









-
Salvia miltiorrhiza is one of the most commonly used Chinese medical herbs. As a representative species of the Lamiaceae, it is widely used in the treatment of cardiovascular and cerebrovascular diseases[1]. Its main active components are phenolic acids and tanshinones, of which phenolic acids consist of salvianolic acid A (Sal A), salvianolic acid B (Sal B), caffeic acid (CA), and rosmarinic acid (RA)[2−4], while tanshinones include dihydrotanshinone (DT), cryptotanshinone (CT), tanshinone I (TI), and tanshinone IIA (TIIA). The above active ingredients have various pharmaceutical values including anti-tumor, antioxidant, and anti-inflammatory effects[5−8]. In recent years, several transcription factors have been reported to participate in and regulate the synthesis of secondary metabolites of S. miltiorrhiza[9]. DNA binding with one finger (Dof) family is a typical transcription factor (TF) family with zinc finger proteins domain, which is unique to plants and plays an important role in modulating plant growth and development[10]. The Dof family has two main regional domains, namely the N-terminal conserved DNA binding domain and the C-terminal transcriptional regulation domain[10]. The N-terminal of the Dof protein is usually a highly conserved C2-C2 zinc finger domain consisting of 50−52 amino acids, and it can bind to the AAAG cis-acting element in the promoter region of the target gene[11]. The DNA binding domain is a key region, that is considered to be a bidirectional domain and can interact with other proteins[11−13]. The transcriptional regulatory domain of the C-terminal region may perform a variety of functions as it interacts with different regulatory proteins to activate the expression of target genes[11,13].
Dof proteins play a vital role in plant carbon and nitrogen metabolism[14,15], abiotic stress[16], hormone regulation[17], flowering control[18], light responses, and others[19]. Dof gene (ZmDof1) was first discovered in Zea mays[20], and it was thought to participate in the process of carbon metabolism by regulating the expression of the C4 photosynthetic phosphoenolpyruvate carboxylase (C4PEPC) gene in Z. mays[21]. JcDof3 interacts with F-box protein to regulate photoperiodic flowering and affect the flowering time[22]. In addition, multiple studies have shown that Dof genes are involved in various environmental changes[23]. OsDof18 is associated with the transport of ammonium salt in rice, thus regulating the utilization efficiency of nitrogen in rice[24] , and it can also restrict the biosynthesis of ethylene and increase prophase primary root elongation[17]. The expression of ThDof1.4 and ThZFP1 of Tamarix ramosissima can increase the content of proline and enhance the scavenging ability of ROS, thus improving the tolerance of Tamarix to salt stress and osmotic stress[25]. In Arabidopsis thaliana, AtCDF3 was highly induced by drought, low temperature, and abscisic acid (ABA), and the overexpression of AtCDF3 in transgenic plants enhanced their tolerance to drought, cold, and osmotic stress[26]. SlDof22 is involved in the production of ascorbic acid and the process of tomato salt stress in tomato[27]. These studies uncovered the importance of Dofs in the life cycle of plants.
Plant hormones are trace compounds involved in the whole process of plant growth and development, and influence the growth and development of plants[28]. ABA, as an important plant hormone can accelerate the shedding of plant organs, and can impact the synthesis of secondary metabolites by stimulating the corresponding transcription factors in plants[9]. In S. miltiorrhiza, ABA can induce the expression of SmbZIP1 leading to the upregulation of SmC4H1 to promote the accumulation of phenolic acids[9]. ABA can also significantly promote the expression of HMGR, FPS, CYP71AV1, and CPR, thus increasing the content of artemisinin in Artemisia annua[29].
In recent years, the Dof gene family has been gradually identified in many plants due to the continuous publication of the high quality of plant genomes. There were 36 Dof genes in Arabidopsis[30], 103 Dof genes in Camelina sativa[31], 34 Dof genes in melon[32], and 51 Dof genes in blueberry[33]. However, the Dof family has not been fully explored in the whole genome of S. miltiorrhiza. Due to the importance of the Dof gene in various physiological processes of plants, it is necessary to study its specific role in S. miltiorrhiza. In the present study, genome and transcriptome data of S. miltiorrhiza were used to identify the Dof genes. Then, multiple sequence matching, evolutionary tree analysis, gene structure, and cis-acting element analysis were systematically investigated in the whole genome of S. miltiorrhiza. To predict the function of SmDofs in regulating the biosynthesis of tanshinones and phenolic acids in S. miltiorrhiza, co-expression analysis of the biosynthetic pathway genes related to tanshinones and phenolic acids and the SmDofs was performed based on the transcriptome data induced by ABA, and then the target gene of candidate SmDofs were validated by the dual luciferase (Dual-LUC) assay. This study enlarges the understanding of the SmDof gene family, and reveal the potential molecular mechanism of SmDofs in regulating the biosynthesis of tanshinones and phenolic acids in S. miltiorrhiza.
-
The genome sequences were downloaded from the S. miltiorrhiza database[34]. Based on the Pfam database (
http://pfam.xfam.org/ ), the Hidden Markov Model (HMM) file of the Dof gene family (PF02701.18) was obtained, and the whole genome of S. miltiorrhiza compared using the HMMER search program in HMMER3.0 software package to obtain the gene sequence of the initial screening[35]. SMART (http://smart.embl-heidelberg.de/ ) and MOTIF Search (www.genome.jp/tools/motif ) are employed to predict the structure of the candidate protein domains. ExPASy (http://web.expasy.org/compute_pi/ ) was used to calculate the sequence length, molecular weight, and isoelectric point[36]. Finally, WoLF PSORT (https://wolfpsort.hgc.jp/ ) was introduced to predict the subcellular localization of the identified Dof proteins[37].Multiple sequence alignment and phylogenetic tree construction
-
The conserved domain of SmDofs protein was studied by multiple sequence alignment using the DNAMAN 7.0 software. The AtDof protein sequences of A. thaliana were downloaded from the TAIR database (
www.arabidopsis.org )[38]. AtDofs and SmDof proteins were analyzed using MEGA 6.0. The phylogenetic tree was constructed using the neighborhood join method (NJ) with the bootstrap value set to 1,000[39].Gene structure analysis and cis-elements of SmDofs promoters
-
The organization of exons, introns, and untranslated regions of the SmDof genes were analyzed using the Gene Structure Display Server (
http://gsds.cbi.pku.edu.cn/ ), and visualized by loading the GFF files of SmDof genes of S. miltiorrhiza to the TBtools (v.2.003) software, which was also used for analyzing and searching for conserved motifs[40]. PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/ ) was introduced to study the cis-acting elements in a length of 1,500-bp in the upstream of the initiation codon of the 31 SmDof genes in S. miltiorrhiza. According to the functional annotations of cis-acting elements, the candidate elements were gathered for further research[41].RNA-seq and qRT-PCR detection
-
Two transcriptome datasets of S. miltiorrhiza, of which one is generated from four tissues including flower, stem, leaf, root, and another is collected from hairy roots induced by ABA, were adopted to analyze the expression level of SmDof genes[42]. TBtools (v.2.003) software was employed to draw a heat map to exhibit the expression level of the SmDof genes derived from transcriptome dataset[40]. To detect the expression profile of candidate SmDof genes, hairy roots of S. miltiorrhiza were treated with 50 μM ABA and collected after treatment for 0-, 0.5-, 1-, 2-, 4-, and 8-h, respectively[7] . The collected samples were quickly placed in liquid nitrogen and stored in the refrigerator at −80 °C for subsequent RNA extraction. Total RNA was extracted from S. miltiorrhiza hairy roots using the Plant Total RNA Extraction Kit (Vazyme Biotech Co., Ltd, China). Meanwhile, the concentration and purity of the extracted RNA was measured by spectrophotometer, and then the RNA integrity was observed by electrophoretic analysis with 1% agarose gel. Reverse transcription was performed with the cDNA Synthesis Kit (Vazyme Biotech Co., Ltd, China), and a total of 100 ng RNA was prepared for cDNA synthesis reaction with a volume of 50 μL[43]. Quantitative primer pairs were designed using the Primer 5.0 software. SuperReal PreMix Plus kit (Vazyme Biotech Co., Ltd, China) was used in ABI Step One Plus real-time PCR System. Quantitative real-time PCR (qPCR) was performed using 10 μL real-time PCR reaction solution, including 1 μL cDNA was used as a template; the upper and downstream primers were 0.2 μL, respectively; 5 μL Taq Pro SYBR qPCR Master Mix and 3.6 μL ddH2O. The PCR reaction conditions were as follows: 95 °C for 15 s, 60 °C for 30 s, 72 °C for 30 s, a total of 40 cycles, each sample was triply repeated. SmActin was used as the internal reference gene to normalize the expression level of Dof genes. The method of 2−ΔΔCᴛ was used to calculate the relative expression level of SmDofs[7].
