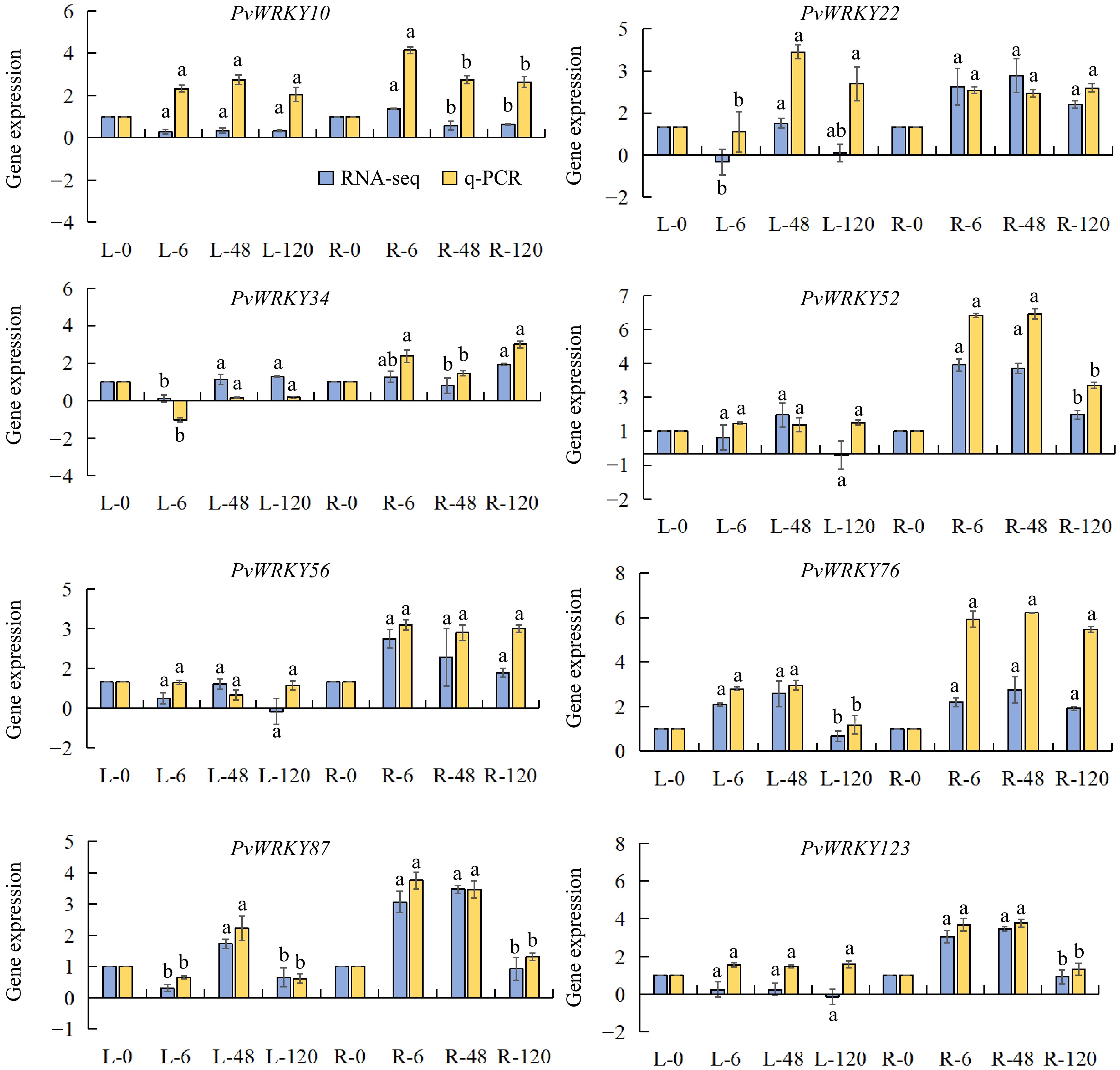
Figure 7.
Expression levels of PvWRKY genes in roots and leaves under salt stress. Error bars represent the standard error of three biological replicates, as calculated from qRT-PCR analysis. Letters indicate no statistically significant differences between means, as determined by ANOVA (p > 0.05). 'L' denotes leaf expression levels, and 'R' denotes root expression levels.