Co-expression analysis
-
The co-expression relationship between the SmDof genes and the biosynthetic genes involved in tanshinones and phenolic acids biosynthesis was resolved. Pearson correlation coefficient > 0.8 and p-value < 0.05 was set as the cutoff. Then, the co-expression relationship was visualized with the Cytoscape software[44].
Subcellular localization
-
To dissect the subcellular localization profiles of SmDof proteins, the open reading fragment (ORF) cDNA sequences of SmDof12 and SmDof29 are amplified and inserted into the vector of PHB-YFP to generate the fusion recombinant of PHB-SmDof12-YFP and PHB-SmDof29-YFP, and then they are transformed into Agrobacterium tumefaciens GV3101 and injected into N. benthamiana leaves for transient transformation, respectively[3]. pHB-YFP was used as the negative control. The transgenic N. benthamiana leaves were cultivated in the dark for 24 h and then transferred to the light for 24 h. YFP signals from infected N. benthamiana leaves were visualized using a high-resolution microscope observation system. The nuclei of epidermal cells of infected N. benthamiana leaves were stained with 4',6-diamidino-2-phenylindole dihydrochloride (DAPI) solution (10 μg/mL) for 2 h before observation.
Dual-luciferase assay
-
To investigate the ability of SmDofs to transcriptionally activate the tanshinones biosynthetic genes, Dual-luciferase (Dual-LUC) assays were performed as previously reported[45]. Each of the recombinant plasmids of PHB-SmDof12-YFP and PHB-SmDof29-YFP was introduced into A. tumefaciens strain GV3101 to be the effector, and PHB-YFP plasmid was used as a negative control. The promoters of PAL and GGPPS were inserted into pGREEN0800 vector as the reporter constructs to drive the expression of the firefly luciferase gene, respectively. The Renilla luciferase gene driven by CaMV 35S promoter was used as an internal control. And then, each of them was co-transformed into A. tumefaciens strain GV3101 with the helper plasmid pSoup19. The reporter strains were mixed with effector strains at a ratio of 1:1 to inject into N. benthamiana leaves. Leaves were collected after 48 h for determination of fluorescence values according to the manufacturer's instructions (Promega, Madison, WI, USA)[9]. Three biological replicates were measured for each sample.
Measuring tanshinones and phenolic acids contents by high-performance liquid chromatography
-
Different tissues including roots, stems, leaves, and flowers of S. miltiorrhiza were collected and dried in an oven. The dried tissues were then ground to powder for compound analysis. Extraction of tanshinones and phenolic acids and high-performance liquid chromatography (HPLC) detection were done as the previous report[6,9]. The total content of tanshinones and phenolic acids were quantified by comparing the standard curves and retention times, with solutions without extracts added as the controls.
Statistical analysis
-
All the detections performed in the present study, including qRT-PCR, HPLC, and Dual-LUC assays, were triply repeated. Gene expression levels, tanshinone contents, and phenolic acid contents were presented as the mean value ± SD. SPSS 16.0 software (SPSS) was employed to analyze statistical significance by single-sample t-test and one-way analysis of variance. p-value < 0.05 was regarded to be statistically significant.
-
The Hidden Markov model (HMM) of the Dof domain (PF02701.18) was employed to search for Dof genes in S. miltiorrhiza. A total of 31 Dof genes were detected, and the gene was named SmDof1-SmDof31, respectively (Supplementary Table S1). The results of Pfam and SMART analysis showed that all of these proteins contained complete Dof domains[23]. The CDS length, protein molecular weight (MW), isoelectric point (pI), and subcellular location of each SmDof gene in S. miltiorrhiza were further analyzed (Table 1). Of the 31 proteins, SmDof25 and SmDof22 had the lowest number of amino acids, decreasing to 168, while SmDof16 had the highest number of amino acids, reaching to 511. The pI of SmDofs ranges from 6.01 (SmDof5) to 10.55 (SmDof17), and the molecular weight ranges from 18,463.7 (SmDof22) to 55,341.6 (SmDof16). Subcellular localization prediction revealed that 27 SmDofs were located in the nucleus, while four SmDofs including SmDof19, 21, 22, and 25 located in chloroplasts (Table 1).
Table 1. Length, molecular weight, isoelectric point, and subcellular localization of 31 SmDof proteins in S. miltiorrhiza.
Gene ID Name Length (aa) MW (Da) pI Subcellar
localizationSMILT016590.1 SmDof1 304 33,168.7 8.66 nucleus SMILT016591.1 SmDof2 246 26,202.9 9.8 nucleus SMILT016651.1 SmDof3 242 26,184.9 8.96 nucleus SMILT021318.1 SmDof4 225 23,560.2 8.6 nucleus SMILT032678.1 SmDof5 224 24,689.4 6.01 nucleus SMILT003591.1 SmDof6 241 25,256.1 4.66 nucleus SMILT009582.1 SmDof7 306 32,436.3 4.69 nucleus SMILT017417.1 SmDof8 301 33,248.9 6.7 nucleus SMILT020107.1 SmDof9 332 36,827.9 7.94 nucleus SMILT023380.1 SmDof10 318 34,176.8 9.72 nucleus SMILT025505.1 SmDof11 283 30,690.9 8.48 nucleus SMILT025760.1 SmDof12 274 30,006.1 8.47 nucleus SMILT028288.1 SmDof13 249 27,303.1 8.99 nucleus SMILT030586.1 SmDof14 334 36,582.9 6.92 nucleus SMILT031093.1 SmDof15 230 23,444.9 8.49 nucleus SMILT000323.1 SmDof16 511 55,341.6 5.23 nucleus SMILT000784.1 SmDof17 265 27,611.4 10.55 nucleus SMILT000789.1 SmDof18 198 22,350 9.04 nucleus SMILT001058.1 SmDof19 190 20,795.1 9.27 chloroplast SMILT001687.1 SmDof20 216 24,023.7 9.28 nucleus SMILT002891.1 SmDof21 268 29,359.3 4.54 chloroplast SMILT004451.1 SmDof22 168 18,463.7 8.83 chloroplast SMILT005491.1 SmDof23 266 29,274.4 9.31 nucleus SMILT007077.1 SmDof24 251 27,427 9.06 nucleus SMILT007580.1 SmDof25 168 18,625.9 9.22 chloroplast SMILT009335.1 SmDof26 191 21,529.9 9.5 nucleus SMILT010473.1 SmDof27 240 24,863.9 7.82 nucleus SMILT012697.1 SmDof28 248 26,508.7 9.28 nucleus SMILT019592.1 SmDof29 283 30,740.7 8.39 nucleus SMILT023561.1 SmDof30 337 35,816.5 9.51 nucleus SMILT024154.1 SmDof31 258 27,841.9 8.06 nucleus Sequence alignment and phylogenetic traits of SmDof proteins
-
To dissect the characteristics of the domain within SmDof proteins, DNAMAN software was employed to conduct multiple amino acid sequence alignment. The results showed that all the SmDof proteins contained a conserved domain in its core sequence, namely CX2CX21CX2C zinc finger structure (Fig. 1). The conserved domain consists of 50 amino acid residues, of them four cysteine residues are relatively conserved within the zinc finger domain in the N-terminal region of SmDof proteins[11].

Figure 1.
Multiple sequence alignment of the 31 SmDof proteins. Different colors represent identical and conserved amino acid residues, and the red box shows the conserved zinc-finger domain.
To further explore the evolutionary relationships among the SmDof genes, a phylogenetic tree of a total of 67 Dof proteins (Supplementary Table S2) in Arabidopsis (36 members) and S. miltiorrhiza (31 members) were constructed. The total number of Dof genes in S. miltiorrhiza and A. thaliana is comparatively secure, and it indicates the conservative features of this gene family. Sixty seven Dof proteins are divided into five groups based on the branch of the tree, Groups I−V (Fig. 2). There are 31 SmDof gene families in S. miltiorrhiza, among them, six SmDofs are distributed in Group I and Group IV, 11 in Group II, and eight in Group III. In Arabidopsis, Groups I to IV contain 7, 0, 0, 10, and 19 Dof genes, respectively. The variable number of the five subgroups is beneficial for us to evaluate the degree of gene expansion or loss during the evolution of the two species.
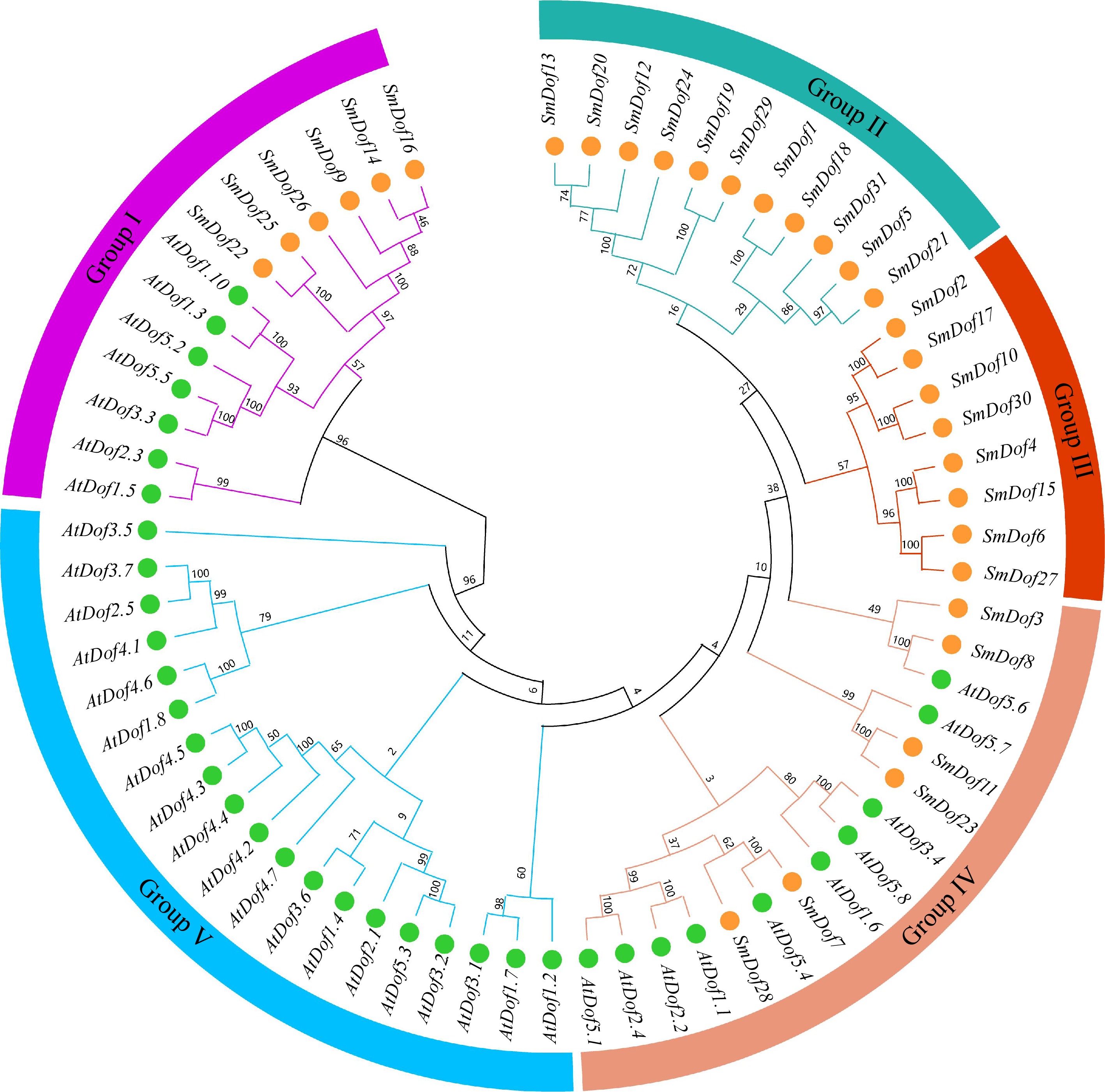
Figure 2.