Prediction of protein interaction network for PvWRKYs
-
To probe the molecular basis and interaction partners potentially involved in salt tolerance regulation by specific PvWRKY transcription factors, a protein-protein interaction (PPI) network was constructed. This network focused on four genes (PvWRKY86, PvWRKY52, PvWRKY22, and PvWRKY123) identified as significantly upregulated in our qRT-PCR experiments. The interaction predictions were derived using the STRING database, leveraging known interactions among the Arabidopsis thaliana homologs of these target PvWRKY proteins.
The constructed PPI network indicated that the selected PvWRKY proteins participate in complex interaction landscapes (Fig. 8; Supplementary Table S7). PvWRKY52 appeared as a significant hub, predicted to interact with core components of the MAPK signaling cascade, including the substrate MKS1[59] and the kinases MPK3 and MPK4[60], which are known mediators of biotic and abiotic stress signals. Interactions were also predicted with SIB1 and SIB2, bZIP transcription factors implicated in disease resistance[61]. This convergence of interactions points towards a potential mechanism where PvWRKY52 contributes to salt tolerance through the modulation of MAPK pathways and downstream gene regulation.
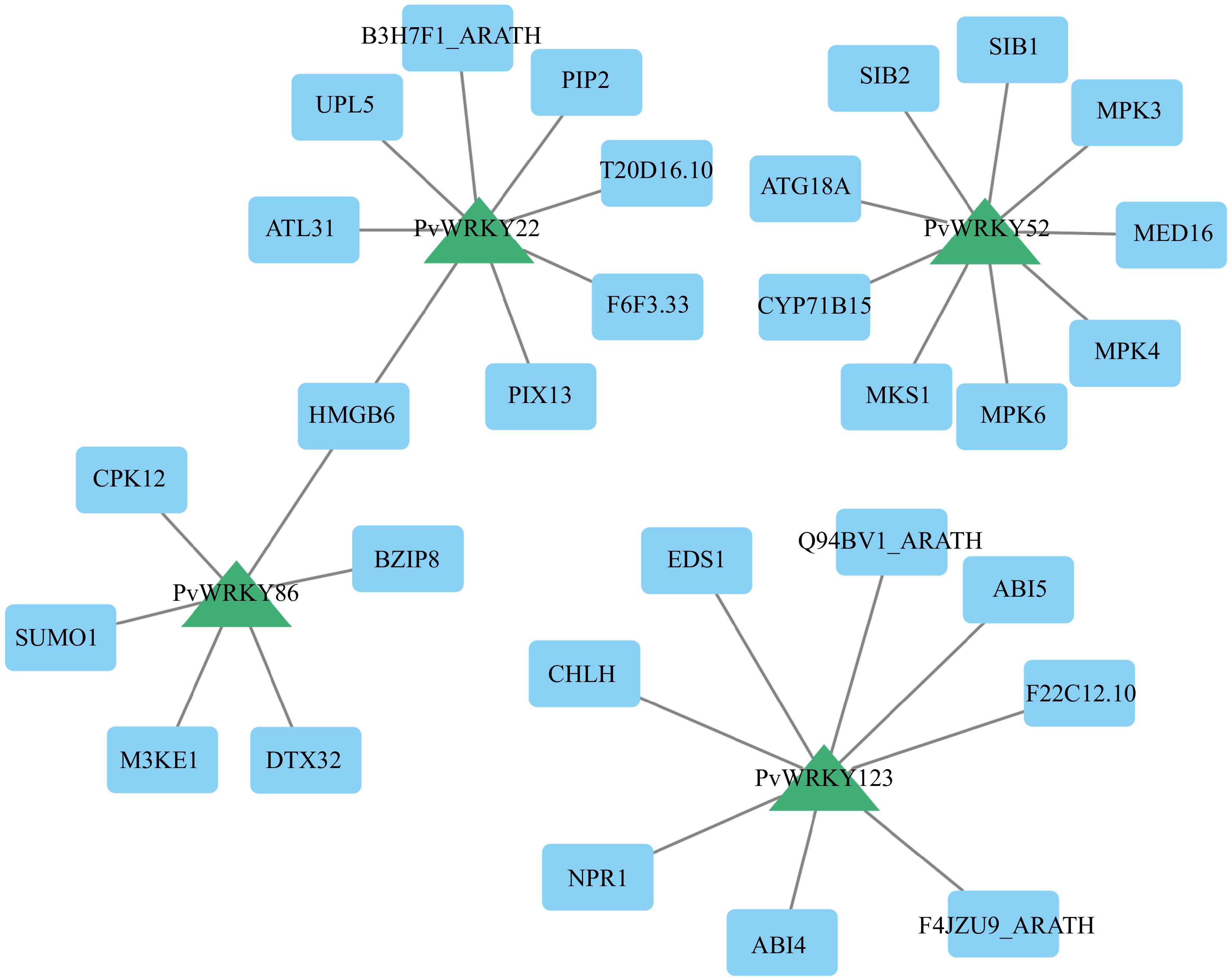
Figure 8.