Evolutionary relationship of SmDof proteins in S. miltiorrhiza and Arabidopsis. Varied colors represent different groups. There were five groups, Groups I−V, with the green circles representing the SmDof proteins of Arabidopsis and the orange circles representing the Dof proteins of S. miltiorrhiza.
Structural feature of SmDof genes
-
To further investigate the functional regions of SmDof proteins, the conserved motif was predicted by the MEME program utilizing a two-component finite mixture model. In total, 15 motifs were identified in all the SmDof proteins, and we found that many groups of SmDofs shared a similar conserved motif. As shown in Fig. 3a and b, motif 1 is included in all SmDof proteins. Among all groups, Group I contained the most SmDof members being consisted of motifs 1, 2, 3, 8, and 15. The common motifs among the SmDof proteins are indicative of conserved evolutionary relatedness and similar biological functions.
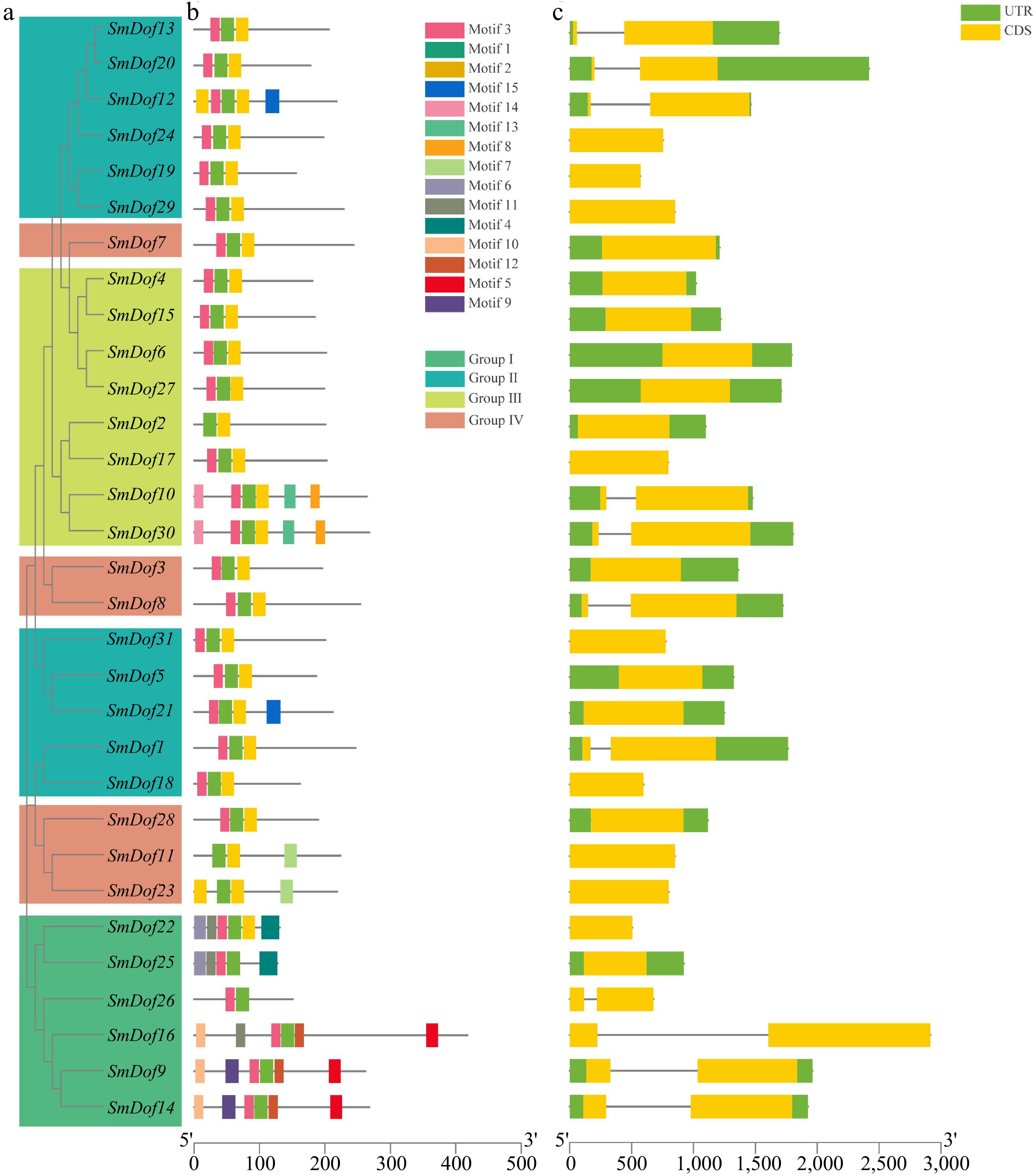
Figure 3.
Phylogeny, conserved motifs and gene structure of SmDof proteins in S. miltiorrhiza. (a) SmDof proteins evolutionary tree. (b) Conserved motifs of the 31 SmDof proteins. Different colors represent 15 different motifs, and the bottom line represents the length of the sequence. (c) Exon/intron structures of SmDofs. Green represents UTRs and yellow represents CDS.
To study the structure of SmDof genes, the full-length cDNA sequences of all SmDof genes with the corresponding genomic DNA were aligned (Fig. 3c). The number of exons in SmDof ranged from 1 to 2. There were no more than two introns in each SmDofs. The variation in the number of exons may indicate that the SmDof genes may have diverse functions related to the medicinal substance biosynthesis, growth, or development in S. miltiorrhiza.
Cis-acting elements in the promoter region of SmDof genes
-
PlantCARE was introduced to analyze the promoter sequence of 31 SmDof genes from the translation initiation site (ATG), and 55 cis-acting elements were identified. Among of them, they were related to plant cell development, plant hormones, environmental stress, and light response, respectively (Fig. 4). The results show that 22 light-responsive elements get the most abundant compared to other elements, and 31 SmDof genes have light-responsive elements like Box 4, MRE, GT1-motif. In addition, 12 cis-acting elements related to plant hormones were identified. In addition, there are five cis-acting elements associated with cell development, like CAAT-box, HD-Zip 1, MBSI, CCAAT-box, and MSA-like. There are four cis-acting elements associated with environmental stress, like TC-rich repeats, AT-rich element, LTR, and MBS (Fig. 4). It is implied that most of the SmDofs may play an important role in response to plant hormones and are light responsive. This is in agreement with the previous studies on Dof gene families in sugarcane, which is thought to be involved in light response, metabolism, and other functions[19].

Figure 4.
Cis-acting elements of SmDof promoters in S. miltiorrhiza. Dof family cis-acting element of S. miltiorrhiza. Different colors represent different classes of cis-acting elements and motifs. Green represents cis-acting elements associated with light, yellow represents cis-acting elements associated with plant hormones, purple represents cis-acting elements associated with cell development, and blue represents environmental stress.
Expression patterns of SmDofs and synthetase genes involved in tanshinones and phenolic acids biosynthesis pathway in various tissues and under ABA treatment
-
To gain a deeper understanding of SmDof expression patterns, four tissues including root, stem, leaf, and flower were collected to measure the total content of tanshinones and phenolic acid by HPLC, and subjected to transcriptome sequencing to investigate the expression of SmDofs. The results showed that the total phenolic acids and tanshinones content were all highest in root compared to other tissues (Fig. 5a), and a total of five genes (SmDof6, 12, 13, 27, 29) were highly expressed in the root, which is harvested in practice as the medicinal tissue[3] (Fig. 5b & Supplementary Table S3).
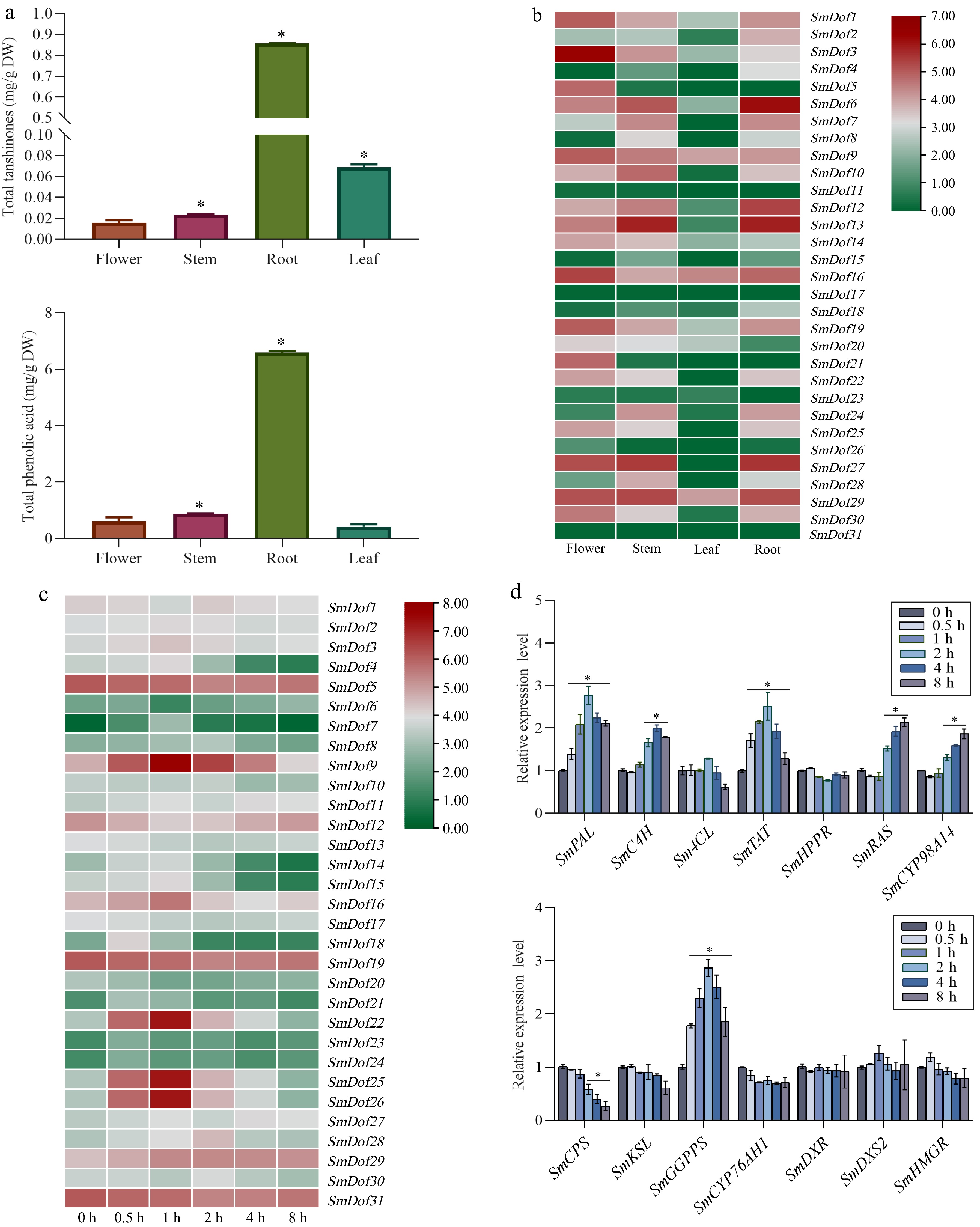
Figure 5.
Expression profiles of SmDof genes and synthetase genes involved in tanshinones and phenolic acids biosynthesis pathway. (a) Contents of tanshinones and phenolic acids in different tissues of S. miltiorrhiza. (b) Expression profiles of SmDof gene in various tissues of S. miltiorrhiza. (c) Expression profile of SmDof genes under the treatment of ABA induction based on the transcriptome dataset. Red and blue boxes indicate high and low expression levels of SmDofs, respectively. (d) Expression profiles of synthetase genes involved in tanshinones and phenolic acids biosynthesis pathway under the induction of exogenous ABA detected by qRT-PCR. Three biological replicates were performed and the mean ± SD was taken, SD was represented by the error line. * indicates significant difference in t-test (*p < 0.05).
To mine the candidate SmDofs in response to ABA treatment, the expression variation of candidate SmDof gene exhibiting at least a 2-fold increase more than the control was set as the cutoff. In total, 11 SmDof genes including SmDof9, 16, 18, 21, 22, 23, 24, 25, 26, 28, and 29 showed an obvious increase compared to the control, among which three SmDofs (SmDof22, 25, and 26) exhibited the highest increase reaching to a 17-fold increase over the control. Whereas, seven SmDof gens including SmDof4, 6, 12, 14, 15, 18, and 20 downregulated the expression levels more than 2-fold than the control (Fig. 5c &Supplementary Table S4). Moreover, qRT-PCR was employed to examine the expression level of synthetase genes involved in the tanshinones and phenolic acids biosynthesis pathway. As shown in Fig. 5d, several genes were revealed including PAL, C4H, TAT, RAS1, and CYP98A14 in phenolic acid biosynthesis pathway and GGPPS in tanshinone biosynthesis pathway upregulated significantly under the induction of exogenous ABA, in particular, PAL, and GGPPS were the most up-regulated. Therefore, the above results provide a valuable dataset for mining functional SmDof genes in regulating medicinal substance metabolite synthesis in S. miltiorrhiza.
Co-expression relationship of SmDofs with the biosynthetic genes involved in tanshinones and phenolic acids biosynthesis in S. miltiorrhiza
-
As reported by Shi et al., ABA can affect the expression of the biosynthetic genes involved in tanshinones and phenolic acids biosynthesis, thereby promoting the medicinal metabolites accumulation in S. miltiorrhiza hairy roots[42]. Therefore, the co-expression relationship between the 31 SmDofs with the biosynthetic genes related to the biosynthesis of tanshinones and phenolic acids in S. miltiorrhiza was dissected. The results showed that 15 SmDofs (including SmDof4, 5, 8, 10, 12, 13, 14, 15, 17, 19, 20, 28, 29, 30, 31) co-expressed with SmRAS, SmHPPR, SmC4H, Sm4CL, SmCYP98A14, SmPAL, or SmTAT genes, respectively, with the Pearson correlation coefficient > 0.8 and p-value < 0.05. Moreover, 15 SmDofs (including SmDof4, 6, 9, 10, 11, 12, 13, 14, 15, 17, 20, 27, 28, 29, 30) exhibited a co-expression pattern with SmCYP76AH1, SmKSL, SmCPS, SmGGPPS, SmDXR, or SmDXS2 genes, respectively, and the correlation coefficient was greater than 0.8. It is noteworthy that SmDof4, 10, 12, 13, 14, 15, 17, 20, 28, 29, 30 not only co-express with tanshinones biosynthetic genes, but also co-express with phenolic acids biosynthetic genes, implying that the above 11 SmDof genes may play a vital role in promoting the accumulation of the above two types of medicinal substances.