The PPI network demonstrates the interactions among various PvWRKY proteins. The diagram was created using a medium confidence threshold of 0.4, with the intensity of node color corresponding to degree values; darker colors represent higher Degree values. Blue rectangles represent PvWRKY proteins, while red circles indicate other interacting proteins.
For PvWRKY22, predicted interactors included proteins linked to protein turnover and stress signaling, such as the E3 ubiquitin ligases UPL5[62] and ATL31 (also involved in C/N balance), alongside HMGB6, a high mobility group protein potentially involved in chromatin dynamics and gene expression control[63]. These associations suggest that PvWRKY22 might influence salt stress responses by participating in the regulation of protein stability and gene accessibility.
PvWRKY123 was linked to pivotal regulators of hormone signaling and defense. Its predicted interactors include ABI4 and ABI5, key transcription factors within the ABA signaling pathway essential for environmental stress adaptation[64], and NPR1, the master regulator of systemic acquired resistance (SAR) and defense responses[65]. This suggests a role for PvWRKY123 at the crossroads of ABA-dependent stress signaling and plant defense mechanisms.
Finally, PvWRKY86 was predicted to associate with the stress-responsive transcription factor BZIP8 and SUMO1, a key component of the SUMOylation machinery that post-translationally modifies proteins, affecting their stability and function[66,67]. These predicted interactions raise the possibility that PvWRKY86's activity is regulated by SUMOylation and that it collaborates with other transcription factors, like BZIP8, to orchestrate the expression of stress-related genes.
-
In this study, a thorough analysis of the PvWRKY gene family in seashore paspalum was performed, identifying 126 members at the whole-genome level. This number was relatively large compared to WRKY gene families in other species. The physicochemical characterization of PvWRKY proteins showed a wide variation in amino acid lengths, molecular weights, isoelectric points (pI), and instability indices. Most of these proteins were hydrophilic and predicted to be unstable, which may facilitate rapid turnover and enable swift responses to environmental stimuli. Subcellular localization predictions indicated that the majority of PvWRKY proteins are located in the nucleus (84.92%), aligning with their function as transcription factors that regulate gene expression[68]. The variation in pI values suggests that PvWRKY proteins may function in diverse cellular environments, potentially participating in various regulatory pathways.
Through the incorporation of AtWRKY proteins, the phylogenetic tree of PvWRKY proteins was divided into three major clades (I–III), aligning with classifications in other plant species. Motif and gene structure analyses further supported these phylogenetic groupings, with closely related genes displaying similar motif patterns and gene structures. The conservation of motifs such as motif 1 and motif 3 across most PvWRKY genes indicates essential functional domains, while the presence of unique motifs in specific subgroups suggests the potential for specialized functions.
Promoter analysis showed that PvWRKY genes contain several cis-acting elements linked to hormone signaling and stress responses. Notably, elements responsive to MeJA and ABA were highly enriched, with the MeJA-responsive element appearing 640 times across 115 genes and ABRE present 562 times in 116 genes. This enrichment suggests that PvWRKY genes are regulated by hormonal signals and play essential roles in mediating responses to abiotic stresses, particularly salt stress. Similar results have been observed in other species, where WRKY genes are involved in ABA signaling pathways and contribute to stress tolerance[69]. Additionally, other stress-related elements, such as MYB-binding sites linked to drought response, indicate that PvWRKY genes may be involved in multiple stress response pathways.
Expression analysis under salt stress conditions revealed that PvWRKY genes exhibit tissue-specific and time-dependent expression patterns. For instance, PvWRKY105, PvWRKY117, and PvWRKY126 were highly expressed in roots, especially after prolonged salt stress, indicating their involvement in root-specific responses to salinity. Gene Ontology (GO) enrichment analysis further supports their functional specialization, as these genes were enriched in biological processes such as root development (GO:0048364) and responses to oxidative stress, aligning with their roles in maintaining cellular homeostasis under prolonged salt exposure (Fig. 8). Conversely, PvWRKY84, PvWRKY58, and PvWRKY90 were predominantly expressed in leaves, suggesting roles in foliar stress responses. These differential expression patterns imply that specific PvWRKY genes are activated in distinct tissues to confer stress tolerance, supporting the concept of functional specialization within gene families[70].