Expression pattern and subcellular localization analysis of SmDof12 and SmDof29 genes
-
Transcriptome dataset and co-expression analysis were integrated to mine the candidate SmDof genes in association with the biosynthesis of tanshinones and phenolic acids in S. miltiorrhiza. By the transcriptome dataset from various tissues, five SmDof genes were found including SmDof6, 12, 13, 27, and 29 all expressed vigorously in the root (Fig. 5b), which is thought to be the main tissue to accumulate the medicinal substances in practice[3]. According to the results of co-expression analysis, SmDof12 had the highest negative correlation coefficient (reaching −0.917) with the SmGGPPS gene related to the biosynthesis of tanshinones. Whereas, SmDof29 got the highest correlation with the SmPAL gene involved in the phenolic acids biosynthetic pathway, with the correlation coefficient of 0.912 (Fig. 6). Those results push us to validate the expression profile of the two SmDof genes. As expected, the expression profiles of SmDof12 and SmDof29 detected by qRT-PCR analysis were consistent with the transcriptomic dataset (Fig. 7a, b & Supplementary Table S5), of which indicated the reliability of the transcriptome dataset derived from four types of tissues and hairy root lines treated with ABA in S. miltiorrhiza.
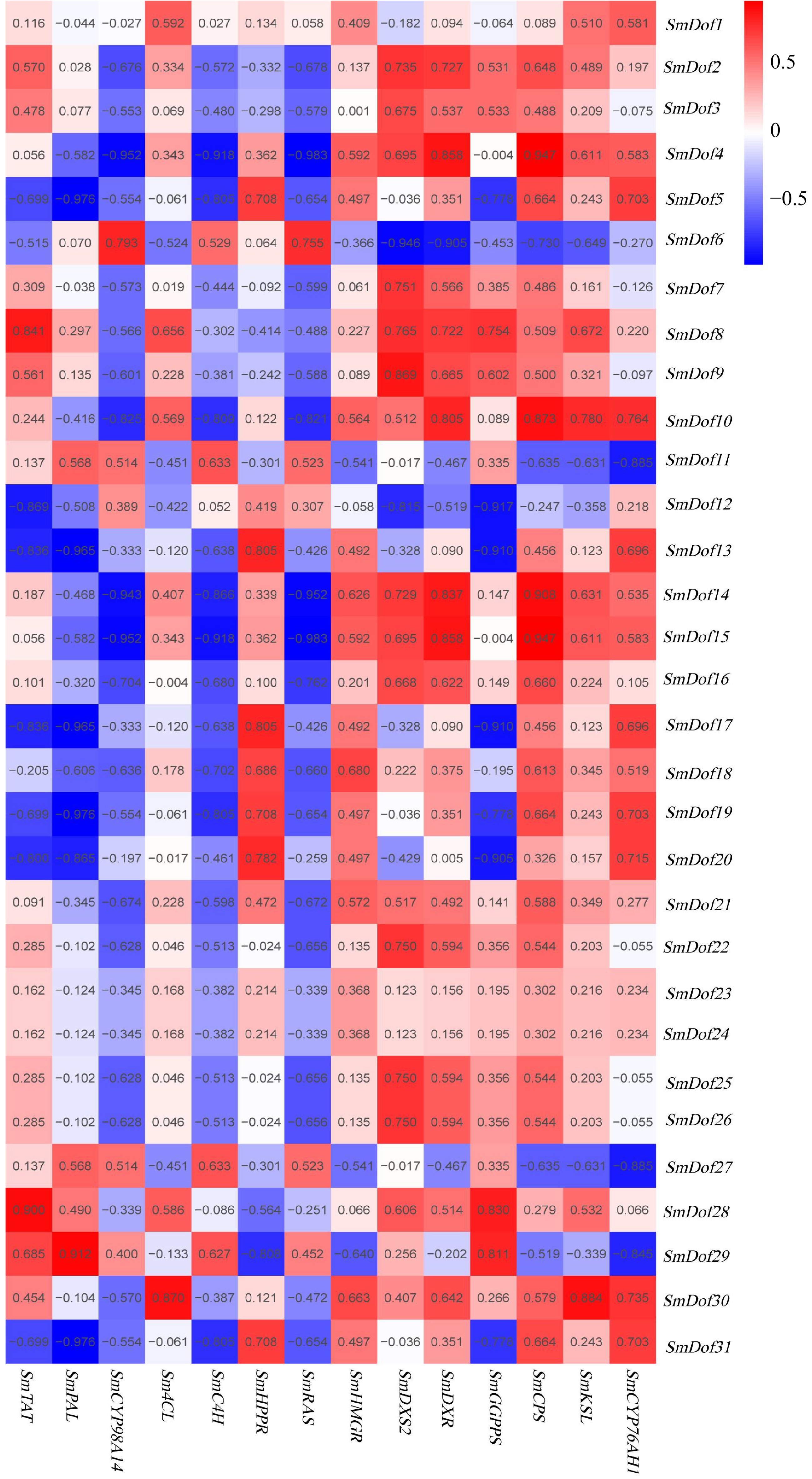
Figure 6.
Co-expression analysis of SmDof genes and the genes involved in the biosynthetic pathway of phenolic acids and tanshinones. R > 0.5 indicates a positive correlation; R < –0.5 indicates a negative correlation. The data were obtained from the ABA transcriptome dataset.
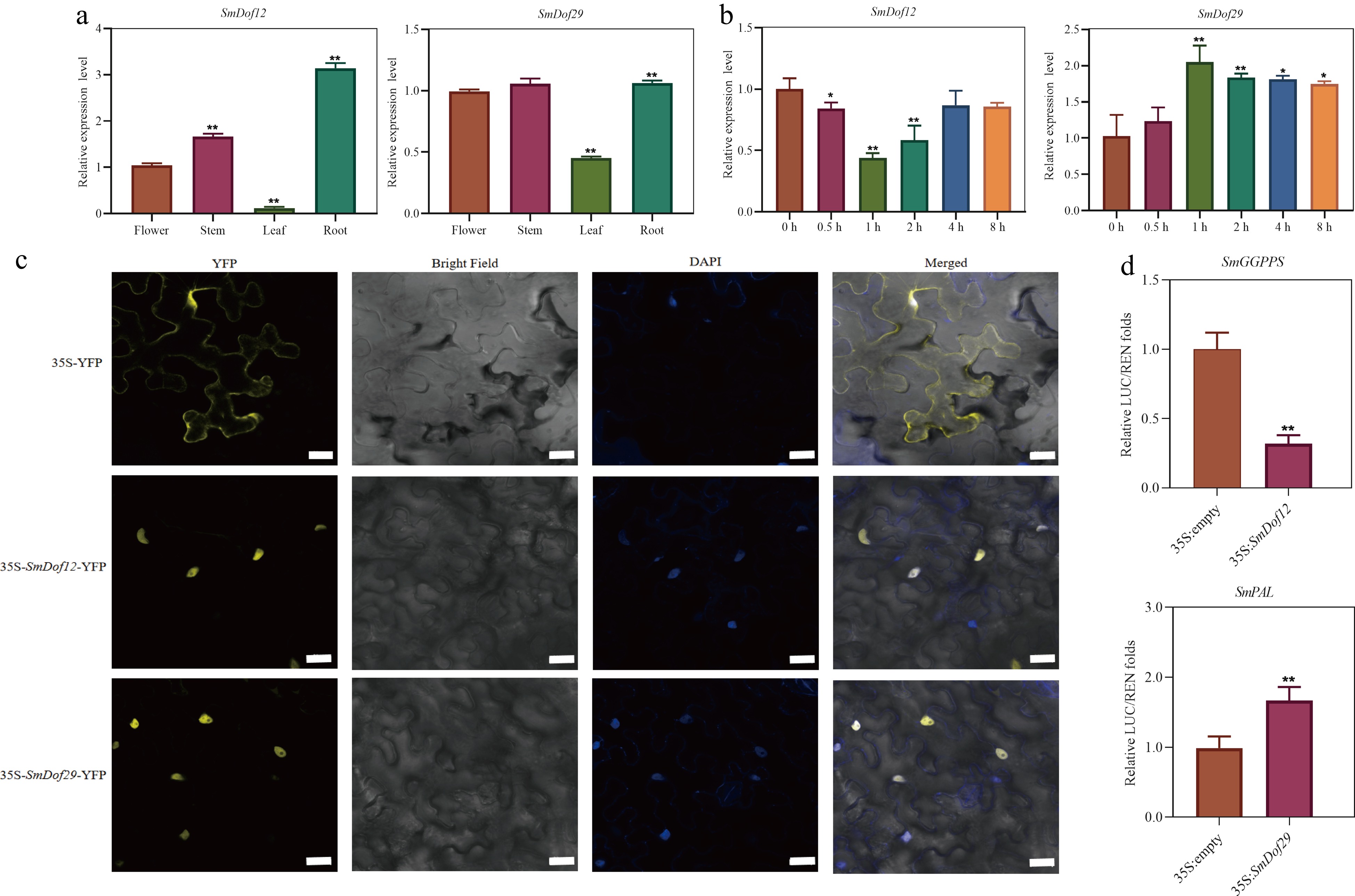
Figure 7.
Functional characterization of SmDof12 and SmDof29. (a) Expression patterns of SmDof12 and SmDof29 in four tissues of S. miltiorrhiza. The fold changes of the relative gene expression level in the other three tissues are all normalized to the expression level in flower. (b) Expression patterns of SmDof12 and SmDof29 in hairy roots of S. miltiorrhiza treated with ABA. The fold changes in the relative gene expression level were all normalized to the control expression without induction at the 0 h time point. (c) Subcellular localization of SmDof12 and SmDof29 in tobacco. 35S-YFP is the control group, yellow is the fluorescence of YFP, and blue is the nucleus. Scale bar = 50 μm. (d) Dual-Luc assay showed that SmDof12 could inhibit the activity of SmGGPPS promoter and SmDof29 could promote the activity of SmPAL promoter. Three biological replicates were performed and the mean ± SD was taken, SD was represented by the error line. * indicates significant difference in t-test (* p < 0.05, ** p < 0.01).
And then, the subcellular localization of the SmDof12 and SmDof29 in epidermal cells from 45-day-old N. benthamiana leaves were studied by transient expression analysis of the two genes fused with YFP, respectively. Robust fluorescence was observed only in the nuclei in 35S-SmDof12-YFP and 35S-SmDof29-YFP, while the 35S-YFP control displayed fluorescence throughout the whole cell (Fig. 7c), suggesting that the SmDof12 and SmDof29 proteins are all localized in the nuclei in S. miltiorrhiza.
Validation of the target genes of SmDof12 and 29 proteins by Dual-LUC assay
-
According to the results of co-expression analysis, it pushes the exploration of the underlying mechanism of SmDof12 and SmDof29 in regulating tanshinones and phenolic acids. By Dual-LUC assay (Fig. 7d), it was revealed that SmDof12 could uniquely inhibit the transcription of the SmGGPPS promoter, leading to a 3-fold decrease compared to the 35S-YFP control, whereas, SmDof29 significantly activated the SmPAL promoter up to 1.69-fold compared to the control. Those results indicated that SmDof12 might inhibit the biosynthesis of tanshinones by decreasing the activity of the SmGGPPS promoter, while SmDof29 activated the transcription of SmPAL to increase the production of phenolic acids in S. miltiorrhiza.
-
Dof genes widely exist in plants and have been validated to participate in diverse biological functions[14,15]. S. miltiorrhiza is a valuable traditional Chinese herbal plant and has been used widely in clinic treatments[1]. Genome-wide identification of SmDof gene lays a foundation for the subsequent study of its function. In the present study, a total of 31 Dof genes were identified in S. miltiorrhiza, and the number of SmDof genes was comparative to that of A. thaliana (36 members)[30], rice (30 members)[32], and tomato (34 members)[46]. The genome size of the above plants varied greatly, but the number of Dof proteins was not related to the size of the genome thus implying its conserved function in the above plant species.
Multiple sequence alignment uncovered the conserved domain within the SmDof proteins in S. miltiorrhiza. Phylogenetic tree construction showed that the SmDofs got low homology with Arabidopsis, and only Group I together with Group IV had more than six Dof genes getting high sequence similarity between S. miltiorrhiza and Arabidopsis. Previous studies have confirmed that phylogenetic analysis can provide a valuable theoretical basis for functional prediction of similar genes in different species[46]. Genes clustering in the same subgroup are relatively conserved in gene structure, gene expression patterns, and functional evolution[47]. By phylogenetic tree construction, it was found that AtDof5.4 and SmDof7 had high homology, and they were grouped into the same branch. Previous studies have verified that AtDof5.4 is a negative regulator modulating cell proliferation and expansion in Arabidopsis[48], so it is speculated that SmDof7 may also have the same function as AtDof5.4. Indeed, SmDof7 got the highest expression level in the stem and root, indicating that SmDof7 might regulate the cell proliferation and expansion in stem and root of S. miltiorrhiza.