The qRT-PCR validation of eight selected PvWRKY genes confirmed the RNA-seq results, showing consistent expression trends. Notably, PvWRKY52 and PvWRKY123 were significantly upregulated in roots during early and mid-term salt stress, suggesting roles in initial stress perception and signal transduction. GO analysis implicated PvWRKY52 in 'MAPK cascade regulation' (aligned with its PPI interaction with MPK3/MPK4), while PvWRKY123 was linked to 'ABA-mediated signaling' (GO:0009738), corroborating its interaction with ABI4/ABI5 in the PPI network. These findings illustrate how GO-based functional prediction complements experimental data, providing a robust framework to hypothesize gene-specific regulatory roles.
The protein-protein interaction (PPI) network predictions provided insights into the molecular mechanisms by which PvWRKY proteins mediate salt stress responses. PvWRKY52 emerged as a central regulator, interacting with key proteins involved in MAPK signaling pathways, such as MKS1, MPK3, and MPK4. These interactions suggest that PvWRKY52 may integrate stress signals via MAPK cascades to regulate downstream gene expression, enhancing salt tolerance, consistent with studies in other species where WRKY proteins interact with MAPKs to modulate stress responses[71]. Similarly, PvWRKY22 interacted with proteins such as UPL5, HMGB6, and ATL31, which are associated with protein degradation, chromatin remodeling, and stress responses. This indicates that PvWRKY22 may regulate protein homeostasis and gene expression under salt stress, contributing to adaptive responses. PvWRKY123 interacted with ABA signaling components like ABI4 and ABI5, as well as the defense regulator NPR1, suggesting its role in integrating hormonal and defense signaling pathways in response to salt stress. The involvement of PvWRKY86 with proteins like BZIP8 and SUMO1 highlights the complexity of regulatory mechanisms, including post-translational modifications affecting protein function and stability[72].
In summary, these results highlight the pivotal roles of the PvWRKY gene family in seashore paspalum's adaptation to salt stress. The expansion of this gene family, coupled with diverse gene structures, motif compositions, and expression patterns, underscores its critical function in saline environment adaptation. Unraveling the specific regulatory mechanisms and functional dynamics of these genes presents promising targets for future in-depth functional characterization and biotechnological applications.
Subsequent research efforts should prioritize the functional validation of key PvWRKY candidates identified in this study. Employing genetic manipulation techniques, such as gene knockout (e.g., using CRISPR/Cas9) or overexpression systems, will be essential to definitively establish their specific contributions to salt tolerance phenotypes. Elucidating the downstream molecular mechanisms requires identifying the direct target genes governed by these PvWRKY factors and mapping their interactions within the broader context of cellular signaling cascades involved in stress adaptation. Moreover, deciphering the upstream regulatory inputs, particularly the influence of hormonal cross-talk and other signaling molecules on PvWRKY expression, will be vital for constructing comprehensive models of the regulatory networks that underpin plant resilience.
-
In this study, a genome-wide characterization of the WRKY transcription factor family in Paspalum vaginatum was performed, identifying 126 PvWRKY genes classified into conserved Groups I–III. Tandem duplications on chromosomes 3 and 9 facilitated gene expansion, while promoter cis-element profiling revealed key stress-responsive motifs (e.g., ABRE and MeJA), indicating their role in salt adaptation. Transcriptional dynamics under salinity highlighted tissue-specific regulators, with PvWRKY105/117/126 enriched in roots and PvWRKY84/58/90 in leaves. Notably, PPI network analysis identified PvWRKY52 as a central regulator linking MAPK signaling (MKS1, MPK3/4) and immune responses (SIB1/2), and PvWRKY123 as a key player in ABA signaling (ABI4/5) and SUMOylation pathways. These findings suggest that targeting PvWRKY52 through CRISPR/Cas9 to enhance MAPK signaling could improve salt tolerance, while engineering PvWRKY123 to modulate ABA-SUMO interactions could boost stress resilience. Future research should focus on validating these interactions, such as through co-immunoprecipitation (Co-IP) assays, and exploring the involvement of PvWRKY genes in regulatory networks like reactive oxygen species (ROS) scavenging. These strategies offer a promising direction for enhancing salt tolerance in crop species.
This work was supported by the National Natural Science Foundation of China (Grant No. 32101423); the Foundation Project of Shandong Natural Science Foundation (Grant No. ZR2021MC066), and the Fundamental Research Funds for the Universities (Grant No. 6631120002).
-
The authors confirm contribution to the paper as follows: experiments conducted: Wu X, Hu X; advice and assistance in the research: Lu W, Sun Q, Wang Y, Qi J, Ren Z, Wu X; manuscript revision: Wu X; design of experiments and manuscript writing: Wu X, Wu X; manuscript revisions and feedback: Tian Z, Wang T. All authors reviewed the results and approved the final version of the manuscript.
-
The RNA-seq dataset mentioned in this article has not yet been published, and the dataset generated or analyzed during the current study can be obtained from the corresponding authors upon reasonable request.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 Primers used in qRT-PCR.
- Supplementary Table S2 Protein sequence of the WRKY gene family in Paspalum vaginatum O. Swartz.
- Supplementary Table S3 Physical and chemical properties of PvWRKY gene family members and prediction of subcellular localization.
- Supplementary Table S4 Motif sequences of PvWRKYs predicted in Betula platyphylla using MEME tools.
- Supplementary Table S5 Analysis of cis-acting elements in the promoters of 126 PvWRKYs in seashore paspalum.
- Supplementary Table S6 Gene Ontology (GO) annotation and enrichment analysis of the PvWRKY gene family in seashore paspalum.
- Supplementary Table S7 Predicted PPI network of PvWRKYs.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wu X, Tian Z, Wang T, Hu X, Sun Q, et al. 2025. Genome-wide identification and expression analysis of WRKY transcription factor family members in seashore paspalum under salt stress. Grass Research 5: e017 doi: 10.48130/grares-0025-0014 

Genome-wide identification and expression analysis of WRKY transcription factor family members in seashore paspalum under salt stress
- Received: 05 March 2025
- Revised: 19 April 2025
- Accepted: 07 May 2025
- Published online: 27 June 2025
Abstract: Although WRKY transcription factors (TFs) are established key regulators of plant stress responses, their contributions to highly salt-tolerant halophytes remain poorly understood. This study presents the first comprehensive genome-wide characterization of the WRKY gene family in the exceptionally salt-tolerant halophyte, seashore paspalum (Paspalum vaginatum). Using HMM profile searches and conserved domain analysis, 126 nonredundant PvWRKY sequences were identified. These were subsequently classified phylogenetically by comparison to Arabidopsis orthologs into established groups: Group I (n = 22), Group II (n = 58, subgroups IIa-IIe), and Group III (n = 46). Protein characterization revealed unstable hydrophilic PvWRKYs predominantly localized to the nucleus (84.92%), consistent with their transcriptional regulatory roles. Promoter cis-element analysis identified enrichment in stress-responsive motifs, with ABA-responsive elements (ABRE; present in 116 genes) and MeJA-responsive elements (detected in 115 genes), highlighting hormonal integration in salt adaptation. Intraspecific collinearity and tandem duplication events on chromosomes 2, 3, 5, and 9 suggested evolutionary expansion via gene duplication. Transcriptome and quantitative reverse transcription (qRT-PCR) analyses revealed spatiotemporal expression dynamics under salt stress: PvWRKY105/117/126 exhibited root-specific upregulation during prolonged stress, whereas PvWRKY84/58/90 were leaf-predominant responders. Functional annotation (GO) linked PvWRKYs to root development (GO:0048364), oxidative stress response, and MAPK signaling, with protein-protein interaction (PPI) networks identifying PvWRKY52 as a central hub interacting with key stress regulators (MKS1, MPK3/MPK4). Additionally, PvWRKY123 showed ABA signaling synergism via ABI4/ABI5 interactions, while PvWRKY86 was associated with SUMOylation-mediated regulation through BZIP8 and SUMO1. This genome-wide exploration of the WRKY family in seashore paspalum emphasizes its regulatory potential in salt adaptation and offers a foundation for future functional analyses.
-
Key words:
- Paspalum vaginatum /
- WRKY transcription factors /
- Halophyte /
- Salt stress /
- Protein interaction networks /
- qRT-PCR