The diverse structure and organization of the Dof genes, is associated with the evolution and functional differentiation of this gene family in certain species[49]. Gene structures analysis of all the Dof genes in S. miltiorrhiza exhibited visible variation between different subgroups, while similar structures were observed within the same subgroup (Fig. 3c). In general, the structure of the 31 SmDof genes was relatively simple and contained one or two exons, of which it was similar to the previous studies on melon[15]. However, 11 SmDof genes (including SmDof 11, 16, 17, 18, 19, 22, 23, 24, 26, 29, 31) had only one intron or even no intron (Fig. 3c). As previously reported, the intron-less genes may be associated with the quick stress response[50].
In previous reports, many Dof genes have been validated to be a key regulatory center involved in secondary metabolic synthesis, abiotic stress response, and hormone regulation pathway[23,51,52]. In grape (Vitis vinifera L.), VyDof8 was validated to be induced by a variety of abiotic stress. Overexpression of VyDof8 in tobacco (Nicotiana tabacum) significantly elevated ABA accumulation and drought tolerance during prolonged droughts compared to the control plants[53]. The expression pattern of candidate genes in a certain tissue or under stress signal is often closely related to the function of these genes[54]. Therefore, in this study, the transcriptome dataset of S. miltiorrhiza was introduced to dissect the expression profiles of SmDof gene families in root, stem, leaf, and flower. Most of the SmDof genes (23 out of 31) are expressed in the root of S. miltiorrhiza. It is speculated that some members of the 23 SmDof genes may be related to the growth and development of S. miltiorrhiza roots. It was also revealed that SmDof3 and 16 are highly expressed in flower (Fig. 5b). As a Dof gene, CDF3 in tomato (Lycopersicon esculentum) getting high gene sequence homology with SmDof3 and 16 were validated to regulate the flowering time through modulating the expression of FT-like genes[55]. Therefore, the research project whether overexpression of SmDof3 or 16 in S. mitiorrhiza has a significant impact on regulating its flowering time or growth is worthy of inquiry. The varied expression profiles of SmDof genes in various tissues provide basic data to explore their functions.
By investigating cis-acting elements within promoters, it indicates that the SmDof genes are related to light response, hormone-related response, cell development, and environmental stress (Fig. 4). In the present study, several SmDof genes (eg. SmDof12 and SmDof29) were found to contain ABA-responsive elements (ACGTG) in putative promoter regions. Through the transcriptome dataset together with gene expression validation detected by qRT-PCR, it was confirmed that SmDof12 and 29 expressed vigorously in S. miltiorrhiza root (Fig. 5b; Fig. 7a, b). Furthermore, SmDof12 and 29 were verified to co-express with the metabolic pathway genes involved in tanshinones or phenolic acids biosynthesis (Fig. 6). This pushes the exploration of the underlying molecular mechanism of how SmDof12 and 29 to regulate the expression of the downstream target gene to modulate the tanshinones and phenolic acids biosynthesis. Subsequently, SmGGPPS and SmPAL were validated to be the target of SmDof12 and SmDof29 by Dual-LUC assay, respectively (Fig. 7d). Through the same strategy, in Scutellariae baicalensis, it is verified that SbNAC25 reduces the synthesis of flavonoid by downregulating the expression of FNSII-2, OMT2, CHI, and F6H2 genes[56]. The fact that the gene expression profile in special tissues and under certain stress treatment detected by transcriptome sequencing and qRT-PCR validation in combination with gene co-expression analysis is a quite valid strategy to mine the candidate regulatory genes and their downstream target genes involved in the biosynthetic pathway of secondary metabolite in many plants[57,58].
-
In the present study, the SmDof gene families in S. miltiorrhiza were characterized based on the whole genome, transcriptome dataset, and qRT-PCR expression analysis. Two SmDof genes (SmDof12 and SmDof29) were mined by gene co-expression strategy from the identified 31 SmDofs, and their target genes of SmDof12 and SmDof29 were validated by Dual-LUC experiments. This study is the first comprehensive analysis of the SmDof gene families in S. miltiorrhiza, and provides valid data for further exploring the underlying molecular mechanism of SmDofs in response to ABA induction. It may also be beneficial to elucidate the diverse biological functions of Dof genes in other plants.
This work was supported by National Natural Science Foundation of China (82373979), Key Scientific and Technological Grant of Zhejiang for Breeding New Agricultural Vareties (2021C02074-3), WenZhou Key Scientific and Technological Innovation Project (ZN2022006) and Zhejiang Provincial Natural Science Foundation of China (LZ24H280002). We appreciate the great experimental support from the Public Platform of Medical Research Center, Academy of Chinese Medical Science, Zhejiang Chinese Medical University.
-
The authors confirm contribution to the paper as follows: study conception and supervision: Zhou W, Kai G, Zhu J; study design: Wang X, Wang Q, Hao S; experiment performance and data analysis: Wang X, Wang Q, Hao S; manuscript suggestions: Wang X, Zhou W; draft manuscript preparation: Wang X, Zhou W. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Xinyu Wang, Qichao Wang, Siyu Hao
- Supplementary Table S1 cDNA sequences of the 31 SmDof genes (5'- 3').
- Supplementary Table S2 Protein sequences of the 31 SmDofs and 36 AtDofs used in the evolutionary tree construction.
- Supplementary Table S3 TPM values of the 31 SmDof genes in different tissues of S. mitiorrhiza derived from the RNA-seq data.
- Supplementary Table S4 TPM values of the 31 SmDof genes derived from the RNA-seq data induced by ABA.
- Supplementary Table S5 Primers used in this study.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang X, Wang Q, Hao S, Zhu J, Kai G, et al. 2024. Genome-wide identification and characterization of Dof gene family in Salvia miltiorrhiza. Ornamental Plant Research 4: e031 doi: 10.48130/opr-0024-0030 

Genome-wide identification and characterization of Dof gene family in Salvia miltiorrhiza
- Received: 06 March 2024
- Revised: 19 September 2024
- Accepted: 01 November 2024
- Published online: 05 December 2024
Abstract: Salvia miltiorrhiza is an important medicinal plant, and its main secondary metabolites are tanshinones and phenolic acids. Dof transcription factors play an irreplaceable role in regulating plant growth and secondary metabolism. However, the characteristics of SmDof genes in S. miltiorrhiza have not yet been studied. Based on the whole genome data of S. miltiorrhiza, the SmDofs family has been systematically explored. A total of 31 Dof members have been identified in S. miltiorrhiza, and they are clustered into five subgroups according to their evolutionary relationships. Co-expression network results indicated that two SmDof genes (SmDof12 and SmDof29) might be involved in modulating the biosynthesis of phenolic acids or tanshinones. To investigate this hypothesis, dual luciferase experiments was introduced to examine the downstream target gene of SmDof12 and SmDof29. Subsequently, it was validated that SmDof12 inhibited the transcription of the SmGGPPS promoter, and SmDof29 significantly activated the SmPAL promoter. The present studies offers important data about the underlying function of SmDof12 and SmDof29 involved in the biosynthesis of phenolic acid or tanshinone, and provides valuable insights into further research of the SmDof gene families in S. miltiorrhiza.
-
Key words:
- S. miltiorrhiza /
- Dof gene family /
- Transcription factor /
- Tanshinone /
- Phenolic acid